

Abstract
The molecular clock mechanism underlies circadian rhythms and is defined by a transcription-translation feedback loop. Bmal1 encodes a core molecular clock transcription factor. Germline Bmal1 knockout mice show a loss of circadian variation in heart rate and blood pressure, and they develop dilated cardiomyopathy. We tested the role of the molecular clock in adult cardiomyocytes by generating mice that allow for the inducible cardiomyocyte-specific deletion of Bmal1 (iCSΔBmal1). ECG telemetry showed that cardiomyocyte-specific deletion of Bmal1 (iCSΔBmal1−/−) in adult mice slowed heart rate, prolonged RR and QRS intervals, and increased episodes of arrhythmia. Moreover, isolated iCSΔBmal1−/− hearts were more susceptible to arrhythmia during electromechanical stimulation. Examination of candidate cardiac ion channel genes showed that Scn5a, which encodes the principle cardiac voltage-gated Na+ channel (NaV1.5), was circadianly expressed in control mouse and rat hearts but not in iCSΔBmal1−/− hearts. In vitro studies confirmed circadian expression of a human Scn5a promoter-luciferase reporter construct and determined that overexpression of clock factors transactivated the Scn5a promoter. Loss of Scn5a circadian expression in iCSΔBmal1−/− hearts was associated with decreased levels of NaV1.5 and Na+ current in ventricular myocytes. We conclude that disruption of the molecular clock in the adult heart slows heart rate, increases arrhythmias, and decreases the functional expression of Scn5a. These findings suggest a potential link between environmental factors that alter the cardiomyocyte molecular clock and factors that influence arrhythmia susceptibility in humans.
Keywords: cardiac excitability, circadian, heart, ion channels, Scn5a, Na+ current
circadian rhythms are approximate 24-h cycles in biology. These rhythms are present at the systems level, the tissue level, the single cell and molecular levels (16, 33, 34, 38). There are several examples of circadian rhythms in the cardiovascular system, with heart rate, blood pressure, and substrate metabolism exhibiting distinct oscillations over time of day (12, 13, 40, 41). The mechanism that underlies circadian function is the molecular clock. The molecular clock is defined, in a simple way, by a transcription-translation feedback mechanism that is composed of the core clock genes Clock, Bmal1, Per1, Per2, Cry1, and Cry2. CLOCK and BMAL1 are transcription factors that heterodimerize and activate transcription of Per1, Per2, Cry1, and Cry2. PER1, PER2, CRY1, and CRY2 form multimers in the cytoplasm of the cell, translocate to the nucleus, and act to inhibit CLOCK:BMAL1 function. This cycle takes ∼24 h and is the fundamental mechanism underlying circadian rhythms. Components of the core clock have also been shown to regulate the expression of genes outside the clock mechanism, and these genes are designated as clock-controlled genes (CCGs). CCGs often encode transcription factors or proteins that control rate-limiting steps in cell physiology (42).
In the last 8 years, research has determined that most, if not all, cells in the body have circadian clock mechanisms (5, 42, 48, 56). Sophisticated studies using the circadian reporter mouse in which luciferase is knocked into the Per2 locus (mPER2:LUC) have allowed investigators to use bioluminescence recording in cell and tissue explants ex vivo to demonstrate that the circadian clocks in peripheral tissues are self-sustaining and can function in a cell-autonomous manner. While the circadian clock has been shown to exist in the heart and other peripheral tissues, we are only just learning what role it plays in cellular function.
Studies in mice and hamsters in which the circadian clocks, in all tissues, are disrupted have provided important data suggesting links between disruption of the molecular clock and cardiac pathology with end points including altered systolic ventricular function, cardiac hypertrophy, and arrhythmogenic events (28, 32, 36, 37). These studies, however, do not distinguish between the role of the clock mechanism in the heart versus the disruption of the central clock. This requires the targeted cardiomyocyte-specific disruption of the molecular clock and, at this stage, there are only two studies that have used this approach (18). In these papers, heart-specific disruption of the molecular clock was accomplished either through overexpression of a mutant CLOCK protein or through targeted deletion of Bmal1. These studies show that disruption of the molecular clock in cardiomyocytes is sufficient to induce significant reductions in heart rate across all times of day, alter substrate metabolism, and contractile function (12, 18).
The purpose of the present study was to use an inducible cardiac-specific deletion of the core clock gene Bmal1 in mice to identify downstream molecular target(s) and cellular phenotypes.1 Echocardiography showed that deletion of Bmal1 resulted in no significant structural or functional changes when compared with control; however, ECG recordings demonstrated prolonged RR and QRS intervals and episodes of premature beats and pauses not seen in control mice. These changes persisted in an ex vivo working heart preparation, suggesting that they reflect a modification in the intrinsic properties of heart. A screen of candidate cardiac ion channel genes in control hearts showed that Scn5a, the major contributor to Na+ current (INa) in ventricular myocytes, followed a circadian pattern of expression, and this pattern was lost in hearts after the deletion of Bmal1. In addition, in vitro analyses showed that the human Scn5a promoter-reporter exhibited a circadian pattern of luciferase activity in C2C12 cells, and it was transactivated by the overexpression of BMAL1 and CLOCK. Importantly, voltage-gated Na+ channel (NaV1.5) expression and macroscopic INa were significantly reduced in the cardiomyocytes after the deletion of Bmal1. Together, these data demonstrate that the cardiomyocyte molecular clock plays a direct role for intrinsic cardiac arrhythmia susceptibility and the functional expression of Scn5a. We conclude that genetic, extrinsic, or environmental factors that disrupt the molecular clock in the heart likely have implications for arrhythmia susceptibility.
MATERIALS AND METHODS
Inducible deletion of Bmal1 in adult cardiomyocytes.
All animal procedures were conducted in compliance with the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) and were approved by the Institutional Animal Care and Use Committee of University of Kentucky. The inducible mouse model [inducible cardiac-specific ΔBmal1 (iCSΔBmal1)] for these studies was bred by crossing the floxed Bmal1 mouse and the cardiac-specific MerCreMer recombinase mouse (Myh6-MerCreMer) (4, 49). The floxed Bmal1 mouse has loxP sites flanking exon 8 and is indistinguishable from wild-type (WT) C57Bl/6 mice. Breeding with the cardiac-specific inducible Cre-recombinase mouse generates offspring that will undergo selective deletion of the bHLH domain of Bmal1 in cardiomyocytes after tamoxifen administration. This inducible animal model allows us to study loss of BMAL1 function in adult cardiomyocytes. Cardiac-specific inducible Bmal1−/− mice (iCSΔBmal1−/−) were generated as follows: The Bmal1flox/flox female was crossed with the cardiac-specific Cre-recombinase male. This yielded an F1 generation of cardiac-specific Cre+/−;Bmal1+/flox mice. Breeding the F1 generation males to the Bmal1flox/flox females resulted in the cardiac-specific Cre+/−; Bmal1flox/flox mice needed for this study. Cre-recombination was activated once the mice reached 12 wk of age by intraperitoneal injections of tamoxifen (2 mg/day) for 5 consecutive days. The concentration and duration of the tamoxifen injections used have been shown to cause effective cardiomyocyte-specific recombination without any obvious long-term (>6 wk) tamoxifen toxicity as assessed by changes in the structure, function, and ECG (data not shown) (8, 20, 24). Parallel control mice (iCSΔBmal1+/+) were generated by injecting Cre+/−;Bmal1flox/flox mice with vehicle (15% ethanol in sunflower seed oil) instead of tamoxifen.
