

Abstract
Participation of connexin 40 (Cx40) in the regulation of renin secretion and in the tubuloglomerular feedback (TGF) component of renal autoregulation suggests that gap junctional coupling through Cx40 contributes to the function of the juxtaglomerular apparatus. In the present experiments, we determined the effect of targeted Cx40 deletion in C57BL/6 and FVB mice on TGF responsiveness. In C57BL/6 mice, stop-flow pressure (PSF) fell from 40.3 ± 2 to 34.5 ± 2 mmHg in wild-type (WT) and from 31 ± 1.06 to 26.6 ± 0.98 mmHg in Cx40−/− mice. PSF changes of 5.85 ± 0.67 mmHg in WT and of 4.3 ± 0.55 mmHg in Cx40−/− mice were not significantly different (P = 0.08). In FVB mice, PSF fell from 37.4 ± 1.5 to 31.6 ± 1.5 mmHg in WT and from 28.1 ± 1.6 to 25.4 ± 1.7 mmHg in Cx40−/−, with mean TGF responses being significantly greater in WT than Cx40−/− (5.5 ± 0.55 vs. 2.7 ± 0.84 mmHg; P = 0.002). In both genetic backgrounds, PSF values were significantly lower in Cx40−/− than WT mice at all flow rates. Arterial blood pressure in the animals prepared for micropuncture was not different between WT and Cx40−/− mice. We conclude that the TGF response magnitude in superficial cortical nephrons is reduced by 30–50% in mice without Cx40, but that with the exception of a small number of nephrons, residual TGF activity is maintained. Thus gap junctional coupling appears to modulate TGF, perhaps by determining the kinetics of signal transmission.
Keywords: micropuncture, stop-flow pressure, genetic background, vascular resistance
besides being affected by multiple systemic factors, the glomerular filtration rate (GFR) is controlled by an intrarenal control mechanism known as tubuloglomerular feedback (TGF). TGF is constructed as a homeostatic feedback loop, in which an increase in NaCl concentration in the tubular fluid passing the apical aspect of macula densa cells is translated into a preglomerular vasoconstriction and a concomitant reduction of single-nephron GFR. While the relationship between the tubular input and the vascular output is well understood, numerous aspects of the juxtaglomerular transmission pathway are still unclear.
Because of the absence of a structural coupling between macula densa/thick ascending limb of Henle's loop (TAL) cells and the underlying mesangium, it has been generally assumed that the activation of TGF by elevated tubular NaCl concentrations is accompanied by the generation of paracrine vasoactive factors within the confines of the juxtaglomerular apparatus (JGA) and that these paracrine factors mediate the modulation of afferent arteriolar tone. There is strong experimental evidence in support of the notion that activation of A1 adenosine receptors (A1AR) by NaCl-dependent increases in juxtaglomerular adenosine levels provides the most important vasoconstrictor input, with angiotensin II acting as a synergistic cofactor. Specific A1AR antagonists such as 1,3-dipropyl-8-cyclopentylxanthine (DPCPX), PSB-36, or KW-3902 markedly attenuate TGF responses, and the genetic ablation of A1AR causes complete loss of TGF function (2, 3, 19, 23, 26). The appearance of adenosine in the JGA interstitium is for the most part the result of dephosphorylation of released ATP by ecto-ATPases and ecto-5-′nucleotidase (4, 20). Enhancement of TGF responsiveness by vascular overexpression of A1AR indicates that adenosine may predominantly target A1AR on afferent arterioles without excluding a role of extravascular receptors (17). In fact, selective deletion of A1AR in smooth muscle cells by cre-lox-mediated recombination markedly attenuated, but did not abolish TGF responses, directly implicating nonvascular A1AR in TGF (13). Nevertheless, the precise location of adenosine generation and the route by which adenosine reaches vascular or extravascular A1AR are not clear.
In an isolated JGA preparation, TGF activation has been shown to cause an increase in cytosolic Ca that spreads across the mesangial cell field and reaches the afferent arteriole (18). Ca propagation was inhibited by heptanol or glycyrrhetinic acid, known inhibitors of gap junctional coupling, suggesting that signal transmission in the JGA may utilize intercellular communication pathways (18). Gap junctions are formed by connexin (Cx) subunits, and several different Cx proteins are expressed in the JGA, including Cx37, Cx40, Cx43, and Cx45 (8). Cx40 is highly expressed in extraglomerular mesangial cells and might therefore be a candidate for the transmission of a depolarization or Ca signal across the JGA (12, 28, 32). Early functional evidence for a role of connexins in the regulation of renal blood flow was provided by the observation that inhibition of connexin formation by connexin-mimetic peptides reduced autoregulatory efficiency (27). The subsequent demonstration of an impairment of the TGF component of autoregulation in Cx40-deficient mice has directly implicated a specific gap junctional protein in the TGF mechanism (9, 25).
