

Abstract
Purpose
Germ cell tumor (GCT) is the most common malignancy in young adult men. Currently, patients are risk-stratified on the basis of clinical presentation and serum tumor markers. The introduction of molecular markers could improve outcome prediction.
Patients and Methods
Expression profiling was performed on 74 nonseminomatous GCTs (NSGCTs) from cisplatin-treated patients (ie, training set) and on 34 similarly treated patients with NSGCTs (ie, validation set). A gene classifier was developed by using prediction analysis for microarrays (PAM) for the binary end point of 5-year overall survival (OS). A predictive score was developed for OS by using the univariate Cox model.
Results
In the training set, PAM identified 140 genes that predicted 5-year OS (cross-validated classification rate, 60%). The PAM model correctly classified 90% of patients in the validation set. Patients predicted to have good outcome had significantly longer survival than those with poor predicted outcome (P < .001). For the OS end point, a 10-gene model had a predictive accuracy (ie, concordance index) of 0.66 in the training set and a concordance index of 0.83 in the validation set. Dichotomization of the samples on the basis of the median score resulted in significant differences in survival (P = .002). For both end points, the gene-based predictor was an independent prognostic factor in a multivariate model that included clinical risk stratification (P < .01 for both).
Conclusion
We have identified gene expression signatures that accurately predict outcome in patients with GCTs. These predictive genes should be useful for the prediction of patient outcome and could provide novel targets for therapeutic intervention.
INTRODUCTION
Germ cell tumor (GCT) is the most commonly diagnosed solid malignancy and is a leading cause of cancer-related mortality and morbidity in men age 18 to 35 years.1 GCTs can be broadly classified into two main subtypes, seminomas (SEMs) and nonseminomas (NSGCTs), on the basis of cellular differentiation.2 Advances in chemotherapy for metastatic disease during the past 30 years applied to both cell types and to early-stage settings have improved the cure rate to greater than 90% of new occurrences.1
Currently, treatment decisions for patients with metastatic disease are made according to the International Germ Cell Cancer Collaborative Group (IGCCCG) guidelines.1 Good-, intermediate-, and poor-risk groups are based on histology (SEM v NSGCT); serum levels of α-fetoprotein (AFP), lactate dehydrogenase (LDH), and human chorionic gonadotropin (HCG); the site of the primary tumor; and the presence or absence of nonpulmonary visceral metastases.1 Greater than 90% of good-risk, 70% to 75% of intermediate-risk, and 40% to 45% of poor-risk patients are cured.1 Because AFP and HCG are biochemical markers of yolk sac and trophoblastic differentiation, respectively, the IGCCCG guidelines represent the addition of tumor biology to anatomy in standard TNM staging; only testis cancer is staged as TNMS.
Although the IGCCCG model is effective at stratifying patients into groups that require more or less chemotherapy, a predictable proportion of patients with metastatic GCTs succumb as a result of the disease. Better markers of chemotherapy resistance are needed. Molecular markers could improve outcome prediction, discover potential targets for therapeutic intervention, and elucidate mechanisms that result in resistance to chemotherapy.
We describe here an expression profiling study on a panel of 74 patients with NSGCTs who were treated with cisplatin-based chemotherapy for the identification of genes predictive of overall survival (OS) and 5-year OS, with validation in 34 independent NSGCT specimens.
PATIENTS AND METHODS
Tumor Specimens
Tumor specimens were collected under institutional review board– approved protocols from treated patients who gave informed consent at Memorial Sloan-Kettering Cancer Center (MSKCC) between 1985 and 2002. Samples that met the following criteria were included in the study: major histology present was NSGCT; patient was treated with cisplatin-based chemotherapy before 2003; and a sufficient quantity of high-quality RNA for labeling could be recovered. The training set consisted of 74 previously profiled NSGCT specimens from 74 different patients (detailed in the Gene Expression Omnibus (GEO) repository at http://www.ncbi.nlm.nih.gov/geo/, data set GSE3218).3,4 Multiple tumors were available for six patients; one was randomly chosen for inclusion in the training set. The validation set consisted of 34 newly profiled NSGCT specimens from 34 different, more recently treated, patients (in the GEO data set GSE10783). The validation set was enriched for poor outcome samples, in that all patients not earlier selected that died as a result of disease were included.
RNA Isolation, Labeling, and Expression Profiling
RNA was isolated, reverse transcribed, and labeled. Then, RNA was hybridized to Affymetrix U133A+B microarrays (Affymetrix, Santa Clara, CA), washed, and imaged as described previously.3,4
Data Processing and Statistical Analyses
Detailed descriptions of data processing and statistical analyses are available (Data Supplement, online only). Briefly, expression values were generated by using the robust multiarray average method5 within Bioconductor,6 and there was no filtering or gene exclusion before developing predictive models. For analysis of 5-year OS, a modification of prediction analysis for microarrays (PAM)7 was used to build a predictive model, which was tested on the validation set. For analysis of OS, a predictive score was developed by using the weighted sum of genes, with the coefficients of the univariate Cox model as the weights.8 The predictive accuracy of the score was quantified by using the concordance index. Survival curves were estimated by using the Kaplan-Meier method, and differences in survival between groups were evaluated by using the log-rank test. For 5-year OS, a multivariate logistic regression model was employed, whereas a multivariate Cox model was used for OS. A significance cutoff of .05 was used for all tests. To identify genes associated with AFP, LDH, and HCG levels or tumor site or chemotherapy, patients were divided into the appropriate groups and were compared with the MaxT function9 within the multitest package in Bioconductor6; genes were significant if they had adjusted P values less than .05. For GoMiner10 analysis, pathways were considered significant if they had P values less than .05.
