

Abstract
The current drug screening models are deficient, particularly in detecting cardiac side effects. Human stem cell-derived cardiomyocytes could aid both early cardiotoxicity detection and novel drug discovery. Work over the last decade has generated human embryonic stem cells as potentially accurate sources of human cardiomyocytes, but ethical constraints and poor efficacy in establishing cell lines limit their use. Induced pluripotent stem cells do not require the use of human embryos and have the added advantage of producing patient-specific cardiomyocytes, allowing both generic and disease- and patient-specific pharmacological screening, as well as drug development through disease modelling. A critical question is whether sufficient standards have been achieved in the reliable and reproducible generation of ‘adult-like’ cardiomyocytes from human fibroblast tissue to progress from validation to safe use in practice and drug discovery. This review will highlight the need for a new experimental system, assess the validity of human induced pluripotent stem cell-derived cardiomyocytes and explore what the future may hold for their use in pharmacology.
LINKED ARTICLES
This article is part of a themed section on Regenerative Medicine and Pharmacology: A Look to the Future. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2013.169.issue-2
Keywords: cardiac, stem cell, embryonic, iPSC, safety, cardiotoxicity, high throughput
Drug discovery and toxicology: the need for a new experimental system
There is a vast attrition in the number of drugs developed for potential therapeutic use during the transition from the laboratory bench to hospital bedside, with 89% of drugs that pass current in vitro and animal model screening tests being withdrawn in the clinical phase (Kola and Landis, 2004). Thirty percent of these failures are due to lack of efficacy, with another 30% due to safety concerns (Kola and Landis, 2004). This failure in early drug toxicity detection is proving extremely time-consuming and costly, and places people's health at risk. Cardiovascular toxicity is a major cause of drug withdrawal during clinical development, accounting for up to 33% of drug failure (MacDonald and Robertson, 2009). Approximately half of these are due to risk of arrhythmias, including QT prolongation and life-threatening polymorphic ventricular tachycardia or torsade de pointes (TdP) (Mandenius et al., 2011). Part of the reason for this failure in early pharmacological screening is that our current testing systems do not accurately replicate human cardiovascular conditions, particularly cardiac electrophysiology, and cannot account for individual variability. There is clearly a need for new and robust experimental systems to elicit organ dysfunction hitherto unmasked by standard toxicity testing.
This failure has been internationally recognized – the European Medicines Association have published guidelines on ‘safety pharmacology studies for human pharmaceuticals’ (ICH 2000). These guidelines highlight the need for further rigorous evaluation of functional and electrophysiological cardiovascular endpoints in drug screening. They stress that new testing models will need to achieve high specificity, sensitivity, reproducibility and predictive power.
A pharmacological agent, during development towards licensing, undergoes a series of safety tests before entering human clinical trials. The current testing model starts with in vitro single cells and then progresses through to ex vivo tissue and organ baths. Both these models would clearly benefit from using authentic human ‘adult’ cardiomyocytes to replicate the complex electrophysiological and mechanical interactions of human myocardium. Unfortunately, human cells are scarce, difficult and costly to harvest and are terminally differentiated with a low proliferative capacity. They have limited time in culture before they de-differentiate, altering structural features such as t-tubules, rendering the cells inadequate to perform the manipulations desired (Mitcheson et al., 1998). Declining transplant numbers, unpredictability over disease aetiology or previous treatment and absence of a true control, with even unused donor hearts frequently showing pathological changes, further highlight the need for a new human in vitro cardiomyocyte model.
Testing then progresses to animal models, but several factors contribute to their poor predictive power. Efforts to model findings in animal cardiomyocytes have shown quantitative differences with significant inter-species variability (Lu et al., 2001). There are differences in receptor subtypes, distribution and signalling across species (Brito-Martins et al., 2008). Furthermore, animal models may be inadequate for some advanced treatments, such as monoclonal antibodies, as these have been developed specifically for human tissue. This has been a problem with monoclonal antibodies in other areas, such as the TGN1412 antibody, which produced unexpected cytokine storms in healthy human subjects (Suntharalingam et al., 2006). Some drugs cause cardiotoxicity with long-term use through moderate pro-apoptotic or stem cell-targeted necrotic damage, and these might not produce effects in animal models because the time course of investigation is too short. Drug cardiotoxicity may only become evident when there is prior underlying deterioration of the myocardium, as when used in an ageing or cardiac-compromised human populations. Cardiovascular deterioration during aging is difficult to reproduce in animal models. Cardiotoxicity is particularly evident when combinations of agents are used; for example, one trial detected cardiac dysfunction of NYHA class III or IV in 27% of the group given an anthracycline, cyclophosphamide and trastuzumab compared with 8% of the group given an anthracycline and cyclophosphamide alone; 13% of the group given paclitaxel and trastuzumab; and 1% of the group given paclitaxel alone (Slamon et al., 2001). Recapitulating the various possible combinations in animal models is difficult and costly. Lastly, the predictability of transgenic mouse models for diseases such as hypertrophic cardiomyopathy and Brugada syndrome are limited due to rodent heart size and electrophysiology, so alternative models are needed for testing disease specific therapies and toxicities (Lian et al., 2010).
Stem cell-derived cardiomyocytes: new model systems for toxicology testing
A general search for improved, humanized cell models for disease has led to a focus on human stem cell derivatives. Human embryonic stem cells (hESC), first developed in 1998, were quickly shown to produce cells with spontaneous contractility and electrophysiological characteristics of cardiomyocytes (Kehat et al., 2001; Mummery et al., 2002). In one respect, they were superior to the adult cardiomyocyte in that they would continue to beat in culture for months and could be readily transfected using plasmid or siRNA constructs (Braam et al., 2008). Since their discovery, efforts in laboratories have characterized human embryonic stem cell-derived cardiomyocytes (hESC-CM) in detail in terms of electrophysiology, calcium handling, receptor response, growth, proliferation and survival, showing similarities to human cardiomyocytes (Reppel et al., 2004; Harding et al., 2007; Liu et al., 2007; Sartiani et al., 2007; Brito-Martins et al., 2008; Habib et al., 2008; Pekkanen-Mattila et al., 2010).