In the present experiments, Cx40-deficient mice were used to assess the function of Cx40 in TGF signaling using micropuncture techniques in the intact kidneys of anesthetized mice. Cx40-deficient mice of two different genetic backgrounds exhibited reduced TGF magnitudes and abnormal kinetics, although the degree of TGF impairment was widely variable between nephrons. Nevertheless, our data suggest that Cx40 gap junctions or hemichannels contribute to signal transmission between macula densa cells and the vasculature of the afferent arteriole. On the other hand, the maintenance of residual TGF activity in most nephrons of Cx40-deficient mice indicates that macula densa and smooth muscle cells are also connected through pathways that do not require intact Cx40 functionality.
METHODS
Animals.
To reduce the possible risk of strain-dependent effects, experiments were performed in animals on a C57BL/6 or FVB genetic background. C57BL/6 wild-type (WT) and Cx40−/− mice originally generated by Kirchhoff et al. were obtained from Dr. C. de Witt (University of Lubeck, Lubeck, Germany) and kept in the animal facility at the University of Regensburg. Animals used were in a weight range between 21 and 28 g. FVB mice were purchased from Charles River (Sulzfeld, Germany) and crossed with C57BL/6 Cx40 null mice for at least four generations before use (weight range 28–37 g). Genotyping was done according to standard protocols using DNA from tail biopsies. The following gene-specific primers were used: Cx40 forward (ggg aga tga gca ggc cga ctt ccg gtg cg), Cx40 reverse (5′-gta ggg tgc cct gga gga caa tct tcc c-3′), neo forward (5′-gga tcg gcc att gaa caa gat gga ttg cac-3′), and neo reverse (5′-ctg atg ctc ttc gtc cag atc atc ctg atc g-3′). Littermates from heterozygous breeding pairs were used in the experiments. Animals were fed a standard diet and kept at a 12:12-h light-dark cycle. Animal care and experimentation were approved by the IACUCs of the University of Regensburg (54-2532.1-11/12) and the National Institutes of Health (NIDDK ASP 058-KDB-10).
Animal preparation.
For micropuncture experiments, mice were anesthetized with 100 mg/kg thiobutabarbital (Inactin) intraperitoneally and 100 mg/kg ketamine subcutaneously. Body temperature was maintained at 37.5°C by placing the animals on an operating table with a servo-controlled heating plate. The trachea was cannulated, and 100% oxygen was blown at a low rate toward the tracheal tube throughout the experiment to maintain arterial oxygen saturation. The left carotid artery was catheterized with hand-drawn polyethylene tubing for continuous measurement of arterial blood pressure. A catheter connected to an infusion pump was inserted into the right jugular vein for an intravenous maintenance infusion of saline at 300 μl/h.
Micropuncture experiments.
Measurements of stop-flow pressure (PSF) in superficial cortical nephrons during perfusion of the loop of Henle were done as described previously (24, 31). When PSF had stabilized, the perfusion rate of the loop of Henle was increased to 30 nl/min and maximum PSF responses were determined. The perfusion rate was then returned to 0 nl/min. Flow conditions were maintained until steady states were achieved. When possible, two such responses were determined successively in each nephron. The perfusion fluid contained (in mM/l) 136 NaCl, 4 NaHCO3, 4 KCl, 2 CaCl2, and 7.5 urea as well as 100 mg/100 ml FD&C green (Keystone, Bellefonte, PA).
GFR.
GFR in conscious mice was measured by single-injection FITC-inulin or FITC-sinistrin clearance (13). FITC-inulin or FITC-sinistrin (gift from Dr. D. Schock-Kusch, University of Heidelberg, Heidelberg, Germany) was injected at 3.7 μl/g body wt into the retroorbital plexus during brief sevoflurane anesthesia. At 3, 7, 10, 15, 35, 55, and 75 min, the mice were placed in a restrainer, and the tail vein was punctured with a 30-G needle for collection of ∼2 μl of blood. A total of 500 nl of plasma was then diluted 1:10 in 500 mmol of HEPES (pH 7.4) and measured against a standard curve, as described (7). Fluorescence was determined in 2 μl of the diluted sample (Nanodrop-ND-3300, Nanodrop Technologies, Wilmington, DE). GFR was calculated using a two-compartment model of two-phase exponential decay (7).
