

Abstract
Angiotensin-converting enzyme 2 (ACE2) catalyzes conversion of ANG II to ANG-(1–7). The present study uses newly established proteomic approaches and genetic mouse models to examine the contribution of alternative renal peptidases to ACE2-independent formation of ANG-(1–7). In situ and in vitro mass spectrometric characterization showed that substrate concentration and pH control renal ANG II processing. At pH ≥6, ANG-(1–7) formation was significantly reduced in ACE2 knockout (KO) mice. However, at pH <6, formation of ANG-(1–7) in ACE2 KO mice was similar to that in wild-type (WT) mice, suggesting alternative peptidases for renal ANG II processing. Furthermore, the dual prolyl carboxypeptidase (PCP)-prolyl endopeptidase (PEP) inhibitor Z-prolyl-prolinal reduced ANG-(1–7) formation in ACE2 KO mice, while the ACE2 inhibitor MLN-4760 had no effect. Unlike the ACE2 KO mice, ANG-(1–7) formation from ANG II in PEP KO mice was not different from that in WT mice at any tested pH. However, at pH 5, this reaction was significantly reduced in kidneys and urine of PCP-depleted mice. In conclusion, results suggest that ACE2 metabolizes ANG II in the kidney at neutral and basic pH, while PCP catalyzes the same reaction at acidic pH. This is the first report demonstrating that renal ANG-(1–7) formation from ANG II is independent of ACE2. Elucidation of ACE2-independent ANG-(1–7) production pathways may have clinically important implications in patients with metabolic and renal disease.
Keywords: renin-angiotensin system, mass spectrometry, angiotensinase C, angiotensin-converting enzyme 2, prolyl endopeptidase
the renin-angiotensin system (RAS) is a key regulator of renal and cardiovascular function. In diabetes and renal disease, angiotensin-converting enzyme (ACE) is activated and ANG II is increased (26, 36). ANG II is produced from ANG I by ACE, while ACE2 catalyzes conversion of ANG II to ANG-(1–7). ANG-(1–7) is a heptapeptide that counterbalances the effects mediated by ANG II (14, 24, 34, 45). ACE2 also cleaves the vasoactive peptides apelin-13 and des-Arg9 bradykinin, as well as the opioid dynorphin A-(1–13); however, the biological role of the resulting metabolites has yet to be completely revealed (9, 46). The high efficiency of ACE2 to degrade ANG II and generate ANG-(1–7) indicates an important role for ACE2 in the management of cardiovascular and renal dysfunction. Therefore, amplification of ACE2 activity has therapeutic potential (6, 31, 33, 55).
While the central roles of ACE and ANG II have been well established since the pioneering work of Skeggs and co-workers (39), ACE2 is a relatively newly recognized participant in the RAS (10, 43). A plethora of recent studies suggest a protective role for ACE2 against renal and cardiovascular disease in animal models of diabetes, hypertension, and heart failure (3, 21, 30, 54, 59, 60). ACE2 delivery, in the form of recombinant protein or virus, attenuates the progression of hypertension (13, 53, 58), cardiovascular disease (22), and diabetes-related complications such as retinopathy and nephropathy (6, 25, 31, 45).
However, contrary to the known function of ACE2, mice deficient in ACE2 have at baseline a normal phenotype, with renal, cardiovascular, and metabolic parameters minimally affected (17, 19, 50, 57, 61). In addition, ANG II levels in kidney, heart, and plasma are similar to those in wild-type (WT) animals (18, 52, 57, 61), and ANG-(1–7) is not reduced (61). Evidence for ANG-(1–7) production in the ACE2 knockout (KO) strain suggests that the peptide may be generated by enzymes other than ACE2 (12, 42, 61). Therefore, we propose that, in the absence of ACE2, other proteases assume part of its role and degrade ANG II to ANG-(1–7) to keep the balance between these two biologically active peptides.
In addition to ACE2, ANG-(1–7) can be synthesized by prolyl endopeptidase (PEP) (51, 61), prolyl carboxypeptidase (PCP) (40), neprilysin (NEP) (51, 56), thimet oligopeptidase (35), and neurolysin (35). However, only PEP and PCP are capable of generating ANG-(1–7) from ANG II, and a contribution of these peptidases to renal ANG metabolism has been indicated in previous studies (16, 44). Therefore, the experimental focus of the present study will be on these two proteolytic enzymes. Here we describe the characterization of possible ACE2 compensatory pathways that are involved in renal ANG II processing. Using sensitive, mass spectrometry (MS)-based enzyme assays that were developed in our laboratory for tissue sections (16) or tissue homogenates (12), we determined if kidneys obtained from animals deficient in ACE2, PEP, or PCP generate ANG-(1–7) from ANG II.1
METHODS
Animals.
Male mice were used at 16 wk of age. ACE2 KO breeding stocks were generously provided by Drs. T. Coffman and S. Gurley (Duke University, Durham, NC) (8, 57). PCPgt/gt animals were prepared by insertion of LacZ into intron 4 for gene interruption, as described previously (2). PEP KO mouse embryos were obtained from the European Mouse Mutant Archive with assistance from Martin D. Fray (Mammalian Genetics Unit, Medical Research Council, Harwell, UK). The targeted allele for Prep<tm1Dgen> is located on mouse chromosome 10:44787020-44878801 bp. Animals were housed at 22°C under a 12:12-h light-dark cycle with ad libitum access to water and standard mouse chow (Harlan Teklad, Madison, WI). All experimental protocols were approved by the institutional Animal Care and Use Committees. For tissue collection, mice were decapitated and kidneys were quickly removed and frozen in liquid nitrogen.
Urinary ANG peptide analysis.
Twenty-four-hour urine samples were obtained as follows: 10 μl of 6 N HCl were added to the urine collection tube before and 12 h after the urine collection was started. Urinary ANG peptides were measured in the laboratory of Dr. K. Bridget Brosnihan (Wake Forest University Medical Center, Winston-Salem, NC). A cocktail of protease inhibitors, including 0.44 mM 1,20-o-phenanthroline monohydrate, 0.12 mM pepstatin, 1 μM rat renin inhibitor, and 1 mM Na p-hydroxymercuribenzoate, was added to the samples. Urine ANG was extracted using Sep-Pak columns and analyzed by radioimmunoassay. ANG II was measured using an antibody obtained from Alpco Diagnostics (Windham, NH), and ANG-(1–7) was measured using an antibody described elsewhere (38). The minimum detectable levels of the assays were 0.8 and 2.8 fmol/ml for ANG II and ANG-(1–7), respectively. Intra- and interassay coefficients of variation were 12% and 22% for ANG II and 8% and 20% for ANG-(1–7).
Imaging MS assessment of in situ enzyme activity.