However, there are problems associated with hESC use. A major criticism is that hESC-CMs resemble fetal rather than adult cardiomyocytes in appearance, structure and function (He et al., 2003; Mummery et al., 2003; Brito-Martins et al., 2008; Caspi et al., 2009; Braam et al., 2010). There remains a low efficacy in establishing hESC lines, especially patient specific hESCs via somatic cell nuclear transfer (Lian et al., 2010). There is a risk of immune rejection with allogenic transplant. Notably, there are complex ethical issues surrounding the use of human embryos. Therefore, although the new stem cell model made some promising steps in the right direction, there remains an unmet clinical need for a valid, efficient and acceptable new screening model.
Discovery of iPSC
In 2006, Takahashi and Yamanaka induced pluripotency in mouse adult fibroblasts by the retroviral transduction of four transcription factors –OCT3/4, SOX2, KLF4 and C-MYC (Takahashi and Yamanaka, 2006). OCT3/4 (Nichols et al., 1998; Niwa et al., 2000) and SOX2 (Avilion et al., 2003) are factors that maintain pluripotency in early embryos and embryonic stem cells, and KLF4 (Li et al., 2005) and C-MYC (Cartwright et al., 2005) are up-regulated in tumour cells, encourage rapid proliferation of embryonic stem cells in culture and maintain the embryonic stem cell phenotype.
The following year, Takahashi et al. developed human induced pluripotent stem cells (hiPSC) from human dermal fibroblasts, using the same four transcription factors (Takahashi et al., 2007). RT-PCR demonstrated the expression of undifferentiated stem cell marker genes in both hiPSCs and hESCs. Western blotting revealed similar protein levels in the two cell lines. DNA microarray analysis showed that the global gene expression patterns were similar, with fewer than 4% of the genes analyzed showing greater than a fivefold difference in expression between hiPSCs and hESCs. These cells demonstrated pluripotency in vitro, differentiating into cells from all three germ cell layers, and maintaining pluripotency in vivo, forming teratomas when injected into SCID mice (Takahashi et al., 2007). Their murine iPSC predecessors demonstrated further evidence of pluripotency displaying chimerism and germ line transmission in murine models (Boland et al., 2009; Kang et al., 2009; Zhao et al., 2009).
Around the same time, Yu and colleagues used slightly different transduction factors (OCT4, SOX2, NANOG and LIN28) to produce hiPSCs (Yu et al., 2007). A number of other groups have replicated this work showing that human somatic cells can be reprogrammed to a pluripotent state by forced expression of a small set of transcription factors (Maherali et al., 2007; Okita et al., 2007; Wernig et al., 2007; Lowry et al., 2008; Park et al., 2008b).
A year later, in 2008, cardiomyocytes were differentiated from mouse iPSCs (Mauritz et al., 2008; Narazaki et al., 2008), and in 2009, beating human cardiomyocytes were derived from hiPSCs via embryoid body (EB) formation (Zhang et al., 2009; Zwi et al., 2009). EBs are formed when iPSCs grown in culture are dispersed into small clumps, transferred to suspension and cultured for 7–10 days. During this time, the cell colonies aggregate to form EBs (Kehat et al., 2001; Xu et al., 2002). These EBs develop spontaneously contracting outgrowths that can be observed microscopically. When dissected out and examined, these spontaneously beating cells have molecular, structural and functional similarities to human cardiomyocytes (Kehat et al., 2001; Xu et al., 2002; Zhang et al., 2009; Zwi et al., 2009). Recently, more directed differentiation methods have been developed using signalling factors responsible for germ layer induction. Activin/Nodal/TGFβ, Wnt and BMP pathways are central to cardiovascular development, and so it was thought that manipulation of these pathways could lead to more efficient cardiogenesis from pluripotent stem cells (Laflamme et al., 2007; Yang et al., 2008; Kattman et al., 2011). Indeed, culture manipulation with Activin A, BMP4 and small molecule Wnt inhibitors induces cardiac mesoderm differentiation in EBs from both hESCs and hiPSCs with up to 60% efficiency (Laflamme et al., 2007; Yang et al., 2008; Kattman et al., 2011; Ren et al., 2011). Recent efforts have sought to improve this efficiency even further – this will be discussed later in this review.
Human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CM) open tantalizing new drug screening, diagnostic and therapeutic opportunities. Not only do they provide a potentially unlimited source of human cardiomyocytes, but they also overcome the ethical hurdles that burden hESC-CMs as they do not require the killing of a human embryo. Furthermore, hiPSC-CMs produce patient-specific cells, making patient targeted therapy a real possibility, whilst surmounting the problem of immune rejection in transplantation. Furthermore, they offer a pharmacological test bed for patient specific phenotypes and genotypes. They open opportunities to study diseases of unknown or complex genetic origin and allow us to replicate diseases with no known animal models. However, as with hESC-CMs, hiPSC-CMs still resemble immature fetal-like cardiomyocytes and therefore their validity as a model for healthy adult cardiomyocytes needs to be rigorously examined. Although it is beyond the remit of this review, it is worth mentioning here that in transplanted populations, hiPSC-CMs present a risk of teratoma formation similar to that posed by hESC-CMs. Furthermore, there is also a theoretical risk of viral reactivation in the host when viral vectors are used to generate iPSCs.