Statistical analysis.
Values are means ± SE, and statistical significance was assessed by paired or unpaired t-test.
RESULTS
TGF.
Using animals with the original C57BL/6 genetic background, useable results were obtained in seven WT and seven Cx40−/− mice. The effects of raising the loop of Henle flow rate to saturating levels on PSF of individual tubules are summarized in Fig. 1A. On average, PSF fell from 40.3 ± 2 to 34.5 ± 2 mmHg in WT (n = 18) and from 31 ± 1.06 to 26.6 ± 0.98 mmHg in Cx40−/− mice (n = 20). These flow-induced reductions of PSF were highly significant in both WT and Cx40−/− mice (P < 0.001). The mean difference between PSF at 0 and 30 nl/min, the response magnitude, did not quite achieve significance between genotypes, averaging 5.85 ± 0.67 mmHg in the WT and 4.3 ± 0.55 mmHg in the Cx40−/− mice (P = 0.08; Fig. 2A). Failure to achieve significance was the result of considerable variability in the response magnitude in both WT and Cx40-deficient animals (Fig. 3A). Duration of TGF responses defined as the time from perfusion onset to attainment of a steady state was not different between WT and Cx40−/− mice (22.8 ± 3.5 vs. 23.9 ± 3 s). Previous experiments have shown that PSF in response to flow stimulation declines at an initial fast and a subsequent slow rate, each contributing about half to the total response magnitude (17). Although differential slope determinations were only possible in a subset of nephrons due to blood pressure instabilities (12 of 18 and 11 of 20 in WT and Cx40−/− mice), the fast and slow slopes of 0.87 ± 0.21 and 0.22 ± 0.05 mmHg/s in WT are similar to those reported previously (17). Cx40 deficiency reduced or abolished the slope differences by reducing the fast slope (to 0.38 ± 0.07 mmHg/s; P = 0.017), while the slow slope was not affected (0.19 ± 0.04 mmHg/s; P = 0.66). A PSF reduction to an essentially single slope can be seen in the example in Fig. 4A. PSF values at zero flow rate were consistently and significantly lower in the Cx40-deficient compared with WT mice (P < 0.001). This observation is indicative of an increased preglomerular vascular resistance in the Cx40-deficient animals since mean arterial blood pressure during micropuncture was similar in WT and Cx40−/− of the C57BL/6 line (89.8 ± 3.14 and 89.4 ± 2.3 mmHg, respectively). To functionally determine whether TGF responsiveness may be modified through availability of A1AR, we compared the effect of adding the A1AR agonist cyclohexyl adenosine (CHA) to the perfusate. Perfusion of the loop of Henle with a perfusate containing CHA (10 μM) increased TGF responses in WT to 8.1 ± 0.9 mmHg (from 5.85 ± 0.66; n = 10; P < 0.05) and to 7.6 ± 0.5 in Cx40−/− (from 4.3 ± 0.55; n = 10; P < 0.001). Cx40−/− mice had a reduction in the fast TGF slope from 0.96 ± 0.18 to 0.28 ± 0.05 mmHg/s (P = 0.003), whereas the slow slopes of 0.28 ± 0.05 and 0.13 ± 0.02 mmHg/s were similar (P > 0.05). Thus, like with standard Ringer perfusion, Cx40 deficiency was associated with a leveling of the slope differential mainly by a marked decrease in the speed of the fast TGF component.
Fig. 1.
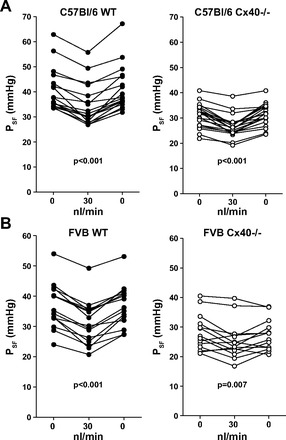
Tubuloglomerular feedback responses of tubular stop flow pressure (PSF) in wild-type control (WT) and connexin 40-deficient mice (Cx40−/−). A: C57BL/6 genetic background. B: FVB genetic background. Lines connect data from individual nephrons at perfusion rates of 0, 30, and 0 nl/min. P values are given for comparisons with the 30 nl/min perfusion rate (paired t-test).
Fig. 2.