Consecutive tissue sections were prepared from fresh frozen kidneys as previously described (16) and incubated with 0.01–1 mM ANG II at 37°C for 5–15 min. Inhibition of renal ANG-(1–7) formation was tested using reaction mixtures spiked with the ACE2 inhibitor MLN-4760 (80 μM; a gift from the former Millennium Pharmaceuticals, Cambridge, MA) or the PCP/PEP inhibitor Z-prolyl-prolinal (ZPP, 0.4–40 μM; Enzo Life Sciences, Farmingdale, NY). The imaging technique is described in detail elsewhere (16). Briefly, a thin-layer chromatography nebulizer was used to spray-coat a matrix consisting of 10 mg/ml α-cyano-4-hydroxycinnamic acid in 60% HPLC-grade methanol, 10% HPLC-grade acetone, and 0.3% sequencing-grade trifluoroacetic acid onto kidney tissue sections. MS images were obtained using an Autoflex III SmartBeam matrix-assisted desorption/ionization-time of flight (MALDI-TOF/TOF) instrument (Bruker Daltonics, Billerica, MA). Spectral analysis was performed with proprietary Bruker imaging software.
In vitro enzyme activity.
ANG-(1–7) formation from ANG II was assessed as previously described with some modifications (12). Kidney homogenate from whole kidney (20 μg of protein) was incubated at 37°C in 50 mM buffer [sodium citrate buffer (pH 4–6), potassium phosphate buffer (pH 6–8), or glycine-NaOH buffer (pH 9–10)] containing 0.02–0.1 mM ANG II, 2 mM PMSF, and 20 μM bestatin. Inhibition of renal ANG-(1–7) formation was tested using reaction mixtures spiked with 80 μM MLN-4760, 0.4–40 μM ZPP, 100 μM captopril, or 100 μM thiorphan. For urinary ANG-(1–7) formation, 24-h urine samples were collected in the presence of 20 μl of inhibitor (Roche Diagnostics, Indianapolis, IN) containing 2.5 mM PMSF. Urine samples were collected every 12 h and kept at 4°C until the 24-h collection was completed. Urine (2 μl) was incubated for 1.5 h at 37°C in 50 mM sodium citrate buffer (pH 5) containing 0.5 μM ANG II, 2 mM PMSF, and 20 μM bestatin. The reaction was stopped by acidification with trifluoroacetic acid. Peptides were purified using micro C18 ZipTips (Millipore, Billerica, MA). Mass spectra were obtained using an Autoflex III SmartBeam MALDI-TOF/TOF instrument. Spectral analysis was performed with proprietary Bruker Flex Analysis software.
MALDI-TOF MS.
The MS was operated with positive polarity in reflectron mode. A total of 2,000 laser shots were acquired randomly for each spot at a laser frequency of 100 Hz. Spectra were mass-calibrated by collection of 200 laser shots of spots containing Bruker peptide calibration standard II consisting of nine peptide standards covering a mass range of 700–3,200 Da.
Immunofluorescence.
Paraffin-embedded kidney sections were deparaffinized in xylene, rehydrated in graded alcohols, and incubated overnight with primary antisera for ACE2, PCP, and PEP: goat anti-ACE2 (1:100 dilution; R & D Systems, Minneapolis, MN), goat anti-PCP [1:100 dilution, as described elsewhere (2)], or rabbit PEP (1:500 dilution; Abcam, Cambridge, MA) polyclonal antibody. Sections were then incubated with secondary antisera: donkey anti-goat labeled with Alexa Fluor 488 (1:100 dilution; Jackson ImmunoResearch, West Grove, PA) or donkey anti-rabbit labeled with Alexa Fluor 568 (1:200 dilution; Invitrogen, Grand Island, NY). Images were obtained using a confocal microscope (model FV300, Olympus, Center Valley, PA).
Western blotting.
Mouse kidneys were processed in cOmplete Lysis-M EDTA-free buffer (Roche Applied Science, Indianapolis, IN) containing 2.5 mM PMSF using a bead homogenizer (Precellys 24, Bertin Technologies, Montigny-le-Bretonneux, France). Total protein content was determined in the supernatant using the Bio-Rad Protein Assay Reagent (Bio-Rad Laboratories, Hercules, CA) with BSA as a standard. Samples (60 μg) were subjected to SDS-PAGE in 10% polyacrylamide gels and transferred to a polyvinylidene difluoride membrane (Bio-Rad Laboratories). Blot membranes were incubated with rabbit anti-ACE2 (1:200 dilution; Santa Cruz Biotechnology, Santa Cruz, CA), goat anti-PCP (1:500 dilution), or rabbit anti-PEP (1:2,000 dilution) polyclonal antibody. Peroxidase-conjugated secondary antibody [anti-rabbit (1:2,000 dilution) or anti-goat (1:5,000 dilution; Santa Cruz Biotechnology) or anti-rabbit (1:3,000 dilution; Abcam)] and enhanced chemiluminescence reagents (Thermo Scientific, Rockford, IL) were used to detect binding of primary antibody with a Fuji LAS3000 system.
Data analysis.
MALDI imaging signals represented in color on pseudocolor images were quantitated as integrated intensity and normalized to the area of the whole kidney section using MetaMorph image analysis software (Molecular Devices, Sunnyvale, CA). Values are means ± SE. Unpaired Student's t-test was used to evaluate the differences between two groups. For more than two groups, repeated-measures two-way ANOVA followed by Bonferroni's multiple comparison test was used. One-way ANOVA compared the difference in ANG-(1–7) formation in PCP WT animals vs. PCPgt/gt and inhibitor-treated PCPgt/gt mice. P < 0.05 was considered statistically significant.
RESULTS
Urinary ANG peptides are undistinguishable in ACE2 WT and ACE2 KO mice.
ANG II and ANG-(1–7) were measured in 24-h urine of ACE2 WT and ACE2 KO mice by radioimmunoassay. After normalization to urinary excretion per day, there was no significant difference in urinary ANG II levels in ACE2 KO compared with ACE2 WT mice (Fig. 1), nor was there a difference in urinary ANG-(1–7).
Fig. 1.

Urinary ANG-(1–7) and ANG II in angiotensin-converting enzyme type 2 (ACE2) wild-type (WT) and ACE2 knockout (KO) mice. Twenty-four-hour urine samples were collected in the presence of 20 μl of 6 N HCl, mixed with protease inhibitor cocktail, subjected to solid-phase extraction, and analyzed by radioimmunoassay. Peptide levels were normalized to excretion per day. Values are means ± SE of 8–9 mice per group. On 2-way ANOVA, there were no differences among the groups.
In situ generation of renal ANG-(1–7) is dependent on ACE2 genotype, ANG II concentration, and incubation time.