Validating hiPSC-CM for use
Before hiPSC-CMs are adopted for high-throughput pharmacological screening, they need to be meticulously validated for use. They need to display similar genomic, proteomic, pharmacological, mechanical and electrophysiological properties to human cardiomyocytes in vivo. As a surrogate marker for this, human cardiomyocytes in vitro can be used as a benchmark for comparison, possibly together with animal models where suitable.
Genes
RT-PCR performed on RNA isolated from undifferentiated hiPSC and differentiating, spontaneously beating EBs demonstrates ontogeny of gene expression that mirrors that in human cardiomyocytes (Tanaka et al., 2009; Zhang et al., 2009; Zwi et al., 2009). Undifferentiated hiPSCs express pluripotency markers OCT3/4, SOX2 and NANOG; and this expression declines as the cells differentiate and mature (Yokoo et al., 2009; Zhang et al., 2009; Zwi et al., 2009). Four to 10 days after EB formation and differentiation, there is an increase in the expression of primitive steak, mesoderm and cardiomesoderm markers (Brachyury and MESP1), followed by expression of cardiac progenitor markers (ISL-1, TBX5) (Yokoo et al., 2009; Zhang et al., 2009; Zwi et al., 2009).
hiPSC-CMs from 10 to 60 days post differentiation express genes for cardiac-specific proteins. These include cardiac-specific transcription factors (NKX2.5, MEF2C and GATA4), genes coding sarcomeric proteins (α and β-myosin heavy chains, myosin light chains and cardiac troponin) and ion channel proteins (sodium channels, L-type calcium channels, rapid and slow delayed rectifier potassium channels, inward rectifier potassium channels, transient outward potassium channels and If‘funny’ channels).
Proteins and structure
Immunofluorescence confirms the presence of proteins expressed by these genes and illuminates their spatial configuration. Antibody immunolabelling of myofilament proteins (α-actinin, myosin heavy chain, troponin I, troponin T and tropomyosin) in hiPSC-CMs reveals some degree of sarcomeric organization (Tanaka et al., 2009; Zhang et al., 2009; Zwi et al., 2009). These striated patterns seen under immunofluorescence are similar to those observed in hESC-CMs (Tanaka et al., 2009; Zhang et al., 2009). Cardiac troponin T appears across the length of the hiPSC-CMs as punctate fibrous striations, α-actinin as fibrous strands and α- and β-myosin heavy chains as both punctate and globular striations (Guo et al., 2011a). The positive staining of multiple myofilament proteins and their pattern of distribution suggests embryonic sarcomeric organization, similar to that in fetal cardiomyocytes but not yet as developed and structured as in adult cardiomyocytes (Germanguz et al., 2011; Itzhaki et al., 2011b), analogous to hESC-CMs. These structures are illustrated in Figure 1.
Figure 1.

Immunofluorescence image showing differentiated human induced pluripotent stem cell-derived cardiomyocytes. Cells are stained positive for cardiac-specific atrial natriuretic factor (cytoplasmic), mitotic marker Ki67 (nuclear) and myosin heavy chain a/β at 30 days after differentiation. Nuclei are stained with DAPI. Courtesy Gabor Foldes, NHLI, Imperial College.
The expression of key calcium handling proteins such as ryanodine receptor (RyR), pan-IP3R and calsequestrin has also been demonstrated in hiPSC-CMs (Germanguz et al., 2011; Itzhaki et al., 2011b).
Immunofluorescence also reveals the nuclear expression of transcription factors NKX2.5 and GATA-4, and localizes ANP to the secretory granules surrounding the nuclei in much the same manner as seen in human cardiomyocytes (Tanaka et al., 2009). Immunostaining of Cx43, a key gap junction protein, shows localization of fluorescence to the junctions between hiPSC-CMs (Zwi et al., 2009). The ion channel proteins (sodium, L-type-calcium and hERG) are also localized to the intermembrane surfaces (Guo et al., 2011a). Although not evidence of function, this anatomical mapping suggests that these channels and gap junctions at least have the potential to be functional.
Function
To investigate whether hiPSC-CMs function as human cardiomyocytes, we need to assess the electrical activity of individual cells and of cell clusters. We need to examine ion channel properties and interactions, excitation–contraction coupling, spontaneous beating frequency and contractility, and study how these features are affected by cardioactive drugs at a range of doses and time periods.
Inotropy and chronotropy
hiPSC-CMs demonstrate an appropriate dose-dependent response to cardioactive drugs in terms of beating frequency and contractility that match responses seen in hESC-CMs (Figures 2 and 3) (Yokoo et al., 2009). The graphs below illustrate the inotropic and chronotropic responses of hiPSC-CMs and hESC-CMs to a variety of cardioactive drugs including sodium channel blockers (Procainamide, Mexilitine, Flecainide), potassium channel blockers (Amiodarone), calcium channel blockers (Verapamil), β-blockers (Propranolol), non-selective α- and β-receptor agonists (adrenaline) and β-agonists (isoproteronol). The effects of these drugs on hESC-CMs have previously been validated against human cardiomyocytes, showing comparable effects at the same range of drug concentrations (Xu et al., 2002; He et al., 2003; Reppel et al., 2004). These responses also match those expected in the clinical setting (Rosen, 1995).
Figure 2.