Recombination specificity.
iCSΔBmal1 mice were treated with either vehicle or tamoxifen at 12 wk of age. Two weeks posttreatment, mice were anesthetized with isoflurane, and heart, diaphragm, liver, lung, abdominal aorta, brain, soleus, and gastrocnemius were collected and immediately frozen in liquid nitrogen for DNA and protein analysis. Genomic DNA was extracted from the above tissues using the DNeasy Blood and Tissue Kit (Qiagen). To assess recombination specificity, PCR was performed with tissue DNA and primers for the recombined and nonrecombined alleles as described by Storch et al. (49). The forward and reverse primers for the nonrecombined allele were the same as the genotyping primers and yielded a 431 bp product. A second forward primer, 5′-CTC CTA ACT TGG TTT TTG TCT GT-3′, was included to detect the recombined allele, which showed a band at 572 bp (49). The PCR reaction was run on a 1.5% agarose gel (0.0005% ethidium bromide) to visualize the DNA products.
Circadian collections.
Sixty-four iCSΔBmal1 mice (mixed sexes) were housed in individual cages in light boxes and entrained to a 12:12-h light-dark cycle (L/D) for 14 days. Mice had ad libitum access to food and water. Following the 2-wk entrainment period, 32 mice were injected with vehicle and 32 with tamoxifen for 5 consecutive days, generating 32 iCSΔBmal1+/+ and 32 iCSΔBmal1−/− mice, respectively. The light schedule was kept the same during injections and for the subsequent 14 days. Two weeks after the last day of injections, mice were released into constant darkness (DD) for 30 h. Mice were euthanized in darkness (dim red light), and heart and liver were collected every 4 h for 28 h (8 time points) and frozen for RNA and protein analysis.
Thirty-two Wistar-Kyoto (WKY) rats were maintained in 12:12-h light-dark cycles, then released into DD. Starting 30 h after entry into DD, heart from three WKY rats was collected every 4 h for 28 h under dim red light (<5 lux). The heart was removed from each rat and frozen in liquid nitrogen.
Electrocardiography telemetry.
In vivo ECG telemetry (Data Sciences International: DSI) was used to evaluate changes in heart rate, RR, and QRS intervals before or after tamoxifen injection. Mice were anesthetized with isoflurane, and transmitter units (PhysioTel ETA-F10; DSI) were implanted in the peritoneal cavity under aseptic conditions. The two ECG leads were secured near the apex of the heart and the right acromion. Mice were housed singly and allowed to recover for 1 wk. Data were recorded for 24 h/day, for 3–4 days every 10 days up to 8 wk after vehicle or tamoxifen treatment. ECG data were collected and analyzed with DSI Dataquest ART4.1 telemetry software. We used a weighted heart rate approach similar to that used by Jeyaraj et al. (28). Average heart rate was calculated for each hour using the Dataquest analysis software. Measurements of ECG parameters were made on three consecutive beats at the first time point in each hour to match the average heart rate for that hour. In addition, we used a custom program written in Matlab (Mathworks) to compute RR intervals. A combination of gradient and threshold method was used to compute RR intervals from the recorded ECG. Briefly, the recorded ECGs were numerically differentiated using a combined smoothening and differentiation filter and a threshold was applied to the absolute values of the differentiated signal to detect R waves. Manual visualization with editing was used to ensure correct detection. Histograms were computed for RR intervals ranging from 50 ms (minimum) to 150 ms (maximum) with a bin width of 10 ms. All RR intervals smaller (greater) than the lower (upper) bounds were included in the lowest and the highest bins.
Working heart preparations.
Isolated ex vivo working heart preparations were produced from iCSΔBmal1+/+ and iCSΔBmal1−/− mice as described previously (21). Baseline conditions of 10 mmHg preload and 55 mmHg afterload were used. The working heart system was modified by an addition of ECG recording. A reference electrode was attached to the left atrial cannula, and a detecting electrode was attached to the surface of the ventricular apex. The heart was paced supraventricularly at 480 beats/min using an isolated stimulator (A365, World Precision Instruments) with the electrodes attached to the right atrium. Baseline rhythm was recorded, and stretch-induced arrhythmia (17, 30) was examined by increasing the preload from 10 mmHg to 12.5, 15, and 20 mmHg at a fixed afterload of 55 mmHg. The ECG recorded from the surface of the heart did not detect clear P waves as those normally recorded in a standard body surface ECG. Therefore, our analysis focused on abnormal rhythms measured from QRS waves and corresponding left ventricle pressure curves. Taking advantage of constant supraventricular pacing, premature ventricular depolarizations or conduction block/delays are readily identified by changes in the RR interval.
RNA isolation.
Total RNA was prepared from frozen tissue samples using TRIzol (Invitrogen) according to the manufacturer's directions. RNA samples were treated with TURBO DNase (Ambion) to remove genomic DNA contamination. Isolated RNA was quantified by spectrophotometry (λ = 260 nm). First-strand cDNA synthesis from total RNA was performed with a mixture of oligo(dT) primer and random hexamers using SuperScript III First-Strand Synthesis SuperMix (Invitrogen). All isolated RNA and cDNA samples were stored at −80°C until further analysis. Real-time quantitative PCR using TaqMan (Applied Biosystems) assays was used to examine the gene expression of core-clock (Bmal1), clock-controlled (Dbp), and ion channel genes (Scn5a, Kcnd2, Kcnj2, Cacna1c, Ncx, Cxn43). The ΔΔCT method was used for the quantification of real-time PCR data in the circadian collections. Gene expression in each sample was shown as the relative value compared with the mean vehicle value in the heart. This enabled us to compare interorgan differences in the expression pattern/amplitude of each gene.
Cotransfection assays.
Cotransfection assays to test the hScn5a promoter were performed following methods described by Wilsbacher et al. (52), using the Period1 (Per1) reporter gene, 6.8Per1-Luc, as a positive control. The hScn5a promoter reporter plasmid was kindly shared by Dr. D. M. Roden (Vanderbilt Univ.,·Nashville, TN; 55). NIH/3T3 fibroblasts (American Type Culture Collection, Manassas, VA) were used because this cell type has been most commonly used to demonstrate transactivation of Per1 reporter gene by overexpression of BMAL1 and CLOCK (23, 29, 31, 52). FuGene 6 was used at a 3:1 (vol/wt) ratio with the total amount of transfected DNA adjusted to 390 ng with empty pcDNA3.1 plasmid. Forty-eight hours after transfection, luminescence of the lysate (20 μl) was measured using the Dual-Luciferase Reporter Assay System (Promega) in a Lumat LB 9507 (EG&G Berthold). Reporter gene expression (hScn5a-Luc or 6.8Per1-Luc) was assessed in triplicate under four conditions: 1) in the absence of expression vectors; 2) cotransfected with pcDNA3.1-mClock and pcDNA3.1-mBmal1; 3) cotransfected with pcDNA3.1-mBmal1 and pcDNA3.1-mClock Δ19; and 4) cotransfected with pcDNA3.1-mClock and pcDNA3.1-mBmal1R91A (57). The pRL null vector was cotransfected in each experiment to provide a control for variations in transfection efficiency. Scn5a is not endogenously expressed in NIH/3T3 fibroblast cells.
Bioluminescent analysis of circadian expression.
The mouse C2C12 skeletal muscle cell line cells were transiently transfected with the luciferase-based reporter gene containing the human Scn5a promoter (hScn5a-Luc). Transfected cells (3 separate transfections) were grown in 35-mm dishes to confluence. Once the cells reached confluence, serum shock (50% FBS for 2 h) was used to synchronize the circadian clocks across the cells (9, 50, 51, 53, 57). Following serum shock, cells were incubated in recording medium containing 0.1 mM luciferin in phenol red free DMEM with 5% fetal bovine serum (FBS) as previously described (53). Bioluminescence was measured at 10-min intervals for 4 days using the Lumicycle (Actimetrics) (53). Both raw and baseline subtracted data were analyzed for circadian characteristics including period and phase using Lumicycle Analysis software. The period length of luminescence for each reporter gene was calculated based on the distances between peaks or troughs over the 24–60 h post-serum shock from the same three transfections. For these studies, the phase of expression was defined, by convention, as the time of the first peak 24 h after serum shock (9, 50, 51, 53, 57).
