

Abstract
Context
Hereditary pancreatitis is the early onset form of chronic pancreatitis that is carried in an autosomal dominant pattern with variable penetrance. While 80% of HP has been shown to be due to a single mutation in the trypsinogen gene PRSS1, a number of HP families have no identified genetic cause for illness, thus no reliable screening options or clear therapy.
Objective
To explore the use of massive parallel DNA sequencing technology to discover the etiology of pancreatitis in a family with idiopathic hereditary pancreatitis.
Design
candidate gene screening and verification within a kindred.
Setting
Prospective cohort study, university based.
Patients or participants
Kindred with idiopathic hereditary pancreatitis.
Interventions
none
Main outcome measures
identification of DNA variants predicted to increase susceptibility to pancreatitis.
Methods
whole exome sequencing (WES) of two distantly related subjects with variant-specific confirmation in the subjects and other family members.
Results
We identified three deleterious genetic changes in the three major pancreatitis associated genes (PRSS1 CNV, SPINK1 c.27delC and CFTR R117H), two of which were carried by each patient. Individual targeted assays confirmed these variations in the two WES patients as well as affected and non-affected pedigree members.
Conclusion
WES was useful for rapid screening of candidate genes linked to pancreatitis. This method opens the door for time- and cost-effective screening of multiple disease-associated genes and modifying factors that associate in different ways to generate a complex genetic disorder.
Keywords: pancreatitis, chronic pancreatitis, cystic fibrosis, Sequence Analysis, DNA, trypsin, cystic fibrosis transmembrane conductance regulator, trypsin Inhibitor, Kazal pancreatic
Introduction
Most hereditary pancreatitis families have gain-of-function mutations in the cationic trypsinogen gene (PRSS1)(1–4). Familial clustering of pancreatitis occurs with various combinations of pancreatic secretory trypsin inhibitor gene (SPINK1) mutations(5) and/or severe or atypical CFTR mutations (CFTR-CF)(6–8). Heterozygous or complex SPINK1, CFTR, chymotrypsinogen C (CTRC), and calcium sensing receptors (CASR), variants are also seen in idiopathic pancreatitis (8, 9). To add to the complexity, there is increasing recognition that rare or private mutations in known pancreatitis susceptibility genes are associated with risk of pancreatitis, as demonstrated for CTRC (10). Finally, copy number variants in PRSS1 increase risk of pancreatitis and must also be considered (11).
As the number of genes and mutations involved in the onset and progression of pancreatitis becomes higher, the time and cost of screening and sequencing specific exons continues to increase. However, a new approach that uses massive parallel sequencing called next generation sequencing (NGS) is becoming standardized, and the cost per patient is rapidly dropping. NGS includes whole genome sequencing, whole exome sequencing (WES) and other methods. Because the cost of WES in our hands is now less than the cost of sequencing CFTR use of this technology is becoming an attractive alternative to classic targeted gene sequencing or mutation specific genotyping for a genetic counseling workup. The work presented in this article explores the feasibility and utility of WES to identify the cause of disease in an HP family.
The J family was composed of three generations, including four living pancreatitis patients and was typical for HP (Figure 1). Direct sequencing of all exons of PRSS1 and exons 2–3 of SPINK1 sequencing resulted in no apparent familial mutation. Screening of CFTR revealed a non-CF causing p.R117H variant (T7), but this did not segregate exclusively with disease. Linkage analysis suggested a possible shared locus on chromosome 12, but the wide variety of phenotypic features (age of onset and symptom severity) confounded definitive phenotyping in key family members. Onset of pancreatitis symptoms was in the teens for both subjects chosen for WES. To determine whether this family had undetected variants in known pancreatitis susceptibility genes, we conducted WES, focusing our analysis on PRSS1 (5 exons), SPINK1 (4 exons), CFTR (27 exons), CTRC (8 exons), and CASR (7 exons).
Figure 1.