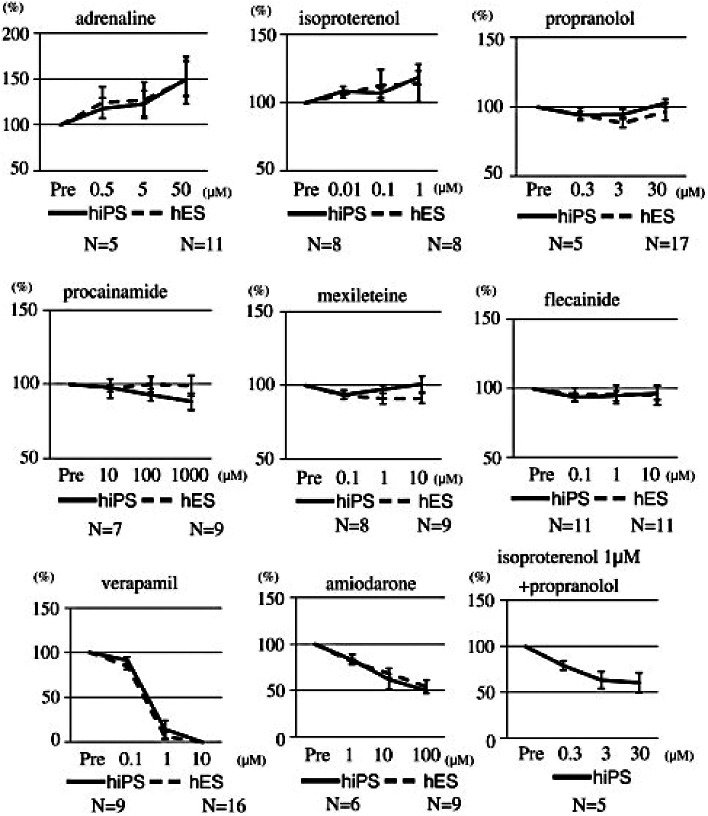
The effects of cardioactive drugs on the beating rates of contractile colonies derived from human iPS cells and human ES cells. Adrenaline, isoproterenol, verapamil, amiodarone and isoproterenol + propranolol had statistically significant effects between pre-drug loading; and the maximum concentration of the drug used in cardiomyocytes was derived from human iPS cells (P < 0.05). There were no statistically significant differences between the concentrations of drugs that elicited effects in human iPS cells and those that elicited effects in human ES cells (Yokoo et al. 2009, Biochem Biophys Res Commun 387: 482–488, with permission).
Figure 3.

The effects of cardioactive drugs on the contractility of contractile colonies derived from human iPS cells and human ES cells. Adrenaline, isoproterenol, procainamide, flecainide, verapamil and isoproterenol + propranolol had statistically significant effects on human iPS cells between pre-drug loading and the maximum concentration of the drug used in cardiomyocytes derived from human iPS cells (P < 0.05). There were no statistically significant differences between the concentrations of drugs that elicited effects in human iPS cells and those that elicited effects in human ES cells (Yokoo et al. 2009, Biochem Biophys Res Commun 387: 482–488, with permission).
Even though stem cell-derived cardiomyocytes are considered to represent a relatively immature stage of development relative to human adult cardiomyocytes, faithful pharmacological responses have now been recorded from over 40 different drugs that include modulators of α-, β1-, β2- and muscarinic-receptors and calcium, potassium and sodium ion channels (Dick et al., 2010). Comparing hESC-CM and hiPSC-CM with human ventricular cardiomyocytes, a strong quantitative similarity has been shown in terms of β1- and β2 adrenoreceptor subtype dependency (Drysdale et al., 2000), unlike all other animal models tested (dog, sheep, rat, rabbit, guinea pig, mouse) (Brito-Martins et al., 2008). Importantly, β1- and β2 adrenoreceptor responses in stem cell-derived cardiomyocytes were better models for those in adult human ventricular cardiomyocytes than those previously found using adult animal cardiomyocytes (Brito-Martins et al., 2008).
Electrophysiological properties
Field potentials (FP) can be recorded from spontaneously contracting EB outgrowths using microelectrode arrays (MEA). This method has been validated previously on mouse and rat ventricular cardiomyocytes (Meiry et al., 2001; Halbach et al., 2003), mouse ESC-CMs (Reppel et al., 2007) and hESC-CMs (Caspi et al., 2009). FP duration correlates with the QT interval in an ECG and has been shown to parallel action potential duration and predict drug effects on depolarization (Caspi et al., 2009). MEAs measure and record the surface electrical activity of cell clusters and are a valuable tool for assessing the electrophysiological properties of cardiomyocyte syncytia. They are easily performed and upscaled; they provide stable recordings that permit dose-dependent relationships and side effects to be elicited. Furthermore, they enable multiple drugs to be tested on the same system as the recordings return to baseline after washing out of the drugs from the external solution (Tanaka et al., 2009).
MEA analyses of hiPSC-CMs reveal FP morphologies similar to those produced by atrial, nodal and ventricular human myocardium (Zhang et al., 2009). Patch clamp analysis of single cells also reveal action potentials of atrial, nodal and ventricular morphologies. The atrial- and ventricular-like cells exhibit a more negative maximum diastolic potential, a higher rate of depolarization and larger amplitudes as compared with nodal-like cells (Ma et al., 2011). The ventricular-like cells also exhibit a plateau phase (phase 2) and accelerated repolarization (phase 3), with longer action potential durations and slower spontaneous beating frequencies (Ma et al., 2011). These action potential morphologies are illustrated in Figure 4 (Ma et al., 2011).
Figure 4.

Action potential morphologies from iPSC-derived cardiomyocytes. From Ma J et al. Am J Physiol Heart Circ Physiol 2011;301: H2006–H2017 with permission. Interval, beat rate interval; MDP, maximum diastolic potential; Peak, peak voltage; Amp, amplitude; dV/dtmax, maximal rate of depolarization, APD, AP duration at different levels of repolarization (APD measured at 10% increments of Amp).