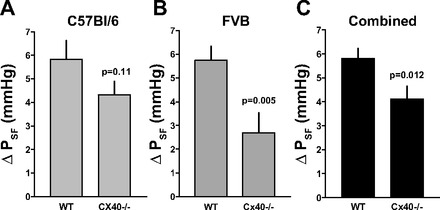
Reduction of tubular stop flow pressure (PSF) in response to a flow increase from 0 to 30 nl/min in WT and Cx40−/− mice on a C57BL/6 (A) or FVB (B) genetic background, and in both strains combined (C). Data are means ± SE. Statistical comparison was by t-test.
Fig. 3.
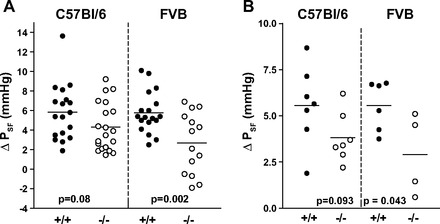
TGF responses in individual nephrons of WT (+/+) and Cx40-deficient mice (−/−) on C57BL/6 (left) and FVB (right) genetic backgrounds. A: TGF responses magnitude in individual nephrons; B: TGF response magnitude in individual mice (averages of 1–4 nephrons/mouse). Mean values are indicated by horizontal lines; a t-test was used for P values.
Fig. 4.
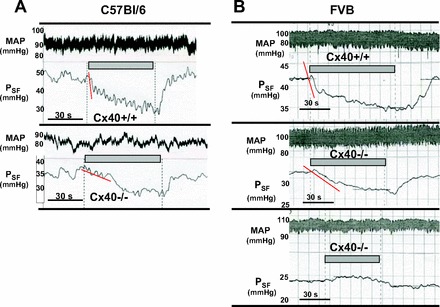
Original recordings of TGF responses of PSF in WT (Cx40+/+) and Cx40−/− mice. A: recordings in 2 nephrons of C57BL/6 mice; arterial blood pressure (MAP) recordings are shown above the PSF traces (top: WT; bottom: Cx40−/−). Periods of loop perfusion at 30 nl/min are indicated by grey bars; onset and offset of perfusion are also indicated by dotted lines. Red lines highlight initial declines of PSF. B: recording in 3 nephrons of FVB mice; traces show arterial blood pressure above PSF (top: WT; middle: Cx40−/−; bottom: Cx40−/−); note steep initial fall of PSF as indicated by red line in WT and a markedly slower initial PSF slope in Cx40−/−. Absence of TGF responses as shown in the third example of this graph was seen in a small number of nephrons.
To reduce the possible impact of strain-dependent effects, the Cx40 null mutation was bred into an FVB genetic background for more than four generations. In these FVB mice, successful micropuncture experiments were performed in six male WT (mean body wt 30 g) and four male Cx40−/− (mean body wt 31.5 g). PSF decreased with loop perfusion from 37.4 ± 1.5 to 31.6 ± 1.5 mmHg in WT (n = 18), whereas it fell from 28.1 ± 1.6 to 25.4 ± 1.7 mmHg in the FVB Cx40−/− animals (n = 14). Data from individual mice are shown in Fig. 1B. Similar to the findings in C57BL/6 mice, PSF at zero flow was significantly lower in the Cx40-deficient mice compared with WT despite the fact that mean arterial blood pressure during micropuncture tended to be higher in Cx40−/− than WT mice (98.4 ± 2.8 vs. 91.3 ± 3.3 mmHg; P = 0.12). The mean TGF response magnitude was 5.75 ± 0.55 mmHg in the FVB WT and 2.7 ± 0.84 in the FVB Cx40−/− mice (P = 0.002; Fig. 2B). Combining the results from both FVB and C57BL/6 strains reveals a significant reduction of the TGF response magnitude from 5.81 ± 0.41 to 4.12 ± 0.52 mmHg (P = 0.012; Fig. 2C). As in the C57BL/6 strain, individual TGF responses showed substantial heterogeneity in both WT and Cx40−/− animals with four nephrons in the latter group displaying inverted responses (Figs. 3B and 4B). This difference does not only stem from differences in individual mice but is also evident in a given mouse, suggesting that the response strength is to a large extent a property of the individual tubule. The duration of responses was not different between WT and Cx40−/− mice (37.6 ± 4.9 s in WT and 44 ± 7.3 s in Cx40−/− mice). Like C57BL/6 mice, TGF responses in FVB mice showed a TGF slope differential with a fast slope of 0.72 ± 0.12 mmHg/s and a slow slope of 0.13 ± 0.02 mmHg/s (slopes could be determined in 13 of 18 tubules). There was a significant reduction in the fast slope to 0.31 ± 0.06 mmHg/s in the FVB Cx40−/− mice (P = 0.03; Fig. 4, A and B), while the slow slope of 0.08 ± 0.01 mmHg/s was not significantly different from WT (slopes could be determined in 7 of 14 tubules). No consistent differences were detected in response onset, the time between changing the perfusion rate and the beginning of the PSF decline.