We previously used and optimized MS imaging to quantify enzymatic ANG II processing to ANG-(1–7) in situ (16). In the present study, ACE2 WT and ACE2 KO kidney sections (12 μm) were incubated at 37°C with ANG II at 0.01–1 mM per section for 5 or 15 min. As expected, ACE2 WT mice formed ANG-(1–7) in a dose- and time-dependent manner (Fig. 2). ACE2 KO mice were also capable of generating ANG-(1–7) from ANG II. At low concentrations of ANG II (≤0.1 mM) and 5 min of incubation, ANG-(1–7) formation was reduced in ACE2 KO mice (Fig. 2A). However, at >0.5 mM ANG II and 15 min of incubation, there was no significant difference in ANG-(1–7) levels between ACE2 WT and ACE2 KO mice (Fig. 2B). It is worth mentioning that these reaction conditions are optimal and the in situ reaction is quick, since at lower concentrations and longer incubation times ANG II and ANG-(1–7) were not detectable.
Fig. 2.

Characterization of renal ANG-(1–7) formation in situ. Kidney sections from ACE2 WT and ACE2 KO mice were incubated with different concentrations of ANG II. Generated ANG-(1–7) was analyzed by in situ imaging mass spectrometry (MS). A: ANG-(1–7) formation at 5 min. Data were collected after 5-min incubation of tissue sections with ANG II. B: ANG-(1–7) formation at 15 min. Data were collected after 15-min incubation of tissue sections with ANG II. Note absence of bars for ANG-(1–7) formation in incubations containing 0.01, 0.05, and 0.1 mM ANG II. Values are means ± SE of 4 mice per group. Effect of genotype, ANG II concentration, and incubation time on ANG-(1–7) formation was analyzed by 2-way ANOVA. *P < 0.01, **P < 0.001, ANG-(1–7) formation in ACE2 WT vs. ACE2 KO mice or ANG-(1–7) formation at 0.5 and 1 mM ANG II vs. 0.01 mM ANG II. #P < 0.05, ##P < 0.01, ANG-(1–7) formation at 5 min vs. 15 min.
In vitro generation of renal ANG-(1–7) in ACE2 WT and ACE2 KO mice is dependent on pH.
An in vitro MS approach was used to characterize the pH dependency of ANG-(1–7) formation in ACE2 WT and ACE2 KO mice. Kidney homogenates were incubated with ANG II in three different buffer systems over a pH range of 4–10. Generation of ANG-(1–7) in ACE2 WT mice was detected from pH 4 to pH 9 (Fig. 3A). At pH 4 and 5, there was no significant difference in ANG-(1–7) generation between ACE2 WT and ACE2 KO mice (Fig. 3A). However, at pH 6–9, ANG-(1–7) formation was significantly decreased in ACE2 KO mice (Fig. 3A). Results suggest that ACE2 is one of the predominant enzymes responsible for ANG-(1–7) formation in the kidney at pH 6–9. It is tempting to speculate that proteolytic enzyme(s) other than ACE2 catalyze(s) this reaction at pH 4–6. Since there is evidence that ANG-(1–7) can be further degraded by renal ACE and NEP (4), we tested the effect of the ACE inhibitor captopril and the NEP inhibitor thiorphan on in vitro ANG II processing in kidney homogenates obtained from ACE2 WT mice at pH 5 and 7. As illustrated in Fig. 3B, both inhibitors had no effect at pH 5 but showed significantly increased renal ANG-(1–7) formation at pH 7.
Fig. 3.

Characterization of renal ANG-(1–7) formation in vitro. A: kidney homogenates (20 μg of protein) from ACE2 WT and ACE2 KO mice were incubated with 0.1 mM ANG II for 30 min at 37°C in 50 mM buffer [sodium citrate buffer (pH 4–6), potassium phosphate buffer (pH 6–8), or glycine-NaOH buffer (pH 9–10)]. ANG peptides were purified and analyzed by MS. B: kidney homogenates from ACE2 WT mice (n = 4) were incubated with 0.1 mM ANG II for 20 min at 37°C in 50 mM citrate buffer (pH 5) or 50 mM phosphate buffer (pH 7) in the presence of 100 μM captopril (an ACE inhibitor) or 100 μM thiorphan (a neprilysin inhibitor). ANG-(1–7) formation at pH 5 and 7 without addition of inhibitors was set at 100% (i.e., control). Values are means ± SE of 3–4 mice per group. *P < 0.05, **P < 0.01, ***P < 0.001, ANG-(1–7) formation at different pH in ACE2 WT vs. ACE2 KO mice (2-way ANOVA). #P < 0.001, ANG-(1–7) formation at pH 5 vs. pH 7 (2-way ANOVA).
The dual PCP-PEP inhibitor ZPP reduces ANG-(1–7) formation in ACE2 KO mice, while the ACE2 inhibitor MLN-4760 has no effect.
The effect of MLN-4760 on renal ANG-(1–7) formation in ACE2 KO mice under conditions of high ANG II concentrations (1 mM, in situ approach) or at pH 5 (in vitro approach) was examined. Under these conditions, incubation with MLN-4760 had no effect on in situ and in vitro renal ANG-(1–7) formation in ACE2 WT and ACE2 KO mice (Fig. 4). Since the ACE2 inhibitor did not block the detected ANG-(1–7)-forming enzyme activity at high substrate concentrations or at pH 5, potential contributions of two other peptidases capable of forming ANG-(1–7) from ANG II, PCP and PEP, were studied using the dual PCP-PEP inhibitor ZPP. At high ANG II concentrations or at pH 5, the in situ and in vitro MS-based assays showed that ZPP significantly inhibited ANG-(1–7) formation from ANG II in ACE2 WT and ACE2 KO mice (Fig. 5). These results suggest involvement of PCP-PEP in renal ANG-(1–7) formation. Comparison of both MS methods revealed a higher efficacy of the in situ approach to detect inhibition of ANG-(1–7) formation by ZPP.
Fig. 4.

Effect of the ACE2 inhibitor MLN-4760 on renal ANG-(1–7) formation in ACE2 WT and ACE2 KO mice at pH 5. A: imaging MS of kidney sections incubated with 1 mM ANG II for 15 min without (control) or with 80 μM MLN-4760 (+MLN). Values are means ± SE of 4 mice per group. On 2-way ANOVA, there were no differences among the groups. B: kidney homogenate (50 μg) was incubated for 30 min in 50 mM citrate buffer (pH 5) containing 0.02 mM ANG II without (control) or with 80 μM MLN-4760 (+MLN), and generated ANG-(1–7) was evaluated using MS. m/z, Mass-to-charge ratio.
Fig. 5.