The presence of functional ion channels can be assessed by the action of cardioactive drugs on the FP morphology. Quinidine, a sodium channel blocker, causes a dose-dependent and reversible decrease in the first FP negative peak, analogous to the action potential upstroke; Verapamil, a calcium channel blocker, causes a dose-dependent and reversible shortening of the FP duration; E-4031, a drug that blocks the Ikr channel (i.e. hERG channel blockade), causes a dose-dependent and reversible prolongation of the FP duration; Chromanol 293B, an Iks channel antagonist, also produces a dose-dependent increase in FP duration (Tanaka et al., 2009; Zwi et al., 2009).
These properties and measurements are nearly identical to those seen in native adult canine ventricular cardiomyocytes (January et al., 1988). Additional studies into drug antagonism of these ion channels have further shown that the current density and activation properties of the channels for INa, ICa, IKr, IKs, IKr, IKi, If and Ito in hiPSC-CMs (Moretti et al., 2010; Ma et al., 2011) are quantitatively similar to those in human cardiomyocytes (Sakakibara et al., 1993; Feng et al., 1996; Pelzmann et al., 1998; Jost et al., 2009).
hiPSC-CMs demonstrate a dose-dependent decrease in AP duration and increase in beating frequency with non-selective β-agonism (isoproterenol) and a dose-dependent decrease in beating frequency with muscarinic agonism (carbamylcholine) at doses between 1 and 10 µmol L−1 (Zwi et al., 2009). In response to the cardiac glycosides digoxin and ouabain, there is a decrease in the sodium spike amplitude, shortening of the FP duration, increase in the calcium wave amplitude and induction of arrhythmic beats (Guo et al., 2011b). This response is similar to that seen in isolated guinea pig hearts, which demonstrate QT shortening, increased contractility and increased arrhythmogenesis (Guo et al., 2011b).
Calcium handling
Intracellular calcium transients require initiation by L-type calcium channels in human cardiomyocytes. These transients have been recorded in hiPSC-CMs using laser confocal microscopy on cells pre-loaded with a fluorescent calcium indicator (Itzhaki et al., 2011b). Removal of extracellular calcium or use of nifedipine, an L-type calcium channel blocker, eliminates these calcium transients. Furthermore, functional RyR-mediated sarcoplasmic reticulum (SR) calcium stores exist in hiPSC-CM, as demonstrated by changes in intracellular calcium transients when stimulated with caffeine or inhibited with ryanodine in a dose-dependent manner (Itzhaki et al., 2011b). Inhibition of the SERCA pump with thapsigargin reduces intracellular calcium transients, through inhibition of re-uptake of calcium into the SR (Itzhaki et al., 2011b). Thapsigargin also causes a decreased calcium response to caffeine. These experiments demonstrate the presence of SERCA-sequestering, RyR-mediated SR calcium stores in hiPSC-CMs, and suggest that intracellular calcium transients depend on both sarcolemal calcium entry via L-type calcium channels and calcium release from intracellular stores, analogous to adult cardiomyocytes (Itzhaki et al., 2011b). hiPSC-CMs also show a dose-dependent decrease in amplitude and frequency of intracellular calcium transients when cultured with either an IP3R antagonist or a PLC inhibitor, implying the presence of a functional IP3 releasable pool of calcium (Itzhaki et al., 2011b). This functional calcium-induced calcium release mechanism is similar to that found in human cardiomyocytes (Cannell et al., 1995; Bers, 2002), though the response of hiPSC-CMs to ryanodine and caffeine is less in magnitude than that of human adult cardiomyocytes (Germanguz et al., 2011).
There is ongoing controversy as to the maturity of the calcium handling pathways in stem cell cardiomyocytes. Initial work on hESC-CMs showed that these cells do not increase in amplitude of contraction in response to increased stimulation, thereby lacking the positive force–frequency response typical of adult cardiomyocytes (Dolnikov et al., 2006). This may be because mature cardiomyocytes rely on internal calcium stores for contraction whereas stem cell-derived cardiomyocytes depend more on external calcium (Dolnikov et al., 2006) as they have underdeveloped SR capacity and t-tubule systems (Satin et al., 2004; Binah et al., 2007; Sedan et al., 2008). They therefore have immature calcium handling within the cell. However, later studies have shown that culture-matured stem cell-derived cardiomyocytes do have a functional SR and do display a positive force–frequency response (Liu et al., 2007; Satin et al., 2008). Forced expression of calsequestrin improves intracellular calcium handling in these cells (Kong et al., 2010). These experiments demonstrate the principle that stem cell cardiomyocytes can be evolved into a more mature phenotype with genetic manipulation, stress induced changes and prolonged time in culture. It is also worth noting that calcium handling in hiPSC-CMs closely resembles that in adult cardiomyocytes from failing human hearts, where regression to the fetal phenotype has occurred.
Application of hiPSC-CM in drug screening
Arrhythmias
Drug-induced QT prolongation is associated with the development of torsade de pointes (TdP) (Carlsson, 2006). Repolarization assays have been recommended by the European Agency for the Evaluation of Medicinal Products in its 1997 document ‘Points to Consider’, with a prolonged QT interval being used as a surrogate end-point for TdP. These assays, however, are poor predictors of drug arrhythmogenicity (Carlsson, 2006; Thomsen et al., 2006b). In particular, the degree of QT prolongation poorly predicts progression to TdP, and other factors such as drug effects on transmural variability of repolarization may play a more important role (Kannankeril and Roden, 2007).