RESULTS
Analysis of 5-Year OS in Patients With GCTs
A total of 108 tumors from patients who received platinum-based chemotherapy were utilized as training (n = 74) and validation (n = 34) sets. Patient and tumor characteristics are listed in Tables 1 and 2. At the time of chemotherapy, the patients' median ages were 29.4 years in the training set (range, 15.0 to 60.5 years) and 28.1 years in the validation set (range, 16.6 to 45.2 years). Both sets were comprised of a diverse panel of tumors that had varying histology (ie, pure embryonal carcinoma, teratoma, yolk sac tumor, choriocarcinoma, and combinations of NSGCT elements) and tumor site (ie, primary testicular, primary mediastinal, and various metastatic sites). Some tumors were obtained before, and others after, chemotherapy treatment. For the binary end point of 5-year OS, 23 patients had died as a result of disease (ie, poor outcome), and 49 were alive at the 5-year cutoff in the training set (ie, good outcome). Two patients were excluded, as they were alive with less than 5 years of follow-up. In the validation set (n = 34), 10 patients died as a result of disease, 20 were alive at the 5-year cutoff, and four had insufficient follow-up times. There were no significant differences between covariates, other than longer follow-up time in the training set. A χ2 test showed that the distribution of good-, intermediate-, and poor-outcome patients in the training and validation sets did not differ significantly (P = .20 for both sets) from the test population used to derive the IGCCCG risk classifier.16
Table 1.
Patient Demographic and Clinical Characteristics and Treatment in Training and Validation Sets
| Characteristic | Training Set (n = 74) |
Validation Set (n = 34) |
P | ||
|---|---|---|---|---|---|
| No. | % | No. | % | ||
| Age, years | |||||
| Median | 29.0 | 28.1 | .36 | ||
| Range | 15.4-65.0 | 16.6-45.2 | |||
| Initial IGCCCG risk group* | .96 | ||||
| Good | 37 | 50 | 18 | 53 | |
| Intermediate | 17 | 23 | 7 | 21 | |
| Poor | 20 | 27 | 9 | 26 | |
| Serum markers† | |||||
| Elevated HCG, mIU/mL | |||||
| % of patients‡ | 21 | 30 | 15 | 48 | .11 |
| > Median | 12.9 | 98.4 | |||
| Range | 2.3-418,700 | 2.5-12,500 | |||
| Elevated AFP, ng/mL | |||||
| % of patients§ | 34 | 48 | 16 | 50 | 1 |
| Median | 328 | 296.1 | |||
| Range | 15.7-51,849 | 19.5-43,038 | |||
| Elevated LDH, U/L | |||||
| % of patients‖ | 30 | 41 | 10 | 31 | .39 |
| Median | 257.5 | 286 | |||
| Range | 202-2,894 | 205-1,540 | |||
| Initial chemotherapy regimen | .72 | ||||
| EP11–13 | 33 | 45 | 18 | 53 | |
| BEP14 | 21 | 28 | 9 | 26 | |
| Other | 20 | 27 | 7 | 21 | |
| BEP + HDCT14 | 0 | 0 | 3 | 9 | |
| VAB615 | 4 | 5 | 0 | 0 | |
| VAB6 + HDCT | 5 | 7 | 0 | 0 | |
| EC13 | 4 | 5 | 0 | 0 | |
| VIP | 3 | 4 | 2 | 6 | |
| VIP + HDCT | 2 | 3 | 1 | 3 | |
| Other | 2 | 3 | 1 | 3 | |
| Response to initial chemotherapy | .27 | ||||
| CR | 40 | 54 | 24 | 71 | |
| IR | 20 | 27 | 6 | 18 | |
| Other | 14 | 19 | 4 | 12 | |
| PR-negative markers | 2 | 3 | 1 | 3 | |
| NA (ie, treated adjuvantly) | 11 | 15 | 3 | 9 | |
| Inevaluable | 1 | 1 | 0 | 0 | |
| Outcome | |||||
| Follow-up, years | |||||
| Median | 9.7 | 6 | < .001 | ||
| Range | 1.9-23.3 | 3.1-13.7 | |||
| OS | .81 | ||||
| Follow-up ≥ 5 years | 72 | 97.3 | 30 | 88.2 | .08 |
| Events (ie, 5-year OS) | 23 | 31.9 | 10 | 33.3 | 1 |
Abbreviations: IGCCCG, International Germ Cell Cancer Collaborative Group; HCG, human chorionic gonadotropin; AFP, α-fetoprotein; LDH, lactate dehydrogenase; EP, etoposide + cisplatin; BEP, bleomycin + etoposide + cisplatin; HDCT, high-dose chemotherapy; VAB6, vinblastine + actinomycin-D + bleomycin + cisplatin + cyclophosphamide; EC, etoposide + carboplatin; VIP, etoposide + ifosfamide + cisplatin; CR, complete response; IR, incomplete response; PR, partial response; NA, not applicable; OS, overall survival.
Characteristics are those at the time of initiation of initial chemotherapy regimen.
For the training set, four patients had HCG levels that were undetermined, three patients had AFP levels that were undetermined, and one had LDH levels that were undetermined. For the validation set, three patients had HCG levels that were undetermined, two patients had AFP levels that were undetermined, and two patients had LDH levels that were undetermined.
Reference range for HCG is ≤ 2.2 mIU/mL.