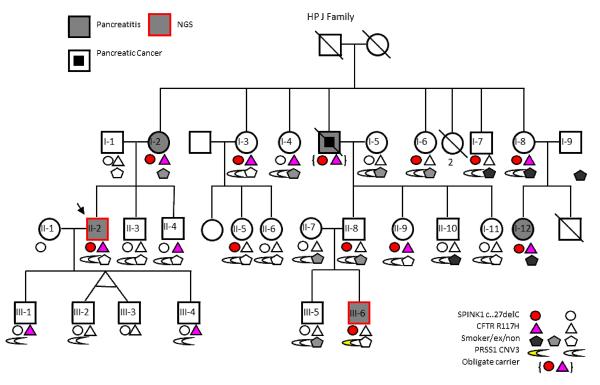
J Family Pedigree and Genotyping/Smoking Data
Methods
Ethics
The study was approved by the Institutional Review Board of the University of Pittsburgh prior to patient contact or ascertainment. All subjects provided written informed consent that conformed to the ethical guidelines of the “World Medical Association Declaration of Helsinki - Ethical Principles for Medical Research Involving Human Subjects”.
Patients
Family J from the Hereditary Pancreatitis (HP) study (12) was selected for NGS using the following criteria: direct sequencing of all exons in PRSS1 and exons 2–3 in SPINK1 were negative for any potential disease-causing mutation; large family with multiple affected family members in different generations; and a pedigree that suggests a complex inheritance pattern or incomplete penetrance. DNA of cases II-1 and III-6 were sent for NGS.
Whole Exome Sequencing
DNA (3ug) was enriched using the Agilent (CA) SureSelect Human All Exon Kit optimized for the SOLiD Sequencing System (AB Life Technologies, CA). This genome-wide approach targets the exons of ~18,000 known genes, covering 38Mb of the human genome. The prepared exome library was emulsion amplified following the manufacturer's directions for the EZBead Amplifier (AB Life Technologies, CA) at a library concentration of 0.3pM. Each sample was sequenced on one quadrant (1/4) of a SOLiD 4 sequencing slide, and we obtained 3.1 and 2.2 Gb of mappable sequence. Color space reads were mapped to the reference human genome (NCBI GRCh37.2) and variants (both SNPs and indels) were called using the CLCbio Genomics Workbench software (v4.6).
Confirmation of WES data
WES results were confirmed by individual assay, testing all samples of Family J with available DNA as well subsets of HP and NAPS cohorts for SPINK1 c.27delC, PRSS1 CNV and CFTR R117H. SPINK1 c.27delC: An individual Taqman® assay was designed for the c.27delC mutation (AB Life Technologies). Data were analyzed using SDS2.3 software (AB Life Technologies). PRSS1 CNV A pre-designed and validated CNV assay located in exon 5 of PRSS1 (AB Life Technologies Hs03192321_cn) was used. PRSS1 copy number was calculated in proportion to RNAseP reference assay (AB Life Technologies cat#4403326): The assays were performed in triplicate and repeated at least once for each sample. Data were analyzed using Copy Caller Software Version 1.0 (AB Life Technologies). CFTR R117H: A multiplex PCR-based SNP assay (sequenom® AB Life Technologies) was used. Primer sequences available on request.
Statistics
No statistical comparisons were performed.
Results
No significant mutations in CTRC, CASR or PRSS1 were identified. We identified a SPINK1 c.27delC (a one-base deletion in exon 1 of SPINK1), shared in a heterozygous state, in both patients. This variant was confirmed by Taqman® (AB Life Technologies) in these two patients, 11 additional family members and was implicated in one obligate carrier. The apparent penetrance of c.27delC for pancreatitis in this family was 42%. This frameshift mutation is expected to cause nonsense mediated decay of the mRNA message, reducing cellular Spink1 protein levels to 50% of normal. SPINK1 c.27delC was previously reported in 2 French HP families with variable penetrance(13). Upon testing 713 subjects from other HP families and 1550 subjects from the North American Pancreatitis Study 2 cohort(14), we found a single additional carrier, a 47-year-old female with recurrent acute pancreatitis, no other identified genetic mutations, and no family history of pancreatic disease.
The CFTR variant R117H was identified in case II-2 but not III-6. R117H is a frequent variant in the general population and has been associated with cystic fibrosis but only when in cis with a T5 allele (15); however, the R117H in this family was not associated with T5. This CFTR variant was confirmed via a multiplex Sequenom® assay and was subsequently identified in 10 additional family members and implicated in 1 obligate carrier. The apparent penetrance of R117H alone for pancreatitis was 44% in this family.
