

Abstract
Aquaporin 11 (AQP11) is a newly described member of the protein family of transport channels. AQP11 associates with the endoplasmic reticulum (ER) and is highly expressed in proximal tubular epithelial cells in the kidney. Previously, we identified and characterized a recessive mutation of the highly conserved Cys227 to Ser227 in mouse AQP11 that caused proximal tubule (PT) injury and kidney failure in mutant mice. The current study revealed induction of ER stress, unfolded protein response, and apoptosis as molecular mechanisms of this PT injury. Cys227Ser mutation interfered with maintenance of AQP11 oligomeric structure. AQP11 is abundantly expressed in the S1 PT segment, a site of major renal glucose flux, and Aqp11 mutant mice developed PT-specific mitochondrial injury. Glucose increased AQP11 protein expression in wild-type kidney and upregulation of AQP11 expression by glucose in vitro was prevented by phlorizin, an inhibitor of sodium-dependent glucose transport across PT. Total AQP11 levels in heterozygotes were higher than in wild-type mice but were not further increased in response to glucose. In Aqp11 insufficient PT cells, glucose potentiated increases in reactive oxygen species (ROS) production. ROS production was also elevated in Aqp11 mutation carriers. Phenotypically normal mice heterozygous for the Aqp11 mutation repeatedly treated with glucose showed increased blood urea nitrogen levels that were prevented by the antioxidant sulforaphane or by phlorizin. Our results indicate an important role for AQP11 to prevent glucose-induced oxidative stress in proximal tubules.
Keywords: proximal tubules, acute kidney injury, protein oligomerization
conventional aquaporins (AQPs) are oligomeric transport protein characterized by the presence of two highly conserved Asn-Pro-Ala (NPA) signature motifs (18, 19). AQP11 is a member of a new aquaporin subgroup AQP11/AQP12, characterized by a poorly conserved NH2-terminal signature motif (17). Previous publications have indicated that AQP11 is a membrane-bound protein of the endoplasmic reticulum (ER), suggesting an important role for AQP11 in ER homeostasis and a role in water translocation to subcellular membranous formations to maintain cytosolic and/or vesicle osmoregulation (10, 17, 20, 38). In the kidney, AQP11 is abundantly expressed in proximal tubule (PT; Refs. 17, 28, 30). We recently identified and characterized a sudden juvenile death syndrome (sjds) mutation of Cys227 to Ser227 in Aqp11sjds/sjds mice. Using genetic complementation analysis, we showed that loss-of-function Cys227Ser mutation caused severe PT damage and kidney injury in Aqp11sjds/sjds mice that die due to kidney failure by the age of 20 days old (28, 29). Cysteine is an important constituent of protein oligomeric formation, and it is plausible that the oligomeric state of cysteine-rich AQP11 determines its function. At present, it remains unclear whether Cys227 is essential in the formation of AQP11 oligomers.
Aqp11sjds/sjds mutants and Aqp11 null mice develop a similar lethal phenotype with vacuole formation in proximal tubular cells (PTC; Refs. 28, 30). The mechanisms of tubular injury in Aqp11-deficient mice remains unclear (10, 17, 20, 38). Recent data indicate that ER disturbance and stress lead to kidney tubular atrophy and injury (11). Our studies provide evidence that the Cys227Ser deactivating mutation causes the alteration of protein oligomerization and induces kidney injury via induction of ER stress and unfolded protein response (UPR). The PT, a major site of glucose transport in the kidney, has emerged as a target for novel therapeutic strategies in the treatment of diabetes and its complications that emphasize the importance of PT in glucose regulation in kidney (3, 12, 33). Hyperglycemia-induced reactive oxygen species (ROS) play a major role in the tubular injury. Genetic factors predisposing PTC to hyperglycemia-induced injury remain uncertain. Our studies provide strong evidence that AQP11 expression is regulated by glucose and that this response is lost in Aqp11 insufficient kidney. The Aqp11 insufficiency in PT predisposed kidney to glucose-induced renal dysfunction that was prevented by antioxidant therapy as well as by inhibition of glucose transport in PT.
MATERIALS AND METHODS
Animals
All experimental procedures were in compliance with the Vanderbilt University Guide for Care and Use of Laboratory Animals (protocol M/11/011). The Aqp11sjds/+ mouse line was generated previously (28). Mice were housed in a pathogen-free veterinary facility accredited by the American Association for the Accreditation of Laboratory Animal Care. Mice were maintained under a controlled 12-h light-dark cycle at a constant temperature of 21 ± 2°C and humidity of 55 ± 10%. Carriers of the sjds mutation were identified by Aqp11sjds allele genotyping using a PCR reaction as described previously (28).
Mice on the high-carbohydrate diet (Test Diet, Richmond, IN) were repeatedly injected intraperitoneally with glucose at 3 g/kg body wt with or without 2 mg/kg body wt antioxidant sulforaphane (SNF; Sigma, St. Louis, MO) for 3 days. SNF was dissolved in DMSO and then diluted to 0.2 mg/ml with sterile saline (1× concentration). Control mice on a normal rodent diet were injected with vehicle solution with or without SNF.
Prolonged pretreatment with phlorizin (Sigma), an inhibitor of renal and intestinal sodium-dependent glucose transport, was performed in mice on the high-carbohydrate diet with intraperitoneal administration of 3 g/kg body wt glucose for 24 days. Phlorizin (0.4 g/kg body wt) was administered subcutaneously every day in two equally divided doses to provide inhibition of glucose transport in the PT. Phlorizin (100×) was prepared in 100% of ethanol and then diluted to 1× solution with saline solution. We did not observed any significant phlorizin-induced side effects in mice including body weight and diarrhea; BUN or plasma creatinine levels were not affected in control mice.
Tissue Homogenates
Frozen tissue was crushed and then mixed with chilled lysis buffer (10× of the original weight of frozen tissue) containing 1× protease inhibitors (7) using a Teflon pestle and a Jumbo Stirrer (Fisher Scientific, Pittsburg, PA). Homogenates were centrifuged at 10,000 g at 4° C for 10 min, and the supernatants were and stored at −80° C. Protein concentration was determined in supernatant by Bradford assay using BSA as standard protein.
Western Blot Analysis
Western blot analysis for detection of heavy chain immunoglobulin binding protein (BiP) was performed on whole kidney tissue homogenates with polyclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) using techniques previously described (13). β-Actin was used as a loading control (Santa Cruz Biotechnology).
Detection of total AQP11 was performed using renal cortex tissue homogenates (3 μg per lane) that were resolved on denaturing SDS-PAGE under reducing conditions with β-mercaptoethanol using NuPAGE 12% Bis-Tris gels (Invitrogen, Carlsbad, CA). Separated proteins were transferred to polyvinylidene difluoride (PVDF) membranes using Tris-glycine transfer buffer at 20 V overnight at room temperature.