Reference range for AFP is ≤ 15 ng/mL.
Reference range for LDH is 60-200 U/L.
Table 2.
Tumor Characteristics in Training and Validation Sets
| Characteristic | Training Set (n = 74) |
Validation Set (n = 34) |
P | ||
|---|---|---|---|---|---|
| No. | % | No. | % | ||
| Tumor site | .15 | ||||
| Primary tumor | 26 | 35 | 18 | 53 | |
| Testis | 17 | 23 | 14 | 41 | |
| Mediastinum | 9 | 12 | 4 | 12 | |
| Metastatic tumor | 48 | 65 | 16 | 47 | |
| RPLN | 36 | 49 | 11 | 32 | |
| Lung | 8 | 11 | 4 | 12 | |
| Other | 4 | 5 | 1 | 3 | |
| Mixed histology | 35 | 47 | 15 | 44 | |
| Seminoma as part of mixed histology | 5 | 7 | 2 | 6 | |
| Teratoma as part of mixed histology | 22 | 30 | 9 | 26 | |
| Pure histology | 39 | 53 | 19 | 56 | |
| Pure teratoma | 16 | 22 | 9 | 26 | |
| Pure or dominant (if mixed) histology | .83 | ||||
| CC | 5 | 7 | 3 | 9 | |
| EC | 19 | 26 | 6 | 18 | |
| TER | 25 | 34 | 12 | 35 | |
| YST | 21 | 28 | 11 | 32 | |
| Trophoblast | 2 | 3 | 2 | 6 | |
| N/A | 2 | 3 | 0 | 0 | |
| Secondary malignant transformation | .10 | ||||
| No. of TER with malignant transformation | 5 | 7 | 6 | 18 | |
| Relationship to chemotherapy | .68 | ||||
| Before | 31 | 42 | 16 | 47 | |
| After | 43 | 58 | 18 | 53 | |
| No. of chemotherapy regimens prior to sample being obtained | .77 | ||||
| 1 | 28 | 38 | 13 | 38 | |
| ≥ 2 | 15 | 17 | 5 | 15 | |
Abbreviations: RPLN, retroperitoneal lymph node; CC, choriocarcinoma; EC, embryonal carcinoma; TER, teratoma; YST, yolk sac tumor.
We used PAM on the training set to develop a classifier for 5-year OS. PAM identified 170 probe sets that represented 140 unique genes (Data Supplement, online only; see Table 3 for a list of the top 20 predictive genes) that predicted outcome with a cross-validated classification rate of 60% (74% of good- and 45% of poor-outcome patients were predicted correctly). Reverse transcriptase polymerase chain reaction analysis confirmed the differential expression of several genes (Appendix Fig A1, online only). When applied to the validation set, the classification rate was 90% (95% CI, 74% to 98%) overall and in both the good- and poor-outcome groups. There were significant differences in survival between patients whom PAM predicted to have good versus poor outcome (P < .001; Fig 1A) and among the IGCCCG risk groups (P < .001; Fig 1B). The gene model was a significant independent predictor of outcome by multivariate logistic regression analysis in the validation set when IGCCCG risk was included as a continuous variable (P = .01). Because intermediate- and poor-risk populations constitute the preponderance of patients who fail treatment, we combined them and applied the PAM model. There was a significant difference in survival between the resulting groups (Fig 1C), which separated those most and least likely to respond to standard therapy. The only good-risk patient to die as a result of disease was predicted by the PAM model to have a poor outcome.
Table 3.
Top 20 Probe Sets for Predicting 5-Year OS
| Probe | Symbol | Location | Good:Poor Fold Change | Poor:Good Fold Change |
|---|---|---|---|---|
| 227662_at | SYNPO2 | chr4q26 | 2.82 | 0.35 |
| 225895_at | SYNPO2 | chr4q26 | 2.47 | 0.41 |
| 212187_x_at | PTGDS | chr9q34.2-q34.3 | 2.75 | 0.36 |
| 235456_at | HIST1H2BD | chr6p21.3 | 0.46 | 2.18 |
| 211748_x_at | PTGDS | chr9q34.2-q34.3 | 2.64 | 0.38 |
| 212592_at | IGJ | chr4q21 | 3.02 | 0.33 |
| 229178_at | LOC145786 | chr15q21.1 | 0.41 | 2.47 |
| 206373_at | ZIC1 | chr3q24 | 0.35 | 2.88 |
| 209447_at | SYNE1 | chr6q25 | 1.71 | 0.58 |
| 214836_x_at | IGKC/IGKV1-5 | chr2p12 | 2.40 | 0.42 |
| 203837_at | MAP3K5 | chr6q22.33 | 1.85 | 0.54 |
| 204446_s_at | ALOX5 | chr10q11.2 | 2.21 | 0.45 |
| 213622_at | COL9A2 | chr1p33-p32 | 0.49 | 2.04 |
| 201953_at | CIB1 | chr15q25.3-q26 | 1.57 | 0.64 |
| 224795_x_at | IGKC/IGKV1-5 | chr2p12 | 3.12 | 0.32 |
| 214677_x_at | IGL@/IGLC1/IGLC2/IGLV3-25/IGLV2-14/IGLJ3 | chr22q11.1-q11.2 | 3.99 | 0.25 |
| 214669_x_at | IGKC | chr2p12 | 2.84 | 0.35 |
| 204914_s_at | SOX11 | chr2p25 | 0.43 | 2.34 |
| 221651_x_at | IGKC/IGKV1-5 | chr2p12 | 3.09 | 0.32 |
| 209374_s_at | IGHM | chr14q32.33 | 2.66 | 0.38 |
Abbreviation: OS, overall survival.