KCNH2-encoded rapid-acting inward rectifying potassium channel or hERG (human Ether-a-go-go) channel inhibition is sometimes used as a surrogate end-point for QT prolongation and TdP. Non-cardiac cell lines over expressing hERG channels are commonly used to screen for hERG channel inhibition (Giorgi et al., 2010). However, not all hERG channel blockers cause QT prolongation or TdP, e.g. verapamil (De Ponti et al., 2002; Meyer et al., 2004), and QT prolongation does not always cause TdP (e.g. ranolazine, alfuzosin, moxifloxacin) (Frothingham, 2001; Extramiana et al., 2005; Thomsen et al., 2006a). Some drugs cause arrhythmias in humans that neither inhibit hERG channels in vitro nor cause QT prolongation in animal models (Demiryürek and Demiryürek, 2005; Chan, 2009).
Drug arrhythmogenic potential is much better assessed in predisposed animal models (Carlsson, 2006; Thomsen et al., 2006b). These pro-arrhythmic animal models confer greater sensitivity and specificity, but these models are expensive and cumbersome, with varying predictive power, and upscaling for use industry is costly. Furthermore, the number and spatial compartmentalization of hERG channels are an important determinant of action potential duration (Zaza, 2010), and these vary across species (Jonsson et al., 2010). It is thought that studying ion current changes in the context of a human ventricular action potential will increase the accuracy of predicting arrhythmias.
hiPSC-CM demonstrate dose and time dependent arrhythmias when challenged with drugs known to cause hERG inhibition, QT prolongation and TdP (astemizole, cisapride, dofetilide, erythromycin, flecainide, quinidine, sotalol, terfenadine and thioridazine) (Guo et al., 2011a). The induced arrhythmias have a characteristic oscillatory impedance pattern typical of TdP, whereas arrhythmias seen with non-TdP arrhythmic drugs such as ouabain and aconitine produce a fibrillation like pattern of impedance (Guo et al., 2011a). On the other hand, drugs that block hERG channels or cause QT prolongation but do not cause TdP clinically (ranolazine, alfuzosin, verapamil, moxifloxacin) do not cause arrhythmias in hiPSC-CMs at therapeutic doses (although supratherapeutic doses of ranolazine and alfuzosin did cause TdP-like arrhythmias in these experiments) (Guo et al., 2011a).
When repolarization is prolonged with hERG channel blockade, early after depolarizations (EADs) are produced by the hiPSC-CMs. EADs are thought to be produced by the arrhythmogenic L-type calcium window current (January 1989) and are a precursor to TdP (Ma et al., 2011). These EADs were abolished when the cells are treated with Nifedipine, a calcium channel antagonist (Guo et al., 2011a).
These results are promising and suggest that hiPSC-CMs may be a faithful model for predicting drug arrhythmogenicity as well as serving as a test bed for discovering new drug treatments for arrhythmias.
Screening of non-cardiac drugs
There is a large market for screening of non-cardiac drugs for cardiac toxicity. This is particularly true in the field of oncology, with chemotherapeutic drugs causing significant cardiac damage: a recent survey of breast cancer patients receiving the monoclonal antibody trastuzumab (Herceptin) reported a 24% rate of congestive heart failure (Wadhwa et al., 2009).
Much of the undetected toxicity of anti-tumour agents is likely to be related to their long term pro-apoptotic or anti-angiogenic effects and anti-proliferative actions on stem cells rather than acute electrophysiological phenomena. Use of hiPSC-CMs for assessing apoptotic, proliferative and hypertrophic activity is much less explored, but assays are being developed using both live and fixed cells (Földes et al., 2011). Proliferative activity, as detected by increase in cell number, Ki67+, phosphorylation of histone H3 and relative distribution of cells in G2 M−1 and G1/G0 phases of cell cycle, can be set up for automated analysis. Assays for hypertrophic changes such as increases in cell size, protein content, sarcomere assembly and ANP/BNP have also been automated and may be useful markers of long-term cardiac toxicity (Földes et al., 2011).
Harnessing genotype variability in iPSC
A wide variety of disease-specific iPSC have been generated, with fibroblasts or bone marrow mesenchymal cells being successfully reprogrammed from patients with ADA-SCID, Gaucher's disease, Duchenne's muscular dystrophy, Becker's muscular dystrophy, Down's syndrome, Parkinson's disease, juvenile diabetes mellitus, Swachman–Bodian–Diamond syndrome, Huntington's disease and Lesch–Nyhan syndrome (Park et al., 2008a).
Of particular interest is the replication of the long-QT syndrome 2 in hiPSC-CMs from a patient with the syndrome (Itzhaki et al., 2011a). As mentioned previously, drug arrhythmogenicity is better assessed in predisposed models. The development of hiPSC-CMs carrying LQT2 syndrome is therefore a very tantalizing model for screening drug arrhythmic potential.
The patient carried a missense mutation in the KCNH2 gene, disrupting the pore-forming region of the hERG channel, thereby reducing Ikr. In the pluripotent stage, the cells displayed ES-like morphology as previously described. These were successfully differentiated into a cardiac lineage, confirmed by gene analysis, immunocytostaining and functional assessment, with all three morphologies of AP generated. LQTS-iPSC-CMs showed a marked prolongation in action potential duration in patch clamp studies, associated with a reduced repolarization velocity in ventricular-like and atrial-like cells but not in nodal-like cells, with comparable results on field potential duration in MEA analysis (Itzhaki et al., 2011a). EADs in atrial and ventricular like cells detected on patch-clamp testing are a harbinger of ventricular arrhythmias in LQT (Marbán, 2002). EADs were seen in 66% of LQTS cells but not in control cells (Itzhaki et al., 2011a). Ectopic activity seen in MEA analysis was observed in 38% of LQTS cells but only 6% of controls. Cisapride, a gastric prokinetic agent and indirect inhibitor of Ikr, withdrawn from the market for increase in mortality due to increase in arrhythmogenicity (Fermini and Fossa, 2003), further prolonged the QT/AP duration in LQTS-iPSC-CMs and increased EADs and ectopic activity (Itzhaki et al., 2011a).