GFR.
GFR measured by FITC-inulin plasma clearance kinetics in conscious C57BL/6 mice averaged 407.5 ± 24 μl/min in WT (n = 12) and 294.2 ± 19 μl/min in Cx40−/− animals (n = 12; P = 0.0012). Body weights and ages were similar between genotypes (WT: 24.8 ± 1.3 g and 20.3 ± 3.1 wk; KO: 23.9 ± 1.3 g and 18.8 ± 2.3 wk). Using FITC-sinistrin clearance in conscious mice of the FVB background, GFR tended to be lower in Cx40−/− than WT mice without reaching significance (379 ± 96 vs. 416 ± 53 μl/min, P = 0.31; n = 9). Systolic blood pressure in animals of the FVB background used for GFR measurements was markedly increased in Cx40-deficient mice compared with WT (143 ± 7 vs. 124 ± 2 mmHg, P = 0.025; n = 5), whereas the heart rate was similar in both genotypes, averaging 723 ± 16 and 706 ± 15 beats/min for Cx40−/− and +/+ mice, respectively (P = 0.40; n = 5).
DISCUSSION
The results of the present study show that global deletion of Cx40 attenuates TGF responses in superficial cortical nephrons in mice but that TGF responsiveness is not fully abolished in the majority of nephrons. Thus intercellular communication pathways through Cx40 appear to modulate TGF response magnitude and dynamics without being an absolute requirement for signal transmission.
Cx40 is a gap junctional protein that in the kidney is expressed in endothelial cells of most renal vessels, intra-and extraglomerular mesangial cells, and renin-producing juxtaglomerular granular cells (8, 29). The availability of Cx40-deficient mice has permitted a detailed evaluation of the role of this particular gap junctional protein in renal function. The observation of dramatic changes in renin cell localization, renin expression, and renin secretion in Cx40-deficient animals has drawn attention to Cx40 as a critical regulator of the function of the JGA (11, 30). Since in addition to regulating renin release the JGA is the site of information transfer from tubules to afferent arterioles, evaluating the role on Cx40 in TGF has been a logical extension of these earlier studies. In this regard, assessments of blood flow autoregulation in intact animals have shown that Cx40−/− mice have a reduced ability to regulate blood flow in response to step blood pressure changes in the time span typical for the TGF component of autoregulation (9). Furthermore, in the perfused juxtamedullary nephron preparation the constrictor response of afferent arterioles to increments of perfusion pressure was blunted in Cx40-deficient mice (25). Prior papillectomy performed to physically disrupt the TGF pathway eliminated the difference between WT and Cx40−/− preparations, indicating that a dysfunctional TGF mechanism was mainly responsible for the impairment of autoregulation (25). Nevertheless, autoregulation is a complex response with a number of functional interactions so that a direct assessment of TGF was felt to be a needed addition to the existing evidence.
Disregarding results in singular nephrons, our results demonstrate that TGF responses in general are not eliminated in mice with null mutations of Cx40. Furthermore, all observations taken together, it also seems well supported that there is a clear attenuation of responses by ∼50% in FVB and 30% in C57BL/6 mice. We realize that these numbers are lower bound estimates of the involvement of gap junctional coupling in TGF since a contribution of other connexins, especially of Cx37, is entirely conceivable. The reasons for the reduced TGF responsiveness is not clear, but it is probably the direct consequence of the absence of Cx40. Our observation that the local administration of the A1AR agonist CHA caused a comparable augmentation of TGF responses in both WT and Cx40−/− mice makes it unlikely that differences in A1AR expression can account for the different TGF responses. Arterial blood pressure can directly affect TGF responses, but pressure was not different in C57BL/6 mice without Cx40 and higher in corresponding FVB mice (1, 5, 21). Furthermore, TGF responses are enhanced by angiotensin II, but angiotensin II levels are likely to be higher, not lower in the Cx40-deficient compared with WT animals (14, 22). An interesting possibility is that an increase in nitric oxide generated by neuronal nitric oxide synthase (nNOS) in macula densa cells contributes to TGF attenuation since the level of macula densa nNOS has been reported to be upregulated in Cx40-deficient mice (10).