Fig 1.
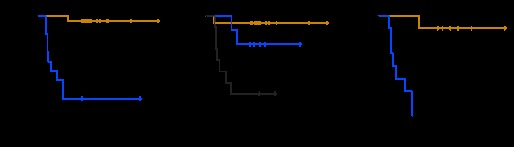
(A) Kaplan-Meier survival curves of patients from validation set (A) who were predicted to have good (gold) or poor (blue) outcome on the basis of genes identified by prediction analysis for microarrays (PAM) to be associated with 5-year overall survival; (B) who had good (gold), intermediate (blue), and poor (gray) International Germ Cell Cancer Collaborative Group (IGCCCG) risk assessment; and (C) who had intermediate and poor IGCCCG risk assessment and good (gold) and poor (blue) PAM prediction.
Analysis of OS in Patients With GCTs
We developed a predictive model for OS by using weighted scores of genes identified by a univariate Cox model. The best model had 10 genes (Table 4), seven of which were also in the PAM list. The model had concordance indexes of 0.66 in the training set and 0.83 in the validation set (95% CI, 0.70 to 0.96). This was similar to the concordance index for the IGCCCG risk stratification in the validation set (0.82; 95% CI, 0.68 to 0.96). When the samples were dichotomized by median score, there was significant separation of the survival curves (P = .002; Fig 2). When included in a multivariate Cox model with IGCCCG risk stratification, both variables were significant predictors of outcome (P = .005 for both).
Table 4.
Top 10 Probe Sets for Predicting OS
| Probe | Symbol | Location | HR |
P |
In 5-Year OS | ||
|---|---|---|---|---|---|---|---|
| Training | Test | Training | Test | ||||
| 212800_at | STX6 | chr1q25.3 | 39.51 | 4.38 | < .0001 | .02 | No |
| 211316_x_at | CFLAR | chr2q33-q34 | 0.11 | 0.22 | < .0001 | .006 | No |
| 213940_s_at | FNBP1 | chr9q34 | 0.15 | 0.06 | < .0001 | .002 | Yes |
| 209898_x_at | ITSN2 | chr2pter-p25.1 | 0.12 | 0.04 | < .0001 | .003 | Yes |
| 209447_at | SYNE1 | chr6q25 | 0.31 | 0.42 | < .0001 | .06 | Yes |
| 203837_at | MAP3K5 | chr6q22.33 | 0.36 | 0.20 | < .0001 | .02 | Yes |
| 212187_x_at | PTGDS | chr9q34.2-q34.3 | 0.54 | 0.44 | < .0001 | .02 | Yes |
| 219076_s_at | PXMP2 | chr12q24.33 | 6.05 | 4.67 | < .0001 | .06 | Yes |
| 219618_at | IRAK4 | chr12q12 | 0.04 | 0.80 | < .0001 | .9 | No |
| 213982_s_at | RABGAP1L | chr1q24 | 0.32 | 0.30 | < .0001 | .02 | Yes |
Abbreviations: OS, overall survival; HR, hazard ratio.
Fig 2.

Kaplan-Meier survival curves of patients from validation set dichotomized on the basis of median score. Gold line indicates scores less than the median; blue line indicates scores greater than the median.
GoMiner Analysis of Predictive Genes
We used GoMiner10 to identify significant pathways that were represented in the predictive gene lists. Genes involved in the immune response were more highly expressed in patients with good outcome, including genes associated with B cells (ie, BLNK, IGHM, IGKC, IGJ, IGL@, IGHA1, IGKV1-5, and IGLV3-25), T cells (ie, PTPRC, SYK, CXCL12, ITGB2), complement activation (ie, C1S, C1R, and C7), and other immune functions (ie, IL6R, IFI16, MNDA, TNFSF13B, and HLA-DPA1). Genes and pathways associated with active development and differentiation, in particular with neural development (ie, BMP7, MDK, NRCAM, OTX2, PCDHB14, PLXNA2, SOX11, and ZIC1), were significantly enriched in tumors from poor-outcome patients, but kidney (eg, BMP7, SOX11, and SALL1) and skeletal (eg, COL2A1, COL9A2) developmental genes also were observed. Good outcome also was associated with neural development pathways, but the genes involved were important in repression of differentiation and proliferation (ie, BHLHB3, FNBP1, and GAS7); BHLHB3 is involved in transcriptional repression and control of proliferation,17 whereas GAS7 is thought to be expressed only in quiescent cells after terminal differentiation.18 This GoMiner analysis suggests that good-outcome genes are associated with immune-related functions and pathways responsible for repression of differentiation, whereas poor-outcome genes are associated with active differentiation processes, most notably neural development.
Gene Expression Associations With IGCCCG Serum Marker Levels
The tumor cohort was analyzed to determine if genes associated with elevated serum tumor marker levels overlapped with the predictive gene lists. The MaxT method9 was used to identify genes highly expressed in patients with elevated serum levels in the training set. No significant genes were associated with elevated LDH,and two genes were associated with elevated HCG (CGA and CGB, which make up the α and β subunits of the HCG hormone); 45 transcripts were associated with elevated AFP (including AFP; Data Supplement, online only), many of which we previously identified as being associated with YST histology.4 Of the 45 AFP-associated and two HCG-associated transcripts, only the serum protein MDK also was found among the outcome-associated genes.