Western blot analysis.
Whole cell lysates were prepared from the left ventricle of frozen hearts from the 58 h in darkness time point (n = 3–4/time point). SDS-PAGE (4–15% separating gel, Bio-Rad) and immunoblotting were carried out with routine protocols. Affinity-purified NaV1.5 polyclonal antibody (Millipore) was visualized with IRDye-conjugated secondary antibody using the Odyssey system (Li-Cor). Each lane contained 50 μg total protein. Normalization for loading and transfer was performed by simultaneously probing blots with the GAPDH monoclonal antibody (Ambion).
Adult cardiomyocyte isolation.
Adult ventricular myocytes were isolated from the mouse heart as described previously at 6–8 wk following injections to avoid any confounding effects of acute tamoxifen toxicity (8, 20, 24, 45). Mice were anesthetized and hearts were rapidly excised and retrogradely perfused at 3 ml/min for 4–8 min at 37°C with a Ca2+-free bicarbonate-based perfusion buffer containing (in mM) 113 NaCl, 4.7 KCl, 0.6 KH2PO4, 1.2 MgSO4, 0.6 NaH2PO4, 5.5 glucose, 12 NaHCO3, 10 KHCO3, 10 HEPES, 0.032 phenol red, 10 2,3-butanedione monoxime, and 30 taurine. The perfusion buffer was gassed with 95% O2-5% CO2 for at least 30 min before use. Enzymatic digestion began with 0.25 mg/ml liberase Blendzyme (Roche), and 12.5 μM CaCl2 was added to the perfusion buffer for ∼13 min until the heart was swollen and pale in color. The heart was then cut from the cannula. Ventricular tissue was placed in a dish with enzyme buffer and gently dissociated for several minutes. After the addition of stop buffer (perfusion buffer containing 10% FBS and 12.5 μM CaCl2), the dissociation continued until large pieces of heart tissue were gently dispersed into the cell suspension. Cells were allowed to sediment by gravity for 10 min, followed by centrifugation at 180 rcf for 1 min. Cells were resuspended in perfusion buffer containing 5% FBS and 12.5 μM CaCl2. External Ca2+ was added incrementally back to the solution to 2.0 mM.
Electrophysiology.
Voltage-clamp was performed with an Axopatch 200B patch-clamp amplifier (Axon Instruments, Foster City, CA) and pClamp10 software (Axon Instruments). INa was recorded using the whole cell patch-clamp technique. Pipette resistances were <1.5 mΩ and series resistance was compensated up to 95%. The electrophysiological recordings were carried out at room temperature with the following bath (extracellular) solution (in mM): 5 NaCl, 90 CsCl, 1 CaCl, 1.2 MgCl, 11 glucose, 10 TEA-Cl, and 5 HEPES (pH 7.3 set with CsOH). The pipette (intracellular) solution contained (in mM) 120 CsF, 20 CsCl, 10 EGTA, 10 TEA-Cl, 1 Na ATP, and 5 HEPES (pH 7.3 set with CsOH). The individual peak current-voltage (I-V) relations were described using the Boltzmann equation: I(V) = Gmax * (V − Erev)/[1 + exp(V½ − V)/k], where Gmax is the maximal conductance, V½ is the voltage for half-maximal activation, and k is the slope factor (mV/e-fold change). To ensure adequate voltage control, only cells with a k > 4 mV/e-fold were used for analyses. Whole cell INa was measured from a holding potential of −140 mV, and cells were pulsed from −80 to 10 mV for 1 s in 10-mV increments.
Statistical analysis.
Results are reported as means ± SE. A two-way ANOVA was used to determine a significant interaction between factors (strain and time; strain and RR interval). If a significant interaction is detected, a Dunnett post hoc comparison was performed. JTK_CYCLE analysis was used to identify and characterize cycling measures in our data sets (26) using the R statistical package (version 2.12.1) to look specifically for 24-h rhythms. Briefly, JTK_CYCLE estimates the amplitude of the most probable period/lag combination by calculating the median sign-adjusted deviation from the median over the first complete cycle. For Western blot and electrophysiological analyses, unpaired t-tests were performed.
RESULTS
Characterization of the mouse model.
The inducible, cardiac-specific Bmal1−/− mouse strain (iCSΔBmal1−/−) was generated by breeding the cardiac-specific αMyHC-MerCre+/−Mer (47) and the Bmal1flox/flox mice (49). Following vehicle (iCSΔBmal1+/+) or tamoxifen (iCSΔBmal1−/−) treatment, we characterized the recombination of DNA isolated from different muscle and nonmuscle tissues. This is the first inducible heart-specific genetic mouse model for Bmal1 disruption. These results are presented in Fig. 1A. Recombination of Bmal1 was cardiac specific and only seen following tamoxifen treatment, confirming the inducible, tissue specificity of these mice (47).
Fig. 1.

The molecular clock is disrupted in cardiac-specific inducible Bmal1−/− (iCSΔBmal1−/−) mouse hearts. A: representative gels of PCR products showing that recombination at the Bmal1 locus occurs specifically in the heart of the iCSΔBmal1−/− mice but not in other tissues. No recombination was seen in iCSΔBmal1+/+. The recombined gene product shows a band at 571 bp, whereas the nonrecombined product shows a band at 431 bp. Dia, diaphragm; Sol, soleus; Gtn, gastrocnemius; Liv, liver; AA, abdominal aorta; Br, brain. B: the circadian expression profile of Bmal1 and Dbp is significantly blunted specifically in the heart of iCSΔBmal1−/− mice. Dark and light bars represent subjective night and day extrapolated from their prior light:dark (L:D) cycle before release in constant darkness (DD). Bar graphs show average amplitude values with 95% confidence intervals as error bars. n = 4/time point; *P < 0.05.
We then carried out a circadian time course collection with vehicle and tamoxifen-treated mice. Heart (ventricular apex) and liver tissue from four iCSΔBmal1+/+ or four iCSΔBmal1−/− mice was collected every 4 h over a 28-h period, RNA was extracted, and real-time PCR was performed to determine expression of Bmal1. To evaluate function of the molecular clock, we also analyzed expression of Dbp, which is a well-defined direct transcriptional target of the molecular clock (referred to as a clock-controlled gene; CCG). The time course collection allowed us to calculate circadian oscillation, period length, and amplitude using JTK_CYCLE analyses (26). Expression of Bmal1 oscillated robustly in the hearts of iCSΔBmal1+/+ mice with ∼10-fold difference from peak to trough. Bmal1 expression oscillated in the hearts of the iCSΔBmal1−/− mice, but the levels were reduced by ∼50% (Fig. 1B), which closely corresponds to the percentage of heart cells that are cardiomyocytes in mice (10). Mean amplitudes with 95% confidence intervals as error bars are graphed in Fig. 1B. Expression of Dbp, a CCG, was antiphase to Bmal1, and levels were significantly decreased in the hearts of iCSΔBmal1−/− mice. We also analyzed expression of Bmal1 and Dbp genes in the liver to validate the specificity of the mouse model system. The pattern and expression level of mRNA for each of these genes was not significantly different between iCSΔBmal1−/− and iCSΔBmal1+/+ in liver tissue. These findings support the recombination data and show that inactivation of Bmal1 in cardiomyocytes results in significant disruption of the molecular clock mechanism and downstream clock-controlled genes in the heart leaving the liver molecular clock unaffected.
