

Abstract
A unified synthetic strategy to access (+)-irciniastatin A (a.k.a. psymberin) and (−)- irciniastatin B, two cytotoxic secondary metabolites, has been achieved. Highlights of the convergent strategy comprise a boron-mediated aldol union to set the C(15)–C(17) syn-syn triad, reagent control to set the four stereocenters of the tetrahydropyran core, and a late-stage Curtius rearrangement to install the acid-sensitive stereogenic N,O-aminal. Having achieved the total synthesis of (+)-irciniastatin A, we devised an improved synthetic route to the tetrahydropyran core (13 steps) compared to the first-generation synthesis (22 steps). Construction of the structurally similar (−)-irciniastatin B was then achieved via modification of a late-stage (−)-irciniastatin A intermediate to implement a chemoselective deprotection/oxidation sequence to access the requisite oxidation state at C(11) of the tetrahydropyran core. Of particular significance, the unified strategy will permit late-stage diversification for analogue development, designed to explore the biological role of substitution at the C(11) position of these highly potent tumor cell growth inhibitory molecules.
Introduction
In 2004 two new potent cytotoxins, (+)-irciniastatin A (1) and (−)-irciniastatin B (2), isolated from the Indo-Pacific marine sponge Ircinia ramose, were discovered by Pettit and coworkers (Figure 1).1 In the same year, a closely related metabolite, (+)- psymberin, was isolated independently by Crews and coworkers from marine sponge Psamminocinia.2 Analysis of these reports suggests that irciniastatin A (1), irciniastatin B (2), and psymberin (1) possessed the same architectural features, including a highly substituted 2,6-trans-tetrahydropyran core, a dihydroisocoumarin, and an N,O-aminal. Crews postulated that both irciniastatin A (1) and psymberin might be identical,2 but unfortunately the NMR spectra of the two congeners were taken in different solvents, thus the exact stereochemical relationship at C(4) and C(8) could not be established. In 2005, De Brabander and colleagues resolved the structural ambiguity with the first total synthesis of psymberin by construction of all four C(4)-C(8) diastereomers of psymberin. This effort not only yielded the absolute configuration of (+)-psymberin, but confirmed that both (+)-irciniastatin A (1) and (+)-psymberin possessed identical chemical structures.3 More recently, De Brabander reported a full account on their first- and second-generation synthesis of (+)-irciniastatin A (1) (a.k.a. psymberin), in conjunction with the synthesis of a series of novel analogues.4
Figure 1.
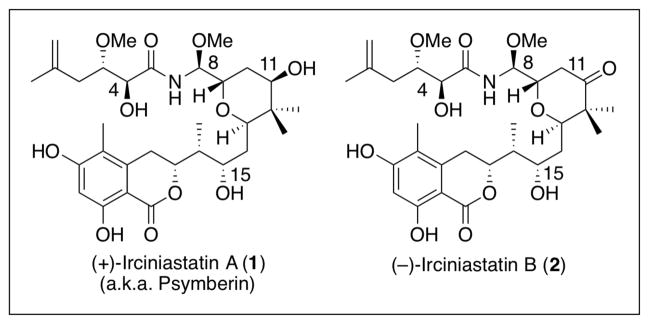
Irciniastatin Family
The discovery that (+)-irciniastatin A (1) (a.k.a. psymberin) and (−)-irciniastatin B (2) display impressive therapeutic properties, including tumor cell growth inhibition at the nanomolar level (0.7 to 0.8 nM),1 in conjunction with the observation by Crews et al., that (+)-irciniastatin A (1) displayed high differential cytotoxicity (>10,000-fold) in the NCI 60 human tumor cell panel, raised the intriguing possibility that the observed cytotoxicity might arise via a novel mode of action.2 Interestingly, even though the chemical structures of (+)-irciniastatin A (1) and (−)-irciniastatin B (2) differ only in the oxidation level at C(11), the ketone congener (2) was reported to be nearly 10 times more active than the alcohol (1) against human pancreas (BXPC-3), breast (MCF-7), and central nervous system (SF268) cancer cell lines.1 Subsequently, a group at Schering-Plough5 reported that (+)-C(11)-deoxy-analogue possesses 3–10 times the cytotoxic activity compared to (1). Taken together, these results suggest that the C(11) hydroxyl group is not critical for potent cytotoxic activity.
In 2010 Usui and coworkers reported that the tumor growth inhibition activity of (+)-irciniastatin A (1) arises from activation of stress-activated protein kinases, such as JNK and p38, that in turn leads to apoptosis.6 Subsequent to this report, De Brabander, in collaboration with Roth, disclosed a forward genetic screen of (+)-irciniastatin A (1) employing C. elegans that demonstrated 1 binds to the ribosome to induce cell death.7 Also of interest, totally synthetic (+)-irciniastatin A (1), from the De Brabander group, did not reveal the high differential cytotoxicity7 previously reported for natural (+)-irciniastatin A (1).2
Given the impressive biological activity, in conjunction with limited natural abundance of the irciniastatins, seven total syntheses3,8–13 of (+)-irciniastatin A (1), including a report from our laboratory,9 have been disclosed since 2004. After DeBrabander’s seminal total synthesis,3 Floreancig’s clever strategy to installing the N,O-aminal to construct (1) proved to be the shortest longest linear sequence to date (14 steps).13 Somewhat more surprising, the first total synthesis of the substantially more active congener, (−)-irciniastatin B (2), was only recently achieved in our laboratory.14 Although several SAR studies have shed some light on the mode of action of (+)- irciniastatin A (1), the biological properties associated with the structural differences of the alcohol (1) and the ketone (2) remain unknown. In particular, we were interested in what role the C(11) substituents in the irciniastatins would play in an SAR study. Towards this end, we report here a full account of the synthesis of both (+)-irciniastatin A (1) and (−)-irciniastatin B (2), including our first-generation synthesis of (+)-irciniastatin A (1), a revised second-generation synthesis of the 2,6-trans-tetrahydropyran core of (+)- irciniastatin A (1), and the subsequent development of a unified strategy, utilizing a latestage selective deprotection/oxidation sequence, which not only led to the first synthesis of (−)-irciniastatin B (2), but also the construction of both C(11) alcohol epimers.
Results/Discussion
A First-Generation Synthesis of (+)-Irciniastatin A (1)
Our strategy to construct (+)-irciniastatin A (1) began with disconnection at the amide linkage, leading to the acid side chain 3 and the Teoc-protected N,O-aminal 4 (Scheme 1). The acid-sensitive N,O-aminal moiety would be installed late in the synthesis, with complete retention of configuration via a Curtius rearrangement, a strategy first developed and successfully exploited in our 2002 synthesis of (+)- zampanolide, bearing a similar N,O-aminal group.15 Disconnection at C(16)-C(17) next provided aldehyde 5 and 2,6-trans-tetrahydropyran 6, which we envisioned would be united via a substrate-controlled aldol reaction. Aryl aldehyde 5 in turn would derive by a [4+2] cycloaddition between known bis-silyl enol ether 716 and allene 8,17 while 2,6-trans-tetrahydropyran 6 would arise via a 6-exo-tet-cyclization of the C(13), hydroxyl with an epoxide that would be installed at the C(8)-C(9). Epoxide 9 in turn would be generated by iterative chemoselective functionalization of diene 10, a synthetic tactic that directs reactivity to the most electron-rich olefin, distal to the electron withdrawing ester group. Synthetic strategies that selectively introduce functionality taking advantage of the difference in electron density of the olefinic linkage along a linear polyene, possessing a terminal electron withdrawing group, have not, as of yet, been widely exploited.18–21
Scheme 1.

Retrosynthetic Analysis
Synthesis of the requisite acid side chain (−)-3 began with methyl ether (+)-11,3 which was constructed from commercially available (+)-isopropylidene glyceraldehyde in a stereocontrolled fashion in two steps; the overall yield was 57% yield (dr > 20:1 Scheme 2). Removal of the acetonide was next achieved by treatment of (+)-11 with aqueous hydrochloric acid. The primary alcohol was then protected chemoselectively as the pivalate ester (+)-12, followed by protection of the secondary alcohol as a SEM ether. Reduction with DIBAL-H then provided primary alcohol (+)-13, which was oxidized via a two-step Parikh-Doering22/Pinnick23 oxidation sequence to provide the desired acid side chain (−)-3.
Scheme 2.
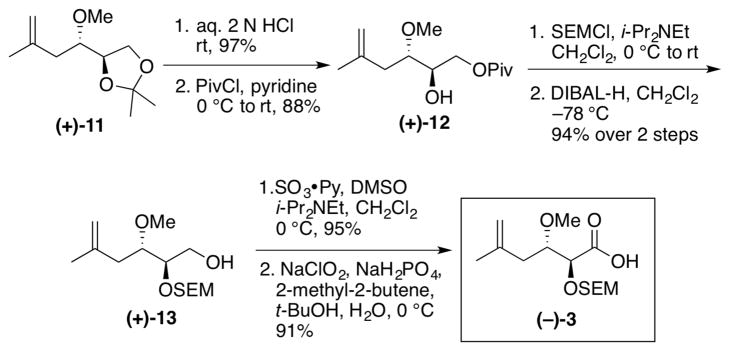
Synthesis of Acid Side Chain (−)-3
The requisite aryl aldehyde 5 was constructed via a Diels-Alder cycloaddition between 1,3-bis(trimethylsiloxy)-1,3-diene 716 and dimethyl-1,3-allene-dicarboxylate 8,17 followed by a fluoride-mediated aromatization to furnish known homopthalate 1424 in 83% yield (Scheme 3). Both phenols were then masked as SEM ethers, followed by chemoselective reduction to furnish aryl aldehyde 5 in an overall yield of 55% for the three-step sequence.
Scheme 3.
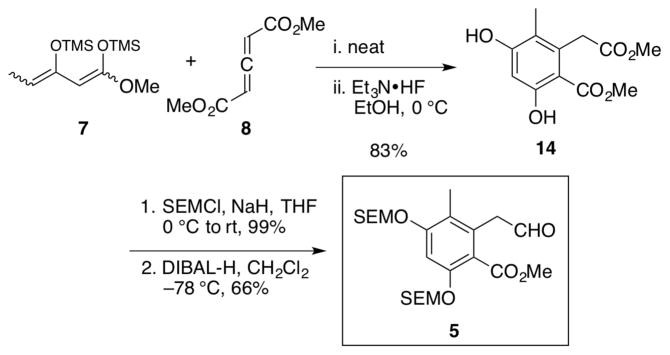
Synthesis of Dihydroisocoumarin Fragment 5
Access to 2,6-trans-tetrahydropyran 6, the core of (+)-irciniastatin A (1), began with commercially available S-epoxybutane (−)-15. Selection of (−)-15 was simply a matter of synthetic convenience, in order to operate with a single diastereomer, as the advanced alcohol would ultimately be oxidized to a ketone (vide infra). Treatment of epoxide (−)-15 with propynyllithium and BF3•OEt2 provided the homopropargylic alcohol,25 which was protected to furnish benzyl ether (−)-16 under acidic conditions. Next, following Negishi’s protocol26 for the in situ generation of the Schwartz reagent from zirconocene dichloride and DIBAL-H, iodination with N-iodosuccinimide furnished the external vinyl iodide (−)-17. Pleasingly, the regioselectivity was excellent (>20:1). Vinyl iodide (−)-17 was then united with methyl acrylate, exploiting a palladium-mediated Heck reaction27 to complete diene (−)-18 in 78% yield.
Installation of the first of the three epoxides to construct the linear precursor of tetrahydropyran 6 entailed chemoselective epoxidation of the most electron-rich olefin in (−)-18 with the Shi fructose-derived catalyst (−)-1928 to furnish (+)-20; excellent diastereoselectivity (14:1) resulted (Scheme 5). Regioselective opening was then achieved with trimethylaluminum29 to provide a single regioisomeric alcohol, which was treated with PMB-Cl to furnish ether (+)-21. Asymmetric epoxidation to introduce the second epoxide required DIBAL-H reduction of ester (+)-21 followed by a Sharpless asymmetric epoxidation.30 Epoxy alcohol (+)-22 resulted as a single diastereomer in 92% yield for the two steps, which was then subjected to Parikh-Doering oxidation conditions;22 the resulting aldehyde was then treated with Horner-Wadsworth-Emmons ylide 23 to provide ester (+)-24 in 89% yield (two steps). Palladium-catalyzed hydrogenolysis31 employing formic acid as the hydrogen source next opened the epoxide in (+)-9, again with excellent chemoselectivity. Subsequent protection of the resulting alcohol as the TBS ether furnished (+)-24 in 92% yield for two steps. The third and final epoxide was installed by reduction of ester (+)-24 with DIBAL-H to reveal the allylic alcohol; Sharpless asymmetric epoxidation,30 this time utilizing (−)-diisopropyl tartrate, furnished epoxide (+)-25 with 20:1 dr. To complete the construction of the linear precursor for tetrahydropyran 6, alcohol (+)-25 was oxidized32 directly to the carboxylic acid, followed by treatment with diazomethane to provide the corresponding methyl ester in 76% yield for the two steps. Oxidative removal of the PMB ether protecting group with DDQ completed the construction of (+)-26, the tetrahydropyran cyclization precursor. The overall yield for the 18-step synthetic sequence was 9.3% yield.
Scheme 5.

Synthesis of Pyran Precursor (+)-26
With (+)-26 in hand, we examined the acid-promoted cyclization to generate the tetrahydropyran core. Baldwin rules33 suggest that both the desired 6-exo-tet and undesired 7-endo-tet cyclization pathways could operate. However, the six-membered ring transition state, in conjunction with the electron-withdrawing nature of the ester, destabilizing the partial cationic character at the α-carbon under Lewis- or Brønsted-acidic conditions, suggested that the tetrahydropyran would predominate. Indeed, treatment with 20 mol % of camphorsulfonic acid (CSA) in methylene chloride achieved the desired 6-exo-tet cyclization pathway, which provided the 2,6-tetrahydropyran (+)-27 in excellent yield (92%), with no trace of the seven-membered ring congener (Scheme 6). Alcohol (+)-27 was then methylated with dimethyl sulfate, followed by palladium-promoted hydrogenolysis to remove the benzyl ether. Dess-Martin periodinane34 oxidation completed construction of the requisite 2,6-trans-tetrahydropyran fragment (+)- 6 in 88% yield for the three steps.
Scheme 6.

Completion of Tetrahydropyran (+)-6
Having constructed the three fragments for the proposed synthesis of (+)- irciniastatin A (a.k.a. psymberin) (1), we turned to the union of tetrahydropyran (+)-6 with aryl aldehyde 5 (Scheme 7), exploiting a substrate-controlled aldol reaction. Generation of the Z-boron enolate of (+)-6, achieved by treatment of (+)-6 with dichlorophenylborane,35 followed by addition of aldehyde 5 furnished (+)-29, the desired syn-aldol product, in 90% yield. The stereochemical outcome was dictated by 1,4-substrate stereoinduction.36 Subsequent chelation-controlled reduction,37 followed by saponification of the methyl ester with concomitant lactonization provided dihydroisocoumarin (+)-30 in 83% yield for the two steps.
Scheme 7.

Fragment Union and Elaboration to Amidation Precursor
At this juncture we called upon a Curtius rearrangement, to install the N,O-aminal (Scheme 7). Acid (+)-30 was converted to the corresponding acyl azide, followed by thermal rearrangement in toluene (ca. 80 °C) to provide the isocyanate, which was intercepted by the addition of 2-trimethylsilylethanol to furnish the desired N,O-aminal, with complete retention of configuration at the methyl ether carbon (NMR). Protection of the remaining free hydroxyl group as the TBS ether was then achieved in 91% yield to complete the construction of advanced amide (+)-4, the coupling partner for acid side chain (−)-3.
Final fragment union of N,O-aminal (+)-4 with side chain (−)-3 required considerable experimentation. Eventually we discovered that deprotonation of the Teoc-protected amine (+)-4 with LiHMDS, followed by addition of the side chain, activated as the pivalate anhydride 31, would lead to the desired amide (+)-32 in 79% yield (Scheme 8).
Scheme 8.

Completion of (+)-Irciniastatin A (1) (a.ka. Psymberin)
Turning our attention to conditions that would achieve global deprotection, while retaining the delicate N,O-aminal moiety, model studies revealed that TAS-F38,39 was the reagent of choice. Treatment of the fully protected irciniastatin A (+)-32 with TAS-F in DMF at 50 °C resulted in two major products (Scheme 8). After purification via preparative TLC, the more polar of the two congeners proved to be (+)-ircinaistatin A (1), while the less polar compound retained one phenolic SEM group. Subjecting the latter to magnesium bromide40 resulted in (+)-irciniastatin A (1), furnishing a combined yield of 74% for the two steps. Pleasingly, the spectral data (1H and 13C NMR) of totally synthetic (+)-irciniastatin A (1) proved to be identical in all respects with the spectra of natural (+)- irciniastatin A (1) reported by Pettit1 and Crews.2 The total synthesis of (+)-irciniastatin A (a.k.a. psymberin) (1) had thus been achieved with a longest linear sequence of 30 steps (ca. 2.2% overall yield).
A Second-Generation Synthesis of (+)-Irciniastatin A (a.k.a. Psymberin)
Although we had achieved the total synthesis of (+)-irciniastatin A (1) (a.k.a. psymberin), construction of the core 2,6-trans-tetrahydropyran required 22 steps and proceeded with an overall yield of only 7.6%. To provide material for future biological evaluation, as well as to access a series of synthetic analogues, an improved second-generation approach to tetrahydropyran (+)-6 would be required. The new strategy to (+)- 6 (Scheme 9) was anticipated to retain the efficient 6-exo-tet cyclization of acyclic epoxide precursor 33, which would be constructed via two reagent-controlled asymmetric transformations from alcohol 34, that in turn would derive via union of aldehyde 35 and ketene acetal 36, exploiting a vinylogous Mukayaima aldol reaction.41 In this strategy, the gem-dimethyl moiety would arise from commercially available 2,2-dimethyl-1,3-propanediol, instead of the epoxide opening strategy employed in our first-generation synthesis. Importantly, the second-generation route would set the requisite stereogenecity in tetrahydropyran (+)-6 via three reagent-controlled asymmetric reactions.
Scheme 9.
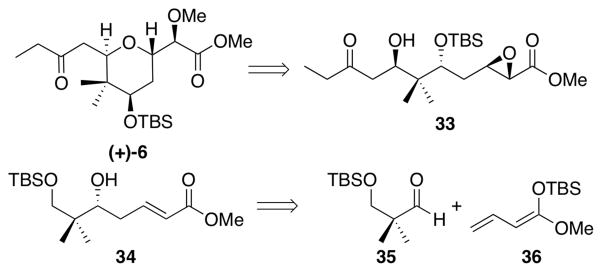
Revised Retrosynthetic Strategy of Tetrahydropyran (+)-6
We began the second-generation synthesis of (+)-6 via monoprotection of commercially available 2,2-dimethyl-1,3-propanediol 37 (Scheme 10), followed by oxidation of the second hydroxyl employing the Parikh-Doering22 protocol to provide aldehyde 35. Treatment of aldehyde 35 and ketene acetal 3639 employing the chiral oxazaborolidinone derived from L-tryptophan, led to a vinylogous Mukaiyama aldol reaction,41 thereby installing the first stereocenter to furnish (+)-34 as a single enantiomer. Mosher’s ester analysis demonstrated the desired (R)-isomer was obtained.42,43 Alcohol (+)-34 was then protected as the TBS ether, followed by reduction of the methyl ester with DIBAL-H to furnish the corresponding allylic alcohol. Asymmetric epoxidation via the Sharpless30 protocol next provided the desired β-epoxide (+)-38 with 13:1 diastereoselectivity, which in turn was converted directly to the corresponding acid via a one-pot TEMPO32 oxidation; subsequent methylation led to methyl ester (+)-39.
Scheme 10.

Second-Generation Synthetic Route towards Tetrahydropyran (+)-6
Chemoselective deprotection of the primary TBS ether was then achieved by treatment of (+)-39 with hydrogen fluoride, buffered with pyridine (Scheme 10). The resulting primary alcohol was oxidized22 to aldehyde (+)-40, and the final stereocenter required for the tetrahydropyran core was introduced via Paterson aldol union44,45 with 2-butanone, employing (−)-B-chlorodiisopinocampheylborane (DIP-Cl) as the chiral Lewis acid. A 5:1 (β:α) diastereomeric mixture resulted. Cyclization employing a catalytic amount of CSA furnished (+)-41 and (−)-42 exclusively via the 6-exo-tet pathway, again without formation of the seven-membered ring construct. Fortunately, the trans- and cis-diastereomers could now be readily separated by column chromatography to yield 2,6-trans-tetrahydropyran (+)-41 in 74% yield for the 2 steps. Methylation of the secondary hydroxyl group was achieved (92% yield) by treatment with Me3•OBF4 and proton sponge [1,8-bis(dimethylamino)naphthalene] to complete the second-generation synthesis of the 2,6-trans-tetrahydropyran core (+)-6. Pleasingly, the new route to (+)-6 proceeded with a longest linear sequence of 13 steps (a 9-step improvement), and with an overall yield of 17.8% yield, thus more than doubling the overall yield for the longest linear sequence compared to the first-generation synthesis.
Total Synthesis of (−)-Irciniastatin B
With an effective route to (+)-irciniastatin A (1), and an improved route to the common tetrahydropyran core, we turned to the construction the biologically more active congener, (−)-irciniastatin B (2). To achieve the requisite ketone oxidation state at C(11), the C(15) secondary hydroxyl in (+)-43, a late-stage intermediate employed in our synthesis of (+)-irciniastatin A (1),9 was envisioned to be protected as a SEM ether, instead of the TBS ether employed earlier (Scheme 11). This protecting group was selected to permit the critical, selective deprotection of the neopentyl secondary TBS ether at C(11). The secondary alcohol would be oxidized to the requisite ketone, followed by global deprotection to provide access to (−)-irciniastatin B (2). Chemical modification of the late stage C(11) ketone would also permit access to a series of analogues possessing varying substitution at C(11), thus enabling a structure activity relationship study (SAR) to be carried out at this key center in the irciniastatin chemotype.
Scheme 11.

Divergent Strategy Towards the Synthesis of (−)-Irciniastatin B
Surprisingly, protection of the C(15) hydroxyl group in (+)-43 as the SEM ether proved particularly challenging. In particular, manipulation of the resultant SEM ether during workup and purification routinely resulted in the unanticipated loss of the phenolic SEM ethers. Attempts at reprotection proved ineffective, even at elevated temperatures. After extensive experimentation with several model systems, we discovered that the phenolic 3,4-dimethoxybenzyl ether (DMB) would prove durable in the late-stage synthetic sequence with the orthogonally protected C(11) TBS ether.