Native gel electrophoresis for detection of AQP11 quaternary structure from WT, Aqp11sjds/sjds, and Aqp11sjds/+ mice was performed using native PAGE 3–8% Tris-acetate gels (Invitrogen, Carlsbad, CA) as previously described (7). This Tris-acetate system allowed visualization of multiple forms of AQP11, whereas Bis-Tris and traditional Tris-glycine systems failed to do so (Atochina-Vasserman E. N. and E. Abramova E., unpublished observations). Equal amounts of total protein (20 μg per lane) were mixed with a cold native Tris-glycine sample buffer before loading. Electrophoresis was run at room temperature at a constant voltage of 150 V for 2 h. To estimate the molecular mass of AQP11 oligomers, Native MARK Unstained Protein Standard (Novex, Life Technologies, Grand Island, NY) was used. The marker with the samples was stained in the cut piece of gel before transfer using Colloidal Blue Staining Kit (Invitrogen). Proteins were transferred to PVDF membranes using Tris-glycine transfer buffer at 20 V overnight at room temperature.
Immunoblotting was performed using primary rabbit polyclonal antibody to the COOH terminus of AQP11 (1:1,000; Alpha Diagnostic, San Antonio, TX). Bands were visualized using enhanced chemiluminescence detection system (GE-Healthcare, Piscataway, NJ) and quantitated by densitometric scanning of exposed films (Eastman Kodak, Rochester, NY) using the Bio-Rad Gel Image System with Quantity One program (Bio-Rad, Hercules, CA). Specificity of AQP11 antibodies was confirmed by preincubation of AQP11 antibodies with an excess of blocking peptide containing the amino acid sequence of AQP11 (Alpha Diagnostic). To test the specificity of the immunoblotting, blots were preincubated with secondary antibodies only. Blots were visualized with exposure time for 10 min and showed no staining signals (data are not shown).
Plasmid Construction, Transfection, and Western Blot Analysis
The Myc-tagged AQP11 construct was made by in-frame subcloning of human AQP11 cDNA into pCMV-Myc (BD Biosciences Clontech, CA). The mutation of the construct was introduced by site-directed mutagenesis (Stratagene, La Jolla, CA). CHO-K1 cells (CCL-61 obtained from ATCC) were transfected at 50–60% confluency using Lipofectamine LTX (Invitrogen).
Transfected CHO-K1 cells were incubated with PBS containing 4% paraformaldehyde at room temperature for 15 min. Subsequently, the cells were lysed in lysis buffer (25 mM Tris pH 7.4, 150 mM NaCl, 2 mM EDTA, 1.5% Nonidet P-40, and Complete protease inhibitor mixture), and the lysate was then mixed with 2× Laemmli sample buffer containing 0.1 M dithiothreitol at 37°C for 20 min. After separation by SDS-PAGE, the protein was transferred to a PVDF membrane and the protein on the membrane associated with antibodies was detected by a Super Signal chemiluminescence detection system (Pierce, Rockford, IL). The anti-Myc antibody was from BD Biosciences Clontech.
Electron Microscopy
Portions of cortex were fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4), processed and embedded in Spurr resin. Transmission electron microscopy was performed using a FEI/Phillips CM12 transmission electron microscope. Electron micrographs were taken at ×6,000 magnification using a systematic uniform random sampling protocol.
Cell Culture
A well-characterized PT epithelial cell line, MCT cells (37), was cultured in 10-mm dishes in 10 ml of DMEM/F-12 medium supplemented with 10% FBS. Cells at ∼60–70% confluence were washed with PBS and then preincubated with or without 200 μM phlorizin for 2 h before overnight incubation with 3–35 mM glucose in DMEM glucose-free medium supplemented with 1% FBS.
Small Interfering RNA Inhibition of Aqp11 Gene Expression in PTC
For silencing Aqp11 gene expression we utilized transfection of MCT cells using synthetic AQP11-specific small interfering (si)RNA sequences that were designed using Stealth RNAi (Invitrogen). Nontargeting control siRNA had a randomly chosen sequence. Cells were cultured in six-well plates in 2 ml of DMEM/F-12 medium supplemented with 10% FBS. The transfection procedures were optimized with respect to relative amounts of siRNAs, transfection agent, and incubation time in accordance with the manufacturer's instructions. Lipofectamine was mixed with a fourfold volume of Opti-MEM I and incubated for 5 min at room temperature. siRNA duplexes were mixed with 100 μl of Opti-MEM I, added to the lipofectamine/Opti-MEM I mix, and incubated for 20 min at room temperature. Cells at ∼50 to 60% confluence were washed with DMEM to remove serum and then transfected with gene-specific and control siRNAs at a concentration of 50 nM in the presence of 100 nM Lipofectamine RNAiMAX (Invitrogen) in Opti-MEM I media for 6 h. Cells were then supplemented with 10% FCS and grown for 24 h until harvesting for analysis.
Analysis of ROS Production
The oxidation-sensitive dye CM-H2DCFDA (Invitrogen-Molecular Probes, Eugene, OR) was used for the measurement of ROS production in PTC. The cells were detached from the plates by trypsinization, washed by PBS and incubated at 37°C with or without of 2 μM CM-H2DCFDA for 30 min, washed with cold PBS, and then analyzed by flow cytometry (23). The differences in mean fluorescent intensity between unstained and CM-H2DCFDA-treated cells were taken as a measure of ROS production. ROS generation was determined by the increase in CM-H2DCFDA fluorescence upon treatment of cells with glucose. Increase in dye oxidation was calculated as the percent increase in mean channel fluorescence of treated cells over untreated cells. Renal ROS production was measured using the horseradish peroxidase-linked Amplex Red fluorescence assay according to the manufacturer's protocol (Invitrogen). Pieces of renal cortex were incubated with 100 μM Amplex Red and 10 U/ml horseradish peroxidase at 37°C during 1 h. With the use of 100-μl samples of media from each sample, fluorescence readings were made in triplicate in a 96-well plate at ex/em = 530/580 nm and values were normalized to dried tissue weight (5).
RT-PCR Analysis of Xbp1 and Aqp11 Gene Expression
RNA was isolated from kidney tissue and converted to cDNA using techniques as previously described (13). Evaluation of transcription factor Xbp1 splicing, which promotes transcription of ER-associated degradation target genes, was performed using methods previously described (13). Primer sequences for Xbp1 were as follows: forward: 5′-CTG GAA AGC AAG TGG TAG A-3′ and reverse: 5′-CTG GGT CCT TCT GGG TAG AC-3′. Two microliters of cDNA were used with a reaction volume of 50 μl. PCR products were run on a 2.5% gel with 398- and 424-bp forms representing spliced and unspliced fragments respectively.
Evaluation of Aqp11 gene expression.
Total RNA was isolated from snap-frozen kidney tissue sections using TRIzol Reagent (Invitrogen) and RNeasy Midi Kit (Qiagen, Valencia, CA). Two micrograms of total RNA from each sample were reversetranscribed by SuperScript II reverse transcriptase in the presence of 40 U/μl RNaseOUT Recombinant Ribonuclease Inhibitor (Invitrogen) for 60 min at 37°C. Sense 5′-CCACCTGGACTCTGACACTGATCTAC-3′ and antisense 5′-CACCTACAGAAGGAGCAAGCCAGTAT-3′ primers from mouse AQP11 (National Center for Biotechnology Information accession no. BAC45005) amplified a 516-bp RT-PCR product during 30 cycles of denaturing at 95°C for 2 min, 95°C for 30 s, annealing at 62°C for 30 s, and extension at 72°C for 60 s. RT-PCR products of mouse AQP11 were separated on a 2% agarose gel.