Because of concerns related to tamoxifen treatment in the heart, we evaluated heart structure and function using noninvasive echocardiography. As others have reported, we did see early changes in heart function (ejection fraction) following tamoxifen treatment, but cardiac function returned to control levels by ∼4 days following tamoxifen treatment. No differences were detected in left ventricular mass, ejection fraction, or fractional shortening at the time selected for our study (data not shown).
Electrical activity in the iCSΔBmal1−/− hearts.
ECG telemetry probes were implanted in mice and data were recorded continuously for 4–6 days. We first compared the heart rates and ECG properties from the iCSΔBmal1+/+ and iCSΔBmal1−/− mice. While the circadian variation in heart rate was preserved in the iCSBmal1−/− mouse (Fig. 2A), the average heart rate was slower (570 ± 14 vs. 607 ± 10 beats/min), the RR interval was longer (Fig. 2B, 107 ± 2.8 vs. 99 ± 1.9 ms), and the mean QRS complex was wider (Fig. 2C, 6.9 ± 0.8 vs. 7.9 ± 0.1 ms; n = 6). Together, these data demonstrate that disruption of the molecular clock slows heart rate and causes changes in ECG parameters.
Fig. 2.

Disruption of the molecular clock alters heart rate and QRS intervals. A: 24-h heart rate measured using telemetry implants. Each point represents the half-hour moving average. Note the preservation of circadian rhythmicity and reduction in heart rate for the iCSΔBmal1−/− mice. BPM, beats/min. B and C: average RR and QRS intervals measured from ECG telemetry traces. Note the reduction in heart rate and the trend for longer RR intervals in the iCSΔBmal1−/− mice. *P < 0.05.
Fig. 6.

Bmal1 and CLOCK transactivate the human Scn5a promoter reporter. A: lumicycle data showing the circadian expression pattern of the hScn5a promoter reporter construct transiently transfected in mouse C2C12 (skeletal muscle) cell line; n = 3. B: luciferase assay results of transfection experiments using either the Per1 reporter gene or the hScn5a reporter gene in mouse fibroblast NIH/3T3 cells (n = 6–10/condition). CLOCK (C) and BMAL1 (B) significantly transactivate Per1 and hScn5a reporter genes (black bars) relative to control transfections (white bars). Activation of the hScn5a reporter is significantly diminished when Bmal1R91A was overexpressed with CLOCK (gray bar) or ClockΔ19 was overexpressed with BMAL1 (vertical striped bar). Values are expressed as means ± SE. *P < 0.05 compared with the hScn5a reporter gene; #P < 0.05 compared with hScn5a reporter gene + CLOCK + BMAL.
To more closely investigate the effect that disruption of the molecular clock has on RR interval, we calculated the percentage of beats as a function RR interval during the light and dark phases for iCSΔBmal1+/+ and iCSΔBmal1−/− mice (Fig. 3A). iCSΔBmal1+/+ mice showed a decrease in the percentage of beats with RR intervals between 80 and 100 ms during the light phase but the iCSΔBmal1−/− mice did not (Fig. 3A). We also calculated the change in the percentage of beats as a function of RR interval over 24 h before and after the administration of vehicle/tamoxifen and disruption of the molecular clock (Fig. 3B). Disruption of the molecular clock decreased the percentage of beats that occurred 80–100 ms and increased the percentage of beats with RR intervals >150 ms (Fig. 3B). The increased number of beats with RR intervals >150 ms reflected an increase in the number of pauses between QRS complexes. One of the powerful aspects of the mouse model is that we were able to evaluate the ECG recording before and after recombination in the same mice (pre-iCSΔBmal1−/− vs. iCSΔBmal1−/− mice). Although pauses were present in the ECG of pre-iCSΔBmal1−/− mice (Fig. 3C), the iCSΔBmal1−/− mice experienced repeating episodes of pauses not seen in the ECGs prior to recombination or in the iCSΔBmal1+/+ mice. These repeating episodes of pauses are similar to the nodal arrhythmias reported in a mouse model of nodal dysfunction (25).
Fig. 3.
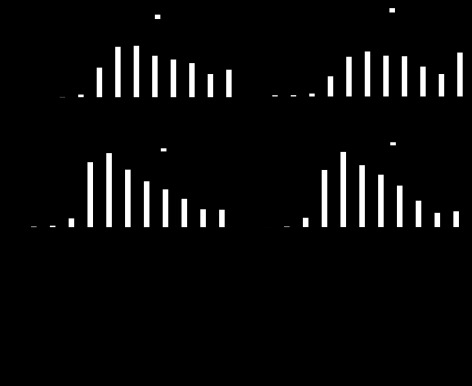
Disruption of the molecular clock alters the diurnal changes in RR and RR variability and increases the incidence of sinoatrial node arrhythmias. A: percentage of beats with the corresponding RR intervals separated by day and night for iCSΔBmal1+/+ and the iCSΔBmal1−/− mice. B: percentage of beats with the corresponding RR intervals over 24 h separated for before (pre-iCSΔBmal1+/+) and after the administration of vehicle or before (pre- iCSΔBmal1−/−) and after disruption of the cardiomyocyte molecular clock. C: representative ECG traces recorded in the pre-iCSΔBmal1−/− and the iCSΔBmal1−/− mice. Mice normally experience isolated pauses, but the iCSΔBmal1−/− mice showed episodes of pauses consistent with sinoatrial node arrhythmias (n = 6 pre-iCSΔBmal1+/+, iCSΔBmal1+/+ and n = 5 pre- iCSΔBmal1−/−, iCSΔBmal1−/−; *P < 0.05).
As a complement to the in vivo telemetry, we tested for altered ventricular cardiac conduction using the isolated working heart preparation. Mechanoelectrical stimulation of the ex vivo heart provides a sensitive way to detect arrhythmia susceptibility (17, 30). For example, stretching the myocardium by increasing the preload from 10 mmHg to 12.5, 15, and 20 mmHg increases the probability of conduction block as measured by changes in the RR interval (21). Hearts were paced from the right atrium at a precise frequency of 8 Hz, so the corresponding RR interval was normally 125 ms (Fig. 4A). Myocardial stretch of the iCSΔBmal1−/− heart preparations showed they were much more sensitive to conduction block than the iCSΔBmal1+/+ hearts as evidenced by the increased presence of conduction block/delay with increasing preload (Fig. 4, A and B, and Table 1). This ex vivo working heart data suggest that disruption of the molecular clock increases the susceptibility to conduction arrhythmias.
Fig. 4.

Disruption of the cardiomyocyte molecular clock stretch-induced arrhythmias in the isolated heart. Stretch-induced conduction block was tested by increasing preload from 10 mmHg to 12.5, 15, and 20 mmHg in iCSΔBmal1+/+ or iCSΔBmal1−/− hearts paced from the right atrium at 8 Hz. A delay in QRS is due to a delay/block of conduction. A: representative sequence of left ventricular pressure (LVP) and the corresponding ventricular depolarizations (QRS) from an isolated iCSΔBmal1+/+ heart. No conduction block was observed at preloads from 10 to 15 mmHg and only 2 out of 7 hearts showed atrioventricular block at a 20 mmHg preload. B: example of block followed by three wide QRS complexes generated from an ectopic foci (note the inversion of the QRS complex). The three wide QRS complexes following the supraventricular conduction block appeared to be generated from a similar source other than the supraventricular stimulus. The iCSΔBmal1−/− mouse hearts showed conduction block at 12.5 mmHg and became more frequent at 15 mmHg preload (3 out of 6) and 20 mmHg (5 out of 6).
Table 1.