Synthesis of the revised aryl fragment 45 thus began with protection of bis-phenol 1424 with DMB-Br (Scheme 12). Reduction of the resulting ester with DIBAL-H furnished the aryl aldehyde 45. From here, the synthetic route continued in similar fashion to the sequence leading to (+)-irciniastatin A (1).9 Aldol union35 between aldehyde 45 and ketone (+)-6 pleasingly furnished β-hydroxyketone (+)-46 in 70% yield, with minor concomitant lactonization (10:1) and excellent diastereoselectivity (>20:1).36 Chelation-controlled reduction37 resulted in a mixture of the desired syn diol and the corresponding lactone (ca. 8:1). The mixture was treated under saponification conditions to provide acid (+)-47 in 69% yield for the 2 steps. With acid (+)-47 in hand, the corresponding acyl azide was generated and subjected to the conditions for the Curtius rearrangement15 to furnish the Teoc-protected N,O-aminal in 67% yield, again with complete retention of stereochemical configuration at C(8). The resulting secondary alcohol was then protected as the SEM ether (+)-48 in 82% yield. Importantly, workup and/or purification proceeded without the formation of undesired side products, compared to the earlier SEM protection strategy.
Scheme 12.

Synthesis of N,O-aminal (+)-48
Achieving the requisite amide union to provide (+)-50 once again proved challenging (Scheme 13). The conditions employed in our earlier (+)-irciniastatin A (1) synthesis,9 involving LiHMDS as the base with the mixed anhydride 31, resulted in low yields (ca. 15%). After significant screening, the conditions employed by Crimmins and coworkers,10 in their synthesis of (+)-irciniastatin A (1), namely the use of i-PrMgCl as the base and acid chloride 49, provided the desired amide (+)-50 in 72% yield.
Scheme 13.
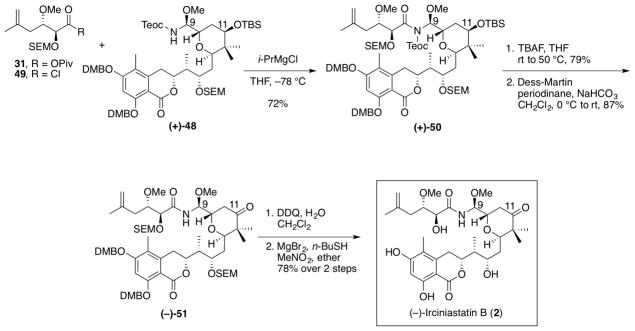
Completion of (−)-Irciniastatin B (2)
Having arrived at the full carbon skeleton of (−)-irciniastatin B (2), we set out to effect selective deprotection of the hindered neopentyl secondary C(11) TBS ether (Scheme 13). The TBS ether (+)-50 was treated with TBAF at room temperature, which resulted in hydrolysis of the Teoc carbamate. Subsequent warming the reaction mixture to 50 °C then led to selective removal of the C(11) TBS group in an overall yield of 79%. Oxidation with Dess-Martin periodinane34 provided ketone (−)-51.
In order to remove the two pairs of protecting groups in (−)-51, and complete the synthesis of (2), a two-stage deprotection protocol was required. Introduction of the ketone moiety in the tetrahydropyran, however, greatly enhances the sensitivity of the molecular structure. For example, treatment of (−)-51 with base readily initiates a retro-Michael/Michael addition, effecting epimerization of C(9) of the tetrahydropyran core,46 while acid treatment results in hydrolysis of the N,O-aminal.47 Fortunately, treatment of ketone (−)-51 with DDQ did provide the desired bis-phenol without observable decomposition. Removal of the two remaining SEM groups with either TAS-F38,39 or TBAF, however, resulted only in complex mixtures, highlighting the base sensitivity of ketone (−)-51. Again, after significant experimentation, we discovered that treatment of (−)-51 with a premixed solution of MgBr2, n-butanethiol, and nitromethane in ether48 removed the SEM ethers cleanly to provide (−)-irciniastatin B (2) in 78% yield for the two steps. Pleasingly, synthetic (−)-irciniastatin B (2) proved to be identical in all regards (1H and 13C NMR) with the spectral data obtained by Pettit and coworkers,1 and thus constituted the first total synthesis of (−)-irciniastatin B (2).
In order to verify the structural relationship of (−)-irciniastatin B (2) with (+)-irciniastatin A (1), we carried out a chemical interconversion of (2) to (1) (Scheme 14). To this end, 2 was treated with NaBH4, which resulted in a mixture (1:1) of (+)-irciniastatin A 1 and epi-C(11)-irciniastatin A (52). The two diastereomers were separated via preparative TLC, and the spectral data of the faster moving diastereomer (TLC) was identical with the spectral data of (+)-irciniastatin A (i.e., 1H, 13C NMR and HRMS), thereby confirming the structural relationship of (+)-irciniastatin A (1) and (−)- irciniastatin B (2).
Scheme 14.

Structural Confirmation of (−)-Irciniastatin B (2) by Chemical Conversion to (+)-Irciniastatin A (1) (a.k.a. Psymberin)
Summary
The evolution of an effective unified synthetic strategy for the construction (+)-irciniastatin A (1) (a.k.a. psymberin) and (−)-irciniastatin B (2) has been achieved. The first-generation synthesis of (+)-ircinastatin A required 30 steps and proceeded in 2.2% overall yield. Of these 30 steps, 22 were required to construct the 2,6-trans-tetrahydropyran, which proceeded in 7.6% overall yield. A significantly improved second-generation synthesis of the 2,6-trans-tetrahydropyran core was subsequently achieved, which now permits an efficient total synthesis of both (+)-irciniastatin A (1) and (−)-irciniastatin B (2), the latter requiring a chemoselective deprotection/oxidation sequence. Finally, the structural relationship of the two similar metabolites has been confirmed via chemical conversion of (−)-irciniastatin B (2) to (+)-irciniastatin A (1) and the corresponding C(11) epimer (52). Importantly, the successful synthesis leading to (−)- irciniastatin B (2) now holds the promise for the elaboration of C(11)-irciniastatin analogues, currently ongoing in our laboratory.
Experimental Section
Materials and Methods
Reactions were carried out in flame-dried or oven-dried glassware under a nitrogen atmosphere unless noted otherwise. Anhydrous diethyl ether (Et2O), tetrahydrofuran (THF), dichloromethane (CH2Cl2) and toluene were obtained from a solvent purification system. All commercially available reagents were used without purification unless otherwise noted. Triethylamine, diisopropylethylamine, and pyridine were freshly distilled from calcium hydride under a nitrogen atmosphere. Reactions were magnetically stirred unless stated otherwise and monitored by thin layer chromatography (TLC) with 0.25 mm Silacycle pre-coated silica gel plates. Silica gel chromatography was performed utilizing ACS grade solvents and silica gel from either Silacycle or Sorbent Technologies.
Infrared spectra were obtained using an FT/IR spectrometer. Optical rotations were obtained using a polarimeter. All melting points were obtained on a melting point apparatus and are uncorrected. 1H NMR spectra (500 MHz field strength) and 13C NMR spectra (125 MHz field strength) were obtained on 500 MHz spectrometer or a cryomagnet (500MHZ/52mm) with a 5 mm dual cryoprobe. Chemical shifts are reported relative to chloroform (δ 7.26) or methanol (δ 4.78) for 1H NMR spectra and chloroform (δ 77.23), methanol (δ 49.3), or benzene (δ 128.0) for 13C spectra. High-resolution mass spectra (HRMS) were measured on an LC-TOF mass spectrometer
Pivalate Ester (+)-12
A 2 N HCl solution (10 mL) was added to acetonide (+)-11 (0.836 g, 4.18 mmol) at rt. After 30 min, the reaction was quenched with sat. NaHCO3 until all gas evolution had ceased. The reaction was then extracted with EtOAc and the combined organic layers were dried over MgSO4 and concentrated. Flash chromatography (40% EtOAc/hexanes) provided diol (+)-53 (0.650 g, 97% yield) as a colorless oil: +21.8 (c 1.5, CHCl3); IR (neat) 3416, 2932, 1646, 1456, 1092, 1050 cm−1; 1H NMR (500 MHz, CDCl3) δ 4.84 (q, J = 1.6 Hz, 1H), 4.80 (m, 1H), 3.82-3.77 (m, 1H), 3.72-3.67 (m, 2H), 3.57-3.53 (ddd, J = 2.4, 6.5, 13.1 Hz, 1H), 3.43 (s, 3H), 2.61 (d, J = 6.7 Hz, 1H), 2.39 (dd, J = 14.0, 6.8 Hz, 1H), 2.32 (dd, J = 7.2, 3.2 Hz, 1 H), 2.18 (dd, J = 14.3, 6.4 Hz, 1H), 1.79 (ap t, J = 1.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 142.49, 113.2, 81.7, 72.8, 63.3, 58.4, 38.9, 22.9; high resolution mass spectrum (CI+) m/z 161.1177 [(M+H)+; calcd for C8H17O3: 161.1178].
To diol (+)-53 (0.243 g, 1.52 mmol) in pyridine (4 mL) was cooled to 0 °C, followed by addition of trimethylacetyl chloride (0.205 mL, 1.67 mmol). The reaction was allowed to warm to rt and after 1 h the reaction was cooled to 0 °C and H2O (5 mL) was added. The reaction was diluted with Et2O (5 mL) and washed successively with 5 mL each of sat. NaHCO3, 1 N HCl, and brine. The organic layer was then dried over MgSO4 and concentrated. Flash chromatography (10% EtOAc/hexanes) provided (+)-12 (0.325 g, 88% yield) as colorless oil: +23.9 (c 1.1, CHCl3); IR (neat) 3467, 2971, 2935, 1728, 1457, 1286, 1163, 1103 cm−1; 1H NMR (500 MHz, CDCl3) δ 4.84 (s, 1H), 4.81 (s, 1H), 4.23 (dd, J = 11.7, 3.5 Hz, 1H), 4.16 (dd, J = 11.6, 6.8 Hz, 1H), 3.92-3.82 (m, 1H), 3.48- 3.39 (m, 1H), 3.40 (s, 3H), 2.39 (d, J = 4.8 Hz, 1H), 2.29 (ddd, J = 19.4, 14.4, 6.2 Hz, 2H), 1.79 (s, 3H), 1.22 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 179.0, 142.6, 113.3, 80.6, 71.4, 65.6, 58.4, 39.0, 38.4, 27.4, 23.0; high resolution mass spectrum (ES+) m/z 245.1743 [(M+H)+; calcd for C13H25O4: 245.1753].
Alcohol (+)-13
A solution of (+)-12 (0.221 g, 0.905 mmol) in CH2Cl2 (3 mL) was cooled to 0 °C and i-Pr2NEt (0.395 mL, 2.5 equiv.) was added, followed by dropwise addition of SEMCl (0.320 mL, 2.0 equiv.). The reaction was allowed to warm to rt and after 2 h, sat. NH4Cl was added. The reaction was extracted with CH2Cl2 (3 × 10 mL) and the combined organic layers were dried over MgSO4 and concentrated. Flash chromatography (5% EtOAc/hexanes) provided SEM ether (−)-54 (338 mg, 99% yield) as colorless oil: −6.3 (c 2.9, CHCl3); IR (neat) 3457, 2925, 1728, 1480, 1283, 1249, 1159, 1107, 1031 cm−1; 1H NMR (500 MHz, CDCl3) δ 4.81 (s, 1H), 4.78 (s, 1H), 4.74 (dd, J = 14.7, 6.9 Hz, 2H), 4.29 (dd, J = 11.8, 3.9 Hz, 1H), 4.11 (dd, J = 11.8, 6.2 Hz, 1H), 3.84-3.79 (m, 1H), 3.68 (ddd, J = 10.1, 6.3, 6.3 Hz, 1H), 3.59 (ddd, J = 10.0, 6.5, 6.5 Hz, 1H), 3.51-3.46 (m, 1H), 3.39 (s, 3H), 2.32-2.19 (m, 2H), 1.77 (s, 3H), 1.19 (s, 9H) 0.99-0.82 (m, 2H), 0.00 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 178.4, 142.7, 113.0, 95.0, 80.3, 76.8, 65.6, 64.0, 58.6, 39.4, 39.0, 27.4, 23.0, 18.2, −1.2; high resolution mass spectrum (ES+) m/z 397.2372 [(M+Na)+; calcd for C19H38O5SiNa: 397.2387].
A solution of SEM ether (−)-54 (0.339g, 0.905 mmol) in CH2Cl2 (4.5 mL) was cooled to −78 °C and DIBAL-H (2.0 mL, 1M in toluene, 2.2 equiv.) was added dropwise. After 5 min, the reaction was quenched with MeOH (0.5 mL). The reaction was allowed to warm to rt before EtOAc (5 mL) and sat. Rochelle’s salt (5 mL) were added. After 1 h, the organic layer transitioned from cloudy to clear. The layers were separated and the aqueous layer extracted with EtOAc (3 × 10 mL). The combined organic layers were dried over MgSO4 and concentrated. Flash chromatography (10% EtOAc/hexanes) provided (+)-13 (0.249 g, 95% yield) as colorless oil: +31.7 (c 1.2, CHCl3); IR (neat) 3457, 2952, 2925, 2892, 1650, 1457, 1378, 1249, 1102, 1054, 1025 cm−1; 1H NMR (500 MHz, CDCl3) δ 4.82 (s, 1H), 4.78 (s, 1H), 4.76 (dd, J = 14.3, 7.0 Hz, 2H), 3.79-3.68 (m, 3H), 3.65-3.56 (m, 2H), 3.49 (ddd, J = 7.6, 4.8, 4.8 Hz, 1H), 3.41 (s, 3H), 3.21 (dd, J = 8.3, 4.2 Hz, 1H), 2.31 (dd, J = 14.4, 7.6 Hz, 1H), 2.21 (dd, J = 14.4, 5.3 Hz, 1H), 1.77 (s, 3H), 0.99-0.92 (m, 2H), 0.02 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 142.6, 113.2, 95.6, 82.6, 81.0, 66.0, 62.7, 58.6, 39.5, 23.0, 18.3, −1.3; high resolution mass spectrum (ES+) m/z 313.1821 [(M+Na)+; calcd for C14H30O4SiNa: 313.1811].
Acid (−)-3
A solution of (+)-13 (0.117 g, 0.404 mmol) in DMSO (0.29 mL, 10 equiv.) and CH2Cl2 (4 mL) was cooled to 0 °C and i-Pr2NEt (0.212 mL, 3 equiv.) was added followed by SO3•pyridine (0.193 g, 3 equiv.) in one portion. After 5 min, brine (10 mL) and H2O (2 mL) were added and the reaction was warmed to rt. The layers were separated and the aqueous layer was extracted CH2Cl2 (3 × 10 mL). The combined organic layers were dried over MgSO4 and concentrated. Flash chromatography (5% EtOAc/hexanes) provided aldehyde (−)-55 (0.113 g, 97%) as a colorless oil: −9.1 (c 0.9, CHCl3); IR (neat) 2952, 2892, 2825, 1732, 1450, 1376, 1249, 1108, 1060, 1029 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.66 (d, J = 1.4 Hz, 1H), 4.85 (s, 1H), 4.84 (s, 1H), 4.80 (dd, J = 19.4, 6.9 Hz, 2H), 4.11 (dd, J = 2.8, 1.5 Hz, 1H), 3.75-3.69 (m, 2H), 3.65 (ddd, J = 9.8, 9.8, 6.9 Hz, 1H), 3.41 (s, 3H), 2.33 (dddd, J = 14.1, 14.1, 14.1, 6.9 Hz, 2H), 1.72 (s, 3H), 0.92 (ddd, J = 10.1, 6.6, 3.3 Hz, 2H), 0.02 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 202.4, 141.7, 114.4, 96.3, 82.5, 81.8, 66.1, 58.1, 38.9, 22.8, 18.2, 1.2; high resolution mass spectrum (ES+) m/z 311.1666 [(M+Na)+; calcd for C14H28O4SiNa: 311.1655].
Aldehyde (−)-55 (0.203 g, 0.704 mmol) was dissolved in t-BuOH (7.5 mL) and H2O (7.5 mL). The solution was cooled to 0 °C followed by addition of 2-methyl-2-butene (6 mL), NaH2PO4•H2O (0.550 g, 5 equiv.), and NaClO2 (0.483 g, 80 wt%, 5 equiv.). After 15 min, the reaction was poured onto sat. NH4Cl (10 mL) and extracted thoroughly with EtOAc. The combined organic layers were dried over MgSO4 and concentrated. Flash chromatography (25% EtOAc/hexanes to 40% EtOAc/hexanes) provided (−)-3 (0.196 g, 92% yield) as a colorless oil: −18.6 (c 1.1, CHCl3); IR (neat) 2953, 2925, 1725, 1649, 1376, 1252, 1110, 1060, 1030 cm−1; 1H NMR (500 MHz, CDCl3) δ 4.83 (s, 1H), 4.81 (s, 1H), 4.78 (s, 2H), 4.39 (d, J = 3.1 Hz, 1H), 3.75 (ddd, J = 8.2, 5.2, 3.2 Hz, 1H), 3.68 (dd, J = 8.5, 8.5 Hz, 2H), 3.43 (s, 3H), 2.39 (dd, J = 14.6, 8.0 Hz, 1H), 2.28 (dd, J = 14.6, 5.2 Hz, 1H), 1.76 (s, 3H), 0.96-0.87 (m, 2H), 0.01 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 175.0, 141.9, 113.5, 95.1, 81.0, 76.2, 66.3, 58.3, 38.7, 22.9, 18.2, −1.3; high resolution mass spectrum (ES+) m/z 327.1614 [(M+Na)+; calcd for C14H28O5SiNa: 327.1604].
Aldehyde 5
A solution of homophthalate 14 (0.305 g, 1.20 mmol) in THF (6 mL) was cooled to 0 °C and NaH (0.106 g, 60 wt%, 2.2 equiv.) was added. After 5 min, SEMCl (0.53 mL, 2.5 equiv.) was added dropwise. The reaction was allowed to warm to rt, and after 30 min the reaction was quenched with slow addition of MeOH (0.5 mL). Sat. NaHCO3 (5 mL) was added and the reaction was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were then dried over MgSO4 and concentrated. Flash chromatography (10% EtOAc/hexanes) provided bis-SEM ether 56 (0.611 g, 99% yield) as a colorless oil: IR (neat) 2952, 2898, 1736, 1597, 1314, 1266, 1250, 1158, 1067 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.94 (s, 1H), 5.22 (s, 2H), 5.17 (s, 2H), 3.85 (s, 3H), 3.77- 3.69 (m, 4H), 3.67 (s, 5H), 2.12 (s, 3H), 0.98-0.91 (m, 4H), 0.00 (s, 9H), 0.00 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 171.1, 168.6, 157.4, 153.8, 132.3, 120.7, 119.0, 101.9, 94.0, 93.4, 66.5, 66.5, 52.2, 52.2, 36.3, 18.3, 18.2, 11.7, −1.2; high resolution mass spectrum (ES+) m/z 537.2303 [(M+Na)+; calcd for C24H42O8Si2Na: 537.2316].
A solution of bis-SEM ether 56 (0.104 g, 0.202 mmol) in CH2Cl2 (2.0 mL) was cooled to −78 °C and DIBAL-H (0.22 mL, 1 M solution in toluene, 1.1 equiv.) was added dropwise over 15 min. After 5 min, the reaction was quenched with MeOH. The reaction was allowed to warm to rt before EtOAc (5 mL) and sat. Rochelle’s salt (5 mL) were added. After 1 h, the organic layer transitioned from cloudy to clear. The layers were separated and the aqueous layer extracted with 3 × 20 mL EtOAc. The combined organic layers were dried over MgSO4 and concentrated. Flash chromatography (10% EtOAc/hexanes) provided 5 (0.64 g, 66% yield) as a light yellow oil: IR (neat) 2953, 2901, 1726, 1595, 1477, 1272, 1251, 1157, 1110, 1065 cm−1; 1H NMR (500 MHz, CDCl3) 9.62 (dd, J = 1.8, 1.8 Hz, 1H), 6.97 (s, 1H), 5.24 (s, 2H), 5.19 (s, 2H), 3.85 (s, 3H), 3.79-3.71 (m, 4H), 3.64 (d, J = 1.8 Hz, 2H), 2.08 (s, 3H), 0.99-0.91 (m, 4H), 0.00 (s, 9H), 0.00 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 198.7, 168.7, 157.7, 154.0, 130.5, 120.8, 119.3, 102.1, 93.9, 93.4, 66.6, 66.6, 52.3, 46.1, 18.3, 18.2, 11.9, −1.2; high resolution mass spectrum (ES+) m/z 507.2210 [(M+Na)+; calcd for C23H40O7Si2Na: 507.2211].
Alkyne (−)-16
To solution of THF (600 mL), cooled to −78 °C, was added propyne (100 g, 4 equiv.) via subsurface cannula to dissolve the gas in the THF. Next, a solution of n-BuLi (570 mL, 2.2 M in hexanes, 2 equiv.) was cooled to −78 °C and added to the propyne solution over 1 h via cannula. After stirring for 1.5 h at −78 °C (S)-1,2-epoxybutane [(−)-15] (45 g, 0.625 mol) was added via cannula over 25 min followed by addition of BF3•OEt2 over 45 min via cannula. After stirring for 1.5 h at −78 °C, the reaction was quenched with sat. NaHCO3 until all gas evolution ceased (ca. 3 h). The layers were separated and the aqueous layer extracted with Et2O (3 × 500 mL). The combined organic layers were dried over MgSO4 and concentrated in a 0 °C bath (to minimize loss of product due to slight volatility) to provide alcohol (+)-57 (55.6 g, 79%) as a colorless oil: +11.0 (c 1.3, CHCl3); IR (neat) 3382, 2964, 2922, 1461, 1435, 1335, 1245, 1112, 1063, 1020 cm−1; 1H NMR (500 MHz, CDCl3) δ 3.69-3.55 (m, 1H), 2.37 (ddddd, J = 16.4, 4.9, 2.5, 2.5, 2.5 Hz, 1H), 2.24 (ddddd, J = 16.4, 7.2, 2.5, 2.5, 2.5 Hz, 1H), 1.95 (d, J = 4.8 Hz, 1H), 1.80 (dd, J = 2.5, 2.5 Hz, 3H), 1.64-1.48 (m, 2H), 0.94 (dd, J = 7.5, 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 78.3, 75.5, 71.7, 29.2, 27.3, 10.1, 3.6; low resolution mass spectrum (ES+) m/z 112.10 [(M+)+; calcd for C7H12O: 112.0888].
To a solution of alcohol (+)-57 (18.5 g, 0.165 mol) in CH2Cl2 (184 mL) and cyclohexane (368 mL) at rt was added benzyl-2,2,2-trichloroacetimidate (50.0 g, 1.2 equiv) followed by dropwise addition of TfOH (0.73 mL, 0.05 equiv.) The solution turned cloudy and light brown as the reaction progressed. After 6 h, hexane (600 mL) was added and the solution stirred 30 min to precipitate solids and then the suspension was filtered. The filtrate was washed with sat. NaHCO3 (200 mL), which was then back extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were then dried over MgSO4 and concentrated. The concentrate was diluted with 10% EtOAc/hexanes (200 mL) to facilitate further precipitation of solids and then filtered. The filtrate was then concentrated and purified by flash chromatography (0.5% EtOAc/hexanes to 1% EtOAc/hexanes) to provide alkyne (−)-16 (30.4 g, 91% yield) as a light yellow oil: − 27.6 (c 1.4, CHCl3); IR (neat) 2964, 2919, 2873, 1496, 1454, 1348, 1207, 1108, 1071, 1028 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.44-7.22 (m, 5H), 4.68 (dab, J = 11.9 Hz, 1H), 4.57 (dab, J = 11.8 Hz, 1H), 3.45 (dddd, J = 6.7, 6.7, 5.1, 5.1 Hz, 1H), 2.45 (ddddd, J = 16.5, 5.1, 2.5, 2.5, 2.5 Hz, 1H), 2.41-2.32 (m, 2H), 1.81 (dd, J = 2.5, 2.5 Hz, 3H), 1.77-1.60 (m, 2H), 0.97 (dd, J = 7.4, 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 138.9, 128.5, 127.9, 127.7, 79.1, 77.2, 76.1, 71.4, 26.8, 23.8, 9.8, 3.7; high resolution mass spectrum (ES+) m/z 202.1354 [(M)+; calcd for C14H19O: 202.1358].