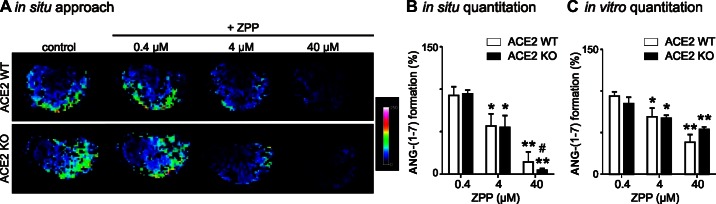
Inhibition of ANG-(1–7) formation with the dual prolyl carboxypeptidase (PCP)-prolyl endopeptidase (PEP) inhibitor Z-prolyl-prolinal (ZPP) at pH 5. A: representative imaging MS of kidney sections incubated with 1 mM ANG II and 0.4–40 μM ZPP for 15 min. B: quantitation of the in situ approach. C: quantitation of in vitro analysis of ANG-(1–7) formation in 30-min incubations containing citrate buffer (pH 5), 0.02 mM ANG II, 50 μg of kidney protein, and 0.4–40 μM ZPP. In B and C, ANG-(1–7) formation without addition of inhibitors was set at 100% (i.e., control). Values are means ± SE of 4 mice per group. *P < 0.01, **P < 0.001, ANG-(1–7) formation in ACE2 WT and ACE2 KO mice in control vs. inhibitor-treated samples (2-way ANOVA). #P < 0.01, in situ vs. in vitro (2-way ANOVA).
Identification of PCP as an alternative renal ANG II-processing enzyme.
To support the enzyme activity data, we used immunofluorescence staining and Western blotting to further analyze the presence and protein expression of renal PCP and PEP in ACE2 WT and ACE2 KO mice. PCP and PEP were localized to glomeruli and tubules in the renal cortex, as was ACE2 (Fig. 6, A–C). Immunofluorescence and Western blot analysis confirmed that ACE2 KO mice were deficient in ACE2 protein (Fig. 6, A and D). PCP and PEP were expressed in kidney cortex, and protein levels of PCP and PEP were unchanged in ACE2 KO mice compared with ACE2 WT mice (Fig. 6, E and F).
Fig. 6.

Immunological characterization of renal ACE2, PCP, and PEP expression in ACE2 WT and ACE2 KO mice. A–C: immunofluorescence staining of ACE2, PCP, and PEP. D–F: Western blot of renal ACE2, PCP, and PEP. Protein levels were normalized to β-actin. Values are means ± SE of 4 mice per group. *P < 0.0001, protein levels in ACE2 WT vs. ACE2 KO mice (unpaired, 2-tailed t-test).
Although ACE2 was depleted (Fig. 7A), ACE2 KO mice were still capable of processing ANG II to ANG-(1–7) at pH 5 (Fig. 7B). To investigate whether renal PEP plays a role in ACE2-independent ANG-(1–7) formation, we examined ANG II processing in PEP WT and PEP KO mouse kidneys at pH 5 and 7. After confirmation by Western blotting that PEP KO mice completely lacked renal PEP protein (Fig. 7C), kidney homogenates were incubated with 0.05 mM ANG II at pH 5 and 7 for 30 min at 37°C and analyzed by MS. ANG-(1–7) formation in PEP WT and PEP KO mice was indistinguishable (Fig. 7D). Finally, PCP WT and PCPgt/gt mouse kidneys were examined for PCP protein expression and ANG II-processing activity at pH 5 and 7. PCPgt/gt animals showed reduced PCP protein levels (54.3 ± 14.3%) and enzyme activity (63.6 ± 7.3%) at pH 5, with no significant difference at pH 7 (Fig. 7, E and F). Addition of ZPP to the PCPgt/gt incubation mixtures produced a further decline in the enzyme activity at pH 5 (Fig. 7F). The in vitro MS assay detected urinary ANG-(1–7) formation at pH 5, which was significantly decreased in PCPgt/gt mice compared with PCP WT mice (Fig. 7G).
Fig. 7.

Expression of renal ACE2, PEP, and PCP and effect of pH on ANG II processing in ACE2 KO, PEP KO, and PCPgt/gt mice. A: Western blot of renal ACE2 in kidney homogenates from ACE2 WT and ACE2 KO mice. B: in vitro analysis of ANG-(1–7) formation in incubations containing 50 mM citrate buffer (pH 5) or phosphate buffer (pH 7), 0.05 mM ANG II, and 20 μg of kidney protein from ACE2 WT or ACE2 KO mice. C: Western blot of renal PEP in kidney homogenates from PEP WT and PEP KO mice. D: in vitro analysis of ANG-(1–7) formation in incubations containing 50 mM citrate buffer (pH 5) or phosphate buffer (pH 7), 0.05 mM ANG II, and 20 μg of kidney protein from PEP WT or PEP KO mice. E: Western blot of renal PCP in kidney homogenates from PCP WT and PCPgt/gt mice. F: in vitro analysis of ANG-(1–7) formation in incubations containing 50 mM citrate buffer (pH 5) or phosphate buffer (pH 7), 0.05 mM ANG II, and 20 μg of kidney protein from PCP WT or PCPgt/gt mice. G: in vitro analysis of urinary ANG-(1–7) formation in incubations containing 50 mM citrate buffer (pH 5), 0.5 μM ANG II, and 2 μl of 24-h urine from PCP WT or PCPgt/gt mice. Values are means ± SE of 4–6 mice per group. *P < 0.05, **P < 0.01, ANG-(1–7) formation at pH 5 and 7 in WT vs. KO (2-way ANOVA). #P < 0.05, γP < 0.001, PCP WT vs. PCPgt/gt + ZPP and PCPgt/gt (1-way-ANOVA). $P < 0.01, PCP WT vs. PCPgt/gt (unpaired, 2-tailed t-test).