Conduction block/delay in iCSΔBmal1+/+ and iCSΔBmal1−/− mice
| Preload, mmHg | iCSΔBmal1+/+ | iCSΔBmal1−/− |
|---|---|---|
| 10 | 0/7 | 0/6 |
| 12.5 | 0/7 | 1/6 |
| 15 | 0/7 | 3/6 |
| 20 | 2/7 | 5/6 |
Conduction block/delay is more common in iCSΔBmal1−/− mice with increased preload. The occurrence of conduction block/delay in the iCSΔBmal1+/+ and the iCSΔBmal1−/− in ex vivo working heart preparations is summarized as the number of hearts showing conduction block/delay over the total number of hearts tested for each group.
Ion channel expression profiles.
The observation that loss of Bmal1 was associated with changes in the intrinsic electrical activity in the absence of structural change prompted the evaluation of the mRNA expression of several ion channels in the hearts of iCSΔBmal1+/+ and iCSΔBmal1−/− mice. We selected genes for evaluation based on the following criteria: they are robustly expressed in both the human and the mouse ventricles; they are important regulators of the cardiac excitability; and changes in their expression and/or function are associated with clinically relevant cardiac arrhythmias. We performed real-time PCR on the RNA from the time course series to analyze Scn5a, Kcnd2, Kcnj2, Cacna1c, Ncx, and Cx43. Using JKT_CYCLE analysis (scanning for period lengths from 22 to 26 h; P < 0.05), we determined that Scn5a, Kcnd2, and Kcnj2 were expressed in a circadian manner (Table 2 and Fig. 5A) in the hearts of the iCSΔBmal1+/+ mice. We next evaluated whether the circadian patterns of expression were altered after loss of Bmal1 and found that the circadian expression pattern for Scn5a and Kcnj2 was lost in the iCSΔBmal1−/− hearts. These data suggest that the cardiomyocyte molecular clock might regulate the expression of Scn5a and Kcnj2 in the hearts of the iCSΔBmal1+/+ mice.
Table 2.
Circadian expression of cardiac ion channel genes
| Gene | iCSΔBmal1+/+ | iCSΔBmal1−/− | WKY |
|---|---|---|---|
| Scn5a | Yes | No | Yes |
| Kcnj2 | Yes | No | No |
| Kcnd3 | Yes | Yes | |
| Cacna1c | No | ||
| Ncx | No | ||
| Cx43 | No |
Real-time PCR was performed on samples from a circadian time course collection using different animal models. Three to four samples were collected every 4 hr for a period of 28 h. Gene expression patterns were analyzed by JTK_CYCLE for circadian pattern of expression. WKY, Wistar Kyoto rats.
Fig. 5.

Scn5a shows a robust circadian oscillation that is lost in iCSΔBmal1−/− mouse hearts. A: the circadian expression profile of Scn5a in the hearts of the iCSΔBmal1+/+ and iCSΔBmal1−/− mice. Dark and light bars represent subjective night and day extrapolated from their prior L:D cycle before release in DD; n = 4/time point. B: Scn5a mRNA is circadian in expression in rat LV; n = 3/time point. WKY, Wistar-Kyoto.
To test whether expression of Scn5a or Kcnj2 was circadian across a different rodent species, we analyzed mRNA expression from a time course collection of hearts from WKY rats (39). We found that Scn5a, but not Kcnj2, was circadianly expressed in the hearts from WKY rats, suggesting a conservation across different species of the relationship between the molecular clock mechanism and expression of Scn5a (Fig. 5B). Thus, the remainder of the study focused on the link between the molecular clock and Scn5a expression and function.
Molecular clock regulation of Scn5a expression.
To study the potential regulatory role of the molecular clock factors BMAL1 and CLOCK on Scn5a expression, we moved to an in vitro system. The 2.7 kb human Scn5a luciferase promoter-reporter construct (hScn5a-Luc), characterized by Yang et al. (55), was used in transfection experiments. The first experiment asked whether the 2.7 kb upstream region of Scn5a was sufficient to direct circadian expression in vitro. For this study, “serum shock,” a technique used to synchronize the molecular clocks across all cultured cells (9, 50, 51, 53, 57), was used. This was coupled with real-time bioluminescence recording collecting light emitted every 10 min for the length of the experiment, in this case, 3 days. The frequency of data collection (every 10 min) for the duration of the experiment (72 h) allows for a very robust statistical analysis of the circadian patterns of luciferase activity and this reflects the regulation of the hScn5a promoter. As seen in Fig. 6A, the hScn5a-Luc reporter construct was sufficient to direct circadian expression of luciferase in the transfected C2C12 cells. (JTK_CYCLE, P = 7.34 × 10−35).
We next tested whether overexpression of the core clock factors CLOCK:BMAL1 in NIH/3T3 mouse fibroblast cells can transcriptionally activate the hScn5a promoter using transient transfection studies. NIH/3T3 fibroblasts were used because this cell type has been most commonly used to demonstrate transactivation of Per1 reporter gene by overexpression of BMAL1 and CLOCK (23, 29, 31, 52). The Per1-Luc construct was used as a positive control and the pRL null vector provided a control for variations in transfection efficiency. Overexpression of CLOCK and BMAL1 transactivated both Per1-Luc and the hScn5a-Luc reporter gene threefold (Fig. 6B). In contrast, overexpression of either of the dominant negative mutant forms of Bmal1 (Bmal1R91A) or Clock (ClockΔ19) did not induce transactivation of the hSch5a-Luc reporter (57). Since all of the control and mutant Bmal1 and Clock cDNAs are expressed by the same plasmid (pcDNA3.1) these results indicate that Scn5a is a clock-controlled gene since transactivation of hScn5a-Luc requires functional CLOCK and BMAL1.
NaV1.5 and INa expression is altered in the iCSΔBmal−/− cardiomyocytes.
Scn5a encodes the principle voltage-gated Na+ channel in the heart (NaV1.5), which is responsible for generating the action potential (AP) in cardiomyocytes and contributes to the conduction of the electrical impulse (3). The half-life of NaV1.5 in adult cardiomyocytes is relatively long (up to 35 h) (35), and as such, it is unlikely to follow a circadian pattern similar to the mRNA. However, we tested whether loss of circadian Scn5a expression was sufficient to alter NaV1.5 levels. Western blot analyses of lysates generated from cardiac tissue from iCSΔBmal1+/+ and iCSΔBmal1−/− mice showed that near the end of the presumptive dark phase (58 h) the relative levels of NaV1.5 were significantly reduced, ∼50%, in iCSΔBmal1−/− mouse hearts (Fig. 7A).
Fig. 7.

Disruption of the cardiomyocyte molecular clock reduces voltage-gated Na+ channel (NaV1.5) levels and Na+ current (INa). A: Western blot data for NaV1.5 in the hearts of iCSΔBmal1+/+ and iCSΔBmal1−/− mice at 58 h in darkness (n = 3/strain; *P < 0.05). B and C: representative families of currents (B) and the mean peak current-voltage (C) relations recorded from iCSΔBmal1+/+ or iCSΔBmal1−/− ventricular cardiomyocytes isolated from mice at 58 h in darkness. The gray and black lines show the robustness of a Boltzmann fit to data. D: iCSΔBmal1−/− ventricular cardiomyocytes had a smaller maximal Na+ conductance (Gmax). *P < 0.05.