Vinyl iodide (−)-17
To a flask protected from light was added bis(cyclopentadienyl) zirconium(IV) chloride (0.313 g, 2.0 equiv.) and THF (1 mL). The solution was cooled to 0 °C and DIBAL-H (1.07 mL, 1 M in hexanes, 2.0 equiv.) was added dropwise. After 30 min at 0 °C, alkyne (−)-16 (0.108g, 0.534 mmol) was dissolved in THF (0.3 mL) and added to the in situ generated Schwartz reagent. The flask and syringe were then flushed with THF (0.3 mL) into the reaction. The reaction flask was then placed in a 50 °C oil bath. After 1 h, the reaction was cooled to 0 °C and N-iodosuccinimide (0.265 g, 2.2 equiv.) was added. After 10 min, the reaction was quenched with sat. NaHCO3 (10 mL) and then filtered through a 1 cm plug of silica gel. The silica gel was rinsed with EtOAc and the layers separated. The aqueous layer was then extracted with EtOAc (3 ×10 mL) and the combined organic layers were dried over MgSO4 and concentrated. Flash chromatography (10% EtOAc/hexanes) provided vinyl iodide (−)-17 (135 mg, 77% yield, >20:1 selectivity) as a colorless oil: −4.4 (c 1.0, CHCl3); IR (neat) 3030, 2963, 2931, 2872, 1454, 1351, 1092, 1065, 1028 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.42-7.27 (m, 5H), 6.24 (dd, J = 7.5, 6.8 Hz, 1H), 4.54 (s, 2H), 3.37 (dddd, J = 5.9, 5.9, 5.9, 5.9 Hz, 1H), 2.39 (s, 3H), 2.36-2.17 (m, 2H), 1.62-1.52 (m, 2H), 0.95 (dd, J = 7.4, 7.4 Hz, 3H).; 13C NMR (125 MHz, CDCl3) δ 138.9, 137.7, 128.6, 127.9, 127.7, 95.3, 79.3, 71.4, 34.8, 27.9, 26.8, 9.9.; high resolution mass spectrum (ES+) m/z 353.0386 [(M+Na)+; calcd for C14H19IONa: 353.0378].
Diene (−)-18
Vinyl iodide (−)-17 (6.02 g, 18.2 mmol) was dissolved in anhydrous DMF (70 mL) in a sealable tube. Methyl acrylate (2.35 g, 1.5 equiv.) was added followed by Pd(OAc)2 (0.817 g, 0.20 equiv.), NaHCO3 (3.06 g, 2.0 equiv.), and Bu4NI (6.73 g, 1.0 equiv.). The tube was flushed with argon, sealed, and heated to 100 °C over 1 h. After 7 h at 100 °C, the reaction was cooled to 0 °C and sat. NH4Cl (100 mL) added. The reaction was then filtered and extracted with EtOAc (3 × 100 mL). The combined organic layers were washed with H2O, sat. NaHCO3, and brine (100 mL each). The organic layer was then dried over MgSO4 and concentrated. Flash chromatography (3% EtOAc/hexanes to 5% EtOAc/hexanes to 8% EtOAc/hexanes) provided diene (−)-18 (4.11 g, 78% yield) as a light yellow oil: −11.7 (c 1.0, CHCl3); IR (neat) 3033, 2963, 2873, 1719, 1624, 1310, 1194, 1169, 1123, 1097 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.34-7.26 (m, 5H), 5.97 (dd, J = 8.0, 8.0 Hz, 1 H), 5.81 (d, J = 15.5 Hz, 1H), 4.53 (s, 3H), 3.77 (s, 3H), 3.47 (ddd, J = 12.0, 6.0, 6.0 Hz, 1H), 2.51-2.43 (m, 2H), 1.78 (s, 3H), 1.57-1.64 (m, 3H), 0.94 (dd, J = 7.5, 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 168.2, 149.9, 138.9, 138.4, 134.3, 128.6, 127.9, 127.8, 115.6, 79.8, 71.4, 51.7, 33.3, 27.0, 12.6, 9.9; high resolution mass spectrum (ES+) m/z 289.1823 [(M+H)+; calcd for C18H25O3: 289.1804].
Epoxide (+)-20
Diene (−)-18 (2.94 g, 10.2 mmol) was dissolved in CH3CN (460 mL). Na2B4O7 buffer (0.05 M, 115 mL) was added followed Bu4NHSO4 (0.425 g) and ketone catalyst (−)-19 (1.32 g, 0.50 equiv.). Oxone (8.77g, 1.4 equiv) was dissolved in Na2EDTA buffer (59 mL, 4 × 10−4 M) and K2CO3 (8.31 g, 5.9 equiv.) was dissolved in H2O (59 mL). The two solutions were then added simultaneously over 3 h via a dual-syringe pump. After the addition was complete, water was added to dissolve any solids that had formed and the reaction extracted with EtOAc (3 × 300 mL). The combined organic layers were washed with brine (200 mL), dried over MgSO4, and concentrated. Flash chromatography (5% EtOAc/hexanes to 8% EtOAc/hexanes) provided epoxide (+)-20 (2.27 g, 73% yield, β:α = 14:1) as a colorless oil: [α]D 20 +2.2 (c 1.7, CHCl3); IR (neat) 2966, 2932, 2876, 1726, 1654, 1436, 1311, 1170, 1094 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.34-7.31 (m, 5H), 6.75 (d, J = 15.5 Hz, 1H), 6.00 (d, J = 16.0 Hz, 1H), 4.6 (dab, J = 11.5 Hz, 1 H), 4.52 (dab, J = 11.5 Hz, 1 H), 3.75 (s, 3H), 3.59-3.55 (m, 1H), 3.00 (dd, J = 6.5, 6.5 Hz, 1H), 1.85 (ddd, J = 14.5, 8.0, 5.5 Hz, 1H), 1.76 (ddd, J = 4.5, 6.5, 4.5 Hz, 1H), 1. 68-1.60 (m, 2H), 1.41 (s, 3H), 0.95 (dd, J = 7.5, 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 166.8, 150.4, 138.8, 128.6, 128.0, 127.9, 121.3, 78.0, 71.6, 63.8, 58.9, 51.9, 33.2, 27.0, 15.7, 9.5; high resolution mass spectrum (ES+) m/z 327.1578 [(M+Na)+; calcd for C18H24O4Na: 327.1572].
PMB ether (+)-21
To a solution of compound (+)-20 (60 mg, 0.20 mmol) in CH2Cl2 (3 mL) was added H2O (21 μL, 6 equiv.) The reaction was cooled to −40 °C and Me3Al (0.98 mL, 10 equiv., 2 M in hexanes) was added. After 30 min, the reaction was warmed to 0 °C and the reaction quenched with slow addition of H2O (1 mL) followed by addition of 1 N HCl (3 mL) to break up the emulsion. The reaction was extracted with CH2Cl2 (3 × 10 mL) and the combined organic layers were dried over MgSO4 and concentrated. Flash chromatography (5% EtOAc/hexanes to 10% EtOAc/hexanes) provided alcohol (+)-58 (53 mg, 84% yield) as a colorless oil: +31.7 (c 1.6, CHCl3); IR (neat) 3490, 2964, 2875, 1723, 1651, 1435, 1314, 1201, 1173, 1060 cm−1; 1H NMR (500 MHz, CDCl3) 7.38-7.26 (m, 5H), 7.04 (d, J = 16.1 Hz, 1H), 5.81 (d, J = 16.1 Hz, 1H), 4.60 (dab, J = 11.6 Hz, 1H), 4.52 (dab, J = 11.6 Hz, 1H), 3.73 (s, 3H), 3.70 (d, J = 8.9, 1H), 3.65-3.59 (m, 1H), 2.75 (s, 1H), 1.91-1.66 (m, 1H), 1.65-1.50 (m, 3H), 1.07 (s, 3H), 1.06 (s, 3H), 0.91 (dd, J = 7.5, 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 167.5, 155.9, 138.6, 128.7, 128.0, 128.0, 119.2, 78.9, 74.5, 71.6, 51.7, 41.6, 34.2, 26.1, 22.9, 22.9, 10.2; high resolution mass spectrum (ES+) m/z 343.1876 [(M+Na)+; calcd for C19H28O4Na: 343.1886].
To a solution of (+)-58 (0.906 g, 2.83 mmol) in CH2Cl2 (9 mL) and cyclohexane (18 mL) at rt was added p-methoxybenzyl-2,2,2-trichloroacetimidate (3.2 g, 4.0 equiv) followed by addition of TfOH (3 μL, 0.01 equiv.). The solution turned cloudy and light brown as the reaction progressed. After 30 min, hexane (20 mL) was added to precipitate solids and the suspension was filtered. The filtrate was washed with sat. NaHCO3 (25 mL), which was then back-extracted with CH2Cl2 (3 × 25 mL). The combined organic layers were then dried over MgSO4 and concentrated. The concentrate was diluted with 10% EtOAc/hexanes (25 mL) to facilitate further precipitation of solids and then filtered. The filtrate was then concentrated and purified by flash chromatography (3% EtOAc/hexanes to 5% EtOAc/hexanes) to provide PMB ether (+)-21 (0.910 g, 73% yield) as a light yellow oil: +43.5 (c 1.6, CHCl3); IR (neat) 2962, 2875, 2360, 2340, 1722, 1613, 1513, 1248, 1173, 1064 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.44-7.28 (m, 5H), 7.21 (d, J = 8.5 Hz, 2H), 7.13 (d, J = 16.0 Hz, 1H), 6.87 (d, J = 8.5 Hz, 2H), 5.83 (d, J = 16.0 Hz, 1H), 4.62 (dab, J = 11.6 Hz, 1H), 4.46 (dab, J = 10.9 Hz, 1H), 4.35 (dd, J = 2.4, 11.8 Hz, 2H), 3.80 (s, 3H), 3.75 (s, 3H), 3.66-3.56 (m, 1H), 3.50 (d, J = 9.7, 1H), 1.72-1.58 (m, 3H), 1.56-1.48 (m, 1H), 1.12 (s, 3H), 1.12 (s, 3H), 0.93 (dd, J = 7.4, 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 167.5, 159.2, 156.5, 139.2, 131.2, 129.2, 128.5, 127.8, 127.6, 118.6, 113.9, 83.0, 76.9, 74.9, 70.0, 55.4, 51.6, 42.7, 36.5, 26.2, 23.4, 23.4, 9.1; high resolution mass spectrum (ES+) m/z 440.2568 [(M+)+; calcd for C27H36O5: 440.2563].
Epoxy alcohol (+)-22
A solution of (+)-21 (1.76 g, 4.0 mmol) in CH2Cl2 (20 mL) was cooled to −78 °C and DIBAL-H (1.06 mL, 1.0 M in hexanes, 2.1 equiv.) was slowly added. After 5 min, the reaction was quenched with MeOH (5 mL) followed by addition of sat. Rochelle’s salt (10 mL). The solution was stirred for 2.5 h and the layers were separated. The aqueous layer was extracted with CH2Cl2 (3 × 20 mL), and the combined organic layers were dried over MgSO4 and then concentrated. Flash chromatography (25 % EtOAc/hexanes) provided allylic alcohol (+)-59 (1.56 g, 95%) as a colorless oil: +65.0 (c 1.0, CHCl3); IR (neat) 3425, 2961, 2931, 2873, 1613, 1513, 1464, 1248, 1087, 1063, 1035 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.41-7.26 (m, 5H), 7.20 (d, J = 8.5 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 5.77 (d, J = 15.8 Hz, 1H), 5.60 (ddd, J = 15.8, 5.9, 5.9 Hz, 1H), 4.60 (dab, J = 11.6 Hz, 1H), 4.46 (dab, J = 10.8 Hz, 1H), 4.34 (dd, J = 2.6, 10.9 Hz, 2H), 4.10 (dd, J = 5.8, 5.8 Hz, 2H), 3.79 (s, 3H), 3.58 (ddd, J = 6.5, 4.2, 2.3 Hz, 1H), 3.40 (dd, J = 10.0, 1.5 Hz, 1H), 1.74-1.53 (m, 3H), 1.48 (ddd, J = 14.5, 10.0, 2.2 Hz, 1H), 1.07 (s, 3H), 1.06 (s, 3H), 0.90 (dd, J = 7.5, 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 159.2, 140.6, 139.3, 131.6, 129.2, 128.6, 127.9, 127.7, 126.5, 113.9, 83.6, 77.2, 74.9, 70.0, 64.4, 55.5, 41.7, 36.5, 26.3, 24.5, 23.8, 9.3; high resolution mass spectrum (ES+) m/z 435.2514 [(M+Na)+; calcd for C26H36O4Na: 435.2512].
To freshly activated 3 Å molecular sieves (0.2 g) in CH2Cl2 (2 mL) was added (+)-DIPT (31.8 μL, 0.12 equiv.). The solution was cooled to −20 °C and Ti(O-i-Pr)4 (37.3 μL, 0.1 equiv.) was added followed by t-BuOOH (0.687 mL, 5.5 M in decane, 3.0 equiv.) The reaction was stirred for 30 min and then allylic alcohol (+)-59 (0.521 g, 1.26 mmol) dissolved in CH2Cl2 (1.5 ml) was added via syringe. The flask and syringe were rinsed with CH2Cl2 (2 × 0.8 mL) into the reaction flask. After 2 h, 10% aq. citric acid (10 mL) was added and the reaction warmed to rt. After 1 h at rt, the reaction was filtered through celite, and the celite washed with CH2Cl2 (10 mL). The layers were separated and the aqueous layer extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were dried over MgSO4 and concentrated. Flash chromatography (25% EtOAc/hexanes) provided epoxy alcohol (+)-22 (0.524 g, 97% yield, d.r. > 20:1) as a colorless oil: +56.9 (c 0.8, CHCl3); IR (neat) 3435, 2964, 2932, 2874, 1612, 1514, 1455, 1248, 1088, 1064, 1034 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.40-7.26 (m, 5H), 7.19 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 4.62 (dab, J = 11.6 Hz, 1H), 4.46 (dab, J = 11.0 Hz, 1H) 4.35 (dd, J = 11.4, 11.8 Hz, 2H), 3.85-3.80 (m, 1H), 3.79 (s, 3H), 3.63-3.52 (m, 2H), 3.52 (dd, J = 10.0, 1.4 Hz, 1H), 3.05 (dd, J = 4.8, 2.6 Hz, 1H), 2.94 (d, J = 2.4 Hz, 1H), 1.82- 1.50 (m, 4H), 0.93 (s, 3H), 0.93 (dd, J = 7.6, 7.6 Hz, 3H), 0.87 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 159.2, 139.2, 131.4, 128.9, 128.6, 128.0, 127.7, 113.9, 82.4, 77.0, 74.6, 70.0, 62.2, 61.4, 55.6, 55.5, 39.2, 36.2, 26.3, 20.4, 19.0, 9.2; high resolution mass spectrum (ES+) m/z 451.2444 [(M+Na)+; calcd for C26H36O5Na: 451.2460].
Ester (+)-9
To a 0 °C solution of (+)-22 (0.233 g, 0.545 mmol) in DMSO (6 mL) and CH2Cl2 (7 mL) was added Et3N (0.76 mL, 10 equiv.) followed by SO3•pyridine (0.347 g, 4 equiv.). After 1.5 h, NaHCO3 (4 mL) was added, the layers separated, and the aqueous layer was extracted 3 × 10 mL Et2O. The combined organic layers were then washed with 1M NaHSO4, sat. NaHCO3, and brine (10 mL each). The organic layer was then dried over MgSO4 and concentrated. Flash chromatography (10% EtOAc/hexanes) provided aldehyde (+)-60 (1.43 g, 99% yield) as a colorless oil: +115.0 (c 1.2, CHCl3); IR (neat) 2964, 2931, 2876, 1728, 1613, 1514, 1464, 1248, 1090, 1064, 1035 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.01 (d, J = 6.1 Hz, 1H), 7.44-7.27 (m, 5H), 7.19 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 4.64 (dab, J = 11.7 Hz, 1H), 4.43 (dab, J = 11.0 Hz, 1H), 4.38 (dab, J = 11.2 Hz, 1H), 4.35 (dab, J = 11.6 Hz, 1H), 3.80 (s, 3H), 3.64-3.58 (m, 1H), 3.56 (d, J = 9.8 Hz, 1H), 3.27 (dd, J = 6.1, 1.8 Hz, 1H), 3.21 (d, J = 1.8, 1H), 1.83-1.49 (m, 4H), 0.95 (s, 3H), 0.93 (dd, J = 7.5, 7.5 Hz, 3H), 0.88 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 199.1, 159.4, 139.1, 131.1, 129.2, 128.6, 127.9, 127.8, 114.0, 82.1, 76.9, 74.8, 70.0, 62.0, 56.9, 55.5, 39.6, 36.2, 26.2, 20.3, 19.1, 9.1; high resolution mass spectrum (ES+) m/z 449.2305 [(M+Na)+; calcd for C26H34O5Na: 449.2304].
A solution of aldehyde (+)-60 (0.445 g, 1.04 mmol) in CH2Cl2 (5 mL) was cooled to 0°C and a solution of carbomethoxy triphenylphosphonium ylide (23) in CH2Cl2 (5 mL) was added over 1 min. The reaction was allowed to warm to rt and after 30 min, the solvent was evaporated. Flash chromatography (8% EtOAc/hexanes) provided ester (+)-9 (0.453 g, 90% yield) as a light yellow oil: +48.7 (c 1.4, CHCl3); IR (neat) 2962, 2876, 1725, 1612, 1513, 1463, 1304, 1249, 1172, 1090, 1064 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.41-7.26 (m, 5H), 7.17 (d, J = 8.5 Hz, 2H), 6.85 (d, J = 8.5 Hz, 2H), 6.66 (dd, J = 15.7, 7.0 Hz, 1H), 6.05 (d, J = 15.7 Hz, 1H), 4.62 (dab, J = 11.6 Hz, 1H), 4.43 (dab, J = 10.9 Hz, 1H), 4.37 (dab, J = 11.2 Hz, 1H), 4.34 (dab, J = 11.8 Hz, 1H), 3.80 (s, 3H), 3.74 (s, 3H), 3.63-3.56 (m, 1H), 3.53 (d, J = 9.2 Hz, 1H), 3.35 (dd, J = 6.9, 1.7 Hz, 1H), 2.88 (d, J = 1.9 Hz, 1H), 1.81-1.50 (m, 4H), 0.94 (s, 3H), 0.93 (dd, J = 7.4, 7.4 Hz, 3H), 0.86 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 166.4, 159.3, 145.6, 139.2, 131.3, 129.0, 128.6, 128.6, 127.9, 127.7, 123.2, 114.0, 82.1, 76.9, 74.6, 70.0, 67.0, 55.5, 53.8, 51.9, 39.9, 36.1, 26.3, 20.7, 18.6, 9.2; high resolution mass spectrum (ES+) m/z 505.2553 [(M+Na)+; calcd for C29H38O6Na: 505.2566].
TBS ether (+)-24
To freshly distilled dioxane (2 mL) was added Pd2(dba)3•CHCl3 (46 mg, 0.05 equiv.) and n-Bu3P (13 μL, 0.06 equiv.). Next, a solution of HCO2H (0.20 mL, 6.0 equiv.) and Et3N (0.25 mL, 2 equiv.) in dioxane (1 mL) was added. This mixture was stirred at rt for 10 min and then a solution of epoxide (+)-9 (0.452 g, 0.937 mmol) in dioxane (1.5 mL) was added via cannula. The flask and cannula were rinsed with dioxane (2 × 1 mL) into the reaction flask. After 4.5 h, the reaction mixture was filtered through a 1 cm plug of silica gel and the silica gel washed with CH2Cl2 (20 mL). The filtrate was concentrated and subjected to flash chromatography (5% EtOAc/hexanes to 10% EtOAc/hexanes to 15% EtOAc/hexanes) to provide alcohol (+)-61 (0.421 g, 93% yield) as a colorless oil: +95.5 (c 0.7, CHCl3); IR (neat) 3464, 2963, 2875, 1722, 1613, 1514, 1249, 1173, 1063, 1036 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.40-7.26 (m, 5H), 7.13 (d, J = 8.2 Hz, 2H), 7.08 (ddd, J = 14.8, 7.2, 7.2 Hz, 1H), 6.83 (d, J = 8.3 Hz, 2H), 5.90 (d, J = 15.6 Hz, 1H), 4.64 (dab, J = 11.7 Hz, 1H), 4.44 (dab, J = 10.7 Hz, 1H) 4.34 (dab, J = 12.0 Hz, 1H), 4.34 (dab, J = 12.0 Hz, 1H), 4.32 (dab, J = 11.1 Hz, 1H), 4.16 (s, 1H), 3.79 (s, 3H), 3.71 (s, 3H), 3.59-3.56 (m, 1H), 3.54 (d, J = 9.2 Hz, 1H), 2.25 (m, 2H), 1.91-1.67 (m, 3H), 1.62 (ddd, J = 14.1, 7.1, 7.1 Hz, 1H), 1.08 (s, 3H), 0.93 (dd, J = 7.4, 7.4 Hz, 3H), 0.83 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.1, 159.5, 148.0, 139.1, 130.4, 129.2, 128.6, 128.0, 127.8, 122.6, 114.1, 86.7, 77.1, 75.3, 75.0, 70.1, 55.5, 51.5, 41.5, 35.8, 35.1, 26.3, 23.3, 20.4, 9.2; high resolution mass spectrum (ES+) m/z 485.2888 [(M+H)+; calcd for C29H41O6: 485.2903].