Quantitative RT-PCR Analysis of Aqp11 Gene Expression
Total RNA was extracted from PTC. Reverse transcription of total RNA was performed using Superscript II reverse transcriptase (Invitrogen) followed by amplification using 2× SYBR-Green PCR Master Mix and sequence-specific primer pairs on an ABI-Prism 7900HYT Sequence Detection System (Applied Biosystems, Foster City, CA). Results were evaluated using SDS version 2.0 software (Applied Biosystems). The calculation of relative change in mRNA was performed with normalization for the housekeeping gene β-actin. Both water blank and nonreverse transcribed RNA samples were used as negative controls.
Immunohistochemistry
Immunohistochemistry for BiP and α-mannosidase-like protein (Edem) was performed using primary BiP and Edem polyclonal antibodies (Santa Cruz Biotechnology) and standard immunoperoxidase techniques on formalin-fixed, paraffin-embeded kidney tissue as previously described (13, 14).
Immunohistochemistry for AQP11 and cleaved caspase-3 was performed on formalin-fixed, paraffin-embeded kidney tissue. Slides with 4-μm tissue sections were briefly incubated with 3% H2O2 to eliminate endogenous peroxidase activity according to a standard immunohistochemical technique (25). Rabbit anti-caspase-3 antibody (1:1,000 dilution, PharMingen, San Diego, CA), DAKO EnVision systems (DAKO, Carpinteria, CA), or rabbit anti-mouse AQP11 antibody [1:200 dilution, a gift from Dr. K. Ishibashi (17)] were applied to the tissue sections and incubated overnight at 4°C. The slides were then washed in PBS and incubated with secondary goat anti-rabbit IgG horseradish peroxidase-conjugated antibodies for 30 min at room temperature, followed by washing and incubation with 3,3-diamino benzidine solution. The slides were counterstained with hematoxylin.
BUN Measurement
Plasma glucose and BUN were measured by an iSTAT analyzer (Heska, Loveland, CO) in 65 μl of whole mouse blood. Plasma creatinine was measured as described previously using HPLC system (Perkin-Elmer, Waltham, MA) (28).
Data Analysis
All data are expressed as means ± SE derived from at least triplicate observations. Chance differences probabilities (P) were calculated using Student's t-test. P < 0.05 was considered to be statistically different.
RESULTS
Induction of ER Stress, UPR, and Apoptosis in Aqp11 Mutant Kidney Injury
Previously, we have shown that the Cys227Ser mutation causes severe injury specifically in PTs in mice by the age of 3 wk (28, 30). Disturbance of ER homeostasis associates with tubular injury in kidney (11). Investigation of the molecular mechanisms of this PT injury revealed the induction of ER stress and UPR activation in Aqp11 mutant kidney as they aged. In kidneys from 1- and 2-wk-old mutant mice compared with wild-type littermates we examined the expression of a well-recognized marker of ER stress, BiP, using Western blot analysis (13, 27). The splicing of downstream UPR target transcription factor Xbp1 was examined using RT-PCR in samples from same mice. At 1 wk of age, before the onset of detectable tubular damage, there was neither increased expression of BiP nor Xbp1 splicing. In contrast, Aqp11 mutant mice had severely injured tubules and developed enhanced BiP expression and Xbp1 splicing at the age of 2 wk (Fig. 1). Spliced Xbp1 translocates to the nucleus and promotes the transcription of ER-associated degradation target genes such as Edem (13, 27). Immunohistochemistry for BiP and Edem revealed enhanced staining in epithelial cells lining injured tubules in 2-wk-old mutant mice. Kidneys with normal histological appearance from 1-wk-old mutant Aqp11sjds/sjds, Aqp11sjds/+, and wild-type mice were negative for BiP and Edem staining (Fig. 2). These findings are consistent with our hypothesis that Aqp11 mutation results in an improper function of ER-resident AQP11 protein and associates with ER stress.
Fig. 1.

Progression of endoplasmic reticulum (ER) stress and unfolded protein response (UPR) in Aqp11sjdsj/sjds mutant mice with age. A: Western blot for binding protein (BiP) from whole kidney lysates from Aqp11sjds/sjds (sjds/sjds), Aqp11sjds/+ (sjds/+), and wild-type (WT) mice. Normalization to β-actin. B: RT-PCR gel demonstrating splice variants for Xbp1 mRNA in kidney samples from same mice.
Fig. 2.

Immunohistochemical staining of ER stress and UPR markers in injured tubules in sections of kidney tissue from Aqp11sjds/sjds (sjds/sjds), Aqp11sjds/+ (sjds/+), and WT mice, using peroxidase-diaminobenzidine procedure. Note positive staining of ER stress marker BiP and a marker of protein degradation in ER α-mannosidase-like protein (Edem) in epithelial cells lining injured proximal tubules in kidney from 2-wk-old mice. Counterstaining with periodic acid-Schiff reagent (magnification: ×400).
ER stress and proapoptotic activation factors induction lead to mitochondrial dysfunction. We examined mitochondria in kidneys from 16-day-old wild-type and Aqp11sjds/sjds mice (Fig. 3, A–C). There was renal PT-specific mitochondrial injury in Aqp11 mutant mice. Severe changes in mitochondrial ultrastructure of the vacuolated and structurally altered PTCs were observed in 60–70% of the mitochondrial population in mutant kidney (Fig. 3B) compared with wild-type littermates (Fig. 3A). In contrast, mitochondrial and cellular integrity was normal in cells from more distal nephron segments, characterized by short stubby microvilli (Fig. 3C), in kidney samples from the same mutant.
Fig. 3.
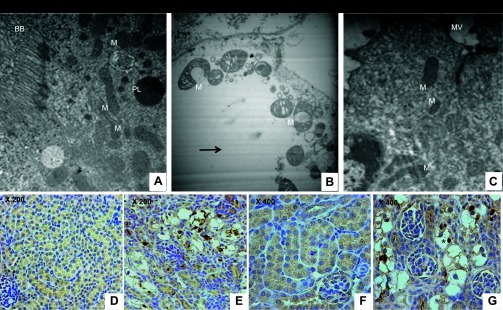
A: representative electron micrograph of mitochondria-rich proximal tubular cell in kidney from 16-day-old WT mouse (BB, brush border; M, mitochondria). B: characteristic cytoplasm vacuolization and severe destruction of mitochondrial material in epithelium in severely injured PT (identified and characterized in previous study; Refs. 28, 29) in kidney from Aqp11sjds/sjds mutant littermate. C: in the same Aqp11sjds/sjds mutant mouse, more distal segment of nephron characterized by short stubby microvilli (MV) showed normal cell and mitochondria ultrastructure. Magnification: ×6,000. Activated caspase-3 immunoreactivity in Aqp11sjds/sjds injured tubules. Representative (n = 4) photomicrographs showing active caspase-3 in WT (D, ×200; F, ×400) and mutant (E, ×200; G, ×400) kidneys from 16-day-old mice. Active caspase-3-positive tubules showed remarkable increases in caspase-3 in cytoplasm and nuclei of injured cells (*: dark brown nuclei). Tumor tissue was used as an independent positive control for activated caspase-3 (data not shown).
Mitochondrial dysfunction and ER stress are known to contribute to activation of caspase-3, a downstream executioner of the ER/mitochondria-mediated cell death, and its translocation from cytosol to nucleus (1). Using immunohistochemical analysis for activated caspase-3, we detected marked elevation of activated caspase-3 in injured tubules, with a strong signal in the nuclei of the damaged cells in Aqp11sjds/sjds mice (Fig. 3, E and G) compared with wild-type littermates (Fig. 3, D and F).