The observed changes of NaV1.5 protein levels suggested that ventricular myocytes isolated from iCSΔBmal1−/− mice might have less INa when compared with ventricular myocytes isolated from iCSΔBmal1+/+ mice (Fig. 7, B and C). We measured whole cell I-V relations of INa from ventricular myocytes isolated near the end of the presumptive dark phase. The individual peak I-V relations were fit using a Boltzmann function to calculate the Gmax and V½ or k for INa activation. The ventricular myocytes isolated from iCSΔBmal1−/− mice had a smaller Gmax compared with the ventricular myocytes isolated from iCSΔBmal1+/+mice (1.31 ± 0.08 μS/pF, n = 12, vs. 1.82 ± 0.14 μS/pF, n = 16, respectively, P < 0.05, Fig. 7D). The mean V½ (iCSΔBmal+/+ = −48.9 ± 1.5 mV, iCSΔBmal1−/− = −50.2 ± 1.4 mV) or k (iCSΔBmal1+/+= 5.2 ± 0.3 mV/e-fold change, iCSΔBmal1−/− = 5.6 ± 0.3 mV/e-fold) was not different. These findings demonstrate that disruption of the cardiomyocyte molecular clock decreases NaV1.5 protein levels and macroscopic INa without affecting NaV1.5 activation gating.
DISCUSSION
Transgenic mouse models that selectively disrupt the molecular clock in a tissue/cell-specific manner have begun to demonstrate the importance of a functioning molecular clock in individual cell populations in the context of a multicellular organ. Until recently, cardiac functions that exhibit a circadian pattern, like heart rate, were thought to be regulated more by extrinsic factors such as neurohumoral factors and less, or not at all, by intrinsic factors. We demonstrate that loss of Bmal1 in adult cardiomyocytes primarily results in an abnormal electrical phenotype, which consists of slower heart rates, prolonged RR and QRS intervals, sinoatrial nodal arrhythmias, and an increased electromechanical sensitivity to conduction block. The observation that disruption of the molecular clock altered the intrinsic electrical properties of the heart in the absence of systemic cues prompted us to focus on ion channel gene expression. We discovered that Scn5a exhibited a circadian pattern of mRNA expression. The combination of data from in vivo and in vitro assays suggests that Scn5a expression is under the regulation of the cardiomyocyte molecular clock. Cardiomyocyte-specific loss of Bmal1 resulted in a loss of the circadian expression of Scn5a, a reduction of NaV1.5 levels, and decreased macroscopic INa.
Consistent with previous reports, our data suggest that the cardiomyocyte molecular clock influences heart rate. A slower heart rate phenotype was also observed in mice engineered to overexpress a dominant negative CLOCK mutation in cardiomyocytes (14, 19).
Similar to iCSΔBmal1−/− mice, the day and night variation in heart rate was blunted in CLOCK mutant mice. However, unlike the CLOCK mutant mice, our data suggest that iCSΔBmal1−/− mice have increased arrhythmia susceptibility. The observation that the increased arrhythmia susceptibility persists in iCSΔBmal1−/− hearts ex vivo, in the absence of acute neurohumoral influences, suggests that the cardiomyocyte molecular clock can directly contribute to arrhythmia susceptibility.
Only recently has the molecular clock been suggested to modulate the arrhythmogenic properties of the heart. Jeyaraj and colleagues (28) recently showed that Kcnd2, which encodes the pore-forming subunit for the fast component of the transient outward K+ current (Ito,f), exhibits a circadian pattern of expression, and this pattern was disrupted in the hearts of germline Bmal1 knockout mice. In agreement with their findings, our screen of candidate cardiac ion channel genes also showed that Kcnd2 followed a circadian pattern of expression in iCSΔBmal1+/+ hearts. However, the circadian pattern of Kcnd2 was not disrupted in iCSΔBmal1−/− hearts, which suggests that the central clock, rather than the cardiomyocyte molecular clock, might regulate the circadian expression of Kcnd2 via neural and/or humoral factors. This raises the intriguing possibility that cardiomyocyte and central clocks can independently regulate the circadian expression of different cardiac ion channels.
The addition of a temporal component to the regulation of Scn5a expression by the cardiomyocyte molecular clock is an exciting new finding. Studies that have generated Scn5a knockout mice show that only the heterozygotes are viable (Scn5a+/−) and their cardiac phenotypes are qualitatively similar to the iCSΔBmal1−/− mice. They show a ∼50% reduction in INa, slower heart rates, and sinoatrial node conduction blocks, and the isolated hearts have an increased susceptibility to arrhythmias (43). The iCSΔBmal1−/− show a 30% reduction in Na conductance, slower heart rates, sinoatrial node arrhythmias, and conduction block. Although both of these models show that mice can readily tolerate relatively large changes in INa, many human patients cannot. Some patients with loss-of-function Scn5a mutations or who have cardiac lesions that decrease INa are at increased risk for ventricular fibrillation (1, 2, 6, 7, 15, 22, 46). A dangerous decrease in INa is predicted to promote reentrant arrhythmias in people by slowing cardiac conduction and enhancing the heterogeneity in cardiac repolarization. The small size and rapid heart rate of mice protect them against reentrant arrhythmias that can cause sudden death. Recent evidence suggests that the functional expression of Scn5a might vary from person to person. These differences are expected to contribute to variable ECG properties in the healthy population and likely underlie patient-specific risk for arrhythmias following mutagenic, myocardial (ischemic), or pharmacological insults that decrease cardiac INa (11, 54, 55). Studies have identified single-nucleotide polymorphisms (SNPs) in the Scn5a promoter that cause variable expression in vitro and are linked to patient-specific ECG changes in response to drugs that block INa (11). Moreover, family-based genetic findings suggest that the presence of SNPs in the Scn5a promoter appears to impact the severity of clinical phenotypes associated with loss-of-function Scn5a mutations (44). As a whole, these studies suggest that transcriptional regulation of Scn5a is an important contributor to cardiac conduction variability and arrhythmia susceptibility in humans. Therefore, our finding that the molecular clock temporally regulates the functional expression of Scn5a might lead to the development of novel therapeutic strategies that manipulate the cardiomyocyte molecular clock.
Generally speaking, proarrhythmic myocardial substrates arise from any one or a combination of the following: fibrosis, hypertrophy, myofiber disorganization, and altered gap junction distribution (27). Recent work from our laboratory demonstrated the presence of dilated cardiomyopathy in the germline Bmal1 knockout mouse (32). These mice also demonstrated increased stretch-induced arrhythmia before any overt structural changes were observed (unpublished observations). These data suggest that myocardial substrate properties might be intrinsically regulated by the cardiomyocyte molecular clock and provide the foundation for future studies examining the role of the cardiomyocyte molecular clock in regulating the myocardial substrate.
In summary, cardiomyocyte-specific deletion of Bmal1 alters heart rate, arrhythmia susceptibility, and the functional expression of Scn5a. Identifying environmental or lifestyle factors that influence the cardiomyocyte molecular clock might provide insight into new strategies that can promote or protect against arrhythmias.
GRANTS
This work was supported by National Institutes of Health Grants RC1-ES-018636 and AR-55246 (to K. A. Esser), R01-HL-098945 (to J. P. Jin), and R01-HL-087039 (to B. P. Delisle) and by American Heart Association Predoctoral Fellowship Award 10PRE3900047 (to M. Lefta).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
E.A.S., K.A.E., and B.P.D. conception and design of the research; E.A.S., M.L., X.Z., D.C.B., H.-Z.F., J.-P.J., and B.P.D. performed the experiments; E.A.S., M.L., X.Z., D.C.B., H.-Z.F., Y.Z., A.P., J.-P.J., and B.P.D. analyzed the data; E.A.S., M.L., H.-Z.F., J.-P.J., K.A.E., and B.P.D. interpreted the results of the experiments; E.A.S., M.L., X.Z., H.-Z.F., J.-P.J., and B.P.D. prepared the figures; E.A.S. drafted the manuscript; E.A.S., A.P., J.-P.J., K.A.E., and B.P.D. edited and revised the manuscript; E.A.S., A.P., J.-P.J., K.A.E., and B.P.D. approved the final version of the manuscript.