DISCUSSION
Our studies characterized renal ANG II processing to ANG-(1–7) by analyzing ANG peptide levels and ANG II metabolism in gene deletion models. First, urinary levels of ANG II and ANG-(1–7) were measured by radioimmunoassay, and no differences were noted between ACE2 KO and ACE2 WT mice. These findings agree with previous reports describing normal ANG II levels in plasma, heart, and kidney of ACE2 KO mice (18, 52, 57, 61). To elucidate whether ANG-(1–7) in ACE2 KO mice originates from ANG II by means of compensating mechanisms, we analyzed the ability of ACE2 KO mice to generate ANG-(1–7) from ANG II. Established MS-based protocols (12) allowed for the enzymatic characterization with respect to genotype, substrate concentration, incubation time, pH, and effect of inhibitors. This type of measurement is not possible with classical, commercially available systems that use nonnatural chromophore or fluorogenic substrates. For example, the substrates Mca-YVADAPK(Dnp) and Mca-APK(Dnp) are commonly used to measure ACE2 activity but are also used to measure activity of other peptidases such as caspase-1 (23, 46). The assay is based on cleavage of an amide bond between the fluorescent [methoxycoumarin (Mca)] and the quencher [dinitrophenyl (Dnp)] group, resulting in an increase in fluorescence. However, such a fluoresence signal could be produced by peptidases other than ACE2. Thus these artificial substrates may not be highly specific, may interact with other enzymes, and can generate conflicting results (16, 20, 46). In contrast, MS assays allow for the use of natural substrates and determination of alternate proteolytic cleavage sites (15). Possible multiple products can be easily detected and differentiated from one another. Therefore, the activity of specific enzymes can be precisely identified. Our laboratory first developed in vitro MS assays for ACE and ACE2 activities in tissue homogenates (11, 12). This was followed by an in situ tissue-imaging technique that allows for the anatomic localization of enzyme activity, showing ACE2 activity primarily in the outer renal cortex (16). The advantages of an MS-based enzyme assay over traditional techniques are its high specificity, sensitivity, and selectivity for the proteolytic products obtained with the endogenous peptide precursor (12, 16). In addition, MS imaging gives information on the spatial localization of enzyme activities within tissue sections (16). We utilized MS techniques to identify and characterize possible ACE2 compensatory pathways that are involved in renal ANG II processing. Kidney sections were analyzed by imaging MS. At >0.1 mM ANG II with 5 or 15 min of incubation, ANG-(1–7) was generated from ANG II to a similar extent in ACE2 KO and ACE2 WT mice. This is the first report to demonstrate renal ANG-(1–7) formation from ANG II that is independent of ACE2. Since the kidney plays a pivotal role in regulation of acid-base homeostasis and small changes in pH can severely affect enzyme activities, we examined the influence of pH on renal ANG-(1–7) formation in ACE2 WT and ACE2 KO mice. We used an in vitro MS approach, testing the processing of ANG II in renal homogenates. In ACE2 WT mice, ANG-(1–7) formation was found at a wide pH range (pH 4–9). In ACE2 KO mice, ANG-(1–7) formation was significantly reduced at pH ≥6 but was not significantly different from ACE2 WT mice at pH 4 and 5. Taken together, results suggest that ACE2 is the predominant enzyme responsible for ANG-(1–7) formation in the kidney under neutral and basic conditions (pH 6–9), while proteolytic enzymes other than ACE2 catalyze this reaction at acidic pH (pH 4–6). The flat pH response at pH 7–9 in ACE2 WT mice is most likely due to quick degradation of formed ANG-(1–7) by other renal peptidases active at neutral and basic conditions. Indeed, NEP and ACE are peptidases known to degrade ANG-(1–7) (4), and addition of NEP or ACE inhibitor to the assay caused a significant increase in ANG-(1–7) signal at pH 7 but had no effect at pH 5. Thiorphan also inhibits ACE at the concentrations used in this assay, which might explain why, at pH 7, both inhibitors increased the ratio of ANG-(1–7) to ANG II to a similar extent. Overall, these experiments indicate that NEP and ACE inhibitors are critical factors that allow for a quantitative comparison between ACE2 and ACE2-independent ANG-(1–7) formation.
The identity of the enzyme(s) catalyzing renal ANG-(1–7) formation in ACE2 KO mice was studied using peptidase inhibitors and genetic mouse models. As previously established for the in situ MS imaging approach (16), the specific ACE2 inhibitor MLN-4760 selectively inhibited ANG-(1–7) formation in ACE2 WT mice at low ANG II concentrations (<0.1 mM). However, at higher ANG II concentrations, MLN-4760 had no effect on ANG-(1–7) formation in ACE2 WT mice. Here, we examined the effect of MLN-4760 on ANG-(1–7) formation in ACE2 KO mice at 1 mM ANG II or at pH 5. As expected, the specific ACE2 inhibitor MLN-4760 failed to reduce ANG-(1–7) formation in ACE2 KO mice in both experiments. In contrast, the dual PCP-PEP inhibitor ZPP, with a Ki of 0.26 μM for PCP (40) and a Ki of 0.35 nM for PEP (5), was effective. ANG-(1–7) formation was reduced to 4–14% by 40 μM ZPP in the in situ approach. When the in vitro method was used, 40 μM ZPP decreased ANG-(1–7) formation to 38–54%. Both experiments suggest a partial contribution of PCP or PEP to renal ANG-(1–7) formation. Western blotting confirmed that PCP and PEP were expressed in ACE2 WT and ACE2 KO mice. PEP has been proposed as a mediator in obesity and eating disorders (29, 49). PCP in the hypothalamus has been demonstrated to regulate α-MSH1–13, and PCPgt/gt mice are lean (47). Unlike ACE2 KO mice, PCPgt/gt mice are constitutively hypertensive and prothrombotic, with vascular inflammation and increased reactive oxygen species (2, 37). Although plasma ANG peptide levels were unchanged (2, 37), renal ANG-(1–7) was increased threefold in PCP-deficient mice (37). This finding is in contrast to ANG-(1–7) levels in whole kidney extracts of ACE2 KO mice, which were not significantly different from ACE2 WT mice (61). It is worth noting that other studies reported significantly lower cortical ANG-(1–7) levels (42) and increased ANG II levels in kidney, heart, and plasma of ACE2 KO mice at baseline (8).
While PEP is most efficient at neutral/basic pH (48), acidic conditions at pH <6 are optimal for PCP (32). This enzymatic characteristic indicates that PCP is the predominant enzyme responsible for ANG II metabolism in ACE2 KO mice. To support this hypothesis, we conducted experiments in PEP KO and PCPgt/gt mice. ANG-(1–7) formation at pH <6 in PEP KO mice was indistinguishable from that in PEP WT mice, while this reaction was reduced in PCPgt/gt animals. Moreover, addition of ZPP to the PCPgt/gt incubations resulted in a further decline of ANG-(1–7) formation at pH 5. These results suggest that PCP is an enzyme catalyzing renal ANG II conversion to ANG-(1–7) independent of ACE2. In fact, a possible involvement of PCP in renal ANG II processing has been identified recently (16) and confirmed by another independent study (44). A contribution of PCP to renal ANG II processing is likely, considering its localization to glomeruli and tubules in the renal cortex, which is similar to previous findings for ACE2 (2, 60). This also fits very well with the MS imaging results. Throughout the current study, MS imaging detected ANG-(1–7) formation exclusively in the renal cortex, regardless of genotype or treatment of samples, showing that the cortex is a key site of ANG-(1–7) formation in the kidney. This is consistent with our previous results (16). Moreover, the response of ZPP inhibition on ANG-(1–7) formation in ACE2 WT and ACE2 KO mice was more effectively revealed by MS imaging than by the in vitro method.