Mitochondria are well-known ROS-generating sites and are important mediators of oxidative stress (15). PTs are rich in mitochondria. It is apparent that mitochondrial dysfunction leads to elevated ROS production in mutant kidney. We found significantly higher levels of superoxide in kidney cortex from 14-day-old Cys227Ser mutation carriers, the age when severe mitochondrial injury appears in PTs. The Cys227Ser mutation carriers showed significantly higher superoxide production in the kidney cortex than wild-type littermates (3.18 ± 0.26 vs. 0.51 ± 0.23 nmol/mg; P < 0.02; n = 4–5 per group). Activation of caspase-3 and increased ROS production may ultimately result in PT injury in mutant mice.
Alteration of AQP11 Protein Oligomeric Structure and Expression in Aqp11 Mutant Kidney
Most of AQPs function as oligomers. Cysteine residues have previously been shown to act as dynamic regulators of oligomerization (7). Protein misfolding triggers ER stress-induced cell death. We hypothesized that that Cys227Ser mutation might affect AQP11 protein oligomeric structure.
Renal cortex homogenates from wild-type, Aqp11sjds/sjds, and Aqp11sjds/+ mice at the age 10–16 days were subjected to native PAGE using Tris-acetate system and an antibody to the COOH terminus of AQP11. As demonstrated in Fig. 4, A, top, two major forms were characteristic for the wild-type AQP11. Previous studies suggested that AQP11 formed multimeric structures similar to AQP4 (6, 26) for which putative tetramer and higher order multimers were detected (26). Based on the position of the AQP11 bands in native PAGE relative to the molecular mass markers (Fig. 4B), both lower (below 480-kDa marker) and higher (below 1,048-kDa marker) forms are likely to represent multimers of AQP11 with differing order of oligomerization. Given the fact that physicochemical properties of AQP11 are yet unknown and that inherent difficulties and inaccuracies are associated with estimation of molecular mass of multimeric proteins using native PAGE, additional studies would be required for identification of the exact size of the AQP11 oligomeric forms and the number of constituent monomers.
Fig. 4.

Panel 1: Cys227Ser mutation modulates AQP11 oligomeric structure in Aqp11sjds/sjds mutant kidney. A: tissue samples containing equal amounts of total protein (20 μg per lane) were subjected to native gel electrophoresis to determine oligomeric structure of AQP11 in renal cortex from Aqp11sjds/sjds (sjds/sjds), Aqp11sjds/+ (sjds/+), and WT mice (top) at the age 10–16 days. Aqp11sjds/sjds mice demonstrated an additional intermediate forms confirming that AQP11 complex is disassembled. Bands were visualized using enhanced chemiluminescence detection system. The same samples (3 μg per lane) were subject to SDS-PAGE under reduced conditions followed by immunoblotting with anti-AQP11 antibody (middle). To monitor equal protein loading the same blot was stripped and re-probed with GAPDH (bottom). B: protein molecular mass marker used to estimate the molecular mass of AQP11 oligomeric structure. Panel 2: Densitometric quantification of total AQP11 monomer bands in reducing SDS-PAGE normalized to GAPDH (C), AQP11 lower forms (D), and intermediate forms (E) in native PAGE. Intensities for both forms were normalized to the total AQP11 values assessed in SDS-PAGE of the same samples. Data presented as a percentage of WT level. Black, light grey, and grey bars are WT, Aqp11sjds/sjds (sjds/sjds), and Aqp11sjds/+ (sjds/+), respectively. Values are presented as means ± SE (n = 4–5 mice per group; statistical data are specifically presented for the samples where both SDS and native PAGE were performed). *P < 0.05, **P < 0.01 vs. WT.
All AQP11 forms were also seen in heterozygous Aqp11sjds/+ samples. In contrast, the lower molecular mass form of AQP11 in Aqp11sjds/sjds mouse kidney was dramatically decreased and an additional major intermediate band representing different oligomeric structure was detected (Fig. 4A, top). Figure 4, D and E, demonstrates statistically significant loss of lower order molecular mass form and an increase of intermediate form in Aqp11sjds/sjds renal cortex. The fact that the proportion of lower order molecular mass form is lowest in homozygous mutants suggests that its loss results in the development of PT injury and that this form is a major functional unit of AQP11. It is worth noting that this lower oligomeric AQP11 form was increased in samples from heterozygoutes suggesting a compensatory transcriptional mechanism.
To confirm that Cys227Ser mutation affects AQP11 oligomerization we expressed Myc-tagged wild-type and mutant human AQP11, Myc-hAQP11, or Myc-hAQP11-Cys227Ser, respectively, in CHO-K1 cells. We previously reported that paraformaldehyde was a useful cross-linker for detection of the oligomeric structure of AQP11 (7). Therefore, we used paraformaldehyde to evaluate the ability of AQP11 and its mutant to assemble the higher order structures. After fixation of cells expressing Myc-hAQP11 or Myc-hAQP11-Cys227Ser, cellular proteins were analyzed by SDS-PAGE and Western blotting using anti-Myc antibodies (Fig. 5A). The analysis of cells expressing Myc-hAQP11 yielded various bands heavier than the monomer at ∼29 kDa providing proof that hAQP11 forms higher order structures. In contrast, the abundance of oligomeric forms was reduced in mutant protein. Thus we concluded that Cys227Ser mutant in AQP11 indeed affects protein oligomerization. Additionally, higher multimeric forms of AQP11 shown in Fig. 4A were missing. Paraformaldehyde has been thought to mainly cross-link proteins whose distance is <3 Å (8). Therefore, if the AQP11 multimers include more loosely interacted proteins, our method may exclude the detection of these multimers. The absence of higher forms of AQP11 multimers after cross-linking may also indicate that the formation of high AQP11 polymers might be induced by different mechanisms as it has been shown for AQP4 (4). Visualization of multimeric structures also might be affected by reducing SDS-PAGE conditions. More detailed identification of multimeric structures requiring an extensive optimization of cross-linking conditions or using alternative cross-linking reagents, was out of the scope of the current study.
Fig. 5.

Cys227Ser mutation in human AQP11 affects protein oligomerization. A: oligomerization state of each protein was evaluated using a chemical cross-linker, paraformaldehyde. Typical examples of oligomerization of Myc-hAQP11 (left) and Myc-hAQP11-Cys227Ser (middle). Right: same as middle at 20-fold longer exposure time. B: densitometric analysis of cross-linked Myc-hAQP11 (black bar) and Myc-hAQP11-Cys227Ser (grey bar). Oligomerization state of each protein was evaluated using the ratio of cross-linked AQP11 to the total AQP11 (monomers plus cross-linked proteins). Values are presented as means ± SE. **P < 0.01 vs. Myc-hAQP11.