To a 0 °C solution of alcohol (+)-61 (0.789 g, 1.63 mmol) in CH2Cl2 (16.3 mL) was added 2,6-lutidine (0.38 mL, 2.0 equiv.) followed by dropwise addition of TBSOTf (0.45 mL, 1.2 equiv.). After 20 min, sat. NaHCO3 (10 mL) was added and the layers separated. The aqueous layer was extracted CH2Cl2 (3 × 15 mL), and the combined organic layers were dried over MgSO4 and concentrated. Flash chromatography (5% EtOAc/hexanes) provided TBS ester (+)-24 (0.965 g, 99% yield) as a colorless oil: +42.1 (c 0.9, CHCl3); IR (neat) 2956, 2931, 2881, 2855, 1725, 1513, 1463, 1249, 1170, 1073 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.37-7.26 (m, 5H), 7.20 (d, J = 8.5 Hz, 2H), 7.06 (ddd, J = 15.7, 8.3, 6.4 Hz, 1H), 6.86 (d, J = 8.5 Hz, 2H), 5.76 (d, J = 15.7 Hz, 1H), 4.64 (dab, J = 11.5 Hz, 1H), 4.47 (dab, J = 10.9 Hz, 1H), 4.33 (dab, J = 10.8 Hz, 1H), 4.32 (dab, J = 11.7 Hz, 1H), 3.80 (s, 3H), 3.73 (s, 3H), 3.65 (dd, J = 6.4, 4.0 Hz, 1H), 3.60-3.55 (m, J = 9.9, 5.5 Hz, 1H), 3.52 (dd, J = 6.5, 5.0 Hz, 1H), 2.51 (ddd, J = 14.9, 4.2, 4.2 Hz, 1H), 2.36 (ddd, J = 15.0, 7.5, 7.5 Hz, 1H), 1.74-1.64 (m, 1H), 1.65-1.58 (m, 1H), 1.56 (dd, J = 5.9, 5.9 Hz, 2H), 0.95 (s, 3H), 0.92 (dd, J = 7.5, 7.5 Hz, 3H), 0.90 (s, 9H), 0.89 (s, 3H), 0.05 (s, 3H), 0.03 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.1, 159.2, 148.6, 139.2, 131.7, 129.0, 128.6, 127.8, 127.7, 122.2, 113.9, 81.3, 77.3, 76.6, 75.0, 70.3, 55.5, 51.5, 44.7, 36.2, 36.1, 26.3, 26.2, 21.0, 20.1, 18.6, 9.2, −3.2, −3.8; high resolution mass spectrum (ES+) m/z 621.3605 [(M+Na)+; calcd for C35H54O6SiNa: 621.3588].
Epoxide (+)-25
A solution of (+)-24 (1.43 g, 2.38 mmol) in CH2Cl2 (12 mL) was cooled to −78 °C and DIBAL-H (1.06 mL, 1.0 M in hexanes, 2.1 equiv.) was added slowly over 5 min. After 2 min, the reaction was quenched with methanol (1 mL) followed by addition of sat. Rochelle’s salt (10 mL). The solution was stirred for 2 h and the layers were separated. The aqueous layer was extracted with CH2Cl2 (3 × 25 mL), and the combined organic layers were dried over MgSO4 and then concentrated. Flash chromatography (5 % EtOAc/hexanes) provided allylic alcohol (+)-62 (1.32 g, 97%) as a colorless oil: +47.7 (c 0.7, CHCl3); IR (neat) 3444, 2958, 2930, 2880, 2855, 1613, 1514, 1463, 1249, 1070 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.41-7.27 (m, 5H), 7.21 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 5.74 (ddd, J = 15.2, 7.2 Hz, 1H), 5.55 (ddd, J = 15.4, 5.8, 5.8 Hz, 1H), 4.64 (dab, J = 11.4 Hz, 1H), 4.50 (dab, J = 10.9 Hz, 1H), 4.35 (dab, J = 10.8 Hz, 1H), 4.34 (dab, J = 11.2 Hz, 1H), 4.03-3.91 (m, 2H), 3.80 (s, 3H), 3.64-3.59 (m, 1H), 3.58 (dd, J = 9.7, 1.3 Hz, 1H), 3.55 (dd, J = 5.3, 5.3 Hz, 1H), 2.41 (ddd, J = 14.5, 5.4 Hz, 1H), 2.19 (ddd, J = 14.0, 6.7 Hz, 1H), 1.75-1.64 (m, 1H), 1.64- 1.49 (m, 3H), 1.47 (dd, J = 6.0, 6.0 Hz, 1H), 0.98 (s, 3H), 0.92 (dd, J = 7.5, 7.5 Hz, 3H), 0.90 (s, 9H), 0.88 (s, 3H), 0.04 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 159.2, 139.1, 131.8, 131.4, 130.7, 129.0, 128.6, 128.1, 127.7, 113.9, 80.9, 77.3, 75.0, 70.5, 63.8, 55.5, 45.0, 36.4, 36.0, 26.3, 26.3, 21.0, 19.9, 18.6, 9.2, −3.1, −3.9; high resolution mass spectrum (ES+) m/z 593.3651 [(M+Na)+; calcd for C34H54O5SiNa: 593.3639].
To freshly activated 3 Å molecular sieves (0.4 g) in CH2Cl2 (4.6 mL) was added (−)- DIPT (58 μL, 0.12 equiv.). The solution was cooled to −20 °C and Ti(Oi-Pr)4 (68 μL, 0.1 equiv.) was added followed by t-BuOOH (1.26 mL, 5.5 M in decane, 3.0 equiv.). The reaction was stirred for 30 min and then allylic alcohol (+)-62 (1.32 g, 2.31 mmol) dissolved in CH2Cl2 (2 ml) was added via cannula. The flask and cannula were rinsed with CH2Cl2 (2 × 1.5 mL) into the reaction flask. After 4 h, 10% aq. citric acid (10 mL) was added and the reaction warmed to rt. After 2 h, the layers were separated and the aqueous layer extracted with CH2Cl2 (3 × 25 mL). The combined organic layers were dried over MgSO4 and concentrated. Flash chromatography (10% EtOAc/hexanes to 15% EtOAc/hexanes) provided epoxide (+)-25 (1.24 g, 92% yield, dr > 20:1) as a colorless oil: +72.4 (c 1.9, CHCl3); IR (neat) 3444, 2957, 2930, 2881, 2856, 1613, 1514, 1463, 1249, 1071 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.39-7.27 (m, 5H), 7.22 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.6 Hz, 2H), 4.61 (dab, J = 11.3 Hz, 1H), 4.50 (dab, J = 10.9 Hz, 1H), 4.37 (dab, J = 10.8 Hz, 1H), 4.31 (dab, J = 11.3 Hz, 1H), 3.80 (s, 3H), 3.78-3.71 (m, 2H), 3.62-3.51 (m, 2H), 3.49 (dd, J = 9.3, 2.0 Hz, 1H), 3.10 (ddd, J = 6.8, 4.7, 2.1 Hz, 1H), 2.70 (dd, J = 4.3, 2.6 Hz, 1H), 1.80-1.64 (m, 3H), 1.64-1.50 (m, 3H), 0.98 (s, 3H), 0.92 (s, 12H), 0.92 (dd, J = 7.4, 7.4 Hz, 3H), 0.11 (s, 3H), 0.10 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 159.2, 139.3, 131.7, 129.0, 128.5, 127.9, 127.6, 113.9, 81.4, 77.3, 75.1, 75.0, 70.0, 61.7, 59.9, 55.5, 53.7, 44.2, 35.9, 35.1, 26.4, 26.2, 20.9, 20.0, 18.6, 9.2, −3.5, − 3.6; high resolution mass spectrum (ES+) m/z 609.3596 [(M+Na)+; calcd for C34H54O6SiNa: 609.3588].
Alcohol (+)-26
Epoxy alcohol (+)-25 was dissolved in CH3CN (12 mL) and then TEMPO (15 mg, 0.08 equiv.) was added followed by pH 7 buffer (12 mL). Next, NaClO2 (0.42 g, 2.5 equiv.) was added in one portion followed by dropwise addition of NaOCl (0.32 mL, 5 wt% solution, 0.2 equiv.). After 1.5 h, anhydrous Na2SO3 (0.49 g, 3.2 equiv.) was added and the reaction stirred for 30 min, upon which, the solution turned from orange to colorless. The reaction was acidified to pH 4 with 10% aq. citric acid solution and then extracted with EtOAc (3 × 25 mL). The combined organic layers were dried over MgSO4 and concentrated.
The unpurified acid was then dissolved in Et2O (24 mL) and cooled to 0 °C. A solution of CH2N2 in Et2O was then added dropwise until gas evolution ceased and the reaction turned light yellow. Argon was bubbled through the reaction mixture for 15 min to remove any excess CH2N2 and then the reaction was concentrated. Flash chromatography (10% EtOAc/hexanes) provided methyl ester (+)-63 (0.557 g, 76% yield, 2 steps) as a colorless oil: +86.8 (c 1.0, CHCl3); IR (neat) 2957, 2931, 2882, 2857, 1754, 1513, 1458, 1249, 1065 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.41-7.27 (m, 5H), 7.21 (d, J = 8.6 Hz, 2H), 6.86 (d, J = 8.7 Hz, 2H), 4.62 (dab, J = 11.5 Hz, 1H), 4.50 (dab, J = 10.9 Hz, 1H), 4.36 (dab, J = 10.9 Hz, 1H), 4.30 (dab, J = 11.4 Hz, 1H), 3.80 (s, 3H), 3.75 (s, 3H), 3.75-3.70 (m, 1H), 3.60-3.53 (m, 1H), 3.49-3.42 (m, 1H), 3.31 (ddd, J = 6.5, 4.5, 1.8 Hz, 1H), 3.04 (d, J = 1.8 Hz, 1H), 1.80 (ddd, J = 14.5, 7.7, 4.5 Hz, 1H), 1.71-1.64 (m, 2H), 1.64-1.56 (m, 1H), 1.54 (dd, J = 7.6, 4.5 Hz, 2H), 0.96 (s, 3H), 0.93-0.90 (m, 15H), 0.12 (s, 3H), 0.09 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 169.9, 159.2, 139.3, 131.6, 129.0, 128.6, 127.8, 127.7, 113.9, 81.4, 77.1, 75.1, 74.9, 70.0, 56.6, 55.5, 54.7, 52.5, 44.2, 35.9, 34.8, 26.4, 26.1, 21.0, 20.0, 18.6, 9.1, −3.6, −3.6; high resolution mass spectrum (ES+) m/z 637.3521 [(M+Na)+; calcd for C35H54O7SiNa: 637.3537].
A solution of (+)-63 (1.32 g, 2.14 mmol) in CH2Cl2 (22 mL) and pH 7 buffer (5.4 mL) was cooled to 0 °C and DDQ (0.542 g, 1.1 equiv) was added in three portions over 1 min. After 45 min, the reaction was diluted with CH2Cl2 (20 mL) and filtered through Celite. The Celite was then washed with sat. NaHCO3 (10 mL). The layers were separated and the aqueous layer extracted with CH2Cl2 (3 × 20 mL). The combine organic layers were then dried with MgSO4 and concentrated. Flash chromatography (5% EtOAc/hexanes until the anisaldehyde eluted then 10% EtOAc/hexanes) provided alcohol (+)-26 (0.998g, 94% yield) as a light yellow oil: +54.3 (c 0.7, CHCl3); IR (neat) 3494, 2957, 2930, 2880, 2855, 1755, 1452, 1254, 1204, 1058 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.42-7.23 (m, 5H), 4.63 (dab, J = 11.6 Hz, 1H), 4.53 (dab, J = 11.6 Hz, 1H) 3.91 (dd, J = 8.8, 3.2 Hz, 1H), 3.84 (s, 1H), 3.82-3.76 (m, 1H), 3.77 (s, 3H), 3.69 (ddd, J = 11.4, 5.9, 5.9 Hz, 1H), 3.32 (ddd, J = 7.9, 3.3, 1.9 Hz, 1H), 3.23 (d, J = 1.8 Hz, 1H), 1.98 (ddd, J = 14.8, 8.2, 3.4 Hz, 1H), 1.69-1.52 (m, 3H), 1.50-1.45 (m, 2H), 0.95 (s, 3H), 0.94-0.91 (m, 12H), 0.72 (s, 3H), 0.16 (s, 3H), 0.15 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 169.6, 139.5, 128.5, 127.9, 127.6, 79.5, 78.2, 72.2, 71.4, 56.6 54.4, 52.7, 40.9, 36.1, 35.2, 27.5, 26.3, 22.5, 20.0, 18.5, 9.9, −3.7, −4.1; high resolution mass spectrum (ES+) m/z 495.3125 [(M+H)+; calcd for C27H47O6Si: 495.3142].
Alcohol (+)-27
To a solution of (+)-26 (0.998 g, 2.02 mmol) in CH2Cl2 (40 mL) was added camphorsulfonic acid (94 mg, 0.2 equiv). After stirring for 5 h at rt, sat. NaHCO3 (20 mL) was added and the reaction mixture stirred for 10 min. The layers were separated and the aqueous layer extracted with CH2Cl2 (4 × 15 mL). The combined organic layers were then dried over MgSO4 and concentrated. Flash chromatography (5% EtOAc/hexanes) provided alcohol (+)-27 (0.917g, 92%) as a colorless oil: +38.1 (c 1.6, CHCl3); IR (neat) 3461, 2956, 2930, 2857, 1741, 1471, 1437, 1360, 1256, 1080 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.44-7.17 (m, 5H), 4.54 (dd, J = 41.3, 11.5 Hz, 2H), 4.23 (dd, J = 5.8, 4.0 Hz, 1H), 4.07 (dd, J = 9.9, 3.5 Hz, 1H), 3.78 (s, 3H), 3.80-3.75 (m, 1H), 3.63 (dd, J = 3.8, 3.8 Hz, 1H), 3.56-3.48 (m, 1H), 2.87 (d, J = 6.2 Hz, 1H), 2.31 (dd, J = 13.0, 13.0 Hz, 1H), 2.06 (ddd, J = 13.4, 10.0, 3.1 Hz, 1H), 1.71-1.48 (m, 3H), 1.36 (ddd, J = 13.6, 3.9, 3.9 Hz, 1H), 1.01 (s, 3H), 0.92 (dd, J = 7.5, 7.5 Hz, 3H), 0.90 (s, 9H), 0.86 (s, 3H), 0.05 (s, 3H), 0.03 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 173.2, 139.5, 128.5, 128.4, 128.0, 127.5, 77.9, 73.8, 73.7, 71.6, 67.6, 52.6, 37.3, 33.0, 30.1, 27.2, 26.5, 26.0, 21.5, 18.2, 9.6, −4.4, −4.9; high resolution mass spectrum (ES+) m/z 517.2970 [(M+Na)+; calcd for C27H46O6SiNa: 517.2962].
Methyl ether (+)-28
A solution of (+)-27 (0.236g, 0.478 mmol) in THF (5 mL) was cooled to 0 °C and NaH (29 mg, 60% in mineral oil, 1.5 equiv.) added in one portion. After 20 min, Me2SO4 (59 μL, 1.3 equiv.) was added dropwise and the reaction allowed to warm to rt. After 1.5 h, the reaction was quenched with sat. NaHCO3 (5 mL). The layers were separated and the aqueous layer extracted with CH2Cl2 (5 × 15 mL). The combined organic layers were then dried over MgSO4 and concentrated. Flash chromatography (3% EtOAc/hexanes to 5% EtOAc/hexanes) provided methyl ether (+)- 28 (0.229g, 94% yield) as a colorless oil: +20.0 (c 2.8, CHCl3); IR (neat) 2956, 2931, 2857, 1751, 1462, 1359, 1256, 1195, 1126, 1083, 1006 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.44-7.14 (m, 5H), 4.57 (dab, J = 11.5 Hz, 1H), 4.51 (dab, J = 11.4 Hz, 1H), 4.08 (ddd, J = 7.1, 4.2, 4.2 Hz, 1H), 3.89 (d, J = 6.4 Hz, 1H), 3.74 (s, 3H), 3.70 (dd, J = 11.3, 1.5 Hz, 1H), 3.59 (dd, J = 6.5, 3.4 Hz, 1H), 3.50-3.44 (m, 1H), 3.39 (s, 3H), 2.08 (dd, J = 12.7, 12.7 Hz, 1H), 1.98 (ddd, J = 13.7, 7.6, 3.5 Hz, 1H), 1.70-1.48 (m, 4H), 1.00 (s, 3H), 0.92 (dd, J = 7.6, 7.6 Hz, 3H), 0.90 (s, 9H), 0.85 (s, 3H), 0.05 (s, 3H), 0.04 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 171.8, 139.8, 128.4, 127.8, 127.4, 83.2, 78.0, 77.9, 73.5, 71.6, 68.4, 58.8, 52.1, 37.9, 33.7, 30.7, 27.4, 26.0, 25.9, 19.2, 18.2, 9.6, −4.3, −4.9; high resolution mass spectrum (ES+) m/z 531.3139 [(M+Na)+; calcd for C28H48O6SiNa: 531.3118].
Tetrahydropyran (+)-6
To a solution of (+)-28 (0.229 g, 0.450 mmol) in EtOAc (4.5 mL) was added 10% Pd/C (0.025 g). The reaction flask was purged with H2 and then a balloon of H2 was attached to the flask. After 5 h at rt, the reaction mixture was filtered through a pad of Celite and the Celite rinsed with CH2Cl2 (10 mL). The reaction was concentrated and then flash chromatography (10% EtOAc/hexanes) provided alcohol (+)- 64 (0.182 g, 97%) as a colorless oil: +12.7 (c 3.6, CHCl3); IR (neat) 3557, 2955, 2930, 2857, 1730, 1463, 1285, 1254, 1127, 1082 cm−1; 1H NMR (500 MHz, CDCl3) δ 4.08 (d, J = 9.0 Hz, 1H), 4.04-4.00 (m, 1H), 3.81 (s, 3H), 3.59-3.53 (m, 2H), 3.53-3.46 (m, 1H), 3.41 (s, 3H), 2.92 (d, J = 4.1 Hz, 1H), 1.93 (ddd, J = 13.8, 4.2, 2.8 Hz, 1H), 1.74 (ddd, J = 13.8, 10.4, 5.7 Hz, 1H), 1.60 (dd, J = 12.3, 12.3 Hz, 1H), 1.53-1.34 (m, 3H), 0.94 (dd, J = 7.4, 7.4 Hz, 3H), 0.89 (s, 12H), 0.83 (s, 3H), 0.05 (s, 3H), 0.04 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 173.1, 81.0, 76.1, 72.6, 72.2, 68.8, 58.8, 52.6, 39.0, 36.5, 30.4, 30.2, 26.0, 23.8, 18.2, 14.2, 10.6, −4.1, −4.8; high resolution mass spectrum (ES+) m/z 441.2642 [(M+Na)+; calcd for C21H42O6SiNa: 441.2649].
A solution of alcohol (+)-64 (0.187g, 0.446 mmol) in CH2Cl2 (4.5 mL) was cooled to 0 °C and NaHCO3 (56 mg, 1.5 equiv.) was added followed by Dess-Martin periodinane (0.568 g, 3 equiv.). After 1 h at 0 °C, H2O, sat. NaHCO3, and CH2Cl2 (5 mL each) were added. The solution was stirred until the organic layer went clear (ca. 30 min). The layers were separated and the aqueous layer extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were then dried over MgSO4 and concentrated. Flash chromatography (10% EtOAc/hexanes) provided tetrahydropyran (+)-6 (0.181g, 97% yield) as a colorless oil: +16.5 (c 1.7, CHCl3); IR (neat) 2954, 2934, 2886, 2858, 1754, 1722, 17.112, 1462, 1361, 1255, 1119, 1073 cm−1; 1H NMR (500 MHz, CDCl3) δ 4.10 (dd, J = 10.6, 2.5 Hz, 1H), 4.04 (ddd, J = 11.9, 4.5, 2.3 Hz, 1H), 3.80 (d, J = 4.5 Hz, 1H), 3.70 (s, 3H), 3.53 (dd, J = 2.7, 2.7 Hz 1H), 3.40 (s, 3H), 2.60-2.38 (m, 3H), 2.23 (dd, J = 14.5, 2.5 Hz, 1H), 1.96 (ddd, J = 14.1, 12.0, 2.4 Hz, 1H), 1.40 (ddd, J = 13.8, 3.1, 2.5 Hz, 1H), 1.04 (dd, J = 7.3, 7.3 Hz, 3H), 0.90 (s, 9H), 0.89 (s, 3H), 0.80 (s, 3H), 0.03 (s, 3H), 0.01 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 210.4, 171.7, 82.5, 77.4, 73.1, 69.8, 58.8, 52.2, 42.6, 38.1, 37.2, 30.3, 26.0, 25.1, 18.2, 17.9, 7.8, −4.2, −4.8; high resolution mass spectrum (ES+) m/z 439.2491 [(M+Na)+; calcd for C21H40O6SiNa: 439.2492].
β-Hydroxy ketone (+)-29
A solution of ketone (+)-6 (0.140 g, 0.336 mmol) in CH2Cl2 (3.4 mL) was cooled to −78 °C and Cl2BPh (52 μL, 1.2 equiv.) was added. After stirring for 20 min, i-Pr2NEt (88 μL, 1.5 equiv.) was added dropwise. The reaction was stirred for 1 h at −78 °C, warmed to 0 °C over 10 min, then stirred for 1 h at 0 °C. After cooling back to −78 °C, aldehyde 5 (0.237g, 1.45 equiv.) was dissolved in CH2Cl2 (1.5 mL) and added to the boron enolate dropwise over 15 min. After 1 h at −78 °C, the reaction was quenched with a 1:1 mixture of MeOH/pH 7 buffer (6 mL). After warming to 0 °C, the reaction was neutralized to pH 7 with pH 8 buffer and stirred for 1 h at 0 °C. The layers were separated and the aqueous layer extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were then dried over MgSO4 and concentrated. Flash chromatography (15% EtOAc/hexanes) provided β-hydroxy ketone (+)-29 (0.273 g, 90% yield, single diastereomer) as a colorless oil: +30.0 (c 0.9, CHCl3); IR (neat) 3467, 2952, 2896, 1731, 1593, 1463, 1253, 1064 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.88 (s, 1H), 5.21 (s, 2H), 5.15 (d, J = 2.3 Hz, 2H), 4.09 (dd, J = 11.1, 5.7 Hz, 1H), 4.07-4.00 (m, 2H), 3.94 (d, J = 6.1 Hz, 1H), 3.87 (s, 3H), 3.78-3.70 (m, 4H), 3.72 (s, 3H), 3.64 (dd, J = 7.8, 3.8 Hz, 1H), 3.43 (d, J = 5.6 Hz, 1H), 3.40 (s, 3H), 3.06 (dd, J = 16.4, 9.4 Hz, 1H), 2.84 (dd, J = 14.2, 3.2 Hz, 1H), 2.75-2.63 (m, 1H), 2.61-2.50 (m, 2H), 2.16 (s, 3H), 1.95 (ddd, J = 13.8, 5.8, 3.9 Hz, 1H), 1.57 (ddd, J = 13.3, 7.8, 4.9 Hz, 1H), 1.19 (d, J = 7.1 Hz, 3H), 0.95 (s, 3H), 0.98-0.92 (m, 4H), 0.90 (s, 9H), 0.84 (s, 3H), 0.04 (s, 6H), 0.01 (s, 9H), 0.00 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 212.5, 171.6, 170.7, 157.8, 153.7, 136.6, 120.3, 119.1, 101.1, 93.9, 93.3, 82.4, 76.7, 73.1, 71.6, 70.1, 66.5, 66.5, 58.8, 53.2, 52.7, 52.2, 42.5, 38.1, 35.9, 30.1, 26.0, 24.9, 18.3, 18.2, 17.6, 11.8, 11.4, −1.2, −4.3, −4.8; high resolution mass spectrum (ES+) m/z 923.4782 [(M+Na)+; calcd for C44H80O13Si3Na: 923.4805].