Treatments were also tested on this model. Nifedipine (L-type calcium channel blocker) and pinacidil (K-ATP opener, increasing potassium outflow) shortened the AP duration, eliminated EADs and triggered beats on patch clamp testing, and shortened FP duration and arrhythmic activity on MEA analysis (Matsa et al., 2011; Itzhaki et al., 2011a). Ranolazine (late sodium channel blocker, and therefore possibly more suited to LQT3 where there is a gain of function sodium channel mutation) had no effect on AP duration/FP duration but decreased the EADs and arrhythmias seen (Itzhaki et al., 2011a; Matsa et al., 2011).
A missense mutation in the L-type calcium channel Cav1.2 leads to QT prolongation in patients with Timothy syndrome. hiPSC-CMs generated from the skin cells of Timothy syndrome patients have revealed ventricular-like cells with irregular electrical activity, excess calcium influx with abnormal calcium transients and prolonged AP duration (Yazawa et al., 2011). Pharmacological blockade of this channel restored normal calcium signalling in these cells (Yazawa et al., 2011).
hiPSC-CMs from a patient with LEOPARD syndrome, a disease predisposing to hypertrophic cardiomyopathy, are larger, with a greater degree of sarcomeric organization and preferential localization of NFATC4 in the nucleus (Carvajal-Vergara et al., 2010). This model has been used to further understand the molecular mechanisms underlying the disease, implicating disruption of the RAS–MAPK pathway as evidenced by up regulation of the phosphoprotein MEK1, an upstream kinase of ERK1/2 (Carvajal-Vergara et al., 2010).
The potential for producing patient-specific lines and its implications on pharmacological screening and drug discovery is vast. These cells provide robust assays for investigating the molecular and cellular mechanisms behind a variety of cardiac diseases. Arrhythmogenicity can be investigated in normal subjects, in heart failure models and in subjects with inherited QT abnormalities. Likewise, hypertrophic insults can be investigated in a variety of genetically predisposing conditions. As has been demonstrated in the models so far, these disease-specific cells also provide a test bed for the development of new treatments.
High-throughput systems
Both efficient, automated cardiogenesis and advanced, integrated bioanalytical techniques are required if high-throughput toxicity screening on hiPSC-CMs is to become a reality.
Industry demands high-quality, uniform, validated cells. Several groups have established protocols to improve the efficiency of isolating cardiomyocytes from beating EBs, including by pipette dissociation of beating clusters (Shinozawa et al., 2012), use of pro-cardiogenic growth factors to stimulate mesoderm induction (e.g. BMP4 and FGF2) (Burridge et al., 2011; Xu et al., 2012), manipulation of Activin/Nodal and Wnt signalling pathways as mentioned earlier (Laflamme et al., 2007; Yang et al., 2008; Kattman et al., 2011; Ren et al., 2011), and supplementing the culture media with chemical promoters of cardiac differentiation (e.g. 20% FBS, PVA and physiological oxygen tension) (Burridge et al., 2011). Use of monolayer cultures, where cardiac progenitor cells are allowed to proliferate and fuse to form single beating sheets, has also increased hiPSC-CM yield (Carpenter et al., 2012). These measures have been adopted in an attempt to remedy the poor differentiation efficiency of hiPSC-CMs of the order of 1–25% and reduce the prolonged time in culture before beating cells are produced (Gai et al., 2009; Zhang et al., 2009). They also aim at producing uniform cell lines and limiting the epigenetic differences between lines (Kim et al., 2010). With optimization, differentiation efficiency can be significantly improved to over 90% of hiPSCs forming beating EBs within 9 days of differentiation (Burridge et al., 2011).
Easily measurable and upscaled endpoints of toxicity have been established. Electrophysiological abnormalities can be detected by MEA analysis, as described earlier. Metabolic activity can be measured by using oxygen uptake as a marker of respiration, detected using luminescence intensity measurements (Papkovsky, 2004). Stress responses can be evaluated by examining gene expression (e.g. IL6/8 release), membrane integrity (CK release) and apoptosis (e.g. TUNEL assay). Surface plasmon resonance can be used to detect cardiac biomarkers, e.g. cardiac troponin T, a clinically valuable marker for cardiomyocyte damage. Multi-wavelength spectroscopy fluorescence can provide real-time information on the protein level of the culture and detect a decrease in this level in the presence of toxic substances due to decrease in cell growth, excretion of proteins from dying cells, and detachment of dying cells from the plate (Mandenius et al., 2011).
Such observations have prompted several companies (e.g. GE-Healthcare, Cellular Dynamics International, Cellartis, Reprocell) to produce stem cell-derived cardiomyocytes for uptake by pharmaceutical companies for integration into their drug safety assessment pipelines. These assays are becoming a commercial reality, with companies such as Cellular Dynamics selling hiPSC-CMs (iCell™) for drug candidate toxicity screening.
Shortcomings
Heterogeneity
hiPSC-CMs are not a homogenous group of cells. Even fully differentiated hiPSC-CMs display heterogeneity in terms of being at various stages of development and maturity. There is also a spectrum of APs demonstrated by these cells, with a continuous range (rather than sharply divided populations) from nodal-like, through to atrial- and ventricular-like APs. Single cell transcriptional profiling confirms heterogeneity between individual hiPSCs (Narsinh et al., 2011). Twenty-eight pluripotency related transcripts and 14 differentiated state related transcripts were measured in hiPSCs and hESCs. In aggregate, they had the same mean levels of transcript expression, but the frequency distribution in hiPSCs was much broader, suggesting greater inter-cell line variability (Narsinh et al., 2011). This heterogeneity impacts on the generation of reproducible results from these cells.