ACKNOWLEDGMENTS
The authors acknowledge Dr. Jonathan Makielski for assistance and commentary in the development of this project. We also thank Dr. Daniel Roden for kindly sharing the human Scn5a promoter reporter construct.
Footnotes
This article is the topic of an Editorial Focus by Edward G. Lakatta, Yael Yaniv, and Victor A. Maltsev (31a).
REFERENCES
- 1.CAST II Investigators Effect of the antiarrhythmic agent moricizine on survival after myocardial infarction. The Cardiac Arrhythmia Suppression Trial II Investigators. N Engl J Med 327: 227–233, 1992 [DOI] [PubMed] [Google Scholar]
- 2.CAST Investigators Preliminary report: effect of encainide, and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. N Engl J Med 321: 406–412, 1989 [DOI] [PubMed] [Google Scholar]
- 3.Abriel H. Roles and regulation of the cardiac sodium channel Nav1.5: recent insights from experimental studies. Cardiovasc Res 76: 381–389, 2007 [DOI] [PubMed] [Google Scholar]
- 4.Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest 100: 169–179, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akhtar RA, Reddy AB, Maywood ES, Clayton JD, King VM, Smith AG, Gant TW, Hastings MH, Kyriacou CP. Circadian cycling of the mouse liver transcriptome, as revealed by cDNA microarray, is driven by the suprachiasmatic nucleus. Curr Biol 12: 540–550, 2002 [DOI] [PubMed] [Google Scholar]
- 6.Amin AS, Asghari-Roodsari A, Tan HL. Cardiac sodium channelopathies. Pflügers Arch 460: 223–237, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amin AS, Tan HL, Wilde AA. Cardiac ion channels in health and disease. Heart Rhythm 7: 117–126, 2010 [DOI] [PubMed] [Google Scholar]
- 8.Andersson KB, Birkeland JA, Finsen AV, Louch WE, Sjaastad I, Wang Y, Chen J, Molkentin JD, Chien KR, Sejersted OM, Christensen G. Moderate heart dysfunction in mice with inducible cardiomyocyte-specific excision of the Serca2 gene. J Mol Cell Cardiol 47: 180–187, 2009 [DOI] [PubMed] [Google Scholar]
- 9.Ballesta A, Dulong S, Abbara C, Cohen B, Okyar A, Clairambault J, Levi F. a combined experimental and mathematical approach for molecular-based optimization of irinotecan circadian delivery. PLoS Comput Biol 7: e1002143, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol 293: H1883–H1891, 2007 [DOI] [PubMed] [Google Scholar]
- 11.Bezzina CR, Shimizu W, Yang P, Koopmann TT, Tanck MW, Miyamoto Y, Kamakura S, Roden DM, Wilde AA. Common sodium channel promoter haplotype in Asian subjects underlies variability in cardiac conduction. Circulation 113: 338–344, 2006 [DOI] [PubMed] [Google Scholar]
- 12.Bray MS, Shaw CA, Moore MW, Garcia RA, Zanquetta MM, Durgan DJ, Jeong WJ, Tsai JY, Bugger H, Zhang D, Rohrwasser A, Rennison JH, Dyck JR, Litwin SE, Hardin PE, Chow CW, Chandler MP, Abel ED, Young ME. Disruption of the circadian clock within the cardiomyocyte influences myocardial contractile function, metabolism, and gene expression. Am J Physiol Heart Circ Physiol 294: H1036–H1047, 2008 [DOI] [PubMed] [Google Scholar]
- 13.Bray MS, Young ME. The role of cell-specific circadian clocks in metabolism and disease. Obes Rev 10: 6–13, 2009 [DOI] [PubMed] [Google Scholar]
- 14.Challet E, Takahashi JS, Turek FW. Nonphotic phase-shifting in Clock mutant mice. Brain Res 859: 398–403, 2000 [DOI] [PubMed] [Google Scholar]
- 15.Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Antzelevitch C, O'Brien RE, Schulze-Bahr E, Keating MT, Towbin JA, Wang Q. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392: 293–296, 1998 [DOI] [PubMed] [Google Scholar]
- 16.Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol 72: 517–549, 2010 [DOI] [PubMed] [Google Scholar]
- 17.Dick DJ, Lab MJ. Mechanical modulation of stretch-induced premature ventricular beats: induction of a mechanoelectric adaptation period. Cardiovasc Res 38: 181–191, 1998 [DOI] [PubMed] [Google Scholar]
- 18.Durgan DJ, Tsai JY, Grenett MH, Pat BM, Ratcliffe WF, Villegas-Montoya C, Garvey ME, Nagendran J, Dyck JRB, Bray MS, Gamble KL, Gimble JM, Young ME. Evidence suggesting that the cardiomyocyte circadian clock modulates responsiveness of the heart to hypertrophic stimuli in mice. Chronobiol Int 28: 187–203, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Durgan DJ, Young ME. The cardiomyocyte circadian clock: emerging roles in health and disease. Circ Res 106: 647–658, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eckardt D, Theis M, Degen J, Ott T, van Rijen HV, Kirchhoff S, Kim JS, de Bakker JM, Willecke K. Functional role of connexin43 gap junction channels in adult mouse heart assessed by inducible gene deletion. J Mol Cell Cardiol 36: 101–110, 2004 [DOI] [PubMed] [Google Scholar]
- 21.Feng HZ, Biesiadecki BJ, Yu ZB, Hossain MM, Jin JP. Restricted N-terminal truncation of cardiac troponin T: a novel mechanism for functional adaptation to energetic crisis. J Physiol 586: 3537–3550, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fukuda K, Davies SS, Nakajima T, Ong BH, Kupershmidt S, Fessel J, Amarnath V, Anderson ME, Boyden PA, Viswanathan PC, Roberts LJ, 2nd, Balser JR. Oxidative mediated lipid peroxidation recapitulates proarrhythmic effects on cardiac sodium channels. Circ Res 97: 1262–1269, 2005 [DOI] [PubMed] [Google Scholar]
- 23.Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, Takahashi JS, Weitz CJ. Role of the CLOCK protein in the mammalian circadian mechanism. Science 280: 1564–1569, 1998 [DOI] [PubMed] [Google Scholar]
- 24.Hall ME, Smith G, Hall JE, Stec DE. Systolic dysfunction in cardiac-specific ligand-inducible MerCreMer transgenic mice. Am J Physiol Heart Circ Physiol 301: H253–H260, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herrmann S, Fabritz L, Layh B, Kirchhof P, Ludwig A. Insights into sick sinus syndrome from an inducible mouse model. Cardiovasc Res 90: 38–48, 2011 [DOI] [PubMed] [Google Scholar]
- 26.Hughes ME, Hogenesch JB, Kornacker K. JTK_CYCLE: an efficient non-parametric algorithm for detecting rhythmic components in genome-scale datasets. J Biol Rhythms 25: 372–380, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Janse M, Vermeulen J, Opthof T, Coronel R, Wilms-Schopman F, Rademaker H, Baartscheer A, Dekker L. Arrhythmogenesis in heart failure. J Cardiovasc Electrophysiol 4: 496–499, 2001 [DOI] [PubMed] [Google Scholar]
- 28.Jeyaraj D, Haldar SM, Wan X, McCauley MD, Ripperger JA, Hu K, Lu Y, Eapen BL, Sharma N, Ficker E, Cutler MJ, Gulick J, Sanbe A, Robbins J, Demolombe S, Kondratov RV, Shea SA, Albrecht U, Wehrens XH, Rosenbaum DS, Jain MK. Circadian rhythms govern cardiac repolarization and arrhythmogenesis. Nature 483: 96–99, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin X, Shearman LP, Weaver DR, Zylka MJ, De Vries GJ, Reppert SM. A molecular mechanism regulating rhythmic output from the suprachiasmatic circadian clock. Cell 96: 57–68, 1999 [DOI] [PubMed] [Google Scholar]