To determine whether Cys227Ser mutation could affect expression of AQP11 protein, renal cortex samples were collected from wild-type, homozygous Aqp11sjds/sjds, and heterozygous Aqp11sjds/+ mice and analyzed by reducing SDS-PAGE and Western blot (Fig. 6). In all samples we observed a ∼29-kDa band consistent with the molecular mass of monomeric AQP11. Figure 6 demonstrated that expression of AQP11 protein in Aqp11sjds/sjds mice was twofold lower when compared with wild type (Fig. 6A), which is consistent with our previous study with AQP11 mRNA levels in mutant type cortex (28). Total AQP11 protein expression in heterozygous Aqp11sjds/+ mice was significantly increased when compared with wild-type mice (Fig. 6B). It is likely that the increased expression of AQP11 is a transcriptional compensation for loss of a functional allele of Aqp11 gene. On the other hand, increased AQP11 transcription of both wild-type and mutant alleles results in accumulation of structurally altered protein within the ER of PTC that might modulate kidney susceptibility to injury in Aqp11sjds/+ mice.
Fig. 6.

AQP11 expression in Aqp11 mutant mice. A: representative blots and densitometric quantification of AQP11 protein expression in renal cortex tissue samples from WT, Aqp11sjds/sjds (sjds/sjds), (A) and Aqp11sjds/+ (sjds/+) mice (B) at 10–16 days of age. Samples of renal cortex tissue were analyzed for total AQP11 protein expression by SDS-PAGE under reduced conditions followed by Western immunoblotting with anti-AQP11 antibody. B: densitometric quantification of AQP11 was normalized to GAPDH and presented as a percentage of WT level. Values are shown as means ± SE. Black bars: WT; light grey bar: Aqp11sjds/sjds (sjds/sjds); dark grey bar: Aqp11sjds/+ (sjds/+); 12–15 mice per group, *P < 0.005, **P < 0.01 vs. WT level (statistical data are presented for all samples where SDS-PAGE was performed).
Glucose-Mediated Regulation of AQP11 Expression
PTs are a major site of glucose flux and ROS production in kidney. Recent studies suggest that PTs might be a triggering site of progression of hyperglycemia-induced oxidative stress and kidney injury (16). However, predisposing factors are yet to be elucidated. We sought to examine whether there is a link between AQP11 and glucose in kidney.
The bulk of the glucose reabsorption and transport along the nephron is mediated by the S1 segment in the PT. We first demonstrated that AQP11 is localized in S1 segment of PT to determine whether AQP11 expression can be regulated by glucose. Immunolabeling of AQP11 was evident in S1 segments. They were identified here as PTs arising from the urinary pole of Bowman's capsular space and its parietal epithelium. No AQP11 immunolabeling was associated with the glomeruli (Fig. 7, A and B).
Fig. 7.

AQP11 protein expression in kidneys from 20-day-old mice. A and B: glomeruli with emerging AQP11 positive convoluted PT (S1 segment) from urinary pole of Bowman's capsule (arrow; magnification: ×400). C: kidney sections from AQP11 knockout (KO) mice at the same age served as a negative control and showed no immunostaining in kidney tissue (magnification: ×400). D: WT kidney section stained with no primary antibody served as a second negative control (magnification: ×400). E: severely deformed AQP11-positive tubules in the Aqp11sjdssjds kidney. F: in WT kidney strong AQP11 immunolabeling is seen in the cortex (CX) and outer stripe of outer medulla (O/M). I/M, inner stripe of outer medulla (n = 5–6 per group; magnification: ×100). Scale bar = 300 μm. Smaller size of mutant kidney section (E) reflects reduced size of kidney in homozygous Aqp11sjds/sjds mutants when compared with kidney from WT littermate (F) and confirms our previously published data (28).
The regulation of AQP11 expression in kidney is largely unknown. Gene and protein expression of AQP11 was examined in the well-characterized kidney PTC line, MCT (37), cultured overnight in glucose-free medium with or without added glucose. Both Aqp11 gene transcripts and protein were increase in glucose-treated cells (Figs. 8 and 9). Phlorizin, an inhibitor of the sodium-glucose cotransporter Sglt abrogated the induction of AQP11 expression by glucose, whereas by itself it did not have any effect on AQP11 expression (Fig. 9).
Fig. 8.

Glucose-induced AQP11 expression. A: real-time quantitative RT-PCR analysis of glucose-induced Aqp11 gene expression in PT cells (PTC) cultured overnight in glucose-free medium supplemented with or without 3–25 mM glucose. Results are expressed as means ± SE, *P < 0.005 vs. untreated cells. All samples were in triplicate and mRNA samples were isolated from each well. B: representative RT-PCR analysis (n = 4) revealed a similar glucose-induced Aqp11 gene expression (516-bp PCR product) in PTC (samples were normalized to total mRNA). Cells were cultured in glucose-free DMEM supplemented with 1% FBS and with or without 3–25 mM glucose overnight.
Fig. 9.

A: representative blots of AQP11 protein expression in PTC treated with indicated concentrations of phlorizin and cultured in glucose-free medium overnight; 20 mM mannitol was used as osmotic control and β-tubulin served as a loading control. B and C: AQP11 protein expression in PTC pretreated with or without 200 μM phlorizin during 2 h prior overnight incubation with 3–35 mM glucose in glucose-free medium. All samples were in quadruplicate and cell samples were collected from each plate. Collected cell samples were analyzed for total AQP11 protein expression by SDS-PAGE under reduced conditions followed by Western immunoblotting with anti-AQP11 antibody. D: densitometric quantification of AQP11 was normalized to β-tubulin and presented as a percentage of WT level. Values are shown as means ± SE. Light grey bars: cells treated with 3–35 mM glucose. Black bars: cells pretreated with 200 μM phlorizin prior incubation with 3–35 mM glucose. *P < 0.05 vs. untreated cells. Data shown are representative of 2 independent experiments.
Aqp11 protein expression in vivo was assessed in kidneys from adult Aqp11sjds/+ heterozygotes and wild-type mice challenged with or without glucose (3 g/kg body wt). Glucose challenge resulted in a significant increase in Aqp11 expression in wild-type kidneys but had minimal effect in heterozygous mice (Fig. 10). In addition, no change was observed in mutant homozygotes treated with high glucose (data are not shown). It is worth noting that because of the mutant allele the level of wild-type AQP11 remains lower in glucose-treated Aqp11sjds/+ mice than in glucose-treated wild-type mice.
Fig. 10.
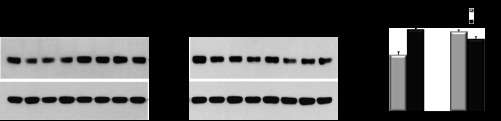
Representative blots of AQP11 protein expression in renal cortex tissue samples from adult WT (A) and Aqp11sjds/+ (sjds/+) (B) mice (5–7 mice per group) intraperitoneally treated with or without 3 g/kg body wt glucose. Samples of renal cortex tissue (3 μg total protein per lane) were analyzed for total AQP11 protein expression by SDS-PAGE under reduced conditions followed by Western immunoblotting with anti-AQP11 antibody. C: densitometric quantification of AQP11 was normalized to GAPDH and presented as a percentage of WT level. Values are shown as means ± SE. Light grey bars: glucose-treated mice. Black bars: untreated mice. Data shown are representative of 3 independent experiments. *P < 0.05; **P < 0.01 vs. WT level.
Inhibition of Aqp11 Gene Expression via RNA Interference in PTC
In PTC glucose cytotoxicity has been suggested to mediate oxidative stress and apoptosis (24). Glucose-induced ROS production is one of the key mediators of kidney stress. To determine whether PTC susceptibility to glucose-induced oxidative stress is directly linked to the Aqp11 insufficiency, we utilized an in vitro approach using siRNA inhibition of Aqp11 gene to partially reduce AQP11 expression in MCT cells, thus mimicking AQP11 insufficiency in heterozygous mice.