Data in Cx40-deficient mice on both genetic backgrounds show significantly lower levels of PSF compared with WT animals regardless of the loop of Henle flow rate. These differences are not a reflection of blood pressure since arterial pressure during micropuncture was not different between genotypes in the C57BL/6 animals and higher in the Cx40-deficient animals on the FVB background. A marked reduction of the baseline diameter of the afferent arteriole close to the vascular pole at constant perfusion pressure has also been observed in juxtamedullary nephron preparations of Cx40-deficient mice (25). It is safe to assume therefore that Cx40-deficiency is associated with an increase of afferent arteriolar resistance near the glomerular entrance. Since PSF differences were found in the absence of macula densa perfusion, the increased resistance is probably not TGF mediated. It is possible that the chronically elevated blood pressure in the Cx40−/− mice causes vascular remodeling, providing a structural reason for the increased resistance. However, acute interference with Cx40 or Cx37 functionality by connexin-mimetic peptides also caused a reduction of renal blood flow and a large increase in renal vascular resistance (6, 27). The increase in renin release and the presumably elevated levels of angiotensin II associated with Cx40 deficiency could provide another reason for increased vasomotor tone. Finally, it has been suggested that inhibition of gap junctional coupling eliminates the tonic vasodilatation exerted by an endothelial-hyperpolarizing factor that is not nitric oxide or a cyclooxygenase product (6). Whatever the reason for the clearly reduced PSF in the Cx40-deficient animals, it is noteworthy that filtration was only moderately lower in the Cx40−/− mice and that previous measurements did not show significant differences in renal blood flow between genotypes (9). Thus vasodilation of other serial resistances in the kidney, for example in interlobular arteries or efferent arterioles, appears to compensate for the constriction of afferent arterioles.
An attempt to interpret the present data is shown in the schematic representation in Fig. 5. This interpretation incorporates the previous demonstration, repeated by two independent groups, that deletion of A1AR completely eliminates TGF responses in mice (2, 26). The most straightforward notion would be that both Cx40-dependent and Cx40-independent components of TGF include a role for A1AR. We propose that interstitial adenosine most likely produced by ATP dephosphorylation interacts with A1AR on mesangial cells and that this triggers afferent constriction by gap junctional transmission, presumably by spreading depolarization and increases in cytosolic Ca (18). Studies in cultured mesangial cells have shown that adenosine increases Ca uptake through a verapamil-sensitive mechanism and that this effect is mediated by activation of A1AR (15, 16). We assume that this component would be Cx40 dependent and would probably constitute the fast component of TGF. The reduction of the speed of the fast TGF component in Cx40-deficient mice in the present study is consistent with this interpretation. We further propose that adenosine can directly interact with A1AR on smooth muscle cells, reaching the vessels by diffusion or generation from ATP near the vasculature. We assume that this component is Cx40 independent, that it is comparably slow, and that it is responsible for the protracted time course of TGF responsiveness. This is consistent with our observation that the duration of TGF-induced changes in PSF in responding nephrons was not different between genotypes. In addition, such a pathway would almost by definition depend on anatomic characteristics so that structural differences between JGAs of different tubules might explain the marked response variability between different nephrons. It has been postulated earlier that adenosine contributes to TGF through effects independent of gap junctional coupling (27).
Fig. 5.
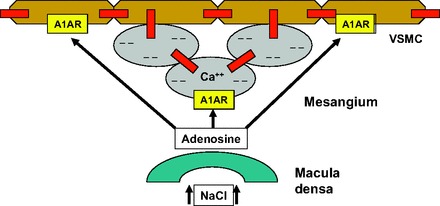
Schematic representation of the proposed role of Cx40 (red boxes) in the TGF response. A Cx40-dependent pathway linking mesangial cells (grey ovals) with the arteriolar smooth muscle cells (brown) is paralleled by a Cx40-independent pathway permitting direct paracrine smooth muscle cell activation. Both pathways are assumed to be activated by A1 adenosine receptors (A1AR) based on the previous demonstration of an absolute requirement of A1AR for TGF responsiveness (2, 25).
In summary, micropuncture determinations of the PSF response to a saturating flow elevation indicate that a deletion of the gap junctional protein Cx40 reduces TGF responsiveness by ∼30% but that a significant residual TGF activity appears to be Cx40 independent. It is possible that a connexin other than Cx40 may be involved in TGF, but an extracellular transmission pathway could be a viable alternative.