Patient III-6 was also found to carry a PRSS1 copy number variant, as demonstrated by the increased average coverage of this gene in the WES platform: coverage across PRSS1 was significantly elevated with respect to other genes in the region (p=0.004, Figure 2A). A pre-designed and validated copy number variant assay targeting PRSS1 (AB Life Technologies Hs03192321_cn) confirmed a statistically significant increased copy number in this patient but in no other family member (Figure 2B). PRSS1 copy number variants have been reported in a small number of HP families around the world and a 50% increase in basal trypsin expression is expected to be causal for pancreatitis, but this is the first report of a single apparent de novo duplication within an established HP family with no other copy number variants. Testing the rest of the HP and NAPS2 subjects for PRSS1 CNV revealed a single additional family with two affected twin sons and a copy number of 3 for each.
Figure 2.

WES and confirmatory CNV of PRSS1
A: PRSS1 coverage of case III-6. Observed coverage of sequencing fragments from WES in reference region (TRB) and PRSS1. Mean coverage of PRSS1 (38.67) was significantly higher than in TRB (20.60), p=0.004 using Welch two sample t-test (95% CI 6.04–30.09). B. PRSS1 copy number variant assay of pedigree 21. PRSS1 copy number is plotted for family members, and an unrelated hereditary pancreatitis PRSS1 R122H case. All samples had the expected copy number of 2, except III-6, with CNV3.
Heterozygous loss-of-function mutations in CFTR, SPINK1, CTRC, and CASR are commonly identified in control populations, with epistasis seen between CFTR and SPINK1 variants(8) and CASR and SPINK1 variants(16). In this family, combined penetrance of 2 or more of these variants was 62.5%.
Smoking and drinking are strong independent risk factors for pancreatitis(17, 18). All but one of the c.27delC/R117H carriers who were affected also self-reported being current or former smokers, while 2/3 of the unaffected c.27delC/R117H carriers were non-smokers. However, alcohol usage did not correlate with disease status: 2/3 of the c.27delC/R117H unaffected carriers were light drinkers, and 2/3 of the affected carriers were never-drinkers, placing all of the subjects well below the risk-threshold for alcohol-associated pancreatitis12. Thus, each affected patient carried unique genetic and environmental factors that individually would not necessarily cause pancreatitis but combined to confer very high risk.
Discussion
These data demonstrate an application of WES in evaluating the entire coding region of 5 confirmed pancreatitis susceptibility genes. This includes discovery of a rare SPINK1 mutation, a common CFTR mutation and a single PRSS1 copy number mutation in a single assay. In addition, other mutations in PRSS1, CTSR and CASR were excluded. Furthermore, any newly reported variant could be quickly checked in these individuals. Finally, the use of this technology in an idiopathic hereditary pancreatitis family was chosen because the risk of rare variants could be validated in other family members. This approach will also be useful in sporadic patients where known variants are identified, or the functional effect of a rare mutation can be determined.
An important application of WES is in the guidance of clinical decision-making, particularly in managing complex chronic inflammatory disorders. In pancreatitis, clinicians are currently limited to broad interventions, such as minimizing pancreatic stimulation or improving drainage. Knowledge of contributory genetic variants could allow them to take advantage of etiology-based treatment strategies being developed that target the acinar (e.g., PRSS1 variants(19)) or duct cells (e.g. CFTR variants(20)). Our use of WES with validation indicated different etiologies in different patients, and therefore different approaches. Also, WES saved significant time at about the same cost compared with direct sequencing and copy number analysis of individual candidate genes1. In addition, individual III-6 now presents completely different risks for disease progression and inheritance as compared to that of the rest of the family, and the genetic risk for pancreatitis for the rest of the third generation of this family can be reported as substantially reduced.
While the standard practice in genotyping HP families is presupposing a single variant is fully responsible for disease, this would have failed in the case of the J family. There is growing evidence the HP families with an absence of a standard (R122H) PRSS1 mutation (and possibly even those with) require much more consideration and have a more complex pathology, with multiple genetic variants and environmental pressures converging into a small family set. As insight into genetic susceptibility to pancreatitis grows the importance of genotyping patients will also grow. We are evaluating the benefit of genotyping patients at the beginning of their clinical evaluation because a positive test immediately completes the diagnostic workup, and may provide direction for etiology-based treatment strategies in the future (e.g. aimed ad the duct cells or aimed at the acinar cell).