Acid (+)-30
A solution (+)-29 (0.116 g, 0.129 mmol) in THF (1.4 mL) and MeOH (0.46 mL) was cooled to −78 °C and Et2BOMe (0.167 mL, 1 M in THF, 1.3 equiv.) was added dropwise. After 25 min, NaBH4 (10 mg, 2.0 equiv.) was added and the reaction was strirred at −78 °C. After 1 h, the reaction was warmed to 0 °C over 10 min and then stirred for 30 min. EtOAc (2 mL) was added followed by H2O (2 mL) and a 1:1 solution of MeOH/30% aq. H2O2 (5 mL). After 1 h, the reaction was extracted with EtOAc (3 × 10 mL) and the combined organic layers were treated with solid Na2S2O3 to destroy any remaining peroxide. The organic layer was then filtered and washed with sat. Na2S2O3, dried over MgSO4, and concentrated. Flash chromatography (15% EtOAc/hexanes to 25% EtOAc/hexanes) provided diol (+)-65 (0.109 g, 95% yield, dr > 20:1) as a colorless oil: +10.3 (c 1.0, CHCl3); IR (neat) 3500, 2953, 2896, 2857, 1734, 1593, 1253, 1157, 1115, 1065 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.87 (s, 1H), 5.21 (s, 2H), 5.16 (s, 2H), 4.16-4.10 (m, 1H), 4.02 (dd, J = 7.5, 4.9 Hz, 1H), 3.97 (d, J = 7.9 Hz, 1H), 3.88 (s, 3H), 3.84 (dd, J = 12.9, 7.1 Hz, 1H), 3.78 (s, 3H), 3.76-3.70 (m, 4H), 3.65 (s, 1H), 3.56- 3.50 (m, 2H), 3.39 (s, 3H), 2.85-2.80 (m, 2H), 2.20 (s, 3H), 1.93 (ddd, J = 13.8, 4.0, 4.0 Hz, 1H), 1.83 (ddd, J = 14.2, 10.3, Hz, 1H), 1.72-1.63 (m, 1H), 1.51 (d, J = 7.0 Hz, 1H), 1.45 (d, J = 14.7 Hz, 1H), 0.98 (d, J = 6.9, 3H), 0.96-0.92 (m, 4H), 0.90 (s, 9H), 0.88 (s, 3H), 0.85 (s, 3H), 0.04 (s, 3H), 0.04 (s, 3H), 0.01 (s, 9H), 0.00 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 171.3, 169.9, 157.2, 153.1, 136.9, 120.2, 119.2, 100.6, 93.6, 93.1, 81.7, 80.9, 76.2, 75.5, 72.1, 71.4, 66.2, 66.2, 58.5, 52.2, 52.2, 41.9, 38.9, 35.8, 33.4, 29.8, 25.7, 24.0, 18.0, 17.9, 15.1, 11.7, 5.8, −1.5, −4.4, −5.0; high resolution mass spectrum (ES+) m/z 925.5000 [(M+Na)+; calcd for C44H82O13Si3Na: 925.4961].
A solution of diol (+)-65 (20.6 mg, 0.023 mmol) was dissolved in MeOH (1.1 mL) and cooled to 0 °C. Next, H2O (8 μL, 20 equiv.) was added followed by LiOH (11 mg, 20 equiv.). The cold bath was removed and the reaction allowed to warm to rt. After 28 h, due to bis-acid formation, the reaction was quenched by diluting with EtOAc and acidified to pH 5 with 5% aq. AcOH. Brine (2 mL) was added and the reaction extracted with EtOAc (5 × 3 mL). The combined organic layers were then dried over MgSO4 and concentrated. Flash chromatography (0.1% AcOH in 30% EtOAc/hexanes to 0.2% AcOH in 60% EtOAc/hexanes) provided acid (+)-30 (17 mg, 87% yield) as a colorless oil: +31.4 (c 1.1, CHCl3); IR (neat) 3467, 2953, 2898, 2858, 1720, 1592, 1250, 1107, 1065 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.95 (s, 1H), 5.30 (dd, J = 20.0, 7.0 Hz, 2H), 5.27 (s, 2H), 4.33 (ddd, J = 12.1, 5.8, 2.5 Hz, 1H), 4.23 (dd, J = 10.9, 6.6 Hz, 1H), 4.15 (d, J = 7.3 Hz, 1H), 3.87-3.79 (m, 3H), 3.78-3.72 (m, 2H), 3.64-3.57 (m, 2H), 3.45 (s, 3H), 3.03 (dd, J = 16.5, 2.4 Hz, 1H), 2.91 (dd, J = 16.4, 12.2 Hz, 1H), 2.24-2.07 (m, 1H), 2.12 (s, 3H), 2.01 (dd, J = 10.6, 7.0 Hz, 1H), 1.97-1.92 (m, 1H), 1.66 (ddd, J = 13.7, 6.8, 4.7 Hz, 1H), 1.54 (d, J = 15.0 Hz, 1H), 1.08 (d, J = 7.0 Hz, 3H), 0.97 (s, 3H), 0.99-0.94 (m, 4H), 0.91 (s, 9H), 0.88 (s, 3H), 0.06 (s, 6H), 0.01 (s, 9H), 0.00 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 172.3, 164.1, 160.1, 159.2, 141.9, 117.45, 109.0, 102.1, 94.4, 93.1, 82.2, 81.9, 79.0, 72.7, 71.9, 69.8, 66.8, 66.8, 58.7, 41.7, 38.7, 33.0, 30.6, 29.5, 26.0, 25.0, 18.3, 18.3, 18.2, 11.4, 9.7, 9.7, −1.2, −1.2, −4.2, −4.8; high resolution mass spectrum (ES+) m/z 879.4569 [(M+Na)+; calcd for C42H76O12Si3Na: 879.4543].
Silyl ether (+)-4
A solution of (+)-30 (54 mg, 0.063 mmol) in acetone (3.2 mL) was cooled to 0 °C and i-Pr2NEt (24 μL, 2.2 equiv.) was added followed by dropwise addition of isobutyl chloroformate (20 μL, 2.4 equiv.). After 45 min at 0 °C, NaN3 (21 mg, 5 equiv.) was dissolved in H2O (0.4 mL) and added to the reaction over 2 min. After 20 min, cold H2O (2 mL) was added and the reaction extracted with cold EtOAc (3 × 5 mL). The combined organic layers were dried thoroughly over MgSO4 and concentrated. The residue was azeotroped with benzene (3 × 5 mL) and placed on the vacuum pump for 30 min. The unpurified acyl azide was dissolved in toluene (3.2 mL), the reaction flask fitted with a reflux condenser, and then heated to 80 °C. After 45 min, 2-trimethylsilyl ethanol (0.181 mL, 20 equiv.) was added through the top of the reflux condenser. After 2 h, the reaction was cooled to rt and the solvent evaporated. Flash chromatography (10% EtOAc/hexanes to 25% EtOAc/hexanes) provided N,O-aminal (+)-43 (45 mg, 74% yield) as a colorless oil: +8.4 (c 0.5, CHCl3); IR (neat) 3501, 3315, 2953, 2901, 2857, 1719, 1595, 1249, 1108, 1064 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.94 (s, 1H), 5.37 (d, J = 10.1 Hz, 1H), 5.31 (dab, J = 7.0 Hz, 1H), 5.27 (dab, J = 6.7 Hz, 1H), 5.26 (s, 1H), 4.89 (dd, J = 10.0, 2.6 Hz, 1H), 4.31 (ddd, J = 11.6, 6.9. 2.4 Hz, 1H), 4.24-4.08 (m, 2H), 3.97 (d, J = 9.4 Hz, 1H), 3.88-3.72 (m, 4H), 3.65 (s, 1H), 3.62 (d, J = 10.4 Hz, 1H), 3.56 (dd, J = 4.9, 3.2 Hz, 1H), 3.36 (s, 3H), 3.10 (dd, J = 16.4, 2.1 Hz, 1H), 2.82 (dd, J = 16.4, 11.9 Hz, 1H), 2.45-2.28 (m, 1H), 2.12 (s, 3H), 1.91-1.80 (m, J = 12.5, 8.0 Hz, 2H), 1.53- 1.37 (m, 2H), 1.11 (d, J = 6.9 Hz, 3H), 1.00 (s, 3H), 0.98-0.93 (m, 6H), 0.90 (s, 9H), 0.88 (s, 3H), 0.05 (s, 3H), 0.04 (s, 3H), 0.02 (s, 9H), 0.00 (s, 9H), 0.00 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 163.5, 159.9, 159.1, 157.2, 141.9, 117.4, 109.4, 102.2, 94.4, 93.1, 83.8, 83.6, 79.4, 77.4, 73.0, 72.7, 68.0, 66.8, 66.7, 63.7, 55.8, 43.4, 38.1, 32.9, 31.1, 29.7, 26.1, 26.0, 18.4, 18.3, 18.2, 17.8, 11.4, 10.2, −1.1, −1.2, −1.3, −4.3, −4.8; high resolution mass spectrum (ES+) m/z 994.5354 [(M+Na)+; calcd for C47H89NO12Si4Na: 994.5360].
A solution of N,O-aminal (+)-43 (19.6 mg, 0.020 mmol) in CH2Cl2 (0.2 mL) was cooled to 0 °C and 2,6-lutidine (9.5 μL, 4.0 equiv.) was added followed by dropwise addition of TBSOTf (10 μL, 2.1 equiv.). After 45 min, CH2Cl2 (1 mL) and sat. NaHCO3 (2 mL) added and the reaction allowed to warm to rt. The layers were separated and the aqueous layer extracted with CH2Cl2 (3 × 5 mL). Preparative TLC (25% EtOAc/hexanes, 500 micron plate) provided silyl ether (+)-4 (20 mg, 91%) as a colorless oil: +36.0 (c 0.8, CHCl3); IR (neat) 2954, 2928, 2896, 2857, 1725, 1594, 1472, 1250, 1108, 1063 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.93 (s, 1H), 5.45 (d, J = 9.7 Hz, 1H), 5.32 (dab, J = 7.0 Hz, 1H), 5.27 (dab, J = 6.7 Hz, 1H), 5.27 (s, 1H), 4.82 (d, J = 9.6 Hz, 1H), 4.23-4.07 (m, 3H), 4.00 (d, J = 8.7 Hz, 1H), 3.88-3.78 (m, 2H), 3.75 (dd, J = 8.7, 7.9 Hz, 2H), 3.58 (dd, J = 3.8, 3.8 Hz, 1H), 3.37 (s, 3H), 3.34 (d, J = 11.6 Hz, 1H), 3.09 (d, J = 15.3 Hz, 1H), 2.64 (dd, J = 16.4, 12.1 Hz, 1H), 2.32-2.22 (m, 1H), 2.12 (s, 3H), 2.03-1.92 (m, 1H), 1.87-1.77 (m, 1H), 1.62 (dd, J = 11.6, 11.6 Hz, 1H), 1.48 (ddd, J = 13.7, 8.2, 8.2 Hz, 1H), 1.07 (d, J = 6.7 Hz, 3H), 0.97 (s, 3H), 0.9-0.93 (m, 6H), 0.91 (s, 9H), 0.86 (s, 3H), 0.80 (s, 9H), 0.09 (s, 3H), 0.07 (s, 3H), 0.06 (s, 3H), 0.01 (s, 3H), 0.00 (s, 9H), 0.00 (s, 18H); 13C NMR (125 MHz, CDCl3) δ 163.5, 159.8, 159.1, 157.2, 141.4, 117.2, 109.5, 102.3, 94.5, 93.1, 84.4, 79.9, 77.7, 77.2, 73.6, 68.9, 67.8, 66.8, 66.7, 63.6, 56.1, 39.9, 37.6, 32.9, 31.7, 29.9, 26.8, 26.2, 26.0, 18.4, 18.3, 18.3, 18.2, 17.8, 11.4, 9.1, −1.1, −1.2, −1.3, −3.3, − 4.2, −4.7, −4.7; high resolution mass spectrum (ES+) m/z 1108.6233 [(M+Na)+; calcd for C53H103NO12Si5Na: 1108.6225].
Amide (+)-32
A solution of acid (−)-3 (47 mg, 4 equiv.) in CH2Cl2 (1.5 mL) was cooled to 0 °C and i-Pr2NEt (30 μL, 1.1 equiv.) was added followed by trimethylacetyl chloride (20 μL, 1.05 equiv.) After 30 min at 0 °C, sat. NH4Cl (3 mL) was added followed by enough H2O to dissolve any solids. The layers were separated and the aqueous layer extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were then dried over MgSO4 and concentrated. The unpurified mixed anhydride 31 was azeotroped with benzene (3 × 3 mL), placed on the vacuum pump for 30 min, and then dissolved in THF (0.4 mL). A solution of (+)-4 (41.5 mg, 0.038 mmol) in THF (0.4 mL) was cooled to −78 °C and LiHMDS (0.152 mL, 0.5M in THF, 2.0 equiv.) was added dropwise over 1 min. The solution was stirred for 30 min at −78 °C and then the solution of mixed anhydride 32 was added dropwise over 5 min. The reaction was stirred at −78 °C for 45 min, then warmed to −60 °C and stirred for 30 min. Sat. NH4Cl (2 mL) was added and the reaction allowed to warm to rt. The reaction was extracted with CH2Cl2 (4 × 5 mL). The combined organic layers were then dried over MgSO4 and evaporated. Preparative chromatography (20% EtOAc/hexanes, 1000 micron plate) provided amide (+)-32 (41.2 mg, 79% yield) as a colorless oil: +40.2 (c 0.4, CHCl3); IR (neat) 2953, 2896, 2857, 1726, 1591, 1467, 1250, 1110, 1063 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.92 (s, 1H), 5.66 (d, J = 4.6 Hz, 1H), 5.34-5.25 (m, 4H), 5.17 (d, J = 4.9 Hz, 1H), 4.77 (d, J = 11.5 Hz, 2H), 4.63 (dd, J = 23.1, 6.6 Hz, 2H), 4.33-4.27 (m, 2H), 4.20 (ddd, J = 12.1, 8.4, 2.0 Hz, 1H), 4.12 (ddd, J = 10.0, 2.9, 2.9 Hz, 1H), 3.90-3.78 (m, 2H), 3.78-3.73 (m, 2H), 3.65-3.59 (m, 1H), 3.59- 3.49 (m, 3H), 3.34 (s, 3H), 3.28 (s, 3H), 3.14 (d, J = 9.6 Hz, 1H), 3.10 (dd, J = 16.5, 2.0 Hz, 1H), 2.93 (dd, J = 16.4, 12.6 Hz, 1H), 2.30 (dd, J = 14.7, 9.0 Hz, 1H), 2.22 (d, J = 12.7 Hz, 1H), 2.19 (s, 3H), 2.07-1.96 (m, 2H), 1.81 (dd, J = 14.0, 10.3 Hz, 1H), 1.75 (s, 3H), 1.69 (ddd, J = 13.2, 10.8, 6.5 Hz, 1H), 1.61 (ddd, J = 13.6, 9.5, 3.2 Hz, 2H), 1.10 (d, J = 6.6 Hz, 3H), 1.10-1.04 (m, 2H), 1.00-0.93 (m, 4H), 0.90 (s, 12H), 0.87-0.80 (m, 2H), 0.83 (s, 3H), 0.78 (s, 9H), 0.05 (s, 15H), 0.04 (s, 3H), 0.01 (s, 9H), 0.00 (s, 9H), −0.02 (s, 3H), −0.05 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 174.9, 163.7, 159.8, 159.1, 154.5, 142.9, 142.1, 117.5, 113.0, 109.5, 102.2, 95.3, 94.5, 93.1, 88.5, 81.3, 80.2, 75.7, 74.9, 73.0, 69.3, 66.8, 66.8, 66.3, 66.1, 58.3, 56.8, 40.7, 39.2, 39.1, 35.3, 30.5, 30.0, 28.7, 26.1, 26.0, 24.2, 23.1, 18.3, 18.3, 18.3, 18.2, 17.8, 13.6, 11.4, 9.1, −1.2, −1.2, −1.3, −1.4, −3.1, − 4.0, −4.5, −4.7.; high resolution mass spectrum (ES+) m/z 1394.7849 [(M+Na)+; calcd for C67H129NO16Si6Na: 1394.7825].
Irciniastatin A (+)-1
Compound (+)-32 (9.8 mg, 7.1 μmol) was dissolved in DMF (0.16 mL) and TASF (30 mg, 15 equiv.) was added. The reaction was heated to 50 °C. After 48 h, the reaction was diluted with EtOAc and sat. NH4Cl (1 mL) was added followed by enough H2O (0.5 mL) to dissolve any solids. The reaction was extracted with EtOAc (4 × 2 mL). The combined organic layers were then dried over MgSO4 and evaporated. Preparative TLC (70% EtOAc/hexanes, 500 micron plate provided (+)-1, irciniastatin A, (1.7 mg) along with a mono-protected compound (SEM group on one of the phenolic oxygens). The mono-protected compound was dissolved in Et2O (0.3 mL) and MeNO2 (20 μL) was added followed by MgBr2 (13 mg, 20 equiv.). After stirring for 15 min at rt, the reaction was diluted with EtOAc (2 mL) and sat. NaHCO3 (1 mL) was added. The reaction was extracted with EtOAc (5 × 1 mL) and the combined organic layers were then dried over MgSO4 and concentrated. Preparative TLC (60% EtOAc/hexanes, 500 micron plate) provided an additional 1.5 mg of the natural product to give a total of 3.2 mg (74% yield, 2 steps) of (+)-1 (irciniastatin A): +21.0 (c 0.1, CHCl3); IR (neat) 3366, 2965, 2928, 2853, 1654, 1515, 1459, 1381, 1254, 1173, 1107, 1067 cm−1; 1H NMR (500 MHz, CD3OD) δ 6.25 (s, 1H), 5.38 (d, J = 8.2 Hz, 1H), 4.74 (s, 1H), 4.72 (s, 1H), 4.49 (ddd, J = 12.2, 5.9, 3.1 Hz, 1H), 4.35 (d, J = 2.6 Hz, 1H), 3.97-3.92 (m, 2H), 3.67 (ddd, J = 9.3, 3.4, 2.7 Hz, 1H), 3.59 (dd, J = 10.9, 4.5 Hz, 1H), 3.50 (dd, J = 10.1, 1.3 Hz, 1H), 3.35 (s, 3H), 3.21 (s, 3H), 3.13 (dd, J = 16.7, 3.1 Hz, 1H), 2.86 (dd, J = 16.7, 12.2 Hz, 1H), 2.35 (dd, J = 14.6, 9.3 Hz, 1H), 2.11 (dd, J = 14.1, 3.6 Hz, 2H), 2.10 (s, 3H), 2.02 (ddd, J = 13.6, 4.3, 2.8 Hz, 1H), 1.91 (ddddd, J = 6.9, 6.9, 6.9, 6.9, 2.7 Hz, 1H), 1.77 (ddd, J = 13.7, 11.1, 6.3 Hz, 2H), 1.72 (s, 3H), 1.68 (ddd, J = 14.5, 3.2, 1.9 Hz, 2H), 1.10 (d, J = 7.0, 3H), 0.97 (s, 3H), 0.90 (s, 3H); 13C NMR (125 MHz, CD3OD) δ 176.3, 172.5, 164.7, 163.8, 144.0, 141.3, 115.4, 113.1, 101.6, 101.5, 82.8, 82.3, 82.1, 79.9, 74.2, 73.6, 73.4, 72.2, 57.8, 56.7, 43.4, 39.9, 38.8, 34.5, 30.6, 29.6, 23.8, 23.0, 14.1, 10.9, 9.3; high resolution mass spectrum (ES+) m/z 632.3038 [(M+Na)+; calcd for C31H47NO11Na: 632.3047].
Aldehyde 35
To a 0 °C solution of diol 37 (0.50 g, 4.8 mmol) in THF (24 mL) was added 60% NaH (0.19 g, 1.0 equiv.) in three equal portions over 5 min. After 10 min, TBSCl (0.72 g, 1 equiv.) dissolved in THF (5 mL) was added dropwise. After 4 h, the reaction was quenched with MeOH (2 mL). Sat. NaHCO3 (15 mL) was added and the reaction extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were then dried over MgSO4 and concentrated. Flash chromatography (10% EtOAc/hexanes) provided TBS ether 66 (0.93 g, 89% yield) as a colorless oil: IR (neat) 3389, 2955, 2929, 2858, 1472, 1254, 1096, 1047 cm−1; 1H NMR (500 MHz, CDCl3) δ 3.46 (s, 4H), 2.86 (dd, J = 5.5, 5.5 Hz, 1H), 0.90 (s, 9H), 0.88 (s, 6H), 0.06 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 73.0, 72.5, 36.6, 26.0, 21.7, 18.4, −5.4; high resolution mass spectrum (CI+) m/z 219.1794 [(M+H)+; calcd for C11H27O2Si: 219.1780].
To a 0 °C solution of TBS ether 66 (1.51 g, 6.91 mmol) in DMSO (4.9 mL, 10 equiv.) and CH2Cl2 (35 mL) was added Et3N (2.89 mL, 3 equiv.) followed by SO3•pyridine (3.25 g, 3 equiv.). After 1 h, brine (20 mL) and H2O (5 mL) were added. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were then dried over MgSO4 and concentrated. Flash chromatography (5% EtOAc/hexanes to 10% EtOAc/hexanes) provided aldehyde 35 (1.43 g, 96% yield) as a light yellow oil: IR (neat) 2956, 2930, 2893, 2858, 1733, 1472, 1257, 1104 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.55 (s, 1H), 3.58 (s, 2H), 1.03 (s, 6H), 0.85 (s, 9H), 0.02 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 206.3, 68.6, 48.3, 26.0, 18.7, 18.4, −5.4; high resolution mass spectrum (CI+) m/z 215.1453 [(M-H)+; calcd for C11H23O2Si: 215.1468].
Alcohol (+)-34
To a suspension of N-Ts-L-tryptophan (5.15 g, 1.2 equiv.) in CH2Cl2 (150 mL) was added Cl2BPh (1.87 mL, 1.2 equiv.) dropwise. After 2 h at rt, the mixture was concentrated in vacuo, n-butyronitrile (50 mL) added, and the solution cooled to −78 °C. A mixture of aldehyde 35 (2.6 g, 12.0 mmol), ketene acetal 36 (3.86 g, 1.5 equiv.), and isopropanol (1.38 mL, 1.5 equiv) in n-butyronitrile (15 mL) was added over 2 h via syringe pump. After an additional 2 h at −78 °C, the solution was warmed to 0 °C over 30 min, followed by addition of sat. NaHCO3 (20 mL) and brine (20 mL). The resulting solution was extracted with Et2O (3 × 30 mL). The combined organic layers were then dried over MgSO4 and evaporated. Flash chromatography (5% EtOAc/hexanes to 10% EtOAc/hexanes to 15% EtOAc/hexanes) provided (+)-34 (2.49 g, 66% yield) as a colorless oil and a single enantiomer, as determined by Mosher ester analysis: +27.9 (c 0.6, CHCl3); IR (neat) 3486, 2953, 2929, 2858, 1726, 1657, 1471, 1258, 1092 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.10 (ddd, J = 15.6, 7.2, 7.2, Hz, 1H), 5.91 (ddd, J = 15.7, 1.5, 1.5, Hz, 1H), 3.75 (d, J = 3.2, 1H), 3.71 (s, 3H), 3.63 (ddd, J = 10.0, 2.9, 2.9 Hz, 1H), 3.49 (d, J = 2.0 Hz, 2H), 2.37 (dd, J = 14.5, 6.8 Hz, 1H), 2.26 (dddd, J = 14.6, 10.0, 7.5, 1.4 Hz, 1H), 0.91 (s, 3H), 0.89 (s, 9H), 0.85 (s, 3H), 0.07 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 167.1, 147.9, 122.6, 78.3, 73.6, 51.6, 38.6, 35.5, 26.0, 22.6, 19.0, 18.3, −5.4, − 5.5; high resolution mass spectrum (ES+) m/z 317.2137 [(M+H)+; calcd for C16H33O4Si: 317.2148].