Gene Expression Changes Associated With Site and Chemotherapy Treatment
We performed analyses to determine if differences between site (primary v metastatic) or treatment (prechemotherapy v postchemotherapy) overlapped with the identified outcome genes. Among all primary (both gonadal and mediastinal) and metastatic tumors, only one gene (ie, TSPY1) was significant, which (on average) was expressed approximately four-fold higher in primary tumors than in metastases. In contrast, 37 probe sets (of 34 genes) showed significant differential expression between prechemotherapy and postchemotherapy samples (Data Supplement, online only). None were in the top 25 predictive genes, which is consistent with the hypothesis that important resistance genes would be seen in representative tumors regardless of time of acquisition. Interestingly, several genes associated with pluripotency (ie, NANOG, DPPA4, UTF1, TERF1, NALP7, and JARID2) were more highly expressed in untreated tumors.
DISCUSSION
We identified predictive gene signatures associated with both OS and 5-year OS that were independent of IGCCCG risk classification as prognostic/predictive factors. Good outcome was associated with two broad gene sets: immune function (in particular, immunoglobulins) and repression of differentiation. Conversely, poor outcome was associated with genes involved in active differentiation. The gene signatures stratified intermediate- and poor-risk patients into highly curable and highly resistant groups. Anecdotally, the gene signature also may have the ability to stratify good-risk patients, because the only good-risk patient who died as a result of disease was predicted to have a poor outcome; however, this pattern clearly will require additional studies to establish.
Although B cells seem to be the likely source of immune gene expression, preliminary studies to determine whether B cells or tumor cells are the source of immunoglobulin gene expression were inconclusive. No discernible differences in the numbers of infiltrating B cells in high- and low-expressing tumors were observed by immunohistochemistry with pan–B-cell markers. However, the immunoglobulins were not significantly expressed in GCT cell lines, and polymerase chain reaction analysis showed that the immunoglobulin gene rearrangements were not clonal in nature (data not shown). There also have been reports that immunoglobulin gene rearrangement and expression can occur early in the ontogeny of germ cells,19 which suggests that the tumors also could express these transcripts. Notably, in a previous study of 19 tumors, expression of immunoglobulin genes also was observed in both EC and YST.20 Interestingly, SEM often shows lymphocytic infiltration, and SEM that we profiled also expressed many of these genes. When our gene predictor was applied to 14 pure SEM specimens (unpublished data), all were correctly predicted to have good outcome, which suggests that the model may also be applicable to SEM. However, we currently lack SEM specimens with poor outcome necessary to rigorously test the model.
Genes predictive of poor outcome were enriched for associations with active differentiation processes, such as kidney, skeletal, and—most prominently—neural differentiation. Although these genes may be only markers of outcome, we believe there may be functional significance to the differentiation signatures. ZIC1, which interacts with GLI proteins in the Hedgehog pathway,21,22 was strongly associated with poor outcome and has been implicated in medulloblastomas and endometrial cancer.23,24 We previously found that induction of differentiation of the pluripotent EC cell line NT2/D1 into neural lineages25 results in resistance (unpublished data) and elevation of ZIC1 expression. Activation of smoothened signaling within the Hedgehog pathway also was evident in these tumors; smoothened plays an essential role during normal CNS development26 and can lead to development of cancer, including brain and skin tumors,21 when aberrantly activated. Interestingly, the neural genotype was observed in all histologic subtypes, even in the absence of a neural phenotype. These results suggest that differentiation, particularly into neural lineages, is associated with poor outcome and may result in cisplatin resistance.
Synaptopodin 2 (SYNPO2), a putative tumor suppressor gene also known as myopodin, had the highest predictive value in the 5-year OS analysis. We recently showed frequent downregulation of SYNPO2 as a result of 4q loss,27 which occurs frequently in GCTs and which is consistent with a possible role as a tumor suppressor. SYNPO2 expression has been reported to suppress tumor growth, its cellular localization is stage dependent, and loss of expression was associated with a poor outcome in both prostate28 and bladder cancers.29
These data imply a relationship between the process of differentiation and chemotherapy resistance in GCT and are consistent with clinical observations. SEM is more sensitive to chemotherapy than NSGCT, and this observation is embedded in the IGCCCG guidelines.1 All teratomas (TERs) are resistant to chemotherapy, and neural and skeletal differentiation are among the most common forms of malignant transformation. AFP and HCG are biochemical markers of yolk sac and trophoblastic differentiation, respectively, and higher values of each are associated with a worse outcome.1 These data implicate somatic, yolk sac, and trophoblastic differentiation pathways in development of drug resistance.
Notably absent from the outcome-associated gene lists were genes involved in DNA repair. Previous studies have postulated that reduced levels of expression of DNA repair genes are responsible for the chemotherapy sensitivity of GCTs,30 but we did not observe enrichment of DNA repair genes in the outcome signatures.
One potential criticism of this analysis is the heterogeneity of the tumor set. This heterogeneity, however, represents the clinical setting that confronts clinicians. GCTs often present with mixed histologies and at different primary sites (ie, gonadal or mediastinal), and only one gene was significantly differentially expressed according to site. Moreover, residual tumors resected after chemotherapy represent those resistant to therapy. Although there were 37 genes that were changed with chemotherapy, none were in the top 25 predictive genes, and many were associated with pluripotency and were more highly expressed in untreated tumors. In a previous study, loss of expression of OCT3/4, a core transcription factor required for pluripotency, was associated with cisplatin resistance.31 These observations are consistent with the hypotheses that undifferentiated GCTs are more sensitive to cisplatin and that residual tumor after cisplatin treatment is depleted of sensitive, pluripotent elements and is enriched for resistant, differentiated elements. We believe that the inherent heterogeneity of GCT enhanced our ability to detect molecular signatures associated with outcome in tumors and accurately reflects those seen in a clinical setting.