In summary, using WES in two members of a family with hereditary pancreatitis, we identified complex etiologies with epistasis between common and rare mutations in CFTR and SPINK1, copy number variants in PRSS1, and a relationship to smoking but not alcohol use. We have thus far limited the study to basic candidate genes for pancreatitis, and even with this temporarily restricted approach, we have located significant genetic lesions that have not been identified through basic techniques. There is a notable decrease in time needed for NGS of our two patients as opposed to direct sequencing and copy number analysis of individual candidate genes, the cost is comparable, and now we have an extensive database in which to mine for additional candidate lesions in this family in the near future. Moving forward, as new susceptibility factors and modifier genes are identified, the patients evaluated by WES can be rapidly and inexpensively screened.
Acknowledgments
The authors thank Kim Stello and Janette Lamb PhD for technical assistance and Michelle Kienholz for technical review and editing of the manuscript. This work was supported by the Wayne Fusaro Pancreatic Cancer Research Fund (DCW), National Institutes of Health (NIH) grants DK061451 (DCW), DK54709 (DCW), and T32DK063922 (JL) from the National Institutes of Diabetes and Digestive and Kidney Diseases (NIDDK). Whole exome target capture, SOLiD next generation sequencing and Sequenom assays were performed at the University of Pittsburgh Genomics and Proteomics Core Laboratories which is supported by Grant Number UL1 RR024153 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the NCRR or NI.
Footnotes
There are no conflicts of interest to disclose.
The cost of candidate gene sequencing in our research laboratory for individual patients is driven by labor cost of sequencing each exon in both directions, and verifying the quality of each sequencing read, in addition to the cost of materials for DNA sequencing. Commercial clinical laboratories (e.g. Ambry Genetics, evaluate for PRSS1, SPINK1, CFTR and CTRC plus CFTR del/dup is ~$3500. WES through our core lab is >$2000 per person, but this data is analyzed by our group and cannot be used for clinical decision making without verification of the variants ina clinical laboratory.
Reference
- 1.Whitcomb DC, Gorry MC, Preston RA, Furey W, Sossenheimer MJ, Ulrich CD, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nature Genetics. 1996;14(2):141–5. doi: 10.1038/ng1096-141. PMID: 8841182. [DOI] [PubMed] [Google Scholar]
- 2.Howes N, Lerch MM, Greenhalf W, Stocken DD, Ellis I, Simon P, et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol. 2004;2(3):252–61. doi: 10.1016/s1542-3565(04)00013-8. PMID 15017610. [DOI] [PubMed] [Google Scholar]
- 3.Rebours V, Boutron-Ruault MC, Schnee M, Ferec C, Le Marechal C, Hentic O, et al. The natural history of hereditary pancreatitis: a national series. Gut. 2009 Jan;58(1):97–103. doi: 10.1136/gut.2008.149179. PMID: 18755888. [DOI] [PubMed] [Google Scholar]
- 4.Joergensen MT, Brusgaard K, Cruger DG, Gerdes AM, Schaffalitzky de Muckadell OB. Genetic, epidemiological, and clinical aspects of hereditary pancreatitis: a population-based cohort study in Denmark. The American journal of gastroenterology. 2010 Aug;105(8):1876–83. doi: 10.1038/ajg.2010.193. PMID: 20502448. [DOI] [PubMed] [Google Scholar]
- 5.Witt H, Luck W, Hennies HC, Classen M, Kage A, Lass U, et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nature Genetics. 2000;25(2):213–6. doi: 10.1038/76088. PMID 10835640. [DOI] [PubMed] [Google Scholar]
- 6.Sharer N, Schwarz M, Malone G, Howarth A, Painter J, Super M, et al. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. New England Journal of Medicine. 1998;339(10):645–52. doi: 10.1056/NEJM199809033391001. PMID 9725921. [DOI] [PubMed] [Google Scholar]