Epoxide (+)-38
To a 0 °C solution of (+)-34 (0.226 g, 0.714 mmol) in CH2Cl2 (7.1 mL) was added 2,6-lutidine (0.164 mL, 2.0 equiv.) followed by dropwise addition of TBSOTf (0.246 mL, 1.5 equiv.). After 1 h, sat. NaHCO3 (5 mL) was added and the layers separated. The aqueous layer was extracted with CH2Cl2 (3 × 10 mL), and the combined organic layers were dried over MgSO4 and concentrated. Flash chromatography (2% EtOAc/hexanes to 5% EtOAc/hexanes) provided TBS ether (+)-67 (0.269 g, 96% yield) as a colorless oil: +3.6 (c 1.0, CHCl3); IR (neat) 2955, 2930, 2886, 2857, 1730, 1657, 1472, 1257, 1090 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.12-6.92 (m, 1H), 5.81 (ddd, J = 15.7, 1.5, 1.5 Hz, 1H), 3.74 (dd, J = 6.4, 4.4 Hz, 1H), 3.72 (s, 3H), 3.28 (dab, J = 9.5 Hz, 1H), 3.26 (dab, J = 9.6 Hz, 1H), 2.48 (dddd, J = 15.0, 6.4, 4.4, 1.7 Hz, 1H), 2.36-2.27 (m, 1H), 0.89 (s, 9H), 0.83 (s, 3H), 0.79 (s, 3H), 0.04 (s, 3H), 0.02 (s, 3H), 0.01 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 167.1, 148.9, 122.0, 75.1, 69.7, 51.6, 41.1, 36.6, 26.2, 26.1, 21.3, 20.9, 18.5, 18.4, −3.5, −4.1, −5.2, −5.3; high resolution mass spectrum (ES+) m/z 431.2997 [(M+H)+; calcd for C22H47O4Si2: 431.3013].
A solution of TBS ether (+)-67 (0.217 g, 0.504 mmol) in CH2Cl2 (10 mL) was cooled to −78 °C and DIBAL-H (1.06 mL, 1.0 M in hexanes, 2.1 equiv.) was slowly added. After 5 min, the reaction was quenched with MeOH (0.5 mL) and allowed to warm to rt. Sat. Rochelle’s salt (10 mL) and EtOAc (10 mL) were added and the solution stirred for 1 h. H2O (10 mL) was then added to dissolve the suspended solids and the layers were separated. The aqueous layer was extracted with EtOAc (3 × 10 mL), and the combined organic layers were dried over MgSO4 and concentrated. Flash chromatography (5 % EtOAc/hexanes to 10% EtOAc/hexanes) provided allylic alcohol (−)-68 (0.237 g, 99%) as a colorless oil: −3.4 (c 1.0, CHCl3); IR (neat) 3327, 2955, 2929, 2892, 2857, 1715, 1472, 1254, 1091, 1006 cm−1; 1H NMR (500 MHz, CDCl3) δ 5.82-5.57 (m, 2H), 4.09 (d, J = 5.7, 2H), 3.64 (dd, J = 6.3, 4.3 Hz, 1H), 3.33 (dab, J = 9.5 Hz, 1H), 3.28 (dab, J = 9.5 Hz, 1H), 2.36 (ddd, J = 14.7, 4.8, 4.8 Hz, 1H), 2.16 (ddd, J = 14.5, 7.1, 7.1 Hz, 1H), 0.89 (s, 21H), 0.80 (s, 3H), 0.04 (s, 3H), 0.02 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 132.3, 130.1, 75.9, 69.9, 64.1, 41.2, 36.5, 26.3, 26.2, 21.4, 20.8, 18.5, 18.4, −3.2, −4.1, − 5.2, −5.3; high resolution mass spectrum (ES+) m/z 403.3065 [(M+H)+; calcd for C21H47O3Si2: 403.3063].
To freshly activated 3 Å molecular sieves (0.2 g) was added (−)-DIPT (15 μL, 0.24 equiv.) in CH2Cl2 (0.5 mL). The solution was cooled to −20 °C and Ti(Oi-Pr)4 (18 μL, 0.2 equiv.) in CH2Cl2 (0.5mL) was added followed by t-BuOOH (0.176 mL, 5 M in decane, 3.0 equiv.) The reaction was stirred for 30 min and then allylic alcohol (−)-68 (0.118 g, 0.293 mmol) dissolved in CH2Cl2 (1 ml) was added via syringe. The flask and syringe were rinsed with CH2Cl2 (0.4 mL) into the reaction flask. After 4 h, 10% aq. citric acid (2 mL) was added and the reaction warmed to rt. After 1 h at rt, brine (3 mL) was added, the layers were separated, and the aqueous layer extracted with EtOAc (3 × 5 mL). The combined organic layers were dried over MgSO4 and concentrated. Flash chromatography (5% EtOAc/hexanes to 10% EtOAc/hexanes to 15% EtOAc/hexanes) provided epoxide (+)-38 (0.108 g, 88% yield, 13:1 inseparable mixture of epoxides) as a colorless oil: +24.3 (c 1.0, CHCl3); IR (neat) 3430, 2955, 2929, 2895, 2857, 1472, 1254, 1091, 1009 cm−1; 1H NMR (500 MHz, CDCl3) δ 3.91 (ddd, J = 12.5, 5.4, 2.4 Hz, 1H), 3.80 (dd, J = 7.4, 3.8 Hz, 1H), 3.60 (ddd, J = 12.5, 7.2, 4.4 Hz, 1H), 3.35 (dab, J = 9.6 Hz, 1H), 3.28 (dab, J = 9.6 Hz, 1H), 3.12 (ddd, J = 6.9, 4.5, 2.3 Hz, 1H), 2.93 (ddd, J = 4.6, 2.4, 2.4 Hz, 1H), 1.80 (dd, J = 7.1, 5.8 Hz, 1H), 1.79-1.72 (m, 1H), 1.65 (ddd, J = 14.6, 7.0, 3.8 Hz, 1H), 0.90 (s, 9H), 0.89 (s, 9H), 0.83 (s, 3H), 0.79 (s, 3H), 0.09 (s, 3H), 0.08 (s, 3H), 0.01 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 73.7, 69.7, 61.9, 60.1, 54.1, 40.7, 35.5, 26.3, 26.1, 21.5, 20.5, 18.6, 18.5, −3.8, −4.0, −5.2, −5.3; high resolution mass spectrum (ES+) m/z 441.2846 [(M+Na)+; calcd for C21H46O4Si2Na: 441.2833].
Ester (+)-39
Alcohol (+)-38 (0.199 g, 0.474 mmol) in CH3CN (4.7 mL) was treated with TEMPO (6 mg, 0.08 equiv.) followed by addition of pH 7 buffer (4.7 mL). The reaction was then treated with NaClO2, followed by dropwise addition of NaOCl (0.18 mL, 5% aq. solution). After 2 h, sodium sulfite (200 mg, 3.3 equiv.) was added and the reaction stirred for 30 min, where the solution turned from a light orange color to colorless. The reaction was acidified to pH 4 with 10% aq. citric acid and extracted with EtOAc (5 × 10 mL). The combined organic layers were dried over MgSO4 and concentrated to yield carboxylic acid which was carried forward without further purification.
The unpurified acid was dissolved in CH2Cl2 (2.4 mL) and MeOH (2.4 mL). The solution was cooled to 0 °C and TMS-diazomethane (0.28 mL, 2 M in Et2O, 1.2 equiv.) was added dropwise until the solution remained yellow in color. Argon gas was bubbled through the reaction for 10 min and then the solvent evaporated. Flash chromatography (5% EtOAc/hexanes) provided ester (+)-39 (0.195 g, 91% yield, 2 steps) as a colorless oil: +15.8 (c 1.0, CHCl3); IR (neat) 2955, 2930, 2887, 2857, 1759, 1472, 1254, 1201, 1089, 1006 cm−1; 1H NMR (500 MHz, CDCl3) δ 3.81 (dd, J = 7.4, 3.8 Hz, 1H), 3.75 (s, 3H), 3.34 (dab, J = 9.6 Hz, 1H), 3.30 (ddd, J = 6.5, 4.5, 1.9 Hz, 1H), 3.28 (dab, J = 9.6 Hz, 1H), 3.22 (d, J = 1.9 Hz, 1H), 1.80 (ddd, J = 14.6, 7.4, 4.5 Hz, 1H), 1.67 (ddd, J = 14.7, 6.8, 3.8 Hz, 1H), 0.90 (s, 9H), 0.88 (s, 9H), 0.83 (s, 3H), 0.78 (s, 3H), 0.10 (s, 3H), 0.08 (s, 3H), 0.01 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 169.9, 73.6, 69.6, 56.9, 54.6, 52.5, 40.7, 35.5, 26.3, 26.1, 21.3, 20.7, 18.6, 18.5, −3.8, −4.0, −5.2, −5.3; high resolution mass spectrum (ES+) m/z 469.2778 [(M+Na)+; calcd for C22H46O5Si2Na: 469.2782].
Aldehyde (+)-40
To bis-silyl ether (+)-39 (72.0 mg, 0.159 mmol) was added a HF•pyridine/pyridine/THF solution (0.64 mL, 10 equiv. HF, stock solution made up of 0.5 mL HF•pyridine, 1.0 mL pyridine, and 5.0 mL THF). After stirring 20 h at rt, sat. NaHCO3 (5 mL) was added slowly. Once all gas evolution had ceased, the reaction was extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were dried over MgSO4 and concentrated. Flash Chromatography (5% EtOAc/hexanes to 10% EtOAc/hexanes to 15% EtOAc/hexanes to 20% EtOAc/hexanes) provided alcohol (+)-69 as a single diastereomer (37.2 mg, 72% yield) as a colorless oil: +46.8 (c 0.5, CHCl3); IR (neat) 3465, 2955, 2930, 2857, 1744, 1446, 1290, 1253, 1205, 1089, 1051, 1004 cm−1; 1H NMR (500 MHz, CDCl3) δ 3.80 (dd, J = 7.4, 3.8 Hz, 1H), 3.77 (s, 3H), 3.59 (d, J = 10.9 Hz, 1H), 3.36-3.27 (m, 2H), 3.26 (d, J = 1.7 Hz, 1H), 2.37 (s, 1H), 1.95 (ddd, J = 14.8, 7.4, 3.9 Hz, 1H), 1.67 (ddd, J = 14.8, 7.3, 3.8 Hz, 1H), 0.99 (s, 3H), 0.91 (s, 9H), 0.81 (s, 3H), 0.14 (s, 3H), 0.14 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 169.6, 76.7, 70.0, 56.6, 54.5, 52.7, 39.6, 35.6, 26.2, 23.0, 21.5, 18.4, −3.9, −4.0; high resolution mass spectrum (ES+) m/z 355.1908 [(M+Na)+; calcd for C16H32O5SiNa: 355.1917].
To a solution of alcohol (+)-69 (0.025 g, 0.075 mmol) in CH2Cl2 (0.75 mL) and DMSO (0.75 mL) at 0 °C was added Et3N (0.105 mL, 10 equiv.) followed by SO3•pyridine (0.047 g, 4 equiv.) in one portion. After 2 h, sat. NaHCO3 (4 mL) was added and the layers separated. The aqueous layer was extracted with CH2Cl2 (3 × 5 mL) and the combined organic layers were dried over MgSO4 and concentrated. Flash chromatography (5% EtOAc/hexanes to 10% EtOAc/hexanes to 20% EtOAc/hexanes) provided aldehyde (+)-40 (23.5 mg, 95% yield) as a light yellow oil: +22.9 (c 0.5, CHCl3); IR (neat) 2955, 2931, 2857, 1754, 1448, 1291, 1254, 1205, 1093 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.56 (s, 1H), 4.06 (dd, J = 7.9, 3.6 Hz, 1H), 3.77 (s, 3H), 3.28 (ddd, J = 7.5, 3.8, 1.9 Hz, 1H), 3.24 (d, J = 1.9 Hz, 1H), 1.90 (ddd, J =14.6, 7.9, 3.8 Hz, 1H), 1.54 (ddd, J = 14.7, 7.5, 3.6 Hz, 1H), 1.06 (s, 3H), 1.02 (s, 3H), 0.88 (s, 9H), 0.13 (s, 3H), 0.08 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 205.8, 169.4, 73.7, 56.0, 54.4, 52.7, 51.3, 35.7, 26.1, 19.3, 18.4, 17.7, −3.9, −4.0; high resolution mass spectrum (ES+) m/z 353.1753 [(M+Na)+; calcd for C16H30O5SiNa: 353.1761].
Tetrahydropyran (+)-41
To a solution of (−)-DIPCl (0.035 g, 1.8 equiv.) in Et2O (0.4 mL) cooled to −78 °C was added Et3N (0.021 mL, 2.5 equiv.) followed by slow dropwise addition of 2-butanone (0.010 mL, 1.8 equiv.). The solution immediately turned cloudy white. After stirring for 2 h at −78 °C, aldehyde (+)-40 (0.020 g, 0.061 mmol) was dissolved in Et2O (0.4 mL) and added dropwise to the boron enolate over 5 min via syringe. The flask and syringe were then rinsed with Et2O (0.4 mL) and the rinse added to the reaction over 2 min. After stirring at −78 °C for 5 h, the solution was warmed to −40 °C over 1 h and held at −40 °C. After 12 h, the reaction was warmed to 0 °C and a 1:1:1 mixture of MeOH/pH 7 buffer/30% aq. H2O2 (1 mL) was added. After 1 h, sat. Na2S2O3 (4 mL) was added very slowly over 30 min to destroy any remaining peroxides. The reaction was then extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over MgSO4 and concentrated. Flash chromatography (10% EtOAc/hexanes to 15% EtOAc/hexanes to 20% EtOAc/hexanes) provided the aldol product (0.020 g, 86% yield) as a 5:1 mixture of diastereomers.
The aldol products were dissolved in CH2Cl2 (1 mL) and CSA (2.3 mg, 0.2 equiv.) was added. After 2 h, sat. NaHCO3 (2 mL) added and the reaction stirred for 5 min. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were dried over MgSO4 and concentrated. Flash chromatography (10% EtOAc/hexanes to 15% EtOAc/hexanes to 20% EtOAc/hexanes) provided (+)-41 (14.8 mg, 60% yield over 2 steps for the major diastereomer) and (−)-42 (2.9 mg, 12% yield over 2 steps for the minor diastereomer).
Major diastereomer (+)- 41
+9.8 (c 0.5, CHCl3); IR (neat) 2954, 2930, 2861, 1741, 1714, 1461, 1388, 1366, 1258, 1074 cm−1; 1H NMR (500 MHz, CDCl3) δ 4.26 (dd, J = 6.3, 4.2 Hz, 1H), 4.15 (ddd, J = 8.0, 3.8, 3.8 Hz, 1H), 4.06 (dd, J = 10.0, 3.5 Hz, 1H), 3.78 (s, 3H), 3.64 (dd, J = 5.0, 3.3 Hz, 1H), 2.93 (d, J = 6.3 Hz, 1H), 2.57 (dd, J = 15.6, 3.6 Hz, 1H), 2.50-2.42 (m, 2H), 2.05 (ddd, J = 13.5, 9.3, 3.2 Hz, 1H), 1.45-1.38 (m, 1H), 1.40-1.36 (m, 1H), 1.05 (dd, J = 7.3, 7.3 Hz, 3H), 1.00 (s, 3H), 0.92 (s, 9H), 0.85 (s, 3H), 0.05 (s, 3H), 0.04 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 210.8, 173.3, 77.7, 73.5, 73.4, 68.8, 52.7, 42.4, 37.5, 36.9, 29.8, 26.0, 25.8, 20.7, 18.2, 7.9, −4.4, −4.8; high resolution mass spectrum (ES+) m/z 425.2330 [(M+Na)+; calcd for C20H38O6SiNa: 425.2336].
Minor diastereomer (−)-42
−38.7 (c 0.8, CHCl3); IR (neat) 3480, 2955, 2930, 2885, 2857, 1741, 1719, 1255, 1096, 1060, 1008 cm−1; 1H NMR (500 MHz, CDCl3) δ 4.24 (dd, J = 6.0, 3.3 Hz, 1H), 4.17 (dd, J = 10.5, 2.3 Hz, 1H), 4.08 (ddd, J = 12.2, 2.7, 2.7 Hz, 1H), 3.75 (s, 3H), 3.55 (dd, J = 2.7, 2.7 Hz, 1H), 2.79 (d, J = 6.1 Hz, 1H), 2.59-2.40 (m, 3H), 2.25 (dd, J = 14.9, 2.4 Hz, 1H), 2.04 (ddd, J = 14.2, 12.2, 2.4 Hz, 1H), 1.22 (ddd, J = 13.4, 3.0, 3.0 Hz, 1H), 1.04 (dd, J = 7.3, 7.3 Hz, 3H), 0.91 (s, 9H), 0.87 (s, 3H), 0.81 (s, 3H), 0.04 (s, 3H), 0.01 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 210.8, 172.9, 76.3, 74.9, 73.9, 73.4, 52.6, 42.8, 37.4, 37.2, 30.0, 26.0, 23.7, 19.7, 18.3, 7.8, −4.4, −4.7; high resolution mass spectrum (ES+) m/z 425.2336 [(M+Na)+; calcd for C20H38O6SiNa: 425.2336].
Tetrahydropyran (+)-6
To a 0 °C solution of (+)-41 (14.8 mg, 0.036 mmol) in CH2Cl2 (0.18 mL) was added proton sponge (95 mg, 12.0 equiv.) followed by Me3•BOF4 (54 mg, 10 equiv). The reaction was then stirred at rt. After 4 h, 10% aq. citric acid (2 mL) added. The layers were separated and the aqueous layer extracted CH2Cl2 (3 × 5 mL). The combined organic layers were washed with 10% aq. citric acid (4 mL) and then brine (4 mL). The organic layer was then dried over MgSO4 and concentrated. Flash chromatography (10% EtOAc/hexanes) provided (+)-6 (14.2 mg, 92% yield) as a colorless oil. All spectral data matched that of the ketone fragment constructed during the first-generation approach: +16.5 (c 1.7, CHCl3); IR (neat) 2954, 2934, 2886, 2858, 1754, 1722, 1712, 1462, 1361, 1255, 1119, 1073 cm−1; 1H NMR (500 MHz, CDCl3) 4.08 (dd, J = 11.3, 6.0 Hz, 1H), 3.99 (dd, J = 9.6, 3.0 Hz, 1H), 3.90 (d, J = 6.4 Hz, 1H), 3.73 (s, 3H), 3.62 (dd, J = 7.5, 3.6 Hz, 1H), 3.40 (s, 3H), 2.91 (dd, J = 15.2, 9.7 Hz, 1H), 2.56- 2.29 (m, 3H), 1.93 (ddd, J = 13.8, 6.1, 3.9 Hz, 1H) 1.60 (ddd, J = 12.7, 9.0, 5.3 Hz, 1H), 1.03 (dd, J = 7.3, 7.3 Hz, 3H), 0.96 (s, 3H), 0.90 (s, 9H), 0.83 (s, 3H), 0.04 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 210.4, 171.7, 82.5, 77.4, 73.1, 69.8, 58.8, 52.2, 42.6, 38.1, 37.2, 30.3, 26.0, 25.1, 18.2, 17.9, 7.8, −4.2, −4.8; high resolution mass spectrum (ES+) m/z 439.2491 [(M+Na)+; calcd for C21H40O6SiNa: 439.2492].
Aldehyde 45
To a solution of bis-phenol 14 (519 mg, 2.043 mmol) in acetone (25.0 mL) was added K2CO3 (1.03 g, 7.452 mmol, 3.7 equiv.) followed by dropwise addition of a solution of 3,4-dimethoxybenzylbromide (2.0 mL, 2.2 M in acetone, 2.2 equiv.). The yellow reaction mixture was stirred for 24 h and quenched with H2O (20 mL). The layers were separated and the aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude mixture was purified via flash chromatography (40% EtOAc: hexanes to 50% EtOAc: hexanes) to provide bis-DMB ether 70 (850 mg, 1.532 mmol, 75%) as a colorless solid: Melting point: 102.0-103.0 °C; IR (neat) 2947, 1732, 1594, 1515, 1459, 1264, 1152, 1025 cm−1; 1H NMR (500 MHz, CDCl3) 6.97-6.81 (m, 6 H), 6.52 (s, 1 H), 4.99 (s, 2 H), 4.95 (s, 2 H), 3.89 (s, 3 H), 3.88 (s, 3 H), 3.88 (s, 3 H), 3.87 (s, 3 H), 3.83 (s, 3 H), 3.69 (s, 2 H), 3.67 (s, 3 H), 2.14 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 171.2, 158.7, 155.2, 149.4, 149.3, 149.1, 148.9, 132.7, 129.6, 129.4, 120.0, 119.7, 119.6, 117.8, 111.3, 111.1, 110.9, 110.1, 98.3, 71.2, 70.7, 56.1, 56.1, 56.1, 56.0, 52.2, 36.2, 11.7; HRMS (ES+) m/z 577.2047 [(M+Na)+; calcd for C30H34O10Na: 577.2050].
To a solution of bis-DMB ether 70 (850 mg, 1.53 mmol) in CH2Cl2 (15.3 mL) was cooled to −78 °C and DIBAL-H (2.1 mL, 1.0 M in hexanes, 1.4 equiv.) was added over 20 min via syringe pump. The reaction mixture was allowed to stir for an additional 5 min before it was quenched by the addition of MeOH (7.0 mL) and was warmed to room temperature, then diluted with EtOAc (2 mL) and a saturated aq. solution of Rochelle’s salt (2 mL). The biphasic reaction mixture was allowed to stir for 1 h at room temperature to allow the organic layer to transition from cloudy to clear. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 3 mL). The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude mixture was purified via flash chromatography (40% EtOAc/hexanes to 50% EtOAc/hexanes) to provide aldehyde 45 (697 mg, 1.33 mmol, 87%) as a colorless solid: Melting point: 122.5-124.0 °C; IR (neat) 2944, 2838, 2726, 1722, 1594, 1515, 1459, 1265, 1153, 1030 cm−1; 1H NMR (500 MHz, CDCl3) 9.65 (t, J = 1.8 Hz, 1 H), 6.97 (d, J = 1.7 Hz, 1 H), 6.94-6.83 (m, 5 H), 6.55 (s, 1 H), 5.02 (s, 2 H), 4.97 (s, 2 H), 3.90 (s, 3 H), 3.89 (s, 9 H), 3.84 (s, 3 H), 3.68 (d, J = 1.6 Hz, 2 H), 2.10 (s, 3 H); 13C NMR (125 MHz, MeOD) δ 198.6, 168.7, 158.9, 155.4, 149.4, 149.4, 149.2, 149.0, 131.0, 129.4, 129.2, 120.1, 119.8, 119.7, 118.0, 111.3, 111.1, 110.0, 110.7, 98.5, 71.2, 70.7, 56.3, 56.1, 56.1, 56.1, 52.3, 46.1, 11.9; HRMS (ES+) m/z 525.2141 [(M+Na)+; calcd for C29H33O9: 525.2125].