There are several likely explanations for the lower classification rate of the training set compared with the validation set. In several instances, we profiled multiple tumors from patients with poor outcome, and one was randomly chosen for the outcome analysis. In three such instances, the patient's outcome was misclassified, but we subsequently observed that other tumors from these patients carried poor outcome signatures; replacement of the original tumor with the second tumor resulted in correct classification of all three patients (data not shown). This suggests that, when multiple tumors are available from a patient, all should be profiled, because one metastasis may carry a poor outcome signature although the others may not. Other unusual occurrences included two good-outcome patients who died after the 5-year cutoff; one was predicted to have poor outcome. Two other good-outcome patients who were predicted by PAM to be poor-outcome patients experienced late relapses after 10 years. Similarly, two good-outcome patients with PAM-predicted poor outcome developed second primary tumors. Because bilateral GCTs are thought to represent separate tumors,1 it is possible that the gene expression signature may have identified a field defect in these patients. Surgical cure is a final confounding problem, because resection of residual disease improves outcome after chemotherapy but cannot be predicted by a gene signature.11,32,33 This may explain why some patients with poor-outcome signatures had good outcomes. In fact, such gene signatures could be an indication for surgical intervention in some scenarios.
Some patients with pure TER specimens had poor outcome signatures; several of these patients died as a result of disease, particularly if secondary malignant transformation was present. Pure TER usually is considered a benign disease, despite its resistant to chemotherapy. Hence, TER should not automatically be viewed as benign disease. We previously have reported wide variations in p53 and Ki67 expression in TER.34 Rather, pure TER may harbor residual, malignant GCT not observed pathologically or may be programmed for malignant transformation. These data set the stage for gene signature studies that may permit more precise decision making regarding surgery after chemotherapy.
In conclusion, we identified gene signatures associated with outcome in patients with GCTs. Our results indicate that signatures representing immune function and repression of differentiation are associated with good outcome, whereas signatures representing active differentiation, particularly into neural lineages, and loss of the pluripotent genotype are associated with poor outcome. Adaptation of subsets of these genes for use in clinical assays could result in improved outcome prediction and risk stratification. We have initiated studies to extend our observations in the following ways: validation of these signatures in independent sets from other institutions; development of a diagnostic subset for use on paraffin tissue; and evaluation of serum MDK expression to determine whether this marker is independent of AFP and HCG levels and is representative of the differentiated state.
Data Supplement
Expression values were generated by using the robust multiarray average method5 within Bioconductor.6 Because the training and validation sets were generated at different times and on different batches of microarrays, each probe set in the validation set was adjusted such that the median expression value was the same as the corresponding probe set in the training set. Follow-up time was calculated as the difference between the start date of chemotherapy and the date of last follow-up. The median and range of follow-up times in Table 1 were calculated only on those patients who were still alive.
For the binary analysis of 5-year OS, a modified version of the PAM algorithm was used.7 The modification was that the shrinkage threshold was a parameter in the cross-validation rather than chosen on all the data. The classification rate was estimated on the basis of 10-fold cross-validation that was repeated 25 times. Because the threshold with the lowest overall misclassification rate gave a trivial result on the test set (ie, all samples were classified as having good outcome), we examined thresholds with slightly higher error rates that contained between 50 and 200 transcripts in the classifier. The models then were applied to the validation set for testing of performance. Survival curves that compared the predicted good- and poor-risk groups were generated by using the Kaplan-Meier method, and the difference in survival between these two groups in the validation set was tested by using the log-rank test. The predictive gene set was included in a multivariate logistic regression model with IGCCCG risk stratification, which was treated as a continuous variable.
For analysis of the OS end point, a predictive score was developed by using the weighted sum of the most significant genes. Specifically, the genes were ordered on the basis of the likelihood ratio test for the univariate Cox model, and the weights were the coefficients in that model.8 Predictive accuracy was measured by using the concordance index. The concordance index could range between 0.5 (ie, random prediction) and 1 (ie, perfect prediction). The best model on the training set was the one with the highest concordance index, and 10-fold cross-validation was used to protect against overfitting. For generation of Kaplan-Meier curves, the test samples were split into two groups on the basis of the median score. Differences in survival between the groups were evaluated by using the log-rank test. For testing in a combined model with IGCCCG risk stratification, the multivariate Cox model was employed. For all analyses, a cutoff for significance of .05 was used.
To identify genes associated with AFP, HDH, and HCG levels, patients were divided into high- and low-expressing serum groups, and then gene expression levels for those groups were compared by using the MaxT function9 within the multitest package in Bioconductor.6 This method was used to adjust for multiple comparisons. MaxT was run with 1,000 permutations to give adjusted P values. Patients were considered to have high serum marker levels by using the following conservative criteria: AFP greater than 100 ng/mL; HCG greater than 100 mIU/mL or HCG, nicked greater than 100 ng/mL; and LDH greater than 400 U/L. Genes were considered significant if they had adjusted P values less than .05.
Supplementary Material
Acknowledgment
We thank the Memorial Sloan-Kettering Cancer Center genome core for hybridization, washing, and imaging of the microarray slides.
Appendix
Fig A1.