GRANTS
This work was supported by the intramural research program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, and by a grant from the Deutsche Forschungsgemeinschaft (SFB699/B7). J. Schnermann was the recipient of a Humboldt Research Award from the Alexander-von-Humboldt Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: M.O, H.C, and J.S. provided conception and design of research; M.O, I.C, I.S, C.E, and J.S. performed experiments; M.O, I.C, I.S, C.E, H.C, and J.S. analyzed data; M.O, I.C, I.S, C.E, H.C, and J.S. approved final version of manuscript; H.C. and J.S. interpreted results of experiments; H.C. and J.S. drafted manuscript; H.C. and J.S. edited and revised manuscript; J.S. prepared figures.
REFERENCES
- 1.Bell PD, Navar LG. Stop-flow pressure feedback responses during reduced renal vascular resistance in the dog. Am J Physiol Renal Fluid Electrolyte Physiol 237: F204–F209, 1979 [DOI] [PubMed] [Google Scholar]
- 2.Brown R, Ollerstam A, Johansson B, Skøtt O, Gebre-Medhin S, Fredholm B, Persson AE. Abolished tubuloglomerular feedback and increased plasma renin in adenosine A1 receptor-deficient mice. Am J Physiol Regul Integr Comp Physiol 281: R1362–R1367, 2001 [DOI] [PubMed] [Google Scholar]
- 3.Carlstrom M, Wilcox CS, Welch WJ. Adenosine A2 receptors modulate tubuloglomerular feedback. Am J Physiol Renal Physiol 299: F412–F417, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castrop H, Huang Y, Hashimoto S, Mizel D, Hansen P, Theilig F, Bachmann S, Deng C, Briggs J, Schnermann J. Impairment of tubuloglomerular feedback regulation of GFR in ecto-5′-nucleotidase/CD73-deficient mice. J Clin Invest 114: 634–642, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen YM, Yip KP, Marsh DJ, Holstein-Rathlou NH. Magnitude of TGF-initiated nephron-nephron interactions is increased in SHR. Am J Physiol Renal Fluid Electrolyte Physiol 269: F198–F204, 1995 [DOI] [PubMed] [Google Scholar]
- 6.De Vriese AS, Van de Voorde J, Lameire NH. Effects of connexin-mimetic peptides on nitric oxide synthase- and cyclooxygenase-independent renal vasodilation. Kidney Int 61: 177–185, 2002 [DOI] [PubMed] [Google Scholar]
- 7.Faulhaber-Walter R, Chen L, Oppermann M, Kim SM, Huang Y, Hiramatsu N, Mizel D, Kajiyama H, Zerfas P, Briggs JP, Kopp JB, Schnermann J. Lack of A1 adenosine receptors augments diabetic hyperfiltration and glomerular injury. J Am Soc Nephrol 19: 722–730, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanner F, Sorensen CM, Holstein-Rathlou NH, Peti-Peterdi J. Connexins and the kidney. Am J Physiol Regul Integr Comp Physiol 298: R1143–R1155, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Just A, Kurtz L, de Wit C, Wagner C, Kurtz A, Arendshorst WJ. Connexin 40 mediates the tubuloglomerular feedback contribution to renal blood flow autoregulation. J Am Soc Nephrol 20: 1577–1585, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krattinger N, Alonso F, Capponi A, Mazzolai L, Nicod P, Meda P, Haefliger JA. Increased expression of renal cyclooxygenase-2 and neuronal nitric oxide synthase in hypertensive Cx40-deficient mice. J Vasc Res 46: 188–198, 2009 [DOI] [PubMed] [Google Scholar]
- 11.Krattinger N, Capponi A, Mazzolai L, Aubert JF, Caille D, Nicod P, Waeber G, Meda P, Haefliger JA. Connexin40 regulates renin production and blood pressure. Kidney Int 72: 814–822, 2007 [DOI] [PubMed] [Google Scholar]
- 12.Kurtz L, Madsen K, Kurt B, Jensen BL, Walter S, Banas B, Wagner C, Kurtz A. High-level connexin expression in the human juxtaglomerular apparatus. Nephron Physiol 116: p1–p8, 2010 [DOI] [PubMed] [Google Scholar]
- 13.Li L, Lai EY, Huang YG, Eisner C, Mizel D, Wilcox CS, Schnermann JB. Renal afferent arteriolar and tubuloglomerular feedback reactivity in mice with conditional deletions of adenosine 1 receptors. Am J Physiol Renal Physiol 303: F1166–F1175, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitchell KD, Navar LG. Enhanced tubuloglomerular feedback during peritubular infusions of angiotensins I and II. Am J Physiol Renal Fluid Electrolyte Physiol 255: F383–F390, 1988 [DOI] [PubMed] [Google Scholar]