β-Hydroxy Ketone (+)-46
To a solution of ketone (+)-6 (114 mg, 0.274 mmol, azeotroped × 3 with benzene, placed under high vacuum overnight) in CH2Cl2 (1.35 mL) was cooled to −78 °C and Cl2BPh (0.05 mL, 0.383 mmol, 1.4 equiv.) was added dropwise. After 20 min, i-Pr2NEt (0.10 mL, 0.574 mmol, 2.1 equiv) was introduced dropwise. After 1 h, the reaction mixture was warmed to 0 °C, where it was stirred for 1 h then cooled back down to −78 °C. Aldehyde 45 (205 mg, 0.391 mmol, 1.5 equiv.) was dissolved in CH2Cl2 (0.7 mL) and was added to the boron enolate solution at −78 °C over 15 min via syringe pump. After 4 h at −78 °C, the reaction mixture was quenched with a 1:1 mixture of MeOH and pH 7 buffer (4 mL). While warming to 0 °C, pH 8 buffer solution was added to neutralize the reaction mixture to pH 7 and the biphasic mixture was stirred for 1 h at 0 °C. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The resulting crude mixture was purified via flash chromatography on deactivated SiO2 (2% v/v triethylamine, 7.5% EtOAc/CH2Cl2 to 10% EtOAc/CH2Cl2) to provide a mixture (ca. 10:1) of β-hydroxy ketone (+)-46 and corresponding lactone (176 mg, 0.192 mmol, 70%) as a colorless foam: +29.8 (c 0.68 CHCl3); IR (neat) 3417, 2924, 2855, 1719, 1590, 1516, 1461, 1264, 1154 cm−1; 1H NMR (500 MHz, CDCl3) Major: δ 6.95-6.91 (m, 3 H), 6.88-6.82 (m, 3 H), 6.49 (s, 1 H), 5.02-4.97 (m, 2 H), 4.95 (s, 2 H), 4.10 (dd, J = 5.3, 10.9 Hz, 1 H), 4.06 (dd, J = 2.9, 9.1 Hz, 1 H), 4.06-4.03 (m, 1 H), 3.94 (d, J = 6.3 Hz, 1 H), 3.89 (s, 3 H), 3.89 (s, 3 H), 3.88 (s, 3 H), 3.88 (s, 3 H), 3.85 (s, 3 H), 3.72 (s, 3 H), 3.64 (dd, J = 3.7, 7.7 Hz, 1 H), 3.46 (d, J = 5.6 Hz, 1 H), 3.41 (s, 3 H), 3.06 (dd, J = 9.4, 16.0 Hz, 1 H), 2.87 (dd, J = 3.4, 14.3 Hz, 1 H), 2.68 (dq, J = 7.1, 5.4 Hz, 1 H), 2.62 (dd, J = 10.1, 14.1 Hz, 1 H), 2.54 (dd, J = 3.0, 16.4 Hz, 1 H), 2.20 (s, 3 H), 1.96 (ddd, J = 3.9, 6.0, 13.9 Hz, 1 H), 1.58 (ddd, J = 5.0, 7.9, 13.3 Hz, 1 H), 1.21 (d, J = 6.9 Hz, 3 H), 0.96 (s, 3 H), 0.90 (s, 9 H), 0.85 (s, 3 H), 0.04 (s, 3 H), 0.04 (s, 3 H); Distinct peaks from minor byproduct: δ 6.54 (s), 5.16 (dab, J = 12.2 Hz), 5.10 (dab, J = 11.8 Hz), 1.33 (d, J = 7.0 Hz); 13C NMR (125 MHz, CDCl3) δ 212.5, 171.5, 170.7, 159.0, 155.2, 149.4, 149.3, 149.1, 148.9, 137.1, 129.5, 129.5, 120.0, 119.8, 119.3, 117.9, 111.3, 111.1, 110.8, 110.7, 97.6, 82.3, 77.4, 76.7, 73.0, 71.6, 71.2, 70.6, 70.1, 58.8, 56.1, 56.1, 56.1, 56.1, 53.2, 52.6, 52.1, 42.4, 38.0, 35.8, 30.0, 26.0, 24.9, 18.2, 11.8, 11.4, −4.3, −4.9; HRMS (ES+) m/z 963.4525 [(M+Na)+; calcd for C50H72O15SiNa: 963.4538].
Acid (+)-47
To a solution of β-hydroxy ketone (+)-46 (203 mg, 0.215 mmol) in THF (2.20 mL) and MeOH (0.73 mL) cooled to −78 °C was added a solution of Et2BOMe (0.31 mL, 1M in THF, 1.4 equiv.). The reaction mixture was stirred for 25 min before NaBH4 (45 mg, 1.189 mmol, 5.4 equiv) was added. After 5.5 h, the reaction was warmed to 0 °C and quenched with a 1:1 mixture of MeOH and pH 7 buffer (4.0 mL) followed by the addition of m-CPBA (0.330 g, 1.31 mmol, 6.0 equiv.) portion-wise. After 30 min, the reaction mixture was warmed to room temperature, and dimethylsulfide was added slowly to quench remaining peroxides. After 10 min, aq. solution of K2CO3 (5 mL, 0.1 N) was added and the layers were separated. The aqueous layer was extracted with EtOAc (3 × 5 mL). The combined organic layers were washed with brine, then dried over MgSO4, filtered and concentrated in vacuo. The crude mixture was purified via flash chromatography (50% EtOAc/hexanes to 60% EtOAc/hexanes then flushed with 10% MeOH/EtOAc) to provide a mixture (ca. 8:1) of diol and lactone (183 mg).
The mixture of diol and lactone was dissolved in MeOH (10.0 mL) and cooled to 0 °C followed by the addition of H2O (70 μL, 3.889 mmol, 20.0 equiv) and LiOH (191 mg, 7.975 mmol, 40.0 equiv). The reaction mixture was allowed to warm to room temperature and after 35 h, the reaction mixture was quenched with 50% aqueous acetic acid solution (5 mL). Brine (6 mL) and H2O (2 mL) were added and the layers were separated. The aqueous layer was extracted with EtOAc (5 × 15 mL). The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. Crude mixture was purified via column chromatography on SiO2 (0.1% acetic acid in 60% EtOAc/hexanes to 0.1% acetic acid in 80% EtOAc/hexanes) to provide acid (+)-47 (136 mg, 0.152 mmol, 69% over two steps) as a colorless foam: +22.4 (c 0.9 CHCl3); IR (neat) 3522, 3058, 2937, 2862, 2862, 1712, 1590, 1515, 1461, 1381, 1261, 1153, 1090 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.33 (d, J = 1.7 Hz, 1 H), 6.92 (m, 3 H), 6.86 (d, J = 8.0 Hz, 1 H), 6.82 (d, J = 8.3 Hz, 1 H), 6.51 (s, 1 H), 5.15 (dab, J = 11.8 Hz, 1 H), 5.09 (dab J = 12.2 Hz, 1 H), 4.99 (s, 2 H), 4.33 (ddd, J = 2.3, 5.5, 8.0 Hz, 1 H) 4.19 (dd, J = 6.0, 11.7 Hz, 1 H), 4.13 (m, 1 H), 3.91 (s, 3 H), 3.89 (s, 3 H), 3.88 (s, 3 H), 3.86 (s, 3 H), 3.85 (d, J = 4.1 Hz, 1 H), 3.60 (d, J = 12.3 Hz, 1 H), 3.57 (dd, J = 4.0, 7.6 Hz, 1 H) 3.41 (s, 3 H), 3.02 (dd, J = 2.7, 17.2 Hz, 1 H), 2.88 (dd, J = 12.3, 16.4 Hz, 1 H), 2.11 (s, 3 H), 2.08 (m, 1 H), 2.01- 1.93 (m, 2 H), 1.66 (ddd, J = 5.1, 8.0, 13.6 Hz, 1 H), 1.56 (app d, J = 14 Hz, 1 H), 1.07 (d, J = 6.9 Hz, 3 H), 0.94 (s, 3 H), 0.89 (s, 9 H), 0.87 (s, 3 H), 0.05 (s, 3 H), 0.04 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 172.3, 164.4, 161.4, 160.4, 149.5, 149.4, 149.2, 148.7, 142.2, 129.6, 128.9, 120.2, 119.0, 116.3, 111.3, 111.0, 110.9, 110.7, 107.7, 97.9, 82.0, 81.8, 79.0, 72.6, 71.6, 71.1, 70.5, 69.8, 58.7, 56.2, 56.1, 41.5, 38.7, 33.1, 30.6, 29.4, 26.0, 25.0, 18.2, 11.3, 9.5, −4..2, −4.8; HRMS (ES+) m/z 919.4287 [(M+Na)+; calcd for C48H68O14SiNa: 919.4276].
SEM-ether (+)-48
A solution of acid (+)-47 (116 mg, 0.129 mmol) in freshly distilled acetone (6.6 mL) was cooled to 0 °C followed by the dropwise addition of i-Pr2NEt (0.05 mL, 0.287 mmol, 2.2 equiv.) and a solution of isobutylchloroformate (0.50 mL, 0.64 M in acetone, 2.4 equiv.). After 1 h, a solution of NaN3 (0.85 mL, 0.78 M in H2O, 5 equiv.) was added to the reaction mixture dropwise. After an additional 20 min at 0 °C, the reaction mixture was diluted with cold H2O (15 mL). The layers were separated and the aqueous layer was extracted with cold EtOAc (3 × 10 mL). The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude acyl azide was azeotroped (benzene x3) and placed under high vacuum (~0.1 mmHg) for 30 min. The acyl azide was dissolved in toluene (6.6 mL) and reaction flask was fitted with a reflux condenser and heated to 80 °C. After 45 min, 2-TMS-ethanol (0.67 mL, 4.674 mmol, 36.2 equiv.) was added via syringe through the top of the condenser. After 5 h at 80 °C, the reaction mixture was cooled to room temperature and concentrated in vacuo. The crude mixture was purified via flash chromatography (40% EtOAc/hexanes to 50% EtOAc/hexanes) to provide N,O-aminal (+)-71 (90 mg, 0.087 mmol, 67%) as a colorless foam: +3.7 (c 0.5, CHCl3); IR (neat) 3502, 3340, 2947, 2859, 1713, 1589, 1515, 1462, 1253, 1151, 1078, 1034, 840 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.34 (d, J = 1.8 Hz, 1 H), 6.93 (m, 3 H), 6.87 (d, J = 8.0 Hz, 1 H), 6.82 (d, J = 8.3 Hz, 1 H), 6.52 (s, 1 H), 5.38, (d, J = 8.8 Hz, 1 H), 5.16 (dab, J = 12.0, 1 H), 5.09 (dab, J = 11.9 Hz, 1 H), 4.99 (s, 2H), 4.89 (d, J = 9.6 Hz, 1 H), 4.31 (ddd, J = 2.2, 6.6, 10.5 Hz, 1 H), 4.19 (ddd, J = 8.3, 10.3, 18.2 Hz, 1 H), 4.12 (m 2 H), 3.98 (d, J = 9.1 Hz, 1 H), 3.92 (s, 3 H), 3.89 (s, 3 H), 3.88 (s, 3 H), 3.87 (m, 1 H), 3.86 (s, 3 H), 3.66 (s, 1 H), 3.62 (d, J =11.5 Hz, 1 H), 3.56 (m, 1 H), 3.35 (s, 3 H), 3.10 (app d, J = 16.5 Hz, 1 H), 2.83 (dd, J = 12.1, 16.0 Hz, 1 H), 2.37 (m, 1 H), 2.12 (s, 3 H), 1.85 (m, 2 H), 1.45-1.41 (m, 2 H), 1.11 (d, J = 6.9 Hz, 3 H), 0.10 (s, 3 H), 0.97 (t, J = 8.6 Hz, 2 H), 0.89 (s, 9 H), 0.87 (s, 3 H), 0.04 (s, 3 H), 0.03 (s, 3 H), 0.01 (s, 9 H); 13C NMR (125 MHz, CDCl3) δ 163.7, 161.2, 160.3, 157.1, 149.6, 149.4, 149.2, 148.7, 142.2, 129.7, 129.0, 120.1, 119.0, 116.2, 111.3, 111.0, 110.9, 110.8, 108.2, 97.9, 83.7, 83.6, 79.3, 77.4, 73.0, 72.7, 71.1, 70.5, 63.7, 56.2, 56.1, 56.1, 55.8, 43.4, 38.0, 32.9, 31.1, 29.7, 26.1, 26.0, 18.1, 17.8, 11.4, 10.1, −1.3, −4.4, −4.8. HRMS (ES+) m/z 1034.5088 [(M+Na)+; calcd for C53H81NO14Si2Na: 1034.5093].
A solution of N,O-aminal (+)-71 (86 mg, 0.085 mmol) in THF (0.6 mL) was cooled to 0 °C followed by the addition of i-Pr2NEt (0.13 mL, 0.746 mmol, 9 equiv.), SEMCl (0.09 mL, 0.509 mmol, 6 equiv.), and TBAI (8 mg, 0.022 mmol, 0.2 equiv.). The reaction mixture was stirred at 0 °C for 15 min and was allowed to warm to room temperature and stirred for 23 h. The reaction mixture was quenched with a saturated aq. solution of NaHCO3 (1 mL). The aqueous layer was extracted with EtOAc (3 × 3 mL). The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude mixture was purified via flash chromatography on SiO2 (35% EtOAc/hexanes to 40% EtOAc/hexanes) to provide SEM ether (+)-48 (0.080 g, 0.070 mmol, 82%) as a colorless foam: +31.6 (c 0.8, CHCl3); IR (neat) 3336, 2952, 2929, 2858, 1716, 1593, 1518, 1464, 1264, 1249, 1160, 1078, 1027, 836 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.33 (d, J = 1.8 Hz, 1 H), 6.98-6.93 (m, 3 H), 6.88 (d, J = 8.7 Hz, 1 H), 6.83 (d, J = 8.3 Hz, 1 H), 6.54 (s, 1 H), 5.60 (bd, J = 9.2 Hz, 1 H), 5.19 (dab, J = 11.7 Hz, 1 H), 5.10 (dab, J = 11.6 Hz, 1 H), 4.98 (s, 2 H), 4.81 (bd, J = 7.9 Hz, 1 H), 4.69-4.59 (m, 2 H), 4.26 (ddd, J = 2.3, 8.4, 11.4 Hz, 1 H), 4.17 (dd, J = 7.5, 10.5 Hz, 1 H), 4.10 (m, 1 H), 3.98-3.94 (m, 2 H), 3.94 (s, 3 H), 3.93 (s, 3 H), 3.89 (s, 3 H), 3.88 (s, 3 H), 3.58 (dd, J = 4.2 Hz, 1 H), 3.54 -3.48 (m, 1 H), 3.44-3.39 (m, 1 H), 3.36 (s, 3 H), 3.34-3.27 (m, 2 H), 2.65 (dd, J = 7.8, 16.0 Hz, 1 H), 2.47 (m, 1 H), 2.15 (s, 3 H), 2.04 (m, 1 H), 1.82 (ddd, J = 2.4, 9.5, 12.9 Hz, 1 H), 1.66 (m, 1 H), 1.51 (ddd, J = 3.9, 8.5, 14.2 Hz, 1 H), 1.13 (d, J = 7.1 Hz, 3 H), 0.98 (s, 3 H), 0.94 (m, 2 H), 0.90 (s, 9 H), 0.87 (s, 3 H), 0.85-0.77 (m, 1 H), 0.71-0.65 (m, 1 H), 0.05 (s, 3 H), 0.04 (s, 3 H), 0.004 (s, 9 H), −0.13 (s, 9 H) ; 13C NMR (125 MHz, CDCl3) δ 163.6, 161.3, 160.4, 157.2, 149.6, 149.5, 149.3, 148.8, 142.2, 129.7, 129.0, 120.2, 119.2, 116.3, 111.4, 111.2, 111.0, 110.9, 108.3, 98.0, 93.6, 84.4, 79.4, 77.4, 75.0, 73.5, 71.2, 70.5, 67.6, 65.7, 63.6, 56.3, 56.2, 56.2, 56.1, 56.0, 39.3, 37.7, 31.7, 29.9, 29.3, 26.3, 26.0, 18.2, 18.1, 17.9, 11.4, 9.9, −1.3, −1.4, −4.3, −4.8. high resolution mass spectrum (ES+) m/z 1164.5879 [(M+Na)+; calcd for C59H95NO15Si3Na: 1164.5907].
Amide (+)-50
To a solution of carboxylic acid (−)-3 (31 mg, 0.102 mmol) in CH2Cl2 was added a solution of pyridine (0.66 mL, 0.62 M in CH2Cl2, 4 equiv.) and a solution of thionyl chloride (0.64 mL, 0.48 M in CH2Cl2, 3 equiv.) and stirred at rt for 2 h. The resulting solution was concentrated under a stream of positive N2 then placed under vacuum (~0.1 mmHg). The crude mixture was dissolved in toluene (0.4 mL) and transferred to an oven-dried vial via cannula transfer (flask rinsed with 2 × 0.4 mL toluene). Crude acid chloride 49 was concentrated in vacuo, dissolved in THF (1.0 mL, 0.1 M), and used in the next step without further purification. To a solution of carbamate (+)-48 (0.030 mg, 0.026 mmol) in THF (0.65 mL) cooled to −78 °C and added a solution of i-PrMgCl (55 uL, 2.0 M in THF, 4 equiv.) over 2 min. The yellow solution was stirred for 30 min at −78 °C then a solution of acid chloride 49 (1.0 mL, 0.1 M in THF, 3.9 equiv.) was added dropwise over 20 min. After 2.5 h, the reaction mixture was quenched with a saturated aq. solution of NaHCO3 (1.5 mL) and warmed to rt. The aqueous layer was extracted with EtOAc (3 × 2 mL) and the combined organic layers were washed with brine, then dried over MgSO4, filtered and concentrated in vacuo. The crude mixture was purified via flash chromatography on deactivated silica gel (1% v/v triethylamine, 25% to 30% EtOAc/hexanes) to furnish amide (+)-50 (27 mg, 0.019 mmol, 72%) as a white foam: +27.0 (c 0.3, CHCl3); IR (neat) 2928, 2856, 1716, 1593, 1518, 1464, 1250, 1160, 1083, 1029, 859, 837 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.34 (d, J = 1.9 Hz, 1 H), 6.97-6.93 (m, 3 H), 6.88 (d, J = 8.0 Hz, 1 H), 6.83 (d, J = 8.2 Hz, 1 H), 6.54 (s, 1 H), 5.62 (d, J = 5.7 Hz, 1 H), 5.17 (d, J = 4.4 Hz, 1 H), 5.16 (d, J = 12.7 Hz, 1 H), 5.10 (d, J = 11.9 Hz, 1 H), 4.98 (s, 2 H), 4.75 (s, 1 H), 4.73 (s, 1 H), 4.67 (dd, J = 6.4, 12.7 Hz, 2 H), 4.59 (dd, J = 7.1, 25.3 Hz, 2 H), 4.35 (m, 1 H), 4.32 (dd, J = 3.6, 6.8 Hz, 1 H), 4.30- 4.25 (m, 2 H), 3.93 (s, 3 H), 3.91 (s, 3 H), 3.90 (s, 3 H), 3.88 (s, 3 H), 3.83 (m, 1 H), 3.64 (m, 1 H), 3.58 (ddd, J = 1.6, 9.8, 13.1 Hz, 2 H), 3.53 (m, 1 H), 3.47-3.38 (m, 2 H), 3.36 (s, 3 H), 3.31 (s, 3 H), 3.24 (dd, J = 1.8, 17.7 Hz, 1 H), 3.20 (m, 1 H), 2.86 (dd, J = 12.4, 15.9 Hz, 1 H), 2.26 (m, 1 H), 2.25 (m, 1 H), 2.21 (s, 3 H), 2.05 (m, 1 H), 1.96 (ddd, J = 4.0, 4.0, 13.7 Hz, 1 H), 1.86 (m, 2 H), 1.73 (s, 3 H), 1.70 (m, 1 H), 1.14 (d, J = 7.6 Hz, 3 H), 1.09 (dd J = 7.1, 9.2 Hz, 2 H), 0.92 (s, 3 H), 0.90 (s, 9 H), 0.86 (s, 3 H), 0.84 (m, 2 H), 0.78 (ddd, J = 5.7, 11.6, 13.6 Hz, 1 H), 0.66 (ddd, J = 5.5, 11.6, 13.6 Hz, 1 H), 0.06 (s, 3 H), 0.05 (s, 3 H), 0.05 (s, 9 H), −0.05 (s, 9 H), −0.15 (s, 9 H) ; 13C NMR (125 MHz, CDCl3) δ 175.2, 163.9, 161.2, 160.4, 154.5, 149.5, 149.4, 149.2, 142.9, 148.7 142.6, 129.7, 129.0, 120.2, 119.1, 116.4, 112.8, 111.2, 111.0, 110.8, 110.7, 108.2, 97.7, 95.1, 94.1, 88.5, 81.1, 79.6, 77.4, 77.0, 75.9, 73.0, 71.2, 70.4, 66.3, 66.0, 65.7, 58.4, 57.0, 56.2, 56.1, 56.1, 56.1, 39.8, 39.1, 38.8, 31.8, 31.1, 29.9, 29.9, 26.1, 24.7, 23.1, 18.2, 18.2, 18.1, 17.7, 11.5, 9.8, −1.3, −1.4, −1.4, −4.1, −4.8. HRMS (ES+) m/z 1450.7513 [(M+Na)+; calcd for C73H121NO19Si4Na: 1450.7508].