Reverse transcriptase polymerase chain reaction validation of outcome associated genes identified by expression profiling. IgJ and SYNPO2 represent genes more highly expressed in patients with good outcome, whereas ZIC1, SOX11, and COL2A1 represent genes more highly expressed in patients with poor outcome. NED, no evidence of disease; DOD, died as a result of disease.
Footnotes
This work was supported by grants from the Lance Armstrong Foundation and by the Memorial Sloan-Kettering Cancer Center Byrne Fund.
Presented in part at the 44th Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, May 30- June 3, 2008; the 6th Copenhagen Workshop on CIS Testis and Germ Cell Cancer, Copenhagen, Denmark, October 26-28, 2006; and the 96th Annual Meeting of the American Association for Cancer Research, Anaheim, CA, April 16-20, 2005.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The author(s) indicated no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
Conception and design: James E. Korkola, Jane Houldsworth, Adam B. Olshen, George J. Bosl, R.S.K. Chaganti
Financial support: James E. Korkola, Jane Houldsworth, George J. Bosl, R.S.K. Chaganti
Administrative support: R.S.K. Chaganti
Provision of study materials or patients: Victor E. Reuter
Collection and assembly of data: James E. Korkola, Darren R. Feldman
Data analysis and interpretation: James E. Korkola, Jane Houldsworth, Darren R. Feldman, Adam B. Olshen, Li-Xuan Qin, Sujata Patil, Victor E. Reuter, George J. Bosl, R.S.K. Chaganti
Manuscript writing: James E. Korkola, Jane Houldsworth, Adam B. Olshen, Li-Xuan Qin, George J. Bosl, R.S.K. Chaganti
Final approval of manuscript: James E. Korkola, Jane Houldsworth, Darren R. Feldman, Adam B. Olshen, Li-Xuan Qin, Sujata Patil, Victor E. Reuter, George J. Bosl, R.S.K. Chaganti
REFERENCES
- 1.Bosl GJ, Motzer RJ. Testicular germ-cell cancer. N Engl J Med. 1997;337:242–253. doi: 10.1056/NEJM199707243370406. [DOI] [PubMed] [Google Scholar]
- 2.Ulbright TM. Germ cell neoplasms of the testis. Am J Surg Pathol. 1993;17:1075–1091. doi: 10.1097/00000478-199311000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Korkola JE, Houldsworth J, Chadalavada RS, et al. Down-regulation of stem cell genes, including those in a 200-kb gene cluster at 12p13.31, is associated with in vivo differentiation of human male germ cell tumors. Cancer Res. 2006;66:820–827. doi: 10.1158/0008-5472.CAN-05-2445. [DOI] [PubMed] [Google Scholar]
- 4.Korkola JE, Houldsworth J, Dobrzynski D, et al. Gene expression-based classification of nonseminomatous male germ cell tumors. Oncogene. 2005;24:5101–5107. doi: 10.1038/sj.onc.1208694. [DOI] [PubMed] [Google Scholar]
- 5.Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 6.Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tibshirani R, Hastie T, Narasimhan B, et al. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci U S A. 2002;99:6567–6572. doi: 10.1073/pnas.082099299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beer DG, Kardia SL, Huang CC, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 9.Westfall PH, Young SS. Resampling-based multiple testing: Examples and methods for p-value adjustment. New York, NY: John Wiley and Sons; 1993. [Google Scholar]
- 10.Zeeberg BR, Feng W, Wang G, et al. GoMiner: A resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kondagunta GV, Bacik J, Bajorin D, et al. Etoposide and cisplatin chemotherapy for metastatic good-risk germ cell tumors. J Clin Oncol. 2005;23:9290–9294. doi: 10.1200/JCO.2005.03.6616. [DOI] [PubMed] [Google Scholar]
- 12.Kondagunta GV, Sheinfeld J, Mazumdar M, et al. Relapse-free and overall survival in patients with pathologic stage II nonseminomatous germ cell cancer treated with etoposide and cisplatin adjuvant chemotherapy. J Clin Oncol. 2004;22:464–467. doi: 10.1200/JCO.2004.07.178. [DOI] [PubMed] [Google Scholar]
- 13.Bajorin DF, Sarosdy MF, Pfister DG, et al. Randomized trial of etoposide and cisplatin versus etoposide and carboplatin in patients with good-risk germ cell tumors: A multi-institutional study. J Clin Oncol. 1993;11:598–606. doi: 10.1200/JCO.1993.11.4.598. [DOI] [PubMed] [Google Scholar]
- 14.Motzer RJ, Nichols CJ, Margolin KA, et al. Phase III randomized trial of conventional-dose chemotherapy with or without high-dose chemotherapy and autologous hematopoietic stem-cell rescue as first-line treatment for patients with poor-prognosis metastatic germ cell tumors. J Clin Oncol. 2007;25:247–256. doi: 10.1200/JCO.2005.05.4528. [DOI] [PubMed] [Google Scholar]
- 15.Bosl GJ, Geller NL, Bajorin D, et al. A randomized trial of etoposide + cisplatin versus vinblastine + bleomycin + cisplatin + cyclophosphamide + dactinomycin in patients with good-prognosis germ cell tumors. J Clin Oncol. 1988;6:1231–1238. doi: 10.1200/JCO.1988.6.8.1231. [DOI] [PubMed] [Google Scholar]