Reduction of Aqp11 gene expression in transfected cells was observed after 24 h (Fig. 11). Aqp11 specific siRNAs reduced mRNA expression with the most effective siRNA T#209 rendering a 62.9 ± 1.2% inhibition (Fig. 11A).
Fig. 11.

A: real-time quantitative (q)RT-PCR showed that siRNAs decrease Aqp11 mRNA in MCT cells. Cells were transfected with T#209, T#241, T#348 Aqp11-specific small interfering (si)RNA duplexes, and nonspecific control siRNA (C siRNA). Whole-cell extracts were analyzed for Aqp11 gene expression by qRT-PCR. Results are means ± SE for 3 replicate determinations for each treatment group (P < 0.05) cells. B: levels of ROS production in the control (C siRNA) and AQP11 siRNA-transfected (T#209) MCT cells treated with (black bars) or without (light gray bars) 3 mM glucose during overnight. Phorbol 12-myristate 13-acetate (PMA)-treated cells were used as a positive control for the induction of reactive oxygen species (ROS) production. Cells were cultured in glucose-free supplemented with or without 3 mM glucose. Values are means ± SE from replicates of 3 different samples (*P < 0.005).
We assessed ROS production in PTC transfected with Aqp11 T#209 or siRNA nontargeting control siRNA using the oxidation-sensitive dye CM-H2DCFDA. Nontransfected cells were incubated with 0.1 mM phorbol 12-myristate 13-acetate (PMA), a stimulator of ROS formation (36), as a positive control. The baseline ROS was increased by 62% in Aqp11 siRNA-transfected cells but was lower than in PMA-stimulated cell (Fig. 11B, light bars). The Aqp11 siRNA-transfected cells were cultured in glucose-free medium and treated with physiologically low 3 mM glucose overnight. We avoided using higher glucose concentrations to prevent glucose-induced PT disturbance in wild-type cells that might mask the effect in Aqp11 insufficient PTC. The Aqp11 siRNA-transfected cells treated with 3 mM glucose showed a fourfold higher ROS production than control glucose-treated cells and more than twofold higher than untreated Aqp11 siRNA-transfected cells (Fig. 11B, black bars). The ROS production in glucose-treated Aqp11 siRNA-transfected cells was comparable with ROS production in normal cells treated with PMA.
Antioxidant SNF and the Inhibitor of Renal Glucose Transport Phlorizin Ameliorate Renal Dysfunction in Glucose-Challenged Aqp11sjds/+ Mutant Mice
Hyperglycemia-induced ROS production is one of the key mediators of kidney injury and dysfunction. To determine whether heterozygous Aqp11sjds/+ insufficient mice were susceptible to renal dysfunction in response to a glucose-mediated stress, we treated these mice with glucose injection at 3 g/kg body wt. Wild-type and Aqp11sjds/+ mice (n = 5–6 per group) were repeatedly injected intraperitoneally with glucose with or without of 2 mg/kg SNF, a well-known antioxidant (32). We found that Aqp11sjds/+ insufficient mice showed a significant increase of BUN values after 3 days of receiving 3 g/kg body wt glucose compared with vehicle Aqp11sjds/+ mice (Fig. 12) and compared with wild-type mice that showed no alteration of kidney function. The effect of glucose was completely abrogated in mice treated with 2 mg/kg body wt SNF. These data establish a direct link between AQP11 insufficiency and susceptibility to glucose-induced oxidative stress in mice.
Fig. 12.

Sulforaphane (SNF), an activator of NRF2-mediated cytoprotection, reduced BUN levels after glucose injections in heterozygous Aqp11sjds/+ mutant (sjds/+) compared with WT mice (6 mice per group). Mice received intraperitoneal injections of 3 mg/kg body wt glucose daily for 3 days with or without SNF (2 mg/kg ip). Blood urea nitrogen (BUN) was assessed after the last glucose injection. Values are shown as means ± SE. *P < 0.05; **P < 0.0005, ***P < 0.001.
After prolonged glucose administration during 24 days in the presence of the nonselective Sglt inhibitor phlorizin (0.4 g/kg body wt), Aqp11sjds/+ mice significantly reduced glucose-mediated BUN levels by day 8 that was persistent until the end of the treatment. Glucose-treated wild-type mice with phlorizin administration showed no change in BUN levels by the end of experiment (Fig. 13).
Fig. 13.

Phlorizin, an inhibitor of sodium-dependent glucose transport, reduced BUN levels after glucose injections in heterozygous Aqp11sjds/+ mutants. Aqp11+/+ (WT; black bars) and Aqp11sjds/+ (JS/+; grey bars) mice received intraperitoneal injections of 3 g/kg body wt glucose with phlorizin subcuntaneous administration (0.4 g/kg body wt) daily for 24 days. BUN was assessed on days 0, 8, and 24 of phlorizin injection. Values are shown as means ± SE. *P < 0.05 vs. glucose-treated mice without phlorizin administration; **P < 0.05 vs. WT mice that received glucose with phlorizin (n = 6 per group).
To confirm that glucose induced renal dysfunction in Aqp11sjds/+mice, we assessed plasma creatinine levels in these mice using HPLC. Glucose loading increased plasma creatinine level to 0.177 ± 0.021 vs. 0.101 ± 0.023 mg/dl in glucose-untreated mice (n = 6 per group; P < 0.01). Phlorizin significantly reduced glucose-stimulated plasma creatinine level to 0.118 ± 0.014 mg/dl (P < 0.05) and did not affect plasma creatinine in mice without glucose loading (0.113 ± 0.014 mg/dl) that was comparable with level in wild-type mice (0.09 ± 0.011 mg/dl). Plasma creatinine level in wild-type mice confirmed our previously published data (28).
DISCUSSION
Mice bearing Cys227Ser mutation in AQP11 present a genetic animal model of PT-specific kidney injury (28). The goal of the current study was to identify the underlying molecular mechanisms and the role of AQP11 insufficiency in predisposing heterozygous mice to kidney injury.
Published data identified AQP11 as residing in the ER (17, 21). The current study demonstrated that AQP11 is an important factor of normal ER functioning, as its inactivating Cys227Ser mutation causes the induction of ER stress and UPR followed by activation of caspase-3, mitochondrial dysfunction, and elevation of ROS production associated with PT injury. This sequence of events mediates Cys227Ser mutation-induced kidney injury via activation of the observed pathways.
Here we report that Cys227 is crucial for the maintenance of oligomerization of AQP11. Our data showing that AQP11 forms various multimeric structures agree with the recent observations that AQP11 may form tetramers similar to the ability of AQP4 to form tetramers and higher order multimers (6, 9, 26). Our data also agree with the recent hypothesis that the Cys227 residue might define functional characteristics of a new AQP11/AQP12 subgroup in the aquaporin superfamily (6, 9). In the mutant Aqp11sjds/sjds kidney, dysfunctional and improperly assembled AQP11 is likely to contribute to aggravation of ER stress in PTC, as indicated by the shortened life span of Aqp11sjds/sjds and compound heterozygous Aqp11sjds/− mice compared with Aqp11 null mice (28).