In addition to a reduced renal ANG-(1–7) formation, MS analysis revealed a decreased urinary ANG-(1–7) synthesis at pH 5 in PCPgt/gt mice, suggesting that enzymatically active PCP is excreted into the urine. We propose that intratubular ANG II is degraded to ANG-(1–7) by ACE2 and/or PCP, thereby preventing activation of cell signaling through luminal ANG II type I receptors. Recent clinical studies closely linked urinary acidity to diabetic patients and metabolic syndrome (1, 27, 28, 41), an association that has also been confirmed in a rat model of type 2 diabetes (7). Acidic urine can indicate changes in the biochemical composition and acidity of the intratubular micromilieu. These alterations, albeit small, can severely affect tubular enzyme activity and, as a result, establish PCP as one of the predominant, intratubular enzymes responsible for ANG II degradation. Therefore, ANG II processing by PCP may have clinical implications in patients with metabolic or renal pathologies.
In conclusion, this study shows that processing of ANG II to ANG-(1–7) is not unilaterally dependent on ACE2. Using novel MS enzyme tests in genetic mouse models, we identified and characterized a possible ACE2 compensatory pathway in kidney that is involved in pH-dependent ANG II processing. We suggest that this ACE2-independent ANG-(1–7)-forming enzyme activity escaped detection, possibly because different methodological approaches were used and the link between this enzyme reaction and its dependency on pH was not recognized. Since change in urinary pH is one of the underlying disturbances in renal and metabolic diseases, renal ANG II-degrading peptidases that prefer acidic conditions might play an important role in these pathologies.
GRANTS
This work was supported by National Institutes of Health Grants R01 HL-093567 (M. Morris and K. M. Elased), F32 DK-093226-01 (N. Grobe), F32 HL-105036 (F. S. Ong), R01 HL-052779, and R21 HL-112666A (A. H. Schmaier), American Heart Association Fellowship 10PRE4330004 (N. M. Weir), and a Cedars-Sinai Medical Center Clinical and Translational Science Institute (CTSI) Clinical Scholars Award (F. S. Ong). The CTSI was supported by National Center for Research Resources Grant UL1 RR-033176 and is now supported by National Center for Advancing Translational Sciences Grant UL1 TR-000124.
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.G., N.M.W., M.M., and K.M.E. are responsible for conception and design of the research; N.G., N.M.W., O.L., and F.S.O. performed the experiments; N.G., N.M.W., O.L., and F.S.O. analyzed the data; N.G., N.M.W., O.L., F.S.O., K.E.B., A.H.S., M.M., and K.M.E. interpreted the results of the experiments; N.G., N.M.W., and O.L. prepared the figures; N.G., M.M., and K.M.E. drafted the manuscript; N.G., N.M.W., O.L., F.S.O., K.E.B., A.H.S., M.M., and K.M.E. edited and revised the manuscript; N.G., N.M.W., O.L., F.S.O., K.E.B., A.H.S., M.M., and K.M.E. approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank Dr. Randall A. Skidgel (University of Illinois, Chicago, IL) for thoughtful discussion. We acknowledge the excellent technical assistance of Mary Key and Dr. Valerie Neff. We thank Dr. K. Bridget Brosnihan (Wake Forest University Medical Center, Winston-Salem, NC) for urinary ANG peptide analysis (National Heart, Lung, and Blood Institute Grant HL-051952).
Footnotes
This article is the topic of an Editorial Focus by Juan Carlos Q. Velez (43a).
REFERENCES
- 1.Abate N, Chandalia M, Cabo-Chan AV, Moe OW, Sakhaee K. The metabolic syndrome and uric acid nephrolithiasis: novel features of renal manifestation of insulin resistance. Kidney Int 65: 386–392, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Adams GN, LaRusch GA, Stavrou E, Zhou Y, Nieman MT, Jacobs GH, Cui Y, Lu Y, Jain MK, Mahdi F, Shariat-Madar Z, Okada Y, D'Alecy LG, Schmaier AH. Murine prolylcarboxypeptidase depletion induces vascular dysfunction with hypertension and faster arterial thrombosis. Blood 117: 3929–3937, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alghamri MS, Weir NM, Anstadt MP, Elased KM, Gurley SB, Morris M. Enhanced angiotensin II-induced cardiac and aortic remodeling in ACE2 knockout mice. J Cardiovasc Pharmacol Ther 18: 138–151, 2013 [DOI] [PubMed] [Google Scholar]
- 4.Allred AJ, Diz DI, Ferrario CM, Chappell MC. Pathways for angiotensin-(1–7) metabolism in pulmonary and renal tissues. Am J Physiol Renal Physiol 279: F841–F850, 2000 [DOI] [PubMed] [Google Scholar]
- 5.Bakker AV, Jung S, Spencer RW, Vinick FJ, Faraci WS. Slow tight-binding inhibition of prolyl endopeptidase by benzyloxycarbonyl-prolyl-prolinal. Biochem J 271: 559–562, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bindom SM, Hans CP, Xia H, Boulares AH, Lazartigues E. Angiotensin I-converting enzyme type 2 (ACE2) gene therapy improves glycemic control in diabetic mice. Diabetes 59: 2540–2548, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bobulescu IA, Dubree M, Zhang J, McLeroy P, Moe OW. Effect of renal lipid accumulation on proximal tubule Na+/H+ exchange and ammonium secretion. Am J Physiol Renal Physiol 294: F1315–F1322, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveira-dos-Santos AJ, da Costa J, Zhang L, Pei Y, Scholey J, Ferrario CM, Manoukian AS, Chappell MC, Backx PH, Yagil Y, Penninger JM. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 417: 822–828, 2002 [DOI] [PubMed] [Google Scholar]
- 9.Danilczyk U, Penninger JM. Angiotensin-converting enzyme II in the heart and the kidney. Circ Res 98: 463–471, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res 87: E1–E9, 2000 [DOI] [PubMed] [Google Scholar]
- 11.Elased KM, Cool DR, Morris M. Novel mass spectrometric methods for evaluation of plasma angiotensin converting enzyme 1 and renin activity. Hypertension 46: 953–959, 2005 [DOI] [PubMed] [Google Scholar]