- 15.Olivera A, Lopez-Rivas A, Lopez-Novoa JM. Adenosine stimulates Ca2+ fluxes and increases cytosolic free Ca2+ in cultured rat mesangial cells. Biochem J 282: 871–876, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olivera A, Tomas M, Lopez-Novoa JM. Effect of adenosine A1 and A2 agonists and antagonists on cAMP and Ca2+ in cultured rat mesangial cells. Am J Physiol Cell Physiol 262: C840–C844, 1992 [DOI] [PubMed] [Google Scholar]
- 17.Oppermann M, Qin Y, Lai EY, Eisner C, Li L, Huang Y, Mizel D, Fryc J, Wilcox CS, Briggs J, Schnermann J, Castrop H. Enhanced tubuloglomerular feedback in mice with vascular overexpression of A1 adenosine receptors. Am J Physiol Renal Physiol 297: F1256–F1264, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peti-Peterdi J. Calcium wave of tubuloglomerular feedback. Am J Physiol Renal Physiol 291: F473–F480, 2006 [DOI] [PubMed] [Google Scholar]
- 19.Ren Y, Arima S, Carretero OA, Ito S. Possible role of adenosine in macula densa control of glomerular hemodynamics. Kidney Int 61: 169–176, 2002 [DOI] [PubMed] [Google Scholar]
- 20.Ren Y, Garvin JL, Liu R, Carretero OA. Role of macula densa adenosine triphosphate (ATP) in tubuloglomerular feedback. Kidney Int 66: 1479–1485, 2004 [DOI] [PubMed] [Google Scholar]
- 21.Schnermann J, Briggs JP. Interaction between loop of Henle flow and arterial pressure as determinants of glomerular pressure. Am J Physiol Renal Fluid Electrolyte Physiol 256: F421–F429, 1989 [DOI] [PubMed] [Google Scholar]
- 22.Schnermann J, Briggs JP. Single nephron comparison of the effect of loop of Henle flow on filtration rate and pressure in control and angiotensin II-infused rats. Miner Electrolyte Metab 15: 103–107, 1989 [PubMed] [Google Scholar]
- 23.Schnermann J, Weihprecht H, Briggs JP. Inhibition of tubuloglomerular feedback during adenosine 1 receptor blockade. Am J Physiol Renal Fluid Electrolyte Physiol 258: F553–F561, 1990 [DOI] [PubMed] [Google Scholar]
- 24.Schnermann JB, Traynor T, Yang T, Huang YG, Oliverio MI, Coffman T, Briggs JP. Absence of tubuloglomerular feedback responses in AT1A receptor-deficient mice. Am J Physiol Renal Physiol 273: F315–F320, 1997 [DOI] [PubMed] [Google Scholar]
- 25.Sorensen CM, Giese I, Braunstein TH, Brasen JC, Salomonsson M, Holstein-Rathlou NH. Role of connexin40 in the autoregulatory response of the afferent arteriole. Am J Physiol Renal Physiol 303: F855–F863, 2012 [DOI] [PubMed] [Google Scholar]
- 26.Sun D, Samuelson LC, Yang T, Huang Y, Paliege A, Saunders T, Briggs J, Schnermann J. Mediation of tubuloglomerular feedback by adenosine: Evidence from mice lacking adenosine 1 receptors. Proc Natl Acad Sci USA 98: 9983–9988, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takenaka T, Inoue T, Kanno Y, Okada H, Hill CE, Suzuki H. Connexins 37 and 40 transduce purinergic signals mediating renal autoregulation. Am J Physiol Regul Integr Comp Physiol 294: R1–R11, 2008 [DOI] [PubMed] [Google Scholar]
- 28.Takenaka T, Inoue T, Kanno Y, Okada H, Meaney KR, Hill CE, Suzuki H. Expression and role of connexins in the rat renal vasculature. Kidney Int 73: 415–422, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Wagner C. Function of connexins in the renal circulation. Kidney Int 73: 547–555, 2008 [DOI] [PubMed] [Google Scholar]
- 30.Wagner C, de Wit C, Kurtz L, Grunberger C, Kurtz A, Schweda F. Connexin40 is essential for the pressure control of renin synthesis and secretion. Circ Res 100: 556–563, 2007 [DOI] [PubMed] [Google Scholar]
- 31.Wright FS, Schnermann J. Interference with feedback control of glomerular filtration rate by furosemide, triflocin, and cyanide. J Clin Invest 53: 1695–1708, 1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang J, Hill CE. Differential connexin expression in preglomerular and postglomerular vasculature: accentuation during diabetes. Kidney Int 68: 1171–1185, 2005 [DOI] [PubMed] [Google Scholar]