The important clinical implications of this study are that AQP11 insufficiency predisposes the kidney to glucose-induced oxidative stress and reduction of kidney function. In the tubular injury, hyperglycemia plays a critical role (22, 31). The inhibition of sodium-dependent glucose transport in PT is emerging as a therapeutic strategy in the treatment of hyperglycemia-induced kidney injury (2, 3, 31, 34, 35). Given that PT is a major site of glucose flux and ROS production, we examined the potential link between AQP11 insufficiency, as a novel factor of kidney injury, glucose, and ROS.
The S1 segment of PT is the major site of glucose re-absorption in kidney. Our results clearly demonstrate the localization of AQP11 in PT S1 segment in mouse kidney. Furthermore, our results establish an important link between glucose and AQP11 expression, demonstrating that wild-type AQP11 expression was upregulated upon exposure to glucose, an effect that could be abrogated by phlorizin, an inhibitor of sodium-dependent glucose transport. The maintenance of proper AQP11 levels appears to be functionally important. In Aqp11sjds/+ mice, AQP11 insufficiency was transcriptionally compensated by elevated expression. The inability of heterozygotes to further increase AQP11 expression might be a factor predisposing to glucose-induced cell injury.
ROS play a major role in the tubular injury induced by hyperglycemia (22, 31). We demonstrated that there is a correlation between higher ROS production and AQP11 insufficiency in mice. This was in agreement with the in vitro studies that showed increased ROS production in Aqp11 insufficient cells that was further augmented by exposure of these cells to even physiological levels of glucose (3 mM). We demonstrated a susceptibility to glucose-induced oxidative stress and renal function impairment in phenotypically normal heterozygous mice (Aqp11sjds/+) compared with wild-type mice, when repeatedly challenged with 3 g/kg body wt glucose, and showed that the increases in BUN were prevented by the antioxidant SNF. The glucose-challenged Aqp11sjds/+ receiving phlorizin therapy also demonstrated significant protection of kidney function after 3 wk of treatment. Neither phlorizin nor SNF affected AQP11 expression by itself indicating that the therapeutic effects of these compounds were mediated by prevention of glucose influx or oxidative stress in PT rather than by alterations in AQP11 expression. Aqp11sjds/+ mice maintained normal blood glucose levels and showed no significant changes in renal morphology during 3 wk of glucose treatment. However, our preliminary observations indicate that after 3–4 mo of sustained hyperglycemia diabetic Aqp11sjds/+ mice developed renal hypertrophy with significantly higher kidney weight-to-body weight index, BUN values, and urinary albumin/creatinine ratio values compared with control and diabetic Aqp11+/+ mice or with nondiabetic Aqp11sjds/+ mice. Histopathological study in kidneys from diabetic Aqp11sjds/+ mice revealed glomerulosclerosis and tubulointerstitial fibrosis (study in progress). The therapeutic effects of phlorizin and SNF in diabetic Aqp11sjds/+mice need to be elucidated in the future studies.
The important clinical implications of this study are that mutation and abnormal functioning of AQP11 in PTs predisposes sjds mutation carriers to renal glucose-induced oxidative stress. This is revealed by renal dysfunction in repeatedly glucose-challenged Aqp11sjds/+ heterozygous mice that were phenotypically normal before the treatment. Recently PTs have been considered as targets for therapy of diabetes and its complications (3, 12, 33). Our animal studies are suggestive that antioxidants and the inhibition of glucose transport therapies might be specifically applicable to the group of diabetic chronic kidney disease patients with AQP11 polymorphisms.
GRANTS
This work was supported by the National Institutes of Health Grants DK-20539 and DK-51265, DK-58404, and DK-62794 (to R. C. Harris); HL-086621 and ES-005022 (to A. J. Gow); HL-105479 (to W. E. Lawson); HL-85317 and HL-92870 (to T. S. Blackwell); UL1 R-024975, DK-79341 (Vanderbilt O′Brien Center); Department of Veterans Affairs (to W. E. Lawson, T. S. Blackwell, and R. C. Harris); and a development grant from the Department of Medicine, Vanderbilt University School of Medicine (to E. E. Tchekneva).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: E.N.A.-V., A.B., E.A., D.-S.C., V.V.P., H.T., S.T., L.F., C.V., S.V.R., S.N., N.S., M.I., and E.E.T. performed experiments; E.N.A.-V., A.B., E.A., D.-S.C., V.V.P., H.T., H.S., L.F., C.V., S.V.R., S.N., N.S., M.I., T.S.B., W.E.L., A.J.G., M.M.D., and E.E.T. analyzed data; E.N.A.-V., H.S., M.I., T.S.B., W.E.L., A.J.G., R.C.H., M.M.D., and E.E.T. interpreted results of experiments; E.N.A.-V., A.B., D.-S.C., V.V.P., H.T., S.T., M.I., M.M.D., and E.E.T. prepared figures; E.N.A.-V., D.-S.C., V.V.P., H.T., M.I., T.S.B., W.E.L., M.M.D., and E.E.T. drafted manuscript; E.N.A.-V., V.V.P., H.T., H.S., C.V., S.V.R., S.N., N.S., M.I., T.S.B., W.E.L., A.J.G., R.C.H., M.M.D., and E.E.T. edited and revised manuscript; E.N.A.-V., A.B., E.A., D.-S.C., V.V.P., H.T., S.T., H.S., L.F., C.V., S.V.R., S.N., N.S., M.I., T.S.B., W.E.L., A.J.G., R.C.H., M.M.D., and E.E.T. approved final version of manuscript; D.-S.C., V.V.P., H.T., S.T., H.S., M.I., T.S.B., W.E.L., M.M.D., and E.E.T. conception and design of research.
ACKNOWLEDGMENTS
We acknowledge the use of the Vanderbilt University Medical Center Research EM Resources. We thank Dr. E. Neilson for support of this project.