- 12.Elased KM, Cunha TS, Gurley SB, Coffman TM, Morris M. New mass spectrometric assay for angiotensin-converting enzyme 2 activity. Hypertension 47: 1010–1017, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Feng Y, Xia H, Cai Y, Halabi CM, Becker LK, Santos RA, Speth RC, Sigmund CD, Lazartigues E. Brain-selective overexpression of human angiotensin-converting enzyme type 2 attenuates neurogenic hypertension. Circ Res 106: 373–382, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giani JF, Munoz MC, Pons RA, Cao G, Toblli JE, Turyn D, Dominici FP. Angiotensin-(1–7) reduces proteinuria and diminishes structural damage in renal tissue of stroke-prone spontaneously hypertensive rats. Am J Physiol Renal Physiol 300: F272–F282, 2011 [DOI] [PubMed] [Google Scholar]
- 15.Greis KD. Mass spectrometry for enzyme assays and inhibitor screening: an emerging application in pharmaceutical research. Mass Spectrom Rev 26: 324–339, 2007 [DOI] [PubMed] [Google Scholar]
- 16.Grobe N, Elased KM, Cool DR, Morris M. Mass spectrometry for the molecular imaging of angiotensin metabolism in kidney. Am J Physiol Endocrinol Metab 302: E1016–E1024, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gurley SB, Allred A, Le TH, Griffiths R, Mao L, Philip N, Haystead TA, Donoghue M, Breitbart RE, Acton SL, Rockman HA, Coffman TM. Altered blood pressure responses and normal cardiac phenotype in ACE2-null mice. J Clin Invest 116: 2218–2225, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gurley SB, Clare SE, Snow KP, Hu A, Meyer TW, Coffman TM. Impact of genetic background on nephropathy in diabetic mice. Am J Physiol Renal Physiol 290: F214–F222, 2006 [DOI] [PubMed] [Google Scholar]
- 19.Gurley SB, Coffman TM. Angiotensin-converting enzyme 2 gene targeting studies in mice: mixed messages. Exp Physiol 93: 538–542, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Guy JL, Jackson RM, Acharya KR, Sturrock ED, Hooper NM, Turner AJ. Angiotensin-converting enzyme-2 (ACE2): comparative modeling of the active site, specificity requirements, and chloride dependence. Biochemistry 42: 13185–13192, 2003 [DOI] [PubMed] [Google Scholar]
- 21.Gwathmey TM, Pendergrass KD, Reid SD, Rose JC, Diz DI, Chappell MC. Angiotensin-(1–7)-angiotensin-converting enzyme 2 attenuates reactive oxygen species formation to angiotensin II within the cell nucleus. Hypertension 55: 166–171, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huentelman MJ, Grobe JL, Vazquez J, Stewart JM, Mecca AP, Katovich MJ, Ferrario CM, Raizada MK. Protection from angiotensin II-induced cardiac hypertrophy and fibrosis by systemic lentiviral delivery of ACE2 in rats. Exp Physiol 90: 783–790, 2005 [DOI] [PubMed] [Google Scholar]
- 23.Kawahara A, Enari M, Talanian RV, Wong WW, Nagata S. Fas-induced DNA fragmentation and proteolysis of nuclear proteins. Genes Cells 3: 297–306, 1998 [DOI] [PubMed] [Google Scholar]
- 24.Liu C, Lv XH, Li HX, Cao X, Zhang F, Wang L, Yu M, Yang JK. Angiotensin-(1–7) suppresses oxidative stress and improves glucose uptake via Mas receptor in adipocytes. Acta Diabetol 49: 291–299, 2012 [DOI] [PubMed] [Google Scholar]
- 25.Liu CX, Hu Q, Wang Y, Zhang W, Ma ZY, Feng JB, Wang R, Wang XP, Dong B, Gao F, Zhang MX, Zhang Y. Angiotensin-converting enzyme (ACE) 2 overexpression ameliorates glomerular injury in a rat model of diabetic nephropathy: a comparison with ACE inhibition. Mol Med 17: 59–69, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luther JM, Brown NJ. The renin-angiotensin-aldosterone system and glucose homeostasis. Trends Pharmacol Sci 32: 734–739, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maalouf NM, Cameron MA, Moe OW, Adams-Huet B, Sakhaee K. Low urine pH: a novel feature of the metabolic syndrome. Clin J Am Soc Nephrol 2: 883–888, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Maalouf NM, Cameron MA, Moe OW, Sakhaee K. Metabolic basis for low urine pH in type 2 diabetes. Clin J Am Soc Nephrol 5: 1277–1281, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maes M, Monteleone P, Bencivenga R, Goossens F, Maj M, van WD, Bosmans E, Scharpe S. Lower serum activity of prolyl endopeptidase in anorexia and bulimia nervosa. Psychoneuroendocrinology 26: 17–26, 2001 [DOI] [PubMed] [Google Scholar]
- 30.Monteiro MB, Senador D, Zhang WF, Morris M, Elased KM. Balance of ACE and ACE2 in diabetes: decreased renal ACE2 activity in hypertensive db/db diabetic mice. Hypertension 52: E91, 2008 [Google Scholar]
- 31.Nadarajah R, Milagres R, Dilauro M, Gutsol A, Xiao F, Zimpelmann J, Kennedy C, Wysocki J, Batlle D, Burns KD. Podocyte-specific overexpression of human angiotensin-converting enzyme 2 attenuates diabetic nephropathy in mice. Kidney Int 82: 292–303, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Odya CE, Marinkovic DV, Hammon KJ, Stewart TA, Erdos EG. Purification and properties of prolylcarboxypeptidase (angiotensinase C) from human kidney. J Biol Chem 253: 5927–5931, 1978 [PubMed] [Google Scholar]
- 33.Oudit GY, Liu GC, Zhong J, Basu R, Chow FL, Zhou J, Loibner H, Janzek E, Schuster M, Penninger JM, Herzenberg AM, Kassiri Z, Scholey JW. Human recombinant ACE2 reduces the progression of diabetic nephropathy. Diabetes 59: 529–538, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel VB, Bodiga S, Fan D, Das SK, Wang Z, Wang W, Basu R, Zhong J, Kassiri Z, Oudit GY. Cardioprotective effects mediated by angiotensin II type 1 receptor blockade and enhancing angiotensin 1–7 in experimental heart failure in angiotensin-converting enzyme 2-null mice. Hypertension 59: 1195–1203, 2012 [DOI] [PubMed] [Google Scholar]
- 35.Rioli V, Kato A, Portaro FC, Cury GK, te Kaat K, Vincent B, Checler F, Camargo AC, Glucksman MJ, Roberts JL, Hirose S, Ferro ES. Neuropeptide specificity and inhibition of recombinant isoforms of the endopeptidase 342416 family: comparison with the related recombinant endopeptidase 342415. Biochem Biophys Res Commun 250: 5–11, 1998 [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez-Iturbe B, Pons H, Herrera-Acosta J, Johnson RJ. Role of immunocompetent cells in nonimmune renal diseases. Kidney Int 59: 1626–1640, 2001 [DOI] [PubMed] [Google Scholar]
- 37.Schadock I, Todiras M, Vilianovich L, Qadri F, Heuser A, Siems WE, Santos RA, Bader M. Contribution of central angiotensin II to the hypertensive phenotype of prolylcarboxypeptidase knockout mice. Hypertension 58: E36, 2011 [Google Scholar]