- 16.International Germ Cell Consensus Classification. A prognostic factor-based staging system for metastatic germ cell cancers: International Germ Cell Cancer Collaborative Group. J Clin Oncol. 1997;15:594–603. doi: 10.1200/JCO.1997.15.2.594. [DOI] [PubMed] [Google Scholar]
- 17.Garriga-Canut M, Roopra A, Buckley NJ. The basic helix-loop-helix protein, sharp-1, represses transcription by a histone deacetylase-dependent and histone deacetylase-independent mechanism. J Biol Chem. 2001;276:14821–14828. doi: 10.1074/jbc.M011619200. [DOI] [PubMed] [Google Scholar]
- 18.Ju YT, Chang AC, She BR, et al. Gas7: A gene expressed preferentially in growth-arrested fibroblasts and terminally differentiated Purkinje neurons affects neurite formation. Proc Natl Acad Sci U S A. 1998;95:11423–11428. doi: 10.1073/pnas.95.19.11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee SS, Fitch D, Flajnik MF, et al. Rearrangement of immunoglobulin genes in shark germ cells. J Exp Med. 2000;191:1637–1648. doi: 10.1084/jem.191.10.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sugimura J, Foster RS, Cummings OW, et al. Gene expression profiling of early- and late-relapse nonseminomatous germ cell tumor and primitive neuroectodermal tumor of the testis. Clin Cancer Res. 2004;10:2368–2378. doi: 10.1158/1078-0432.ccr-03-0341. [DOI] [PubMed] [Google Scholar]
- 21.Rubin LL, de Sauvage FJ. Targeting the Hedgehog pathway in cancer. Nat Rev Drug Discov. 2006;5:1026–1033. doi: 10.1038/nrd2086. [DOI] [PubMed] [Google Scholar]
- 22.Koyabu Y, Nakata K, Mizugishi K, et al. Physical and functional interactions between Zic and Gli proteins. J Biol Chem. 2001;276:6889–6892. doi: 10.1074/jbc.C000773200. [DOI] [PubMed] [Google Scholar]
- 23.Michiels EM, Oussoren E, Van Groenigen M, et al. Genes differentially expressed in medulloblastoma and fetal brain. Physiol Genomics. 1999;1:83–91. doi: 10.1152/physiolgenomics.1999.1.2.83. [DOI] [PubMed] [Google Scholar]
- 24.Wong YF, Cheung TH, Lo KW, et al. Identification of molecular markers and signaling pathway in endometrial cancer in Hong Kong Chinese women by genome-wide gene expression profiling. Oncogene. 2007;26:1971–1982. doi: 10.1038/sj.onc.1209986. [DOI] [PubMed] [Google Scholar]
- 25.Houldsworth J, Heath SC, Bosl GJ, et al. Expression profiling of lineage differentiation in pluripotential human embryonal carcinoma cells. Cell Growth Differ. 2002;13:257–264. [PubMed] [Google Scholar]
- 26.Fuccillo M, Joyner AL, Fishell G. Morphogen to mitogen: The multiple roles of hedgehog signalling in vertebrate neural development. Nat Rev Neurosci. 2006;7:772–783. doi: 10.1038/nrn1990. [DOI] [PubMed] [Google Scholar]
- 27.Korkola JE, Heck S, Olshen AB, et al. In vivo differentiation and genomic evolution in adult male germ cell tumors. Genes Chromosomes Cancer. 2008;47:43–55. doi: 10.1002/gcc.20504. [DOI] [PubMed] [Google Scholar]
- 28.Yu YP, Luo JH. Myopodin-mediated suppression of prostate cancer cell migration involves interaction with zyxin. Cancer Res. 2006;66:7414–7419. doi: 10.1158/0008-5472.CAN-06-0227. [DOI] [PubMed] [Google Scholar]
- 29.Sanchez-Carbayo M, Schwarz K, Charytonowicz E, et al. Tumor suppressor role for myopodin in bladder cancer: Loss of nuclear expression of myopodin is cell-cycle dependent and predicts clinical outcome. Oncogene. 2003;22:5298–5305. doi: 10.1038/sj.onc.1206616. [DOI] [PubMed] [Google Scholar]
- 30.Masters JR, Koberle B. Curing metastatic cancer: Lessons from testicular germ-cell tumours. Nat Rev Cancer. 2003;3:517–525. doi: 10.1038/nrc1120. [DOI] [PubMed] [Google Scholar]
- 31.Mueller T, Mueller LP, Luetzkendorf J, et al. Loss of Oct-3/4 expression in embryonal carcinoma cells is associated with induction of cisplatin resistance. Tumour Biol. 2006;27:71–83. doi: 10.1159/000092324. [DOI] [PubMed] [Google Scholar]
- 32.Stephenson AJ, Sheinfeld J. The role of retroperitoneal lymph node dissection in the management of testicular cancer. Urol Oncol. 2004;22:225–233. doi: 10.1016/j.urolonc.2004.04.029. discussion 234-235. [DOI] [PubMed] [Google Scholar]
- 33.Toner GC, Panicek DM, Heelan RT, et al. Adjunctive surgery after chemotherapy for nonseminomatous germ cell tumors: Recommendations for patient selection. J Clin Oncol. 1990;8:1683–1694. doi: 10.1200/JCO.1990.8.10.1683. [DOI] [PubMed] [Google Scholar]
- 34.Mazumdar M, Bacik J, Tickoo SK, et al. Cluster analysis of p53 and Ki67 expression, apoptosis, alpha-fetoprotein, and human chorionic gonadotrophin indicates a favorable prognostic subgroup within the embryonal carcinoma germ cell tumor. J Clin Oncol. 2003;21:2679–2688. doi: 10.1200/JCO.2003.03.136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.