Ketone (−)-51
To a solution of amide (+)-50 (7.0 mg, 0.005 mmol) in THF (0.1 mL) was added a solution of TBAF (15 uL, 1 M in THF, 3.0 equiv.). The yellow solution was stirred at rt for 1 h then warmed to 50 °C. After 19 h, additional TBAF (10 uL, 0.010 mmol, 2.0 equiv.) was added. The reaction mixture was stirred at 50 °C for an additional 23 h, then cooled to rt, quenched with water and extracted with EtOAc (4 × 0.5 mL). The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude mixture was purified via flash chromatography on SiO2 (40% EtOAc/hexanes to 60% EtOAc/hexanes to 70% EtOAc/hexanes) to furnish alcohol (−)-72 (4.5 mg, 0.004 mmol, 79%) as a colorless oil: −3.2 (c 0.3, CHCl3); IR (neat) 3426, 2951, 2835, 1715, 1685, 1593, 1517, 1265, 1248, 1160, 1028 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.32 (d, J = 1.6 Hz, 1 H), 7.25 (d, J = 9.4 Hz, 1 H), 6.97-6.92 (m, 3 H), 6.87 (d, J = 8.8 Hz, 1 H), 6.83 (d, J = 8.0 Hz, 1 H), 6.53 (s, 1 H), 5.16 (dab, J = 11.7 Hz, 1 H), 5.09 (dab, J = 11.8 Hz, 1 H), 5.09 (dd, J = 2.5, 5.6 Hz, 1 H), 4.98 (s, 2 H), 4.83 (d, J = 6.6 Hz, 1 H), 4.77 (s, 1 H), 4.76 (s, 1 H), 4.71 (dab, J = 6.3 Hz, 1 H), 4.70 (dab, J = 6.8 Hz, 1 H), 4.60 (d, J = 7.7 Hz, 1 H), 4.40 (d, J = 2.1 Hz, 1 H), 4.27 (ddd, J = 1.9, 7.8, 11.9 Hz, 1 H), 4.08 (m, 1 H), 3.99 (m, 1 H), 3.93 (s, 3 H), 3.90 (s, 3 H), 3.89 (s, 3 H), 3.87 (s, 3 H), 3.74 (m, 2 H), 3.67 (dd, J = 4.5, 4.5 Hz, 1 H), 3.58 (ddd, J = 6.5, 10.3, 10.3 Hz, 1 H), 3.50-3.43 (m, 2 H), 3.37 (s, 3 H), 3.32 (s, 3 H), 3.27 (dd, J = 2.2, 16.7 Hz, 1 H), 2.65 (dd, J = 12.7, 16.4 Hz, 1 H), 2.46 (m, 1 H), 2.37 (dd, J = 8.5, 14.5 Hz, 1 H), 2.19 (dd, J = 4.7, 14.8 Hz, 1 H), 2.14 (s, 3 H), 2.10 (ddd, J = 2.6, 8.4, 8.4 Hz, 1 H), 2.01 (m, 1 H), 1.81 (ddd, J = 2.54, 9.4, 13.5 Hz, 1 H), 1.73 (m, 1 H), 1.70 (s, 3 H), 1.56 (dd, J =4.7, 9.6 Hz, 1 H), 1.54 (dd, J = 5.0, 9.3 Hz, 1 H), 1.15 (d, J = 6.7 Hz, 3 H), 1.00 (s, 3 H), 0.94 (s, 3 H), 0.91 (m, 2 H), 0.85-0.79 (m, 1 H), 0.71-0.69 (m, 1 H), 0.01 (s, 9 H), −0.13 (s, 9 H); 13C NMR (125 MHz, CDCl3) δ 171.2, 163.9, 161.3, 160.4, 149.5, 149.4, 149.2, 148.7, 142.4, 142.2, 129.6, 128.9, 120.2, 119.2, 116.7, 113.0, 111.2, 111.0, 110.9, 110.7, 108.1, 97.7, 94.8, 94.4, 81.8, 81.4, 79.6, 77.4, 75.5, 72.9, 71.1, 70.5, 68.0, 66.2, 65.6, 58.0, 56.3, 56.2, 56.2, 56.1, 56.1, 39.2, 38.3, 37.3, 30.7, 30.0, 29.9, 29.5, 26.0, 22.9, 19.4, 18.2, 18.1, 11.4, 9.6, − 1.2, −1.4; HRMS (ES+) m/z 1192.6072 [(M+Na)+; calcd for C61H95NO17Si2Na: 1192.6036].
To a solution of alcohol (−)-72 (3.5 mg, 0.003 mmol) in CH2Cl2 (0.05 mL) was added NaHCO3 (4.2 mg, 16.6 equiv.). The reaction mixture was cooled to 0 °C and Dess-Martin periodinane (6.0 mg, 0.015 mmol, 5.0 equiv.) was added and the resulting mixture was stirred for 3 h. Reaction was quenched with a saturated aq. solution of NaHCO3 and extracted with EtOAc (4 × 0.5 mL). The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude mixture was purified via flash chromatography on SiO2 (40% EtOAc/hexanes to 50% EtOAc/hexanes) to furnish ketone (−)-51 (3.0 mg, 0.0026 mmol, 87%) as a colorless oil: −14.5 (c 0.2, CHCl3); IR (neat) 3403, 2951, 2928, 2835, 1713, 1687, 1593, 1517, 1463, 1265, 1248, 1159, 1080.9, 1029 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.31 (ap s, 1 H), 7.27 (ap s, 1 H), 6.97 (d, J = 8.4 Hz, 1 H), 6.93 (m, 2 H), 6.88 (d, J = 8.4 Hz, 1 H), 6.83 (d, J = 8.2 Hz, 1 H), 6.55 (s, 1 H), 5.18 (dab, J = 12.0 Hz, 1 H), 5.11 (dab, J = 11.7 Hz, 1 H), 5.09 (d, J = 9.8 Hz, 1 H), 4.98 (s, 2 H), 4.83 (dab, J = 6.4 Hz, 1 H), 4.81 (s, 1 H), 4.79 (s, 1 H), 4.73 (dab, J = 6.6 Hz, 1 H), 4.65 (dab, J = 7.0 Hz, 1 H), 4.61 (dab, J = 7.0 Hz, 1 H), 4.38 (d, J = 2.0 Hz, 1 H), 4.29-4.23 (m, 2 H), 3.99 (m, 1 H), 3.93 (s, 3 H), 3.90 (s, 3 H), 3.89 (s, 3 H), 3.88 (s, 3 H), 3.75 (m, 1 H), 3.72 (m, 1 H), 3.56 (ddd, J = 6.4, 10.0, 10.0 Hz, 1 H), 3.51- 3.42 (m, 2 H), 3.40 (s, 3 H), 3.35 (aps, 1 H), 3.34 (s, 3 H), 3.21 (d, J = 15.9 Hz, 1 H), 2.68 (dd, J = 12.7, 16.4 Hz, 1 H), 2.60 (dd, J = 11.4, 14.9 Hz, 1 H), 2.38 (dd, J = 8.9, 14.9 Hz, 1 H), 2.27 (dd, J = 3.8, 10.9, 1 H), 2.24 (dd, J = 5.2, 12.3, 1 H), 2.14 (s, 3 H), 2.17 - 2.09 (m, 1 H), 1.86-1.75 (m, 2 H), 1.73 (s, 3 H), 1.25 (s, 3 H), 1.16 (d, J = 6.6 Hz, 3 H), 1.01 (s, 3 H), 0.96-0.86 (m, 2 H), 0.82-0.76 (m, 1 H), 0.74-0.68 (m, 1 H), 0.01 (s, 9 H), − 0.10 (s, 9 H); 13C NMR (125 MHz, CDCl3) δ 210.8, 171.4, 163.7, 161.4, 160.4, 149.5, 149.4, 149.3, 148.7, 142.2, 141.8, 129.5, 128.8, 120.3, 119.2, 116.1, 113.3, 111.2, 111.0, 110.9, 110.7, 108.0, 97.8, 95.0, 94.8, 81.7, 81.4, 79.8, 79.2, 77.6, 74.6, 72.8, 71.1, 70.5, 66.3, 65.8, 58.1, 56.4, 56.2, 56.2, 56.1, 56.1, 49.6, 39.6, 38.8, 38.3, 30.2, 29.9, 24.8, 23.0, 19.5, 18.2, 18.1, 11.4, 9.7, −1.2, −1.4; high resolution mass spectrum (ES+) m/z 1190.5880 [(M+Na)+; calcd for C61H93NO17Si2Na: 1190.5880].
(−)-Irciniastatin B (2)
To a solution of fully protected irciniastatin B (−)-51 (5.0 mg, 0.0043 mmol) in CH2Cl2 (0.05 mL) and H2O (15 uL) was added a suspension of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (0.1 mL, 0.33 M in CH2Cl2, 8 equiv.). After 24 h, the reaction mixture was quenched with a saturated aq. solution of NaHCO3 and extracted with EtOAc (5 × 0.5 mL). The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude mixture was purified via flash chromatography (40% EtOAc/hexanes) to afford a mixture (1:2) of desired bis-phenol and 3,4- dimethoxybenzalehyde respectively.
The mixture was treated with a stock solution of MgBr2/n-BuSH/MeNO2 in Et2O (0.21 mL: 25 equiv MgBr2, 25 equiv n-BuSH, stock solution made up of 75.4 mg MgBr2, 44 μL n-BuSH, 82 μL, MeNO2 and 0.82 mL Et2O). After 10 h, the reaction mixture was diluted with EtOAc, and quenched with a saturated aq. solution of NaHCO3 and extracted with EtOAc (5 × 0.5 mL). The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude mixture was purified via flash chromatography with water washed SiO2 [50 g of SiO2 washed with H2O (500 mL) then MeOH (500 mL) then EtOAc (500 mL) then hexanes (500 mL) and dried under vacuum overnight, then deactivated with 5% v/v triethylamine, 35% EtOAc/hexanes to 80% EtOAc/hexanes] to afford (−)-irciniastatin B (2) (2.0 mg, 0.0033 mmol, 78% over two steps) as a colorless solid: −28.7 (c 0.2, MeOH) IR (neat) 3356, 2925, 2873, 1710, 1651, 1612, 1510, 1461, 1380, 1266, 1174, 1103 cm−1; 1H NMR (500 MHz, CDCl3) δ 11.11 (s, 1 H), 7.37 (d, J = 10.4 Hz, 1 H), 6.64 (bs, 1 H), 6.30 (s, 1 H), 5.20 (dd, J = 6.4 Hz, 10.1 Hz, 1 H), 4.82 (s, 1 H), 4.79 (s, 1 H), 4.55 (ddd, J = 4.2, 4.2, 11.8 Hz, 1 H), 4.47 (ap t, J = 2.9 Hz, 1 H), 4.21 (ap q, J = 6.4 Hz, 1 H), 4.09 (d, J = 8.4 Hz, 1 H), 4.00 (dd, J = 1.8, 11.2 Hz, 1 H), 3.79-3.77 (m, 1 H), 3.77 (bs, 1H), 3.65 (bs, 1 H), 3.39 (s, 3 H), 3.36 (s, 3 H), 2.94- 2.83 (m, 2 H), 2.67 (ap d, J = 6.4 Hz, 2 H), 2.36 (dd, J = 9.4, 14.5 Hz, 1 H), 2.14 (dd, J = 3.7, 14.5 Hz, 1 H), 2.04 (s, 3 H), 1.91 (1 H, m), 1.84 (ddd, J = 10.1, 14.6, 24.8 Hz, 1 H), 1.75 (s, 3 H), 1.59 (ap d, J = 14.0 Hz, 1 H), 1.16 (s, 3 H), 1.11 (d, J = 7.2 Hz, 3 H), 1.10 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 210.3, 173.2, 170.7, 162.5, 161.3, 142.1, 139.9, 113.5, 113.3, 101.7, 101.5, 83.2, 80.5, 80.5, 80.3, 73.8, 72.7, 72.4, 57.9, 56.6, 49.6, 42.8, 38.8, 37.4, 33.0, 28.3, 22.8, 22.3, 19.4, 10.7, 9.2; HRMS (ES+) m/z 608.3058 [(M+1)+; calcd for C31H46NO11: 608.3071].
(+)-Irciniastatin A (1) and epi-C(11)-Irciniastatin A (52)
To neat (−)-irciniastatin B (2) (1 mg, 1.6 μmol) was treated with a solution of NaBH4 (0.1 mL, 0.024 M in MeOH, 1.5 equiv.) at 0 °C. After 15 min, the reaction mixture was quenched with a saturated aq. solution of NaHCO3 (0.4 mL) and extracted with EtOAc (3 × 0.5 mL). The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude mixture (1:1) of (+)-1 and 52 was purified via preparatory TLC (70% EtOAc/hexanes, 250 micron SiO2 plate) to provide (+)-irciniastatin A (1) (0.3 mg, 0.5 μmol 31%) and epi-C(11)-irciniastatin A (52) (0.3 mg, 0.5 μmol, 31%). All spectral data of (+)-irciniastatin A (1) matched to the synthetic sample from our first-generation approach. Characterization data for (+)-irciniastatin A (1): 1H NMR (500 MHz, MeOD) δ 6.24 (s, 1 H), 5.39 (d, J = 8.3 Hz, 1 H), 4.74 (s, 1 H), 4.71 (s, 1 H), 4.51-4.47 (ddd, J = 3.0, 5.9, 12.0 Hz, 1 H), 4.35 (d, J = 2.6 Hz, 1 H), 3.94 (m, 2 H), 3.67 (ddd, J = 2.6, 3.5, 9.5 Hz, 1 H), 3.60 (dd, J = 4.4, 10.9 Hz, 1 H), 3.50 (dd, J = 2.0, 10.1 Hz, 1 H), 3.35 (s, 3 H), 3.23 (s, 3 H), 3.13 (dd, J = 3.3, 16.7 Hz, 1 H), 2.86 (dd, J = 12.0, 16.6 Hz, 1 H), 2.35 (dd, J = 9.4, 14.8 Hz, 1 H), 2.11 (m, 1 H), 2.10 (s, 3 H), 2.02 (ddd, J = 2.6, 4.5, 13.4 Hz, 1 H), 1.91 (m, 1 H), 1.86-1.74 (m, 2 H), 1.72 (s, 3 H), 1.68 (ddd, J = 2.1, 3.8, 14.6 Hz, 1 H), 1.10 (d, J = 7.1 Hz, 3 H), 0.97 (s, 3 H), 0.90 (s, 3 H); 13C NMR (125 MHz, MeOD) Observable peaks δ 176.3, 172.5, 163.9, 144.0, 141.2, 115.5, 113.1, 101.6, 82.8, 82.3, 82.1, 79.9, 73.6, 73.3, 72.1, 57.8 56.7, 43.4, 39.9, 38.8, 34.5, 30.6, 29.6, 23.8, 23.0, 14.0, 11.0, 9.3; HRMS (ES+) m/z 632.3033 [(M+1)+; calcd for C31H47NO11Na: 632.3047]. Characterization data for epi-C(11)-irciniastatin A (52): 1H NMR (500 MHz, MeOD) Observable peaks δ 6.25 (s, 1 H), 5.25 (d, J = 3.6 Hz, 1 H), 4.76 (s, 1 H), 4.73 (s, 1 H), 4.51-4.47 (ddd, J = 3.1, 6.8, 12.2 Hz, 1 H), 4.37 (d, J = 2.8 Hz, 1 H), 4.04 (m, 1 H), 3.97 (dd, J = 4.0, 13.2 Hz, 1 H), 3.78 (dd, J = 3.1, 11.8 Hz, 1 H), 3.73-3.67 (m, 2 H), 3.33 (s, 3 H), 3.17 (dd, J = 3.3, 16.7 Hz, 1 H), 2.83 (dd, J = 11.9, 16.2 Hz, 1 H), 2.35 (dd, J = 9.7, 15.0 Hz, 1 H), 2.12 (dd, J = 3.9, 14.2 Hz, 1 H), 2.09 (s, 3 H), 1.96 (m, 2 H), 1.79 (m, 1 H), 1.73 (s, 3 H), 1.63 (m, 2 H), 1.12 (d, J = 7.0 Hz, 3 H), 1.01 (s, 3 H), 0.93 (s, 3 H); 13C NMR (125 MHz, MeOD) Observable peaks δ 176.0, 172.6, 164.7, 163.8, 144.0, 141.2, 115.4, 113.2, 101.5, 101.5, 83.0, 82.5, 73.0, 72.7, 72.0, 57.9, 56.7, 42.8, 39.0, 38.7, 30.9, 29.6, 23.1, 22.8, 21.3, 10.9, 9.7; HRMS (ES+) m/z 632.3029 [(M+1)+; calcd for C31H47NO11Na: 632.3047].
Supplementary Material
Scheme 4.

Synthesis of Diene (−)-18
Acknowledgments
Financial support was provided by the National Institute of Health (Institute of General Medical Sciences) through Grant No. CA-19033. We thank Drs. George Furst and Rakesh Kohli (University of Pennsylvania for help in obtaining the high resolution NMR and mass spectral data, respectively. The authors thank the generous help of Professor G. R. Pettit (Arizona State University) for providing the 1H and 13C NMR spectra for (−)- irciniastatin B.
Footnotes
Supporting Information. 1H and 13C spectral data of all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Pettit GR, Xu JP, Chapuis JC, Pettit RK, Tackett LP, Doubek DL, Hooper JNA, Schmidt JM. J Med Chem. 2004;47:1149–1152. doi: 10.1021/jm030207d. [DOI] [PubMed] [Google Scholar]
- 2.Cichewicz RH, Valeriote FA, Crews P. Org Lett. 2004;6:1951–1954. doi: 10.1021/ol049503q. [DOI] [PubMed] [Google Scholar]
- 3.Jiang X, García-Fortanet J, De Brabander JK. J Am Chem Soc. 2005;127:11254–11255. doi: 10.1021/ja0537068. [DOI] [PubMed] [Google Scholar]
- 4.Feng Y, Jiang X, De Brabander JK. J Am Chem Soc. 2012;134:17083–17093. doi: 10.1021/ja3057612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang X, Shao N, Huryk R, Palani A, Aslanian R, Seidel-Dugan C. Org Lett. 2009;11:867–870. doi: 10.1021/ol802772s. [DOI] [PubMed] [Google Scholar]
- 6.Chinen T, Nagumo Y, Watanabe T, Imaizumi T, Shibuya M, Kataoka T, Kanoh N, Iwabuchi Y, Usui T. Toxicol Lett. 2010;199:341–346. doi: 10.1016/j.toxlet.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 7.Wu CY, Feng Y, Cardenas ER, Williams N, Floreancig PE, De Brabander JK, Roth MG. J Am Chem Soc. 2012;134:18998–19003. doi: 10.1021/ja3057002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang X, Shao N, Palani A, Aslanian R, Buevich A. Org Lett. 2007;9:2597–2600. doi: 10.1021/ol071068n. [DOI] [PubMed] [Google Scholar]
- 9.Smith AB, III, Jurica JA, Walsh SP. Org Lett. 2008;10:5625–5628. doi: 10.1021/ol802466t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crimmins MT, Stevens JM, Schaaf GM. Org Lett. 2009;11:3990–3993. doi: 10.1021/ol901655e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watanabe T, Imaizumi T, Chinen T, Nagumo Y, Shibuya M, Usui T, Kanoh N, Iwabuchi Y. Org Lett. 2010;12:1040–1043. doi: 10.1021/ol1000389. [DOI] [PubMed] [Google Scholar]
- 12.Byeon SR, Park H, Kim H, Hong J. Org Lett. 2011;13:5816–5819. doi: 10.1021/ol2024289. [DOI] [PubMed] [Google Scholar]
- 13.Wan S, Wu F, Rech JC, Green ME, Balachandran R, Horne WS, Day BW, Floreancig PE. J Am Chem Soc. 2011;133:16668–16679. doi: 10.1021/ja207331m. [DOI] [PubMed] [Google Scholar]
- 14.An C, Hoye AT, Smith AB., III Org Lett. 2012;14:4350–4353. doi: 10.1021/ol301783p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith AB, III, Safonov IG, Corbett RM. J Am Chem Soc. 2002;124:11102–11113. doi: 10.1021/ja020635t. [DOI] [PubMed] [Google Scholar]
- 16.Yamamoto K, Suzuki S, Tsuji J. Chem Lett. 1978;7:649–652. [Google Scholar]
- 17.Byrson TA, Dolak TM. Org Synth. 1977;57:62. [Google Scholar]
- 18.Forbes JE, Pattenden G. Tetrahedron Lett. 1987;28:2771–2774. [Google Scholar]
- 19.Ebenezer W, Pattenden G. Tetrahedron Lett. 1992;33:4053–4056. [Google Scholar]
- 20.Hunter TJ, O’Doherty GA. Org Lett. 2002;4:4447–4450. doi: 10.1021/ol0269502. [DOI] [PubMed] [Google Scholar]
- 21.Smith AB, III, Walsh SP, Frohn M, Duffey MO. Org Lett. 2004;7:139–142. doi: 10.1021/ol047792c. [DOI] [PubMed] [Google Scholar]
- 22.Parikh JR, Doering WE. J Am Chem Soc. 1967;89:5505–5507. [Google Scholar]
- 23.Bal BS, Childers WE, Pinnick HW. Tetrahedron. 1981;37:2091–2096. [Google Scholar]
- 24.Langer P, Kracke B. Tetrahedron Lett. 2000;41:4545–4547. [Google Scholar]
- 25.Duan JJW, Smith AB., III J Org Chem. 1993;58:3703–3711. [Google Scholar]
- 26.Huang Z, Negishi E-i. Org Lett. 2006;8:3675–3678. doi: 10.1021/ol061202o. [DOI] [PubMed] [Google Scholar]
- 27.Fischetti W, Mak KT, Stakem FG, Kim J, Rheingold AL, Heck RF. J Org Chem. 1983;48:948–955. [Google Scholar]
- 28.Wang ZX, Tu Y, Frohn M, Zhang JR, Shi Y. J Am Chem Soc. 1997;119:11224–11235. [Google Scholar]
- 29.Ishibashi N, Miyazawa M, Miyashita M. Tetrahedron Lett. 1998;39:3775–3778. [Google Scholar]
- 30.Katsuki T, Sharpless KB. J Am Chem Soc. 1980;102:5974–5976. [Google Scholar]
- 31.Oshima M, Yamazaki H, Shimizu I, Nisar M, Tsuji J. J Am Chem Soc. 1989;111:6280–6287. [Google Scholar]
- 32.Zhao MM, Li JEM, Song ZJ, Taschaen DM. Org Synth. 2005:195. [Google Scholar]
- 33.Baldwin JE. J Chem Soc, Chem Commun. 1976:734–736. [Google Scholar]
- 34.Dess DB, Martin JC. J Org Chem. 1983;48:4155–4156. [Google Scholar]
- 35.Hamana H, Sasakura K, Sugasawa T. Chem Lett. 1984;13:1729–1732. [Google Scholar]
- 36.Evans DA, Calter MA. Tetrahedron Lett. 1993;34:6871–6874. [Google Scholar]
- 37.Chen KM, Hardtmann GE, Prasad K, Repič O, Shapiro MJ. Tetrahedron Lett. 1987;28:155–158. [Google Scholar]
- 38.Noyori R, Nishida I, Sakata J, Nishizawa M. J Am Chem Soc. 1980;102:1223–1225. [Google Scholar]
- 39.Scheidt KA, Chen H, Follows BC, Chemler SR, Coffey S, Roush WR. J Org Chem. 1998;63:6436–6437. [Google Scholar]
- 40.Vakalopoulos A, Hoffmann HMR. Org Lett. 2000;2:1447–1450. doi: 10.1021/ol0057784. [DOI] [PubMed] [Google Scholar]
- 41.Simsek S, Horzella M, Kalesse M. Org Lett. 2007;9:5637–5639. doi: 10.1021/ol702640w. [DOI] [PubMed] [Google Scholar]
- 42.Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–4096. [Google Scholar]
- 43.Hoye TR, Jeffrey CS, Shao F. Nat Protoc. 2007;2:2451–2458. doi: 10.1038/nprot.2007.354. [DOI] [PubMed] [Google Scholar]
- 44.Paterson I, Goodman JM. Tetrahedron Lett. 1989;30:997–1000. [Google Scholar]
- 45.Paterson I, Goodman JM, Anne Lister M, Schumann RC, McClure CK, Norcross RD. Tetrahedron. 1990;46:4663–4684. [Google Scholar]
- 46.For example of base catalyzed epimerization of trans-tetrahydropyran see: Allevi P, Anastasia M, Ciuffreda P, Fiecchi A, Scala A. J Chem Soc, Perkin Trans 1. 1989:1275–1280.
- 47.For an example of acid catalyzed hydrolysis of N,O-aminal see: Mori M, Uozumi Y, Ban Y. J Chem Soc, Chem Commun. 1986:841–842.
- 48.Denmark SE, Kobayashi T, Regens CS. Tetrahedron. 2010;66:4745–4759. doi: 10.1016/j.tet.2010.03.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.