- 7.Cohn JA, Friedman KJ, Noone PG, Knowles MR, Silverman LM, Jowell PS. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. New England Journal of Medicine. 1998;339(10):653–8. doi: 10.1056/NEJM199809033391002. PMID 9725922. [DOI] [PubMed] [Google Scholar]
- 8.Schneider A, Larusch J, Sun X, Aloe A, Lamb J, Hawes R, et al. Combined Bicarbonate Conductance-Impairing Variants in CFTR and SPINK1 Variants Are Associated With Chronic Pancreatitis in Patients Without Cystic Fibrosis. Gastroenterology. 2011 Jan;140(1):162–71. doi: 10.1053/j.gastro.2010.10.045. PMID: 20977904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whitcomb DC. Genetic aspects of pancreatitis. Annu Rev Med. 2010;61:413–24. doi: 10.1146/annurev.med.041608.121416. PMID: 20059346. [DOI] [PubMed] [Google Scholar]
- 10.Masson E, Chen JM, Scotet V, Le Marechal C, Ferec C. Association of rare chymotrypsinogen C (CTRC) gene variations in patients with idiopathic chronic pancreatitis. Hum Genet. 2008 Feb;123(1):83–91. doi: 10.1007/s00439-007-0459-3. PMID: 18172691. [DOI] [PubMed] [Google Scholar]
- 11.Masson E, Le Marechal C, Delcenserie R, Chen JM, Ferec C. Hereditary pancreatitis caused by a double gain-of-function trypsinogen mutation. Hum Genet. 2008 Jun;123(5):521–9. doi: 10.1007/s00439-008-0508-6. PMID: 18461367. [DOI] [PubMed] [Google Scholar]
- 12.Applebaum-Shapiro SE, Finch R, Pfützer RH, Hepp LA, Gates L, Amann S, et al. Hereditary Pancreatitis in North America: The Pittsburgh - Midwest Multi-Center Pancreatic Study Group Study. Pancreatology. 2001;1(5):439–43. doi: 10.1159/000055844. PMID 12120221. [DOI] [PubMed] [Google Scholar]
- 13.Le Marechal C, Chen JM, Le Gall C, Plessis G, Chipponi J, Chuzhanova NA, et al. Two novel severe mutations in the pancreatic secretory trypsin inhibitor gene (SPINK1) cause familial and/or hereditary pancreatitis. Hum Mutat. 2004 Feb;23(2):205. doi: 10.1002/humu.9212. PMID: 14722925. [DOI] [PubMed] [Google Scholar]
- 14.Whitcomb DC, Yadav D, Adam S, Hawes RH, Brand RE, Anderson MA, et al. Multicenter approach to recurrent acute and chronic pancreatitis in the United States: the North American Pancreatitis Study 2 (NAPS2) Pancreatology. 2008;8(4–5):520–31. doi: 10.1159/000152001. PMID: 18765957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thauvin-Robinet C, Munck A, Huet F, Genin E, Bellis G, Gautier E, et al. The very low penetrance of cystic fibrosis for the R117H mutation: a reappraisal for genetic counselling and newborn screening. J Med Genet. 2009 Nov;46(11):752–8. doi: 10.1136/jmg.2009.067215. PMID: 19880712. [DOI] [PubMed] [Google Scholar]
- 16.Felderbauer P, Klein W, Bulut K, Ansorge N, Dekomien G, Werner I, et al. Mutations in the calcium-sensing receptor: a new genetic risk factor for chronic pancreatitis? Scand J Gastroenterol. 2006 Mar;41(3):343–8. doi: 10.1080/00365520510024214. PMID: 16497624. [DOI] [PubMed] [Google Scholar]
- 17.Talamini G, Bassi C, Falconi M, Frulloni L, Di Francesco V, Vaona B, et al. Cigarette smoking: an independent risk factor in alcoholic pancreatitis. Pancreas. 1996;12(2):131–7. PMID 8720658. [PubMed] [Google Scholar]
- 18.Yadav D, Hawes RH, Brand RE, Anderson MA, Money ME, Banks PA, et al. Alcohol consumption, cigarette smoking, and the risk of recurrent acute and chronic pancreatitis. Arch Intern Med. 2009 Jun 8;169(11):1035–45. doi: 10.1001/archinternmed.2009.125. PMID: 19506173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morinville VD, Lowe ME, Elinoff BD, Whitcomb DC. Hereditary pancreatitis amlodipine trial: a pilot study of a calcium-channel blocker in hereditary pancreatitis. Pancreas. 2007 Nov;35(4):308–12. doi: 10.1097/mpa.0b013e318120023a. PMID: 18090235. [DOI] [PubMed] [Google Scholar]
- 20.Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010 Nov 18;363(21):1991–2003. doi: 10.1056/NEJMoa0909825. PMID: 21083385. [DOI] [PMC free article] [PubMed] [Google Scholar]