- 30.Kamkin A, Kiseleva I, Wagner KD, Leiterer KP, Theres H, Scholz H, Günther J, Lab MJ. Mechano-electric feedback in right atrium after left ventricular infarction in rats. J Mol Cell Cardiol 32: 465–477, 2000 [DOI] [PubMed] [Google Scholar]
- 31.Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, Maywood ES, Hastings MH, Reppert SM. mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell 98: 193–205, 1999 [DOI] [PubMed] [Google Scholar]
- 31a.Lakatta EG, Yaniv Y, Maltsev VA. Minding the gaps that link intrinsic circadian clock within the heart to its intrinsic ultradian pacemaker clocks. Focus on “The cardiomyocyte molecular clock, regulation of Scn5a, and arrhythmia susceptibility.” Am J Physiol Cell Physiol (March 13, 2013). doi:10.1152/ajpcell.00072.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lefta M, Campbell KS, Feng HZ, Jin JP, Esser KA. Development of dilated cardiomyopathy in Bmal1-deficient mice. Am J Physiol Heart Circ Physiol 303: H475–H485, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lowrey PL, Takahashi JS. Mammalian circadian biology: elucidating genome-wide levels of temporal organization. Annu Rev Genomics Hum Genet 5: 407–441, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lowrey PL, Takahashi JS. Genetics of circadian rhythms in mammalian model organisms. Adv Genet 74: 175–230, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maltsev VA, Kyle JW, Mishra S, Undrovinas A. Molecular identity of the late sodium current in adult dog cardiomyocytes identified by Nav1.5 antisense inhibition. Am J Physiol Heart Circ Physiol 295: H667–H676, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martino TA, Oudit GY, Herzenberg AM, Tata N, Koletar MM, Kabir GM, Belsham DD, Backx PH, Ralph MR, Sole MJ. Circadian rhythm disorganization produces profound cardiovascular and renal disease in hamsters. Am J Physiol Regul Integr Comp Physiol 294: R1675–R1683, 2008 [DOI] [PubMed] [Google Scholar]
- 37.Martino TA, Tata N, Belsham DD, Chalmers J, Straume M, Lee P, Pribiag H, Khaper N, Liu PP, Dawood F, Backx PH, Ralph MR, Sole MJ. Disturbed diurnal rhythm alters gene expression and exacerbates cardiovascular disease with rescue by resynchronization. Hypertension 49: 1104–1113, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Menaker M, Takahashi JS, Eskin A. The physiology of circadian pacemakers. Annu Rev Physiol 40: 501–526, 1978 [DOI] [PubMed] [Google Scholar]
- 39.Miyazaki M, Schroder E, Edelmann SE, Hughes ME, Kornacker K, Balke CW, Esser KA. Age-associated disruption of molecular clock expression in skeletal muscle of the spontaneously hypertensive rat. PLos One 6: e27168, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Molnar J, Zhang F, Weiss J, Ehlert FA, Rosenthal JE. Diurnal pattern of QTc interval: how long is prolonged?: possible relation to circadian triggers of cardiovascular events. J Am Coll Cardiol 27: 76–83, 1996 [DOI] [PubMed] [Google Scholar]
- 41.Okutucu S, Karakulak UN, Sahiner L, Aytemir K, Demiri E, Evranos B, Fatihoglu SG, Kaya EB, Kabakci G, Tokgozoglu L, Ozkutlu H, Oto A. The relationship between circadian blood pressure pattern and ventricular repolarization dynamics assessed by QT dynamicity. Blood Press Monit 17: 14–19, 2012 [DOI] [PubMed] [Google Scholar]
- 42.Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, Hogenesch JB. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 109: 307–320, 2002 [DOI] [PubMed] [Google Scholar]
- 43.Papadatos GA, Wallerstein PM, Head CE, Ratcliff R, Brady PA, Benndorf K, Saumarez RC, Trezise AE, Huang CL, Vandenberg JI, Colledge WH, Grace AA. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci USA 99: 6210–6215, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park JK, Martin LJ, Zhang X, Jegga AG, Benson DW. Genetic variants in SCN5A promoter are associated with arrhythmia phenotype severity in patients with heterozygous loss-of-function mutation. Heart Rhythm 9: 1090–1096, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schroder E, Magyar J, Burgess D, Andres D, Satin J. Chronic verapamil treatment remodels ICa,L in mouse ventricle. Am J Physiol Heart Circ Physiol 292: H1906–H1916, 2007 [DOI] [PubMed] [Google Scholar]
- 46.Shaw RM, Rudy Y. Electrophysiologic effects of acute myocardial ischemia: a theoretical study of altered cell excitability and action potential duration. Cardiovasc Res 35: 256–272, 1997 [DOI] [PubMed] [Google Scholar]
- 47.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res 89: 20–25, 2001 [DOI] [PubMed] [Google Scholar]
- 48.Storch KF, Lipan O, Leykin I, Viswanathan N, Davis FC, Wong WH, Weitz CJ. Extensive and divergent circadian gene expression in liver and heart. Nature 417: 78–83, 2002 [DOI] [PubMed] [Google Scholar]
- 49.Storch KF, Paz C, Signorovitch J, Raviola E, Pawlyk B, Li T, Weitz Charles J. Intrinsic circadian clock of the mammalian retina: importance for retinal processing of visual information. Cell 130: 730–741, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tamanini F. Manipulation of mammalian cell lines for circadian studies. Methods Mol Biol 362: 443–453, 2007 [DOI] [PubMed] [Google Scholar]
- 51.Tong X, Muchnik M, Chen Z, Patel M, Wu N, Joshi S, Rui L, Lazar MA, Yin L. Transcriptional repressor E4-binding protein 4 (E4BP4) regulates metabolic hormone fibroblast growth factor 21 (FGF21) during circadian cycles and feeding. J Biol Chem 285: 36401–36409, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilsbacher LD, Yamazaki S, Herzog ED, Song EJ, Radcliffe LA, Abe M, Block G, Spitznagel E, Menaker M, Takahashi JS. Photic and circadian expression of luciferase in mPeriod1-luc transgenic mice in vivo. Proc Natl Acad Sci USA 99: 489–494, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamazaki S, Takahashi JS. Real-time luminescence reporting of circadian gene expression in mammals. Methods Enzymol 393: 288–301, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang P, Koopmann TT, Pfeufer A, Jalilzadeh S, Schulze-Bahr E, Kaab S, Wilde AA, Roden DM, Bezzina CR. Polymorphisms in the cardiac sodium channel promoter displaying variant in vitro expression activity. Eur J Hum Genet 16: 350–357, 2008 [DOI] [PubMed] [Google Scholar]
- 55.Yang P, Kupershmidt S, Roden DM. Cloning and initial characterization of the human cardiac sodium channel (SCN5A) promoter. Cardiovasc Res 61: 56–65, 2004 [DOI] [PubMed] [Google Scholar]
- 56.Zambon A, McDearmon E, Salomonis N, Vranizan K, Johansen K, Adey D, Takahashi J, Schambelan M, Conklin B. Time- and exercise-dependent gene regulation in human skeletal muscle. Genome Biol 4: R61, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang X, Patel SP, McCarthy JJ, Rabchevsky AG, Goldhamer DJ, Esser KA. A non-canonical E-box within the MyoD core enhancer is necessary for circadian expression in skeletal muscle. Nucleic Acids Res 40: 3419–3430, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]