REFERENCES
- 1. Allen DA, Harwood S, Varagunam M, Raftery MJ, Yaqoob MM. High glucose-induced oxidative stress causes apoptosis in proximal tubular epithelial cells and is mediated by multiple caspases. FASEB J 17: 908–910, 2003 [DOI] [PubMed] [Google Scholar]
- 2. Bagby SP. Diabetic nephropathy and proximal tubule ROS: challenging our glomerulocentricity. Kidney Int 71: 1199–1202, 2007 [DOI] [PubMed] [Google Scholar]
- 3. Chao EC, Henry RR. SGLT2 inhibition–a novel strategy for diabetes treatment. Nat Rev Drug Discov 9: 551–559, 2010 [DOI] [PubMed] [Google Scholar]
- 4. Crane JM, Lam C, Rossi A, Gupta T, Bennett JL, Verkman AS. Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin-4 M1/M23 isoforms and orthogonal arrays. J Biol Chem 286: 16516–16524, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension 49: 717–727, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gorelick DA, Praetorius J, Tsunenari T, Nielsen S, Agre P. Aquaporin-11: a channel protein lacking apparent transport function expressed in brain. BMC Biochem 7: 14, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guo CJ, Atochina-Vasserman EN, Abramova E, Foley JP, Zaman A, Crouch E, Beers MF, Savani RC, Gow AJ. S-nitrosylation of surfactant protein-D controls inflammatory function. PLoS Biol 6: e266, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hannah MJ, Weiss U, Huttner WB. Differential extraction of proteins from paraformaldehyde-fixed cells: lessons from synaptophysin and other membrane proteins. Methods 16: 170–181, 1998 [DOI] [PubMed] [Google Scholar]
- 9. Ikeda M, Andoo A, Shimono M, Takamatsu N, Taki A, Muta K, Matsushita W, Uechi T, Matsuzaki T, Kenmochi N, Takata K, Sasaki S, Ito K, Ishibashi K. The NPC motif of aquaporin-11, unlike the NPA motif of known aquaporins, is essential for full expression of molecular function. J Biol Chem 286: 3342–3350, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ishibashi K, Kobayashi K, Sohara E, Sasaki S, Saihara Y. Aquaporin-11(AQP11) is important for cytosolic osmoregulation. J Am Soc Nephrol 17: 539A, 2006 [Google Scholar]
- 11. Kimura K, Jin H, Ogawa M, Aoe T. Dysfunction of the ER chaperone BiP accelerates the renal tubular injury. Biochem Biophys Res Commun 366: 1048–1053, 2008 [DOI] [PubMed] [Google Scholar]
- 12. Komoroski B, Vachharajani N, Feng Y, Li L, Kornhauser D, Pfister M. Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus. Clin Pharmacol Ther 85: 513–519, 2009 [DOI] [PubMed] [Google Scholar]
- 13. Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, Newcomb DC, Jones BR, Roldan J, Lane KB, Morrisey EE, Beers MF, Yull FE, Blackwell TS. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci USA 108: 10562–10567, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, Blackwell TR, Xu C, Markin C, Ware LB, Miller GG, Loyd JE, Blackwell TS. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol 294: L1119–L1126, 2008 [DOI] [PubMed] [Google Scholar]
- 15. Leinninger GM, Edwards JL, Lipshaw MJ, Feldman EL. Mechanisms of disease: mitochondria as new therapeutic targets in diabetic neuropathy. Nat Clin Pract Neurol 2: 620–628, 2006 [DOI] [PubMed] [Google Scholar]
- 16. Magri CJ, Fava S. The role of tubular injury in diabetic nephropathy. Eur J Intern Med 20: 551–555, 2009 [DOI] [PubMed] [Google Scholar]
- 17. Morishita Y, Matsuzaki T, Hara-chikuma M, Andoo A, Shimono M, Matsuki A, Kobayashi K, Ikeda M, Yamamoto T, Verkman A, Kusano E, Ookawara S, Takata K, Sasaki S, Ishibashi K. Disruption of aquaporin-11 produces polycystic kidneys following vacuolization of the proximal tubule. Mol Cell Biol 25: 7770–7779, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Murata K, Mitsuoka K, Hirai T, Walz T, Agre P, Heymann JB, Engel A, Fujiyoshi Y. Structural determinants of water permeation through aquaporin-1. Nature 407: 599–605, 2000 [DOI] [PubMed] [Google Scholar]
- 19. Nielsen S, Frokiaer J, Marples D, Kwon TH, Agre P, Knepper MA. Aquaporins in the kidney: from molecules to medicine. Physiol Rev 82: 205–244, 2002 [DOI] [PubMed] [Google Scholar]
- 20. Nozaki K, Ishii D, Ishibashi K. Intracellular aquaporins: clues for intracellular water transport? Pflügers Arch 456: 701–707, 2008 [DOI] [PubMed] [Google Scholar]
- 21. Okada S, Misaka T, Tanaka Y, Matsumoto I, Ishibashi K, Sasaki S, Abe K. Aquaporin-11 knockout mice and polycystic kidney disease animals share a common mechanism of cyst formation. FASEB J 22: 3672–3684, 2008 [DOI] [PubMed] [Google Scholar]
- 22. Romagnani P, Kalluri R. Possible mechanisms of kidney repair. Fibrogenesis Tissue Repair 2: 3, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, Dikov MM, Feoktistov I. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J Immunol 187: 6120–6129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Samikkannu T, Thomas JJ, Bhat GJ, Wittman V, Thekkumkara TJ. Acute effect of high glucose on long-term cell growth: a role for transient glucose increase in proximal tubule cell injury. Am J Physiol Renal Physiol 291: F162–F175, 2006 [DOI] [PubMed] [Google Scholar]
- 25. Schneider A, Zhang Y, Zhang M, Lu WJ, Rao R, Fan X, Redha R, Davis L, Breyer RM, Harris R, Guan Y, Breyer MD. Membrane-associated PGE synthase-1 (mPGES-1) is coexpressed with both COX-1 and COX-2 in the kidney. Kidney Int 65: 1205–1213, 2004 [DOI] [PubMed] [Google Scholar]
- 26. Sorbo JG, Moe SE, Ottersen OP, Holen T. The molecular composition of square arrays. Biochemistry 47: 2631–2637, 2008 [DOI] [PubMed] [Google Scholar]
- 27. Tanjore H, Blackwell TS, Lawson WE. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 302: L721–L729, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tchekneva EE, Khuchua Z, Davis LS, Kadkina V, Dunn SR, Bachman S, Ishibashi K, Rinchik EM, Harris RC, Dikov MM, Breyer MD. Single amino acid substitution in aquaporin 11 causes renal failure. J Am Soc Nephrol 19: 1955–1964, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tchekneva EE, Rinchik EM, Dikov MM, Godfrey V, Klebig M, Kadkina V, Mohamed Y, Breyer MD. Genetic complementation of sudden juvenile death syndrome (sjds), a recessive ENU-induced mutation on Ch7 is revealed by the compound heterozygous AQP11-/sjds genotype. J Am Soc Nephrol 17: 777A, 2006 [Google Scholar]
- 30. Tchekneva EE, Rinchik EM, Dunn SR, Bachman S, Polosukhina D, Kadkina V, Breyer MD. Proximal tubule injury and sudden juvenile death syndrome (sjds) in mutant mice carrying a recessive ENU-induced mutation on chromosome 7. J Am Soc Nephrol 16: 141A, 2005 [Google Scholar]
- 31. Thomas MC, Burns WC, Cooper ME. Tubular changes in early diabetic nephropathy. Adv Chronic Kidney Dis 12: 177–186, 2005 [DOI] [PubMed] [Google Scholar]
- 32. Thornalley PJ, Rabbani N. Dietary and synthetic activators of the antistress gene response in treatment of renal disease. J Ren Nutr 22: 195–202, 2012 [DOI] [PubMed] [Google Scholar]
- 33. Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol 300: R1009–R1022, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vallon V, Sharma K. Sodium-glucose transport: role in diabetes mellitus and potential clinical implications. Curr Opin Nephrol Hypertens 19: 425–431, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vallon VK. Pathophysiology of the diabetic kidney. Compr Physiol 1: 1–58, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Whittington K, Harrison SC, Williams KM, Day JL, McLaughlin EA, Hull MG, Ford WC. Reactive oxygen species (ROS) production and the outcome of diagnostic tests of sperm function. Int J Androl 22: 236–242, 1999 [DOI] [PubMed] [Google Scholar]
- 37. Wolf G, Neilson EG. Angiotensin II induces cellular hypertrophy in cultured murine proximal tubular cells. Am J Physiol Renal Fluid Electrolyte Physiol 259: F768–F777, 1990 [DOI] [PubMed] [Google Scholar]
- 38. Yakata K, Tani K, Fujiyoshi Y. Water permeability and characterization of aquaporin-11. J Struct Biol 174: 315–320, 2011 [DOI] [PubMed] [Google Scholar]