- 38.Senanayake P, Moriguchi A, Kumagai H, Ganten D, Ferrario CM, Brosnihan KB. Increased expression of angiotensin peptides in the brain of transgenic hypertensive rats. Peptides 15: 919–926, 1994 [DOI] [PubMed] [Google Scholar]
- 39.Skeggs LT, Jr, Kahn JR, Shumway NP. The preparation and function of the hypertensin-converting enzyme. J Exp Med 103: 295–299, 1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan F, Morris PW, Skidgel RA, Erdös EG. Sequencing and cloning of human prolylcarboxypeptidase (angiotensinase C). Similarity to both serine carboxypeptidase and prolylendopeptidase families. J Biol Chem 268: 16631–16638, 1993 [PubMed] [Google Scholar]
- 41.Taylor EN, Curhan GC. Body size and 24-hour urine composition. Am J Kidney Dis 48: 905–915, 2006 [DOI] [PubMed] [Google Scholar]
- 42.Tikellis C, Bialkowski K, Pete J, Sheehy K, Su Q, Johnston C, Cooper ME, Thomas MC. ACE2 deficiency modifies renoprotection afforded by ACE inhibition in experimental diabetes. Diabetes 57: 1018–1025, 2008 [DOI] [PubMed] [Google Scholar]
- 43.Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 275: 33238–33243, 2000 [DOI] [PubMed] [Google Scholar]
- 43a.Velez JC. Prolyl carboxypeptidase: a forgotten kidney angiotensinase. Focus on “Identification of prolyl carboxypeptidase as an alternative enzyme for processing of renal angiotensin II using mass spectrometry.” Am J Physiol Cell Physiol (April 3, 2013). doi:10.1152/ajpcell.00081.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Velez JC, Ierardi JL, Bland AM, Morinelli TA, Arthur JM, Raymond JR, Janech MG. Enzymatic processing of angiotensin peptides by human glomerular endothelial cells. Am J Physiol Renal Physiol 302: F1583–F1594, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verma A, Shan ZY, Lei B, Yuan LH, Liu X, Nakagawa T, Grant MB, Lewin AS, Hauswirth WW, Raizada MK, Li QH. ACE2 and ANG-(1–7) confer protection against development of diabetic retinopathy. Mol Ther 20: 28–36, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, Godbout K, Parsons T, Baronas E, Hsieh F, Acton S, Patane M, Nichols A, Tummino P. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem 277: 14838–14843, 2002 [DOI] [PubMed] [Google Scholar]
- 47.Wallingford N, Perroud B, Gao Q, Coppola A, Gyengesi E, Liu ZW, Gao XB, Diament A, Haus KA, Shariat-Madar Z, Mahdi F, Wardlaw SL, Schmaier AH, Warden CH, Diano S. Prolylcarboxypeptidase regulates food intake by inactivating α-MSH in rodents. J Clin Invest 119: 2291–2303, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ward PE, Bausback HH, Odya CE. Kinin and angiotensin metabolism by purified renal post-proline cleaving enzyme. Biochem Pharmacol 36: 3187–3193, 1987 [DOI] [PubMed] [Google Scholar]
- 49.Warden CH, Fisler JS, Espinal G, Graham J, Havel PJ, Perroud B. Maternal influence of prolyl endopeptidase on fat mass of adult progeny. Int J Obes (Lond) 33: 1013–1022, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weir NM, Salem E, Grobe N, Yan ZY, Morris M, Elased KM. Renal angiotensin converting enzyme 2 protects kidney function during chronic ANG II infusion in mice. Hypertension 58: E79, 2011 [Google Scholar]
- 51.Welches WR, Brosnihan KB, Ferrario CM. A comparison of the properties and enzymatic activities of three angiotensin processing enzymes: angiotensin converting enzyme, prolyl endopeptidase and neprilysin. Life Sci 52: 1461–1480, 1993 [DOI] [PubMed] [Google Scholar]
- 52.Wong DW, Oudit GY, Reich H, Kassiri Z, Zhou J, Liu QC, Backx PH, Penninger JM, Herzenberg AM, Scholey JW. Loss of angiotensin-converting enzyme-2 (Ace2) accelerates diabetic kidney injury. Am J Pathol 171: 438–451, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wysocki J, Ye M, Rodriguez E, Gonzalez-Pacheco FR, Barrios C, Evora K, Schuster M, Loibner H, Brosnihan KB, Ferrario CM, Penninger JM, Batlle D. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: prevention of angiotensin II-dependent hypertension. Hypertension 55: 90–98, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wysocki J, Ye M, Soler MJ, Gurley SB, Xiao HD, Bernstein KE, Coffman TM, Chen S, Batlle D. ACE and ACE2 activity in diabetic mice. Diabetes 55: 2132–2139, 2006 [DOI] [PubMed] [Google Scholar]
- 55.Xiao L, Gao L, Lazartigues E, Zucker IH. Brain-selective overexpression of angiotensin-converting enzyme 2 attenuates sympathetic nerve activity and enhances baroreflex function in chronic heart failure. Hypertension 58: 1057–1065, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamamoto K, Chappell MC, Brosnihan KB, Ferrario CM. In vivo metabolism of angiotensin I by neutral endopeptidase (EC 3.42411) in spontaneously hypertensive rats. Hypertension 19: 692–696, 1992 [DOI] [PubMed] [Google Scholar]
- 57.Yamamoto K, Ohishi M, Katsuya T, Ito N, Ikushima M, Kaibe M, Tatara Y, Shiota A, Sugano S, Takeda S, Rakugi H, Ogihara T. Deletion of angiotensin-converting enzyme 2 accelerates pressure overload-induced cardiac dysfunction by increasing local angiotensin II. Hypertension 47: 718–726, 2006 [DOI] [PubMed] [Google Scholar]
- 58.Yamazato Y, Ferreira AJ, Hong KH, Sriramula S, Francis J, Yamazato M, Yuan L, Bradford CN, Shenoy V, Oh SP, Katovich MJ, Raizada MK. Prevention of pulmonary hypertension by angiotensin-converting enzyme 2 gene transfer. Hypertension 54: 365–371, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ye M, Wysocki J, Naaz P, Salabat MR, LaPointe MS, Batlle D. Increased ACE 2 and decreased ACE protein in renal tubules from diabetic mice: a renoprotective combination? Hypertension 43: 1120–1125, 2004 [DOI] [PubMed] [Google Scholar]
- 60.Ye M, Wysocki J, William J, Soler MJ, Cokic I, Batlle D. Glomerular localization and expression of angiotensin-converting enzyme 2 and angiotensin-converting enzyme: implications for albuminuria in diabetes. J Am Soc Nephrol 17: 3067–3075, 2006 [DOI] [PubMed] [Google Scholar]
- 61.Zhong J, Guo D, Chen CB, Wang W, Schuster M, Loibner H, Penninger JM, Scholey JW, Kassiri Z, Oudit GY. Prevention of angiotensin II-mediated renal oxidative stress, inflammation, and fibrosis by angiotensin-converting enzyme 2. Hypertension 57: 314–322, 2011 [DOI] [PubMed] [Google Scholar]