Much work on validating hiPSC-CMs has been done by comparing them to previously validated hESC-CMs. This seems reasonable as iPSC-CMs and hESC-CMs have similar expressions of stem cell proteins, proliferation rate, ability to form EBs and capacity for differentiation (Takahashi et al., 2007). Their differentiation efficiency, contraction rates, cardiomyocyte marker genes and electrophysiological properties are also comparable (Gai et al., 2009; Zhang et al., 2009). The sarcomeric structures in hiPSC-CMs have been shown to be immature but functional (Zwi et al., 2009), with an indistinguishable ultrastructure from hESC-CMs (Zhang et al., 2009). However, groups have noted differences in their gene expression signatures, and these differences are maintained in their miRNA expression patterns (Chin et al., 2009; Gupta et al., 2010). Sub-chromosomal genomic differences analyzed using array-comparative genomic hybridization, gene expression differences analyzed by RNA profiling, difference in non-coding RNAs by miRNA profiling, and epigenetic changes analyzed by histone modification profiling have revealed a characteristic gene expression signature shared between iPSC lines that differs slightly from ESC lines (Chin et al., 2009; Gupta et al., 2010). Differences were diminished, but not completely eliminated when late passage hiPSCs were compared with hESCs. These differences mainly arose from insufficient induction of hESC genes or insufficient suppression of fibroblast genes in the hiPSC lines (Chin et al., 2009; Gupta et al., 2010). Alterations in DNA methylation patterns have also been noted (Deng et al., 2009). On the other hand, there is no convincing difference in structure or function between hiPSCs and hESCs as a result of their different transcriptomes (Deng et al., 2009).
Use of retroviridae and lentiviridae in reprogramming introduces a potential risk of viral reactivation and is associated with altered gene expression due to residual expression of reprogramming factors (Kaji et al., 2009; Soldner et al., 2009; Woltjen et al., 2009). Methods have developed to remove these viral vectors after reprogramming. For example, using cre-recombinase excisable viruses has generated hiPSCs with global gene expression profiles that more closely resemble hESCs than their genetically identical hiPSC counterparts (Soldner et al., 2009). Other non-integrating approaches include using episomes (Yu et al., 2009), adenoviruses (Stadtfeld et al., 2008), RNA viruses (Fusaki et al., 2009) and plasmid transfection (Okita et al., 2008; Gonzalez et al., 2009). All these approaches confer varying degrees of success in limiting genetic modification, although vector sub-fragments are rarely completely eradicated (Kaji et al., 2009). Furthermore, these methods are often extremely labour intensive and inefficient (Saha and Jaenisch, 2009).
Fetal phenotype
Another limitation of hiPSC-CMs is that they retain certain stem-cell and fetal characteristics. There is persistent transgenic expression of OCT4 and NANOG in hiPSCs even in micro dissected beating clusters at 60 days post differentiation (Zhang et al., 2009). Nevertheless, protein levels of OCT4 and NANOG, as measured by immunolabelling, are undetectable. This suggests down-regulation in the expression of these proteins despite some persistence in mRNA expression, possibly mediated by miRNA regulation (Zhang et al., 2009).
Quantitative real-time PCR has been used to compare RNA levels between different sources of cardiomyocytes. Commercially available hiPSC-CMs (iCells), after being thawed and cultured for three to five days, were compared with human fetal and human adult cardiomyocytes (Guo et al., 2011a). The expression levels of genes for sarcomeric proteins in hiPSC-CMs more closely resembled those in fetal cardiomyocytes. Specifically, expression of MYL2, MYH7, TNNI3, SCN5A and KCNH2 more closely resembled fetal levels; whereas expression of MYH6 resembled adult levels. The expression of MYL7, MYH7 and TNNI3 was less than that seen in both fetal and adult cardiomyocytes (Guo et al., 2011a).
In terms of their structure, both hESC-CMs and hiPSC-CMs phenotypically resemble the fetal mycocardium (Cao et al., 2008; Xu et al., 2009). With maturation, their myofibrils align and organize and they more closely resemble the adult phenotype, but still appear immature (Snir et al., 2003). Figure 5 is a slide of hiPSC-CMs illustrating sarcomeric structures with varying degrees of organization seen within the same preparation.
Figure 5.
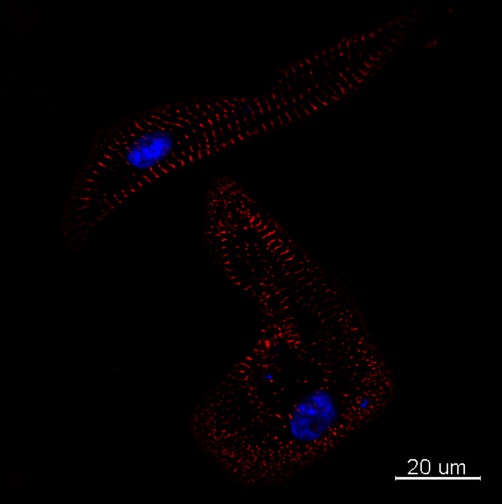
iCell-CMs (Cellular Dynamics International) were cultured for 2 weeks in iCell cardiomyocytes maintenance medium (Cellular Dynamics International), fixed with paraformaldehyde (4%) and immunolabelled with primary antibodies against mouse sarcomeric α-actinin (Sigma, A7811, 1:150). The secondary antibody was Alexa Fluor 594 goat anti mouse IgG (Invitrogen, 1:800). Nuclei were stained with DAPI. Scale bars: 20 µm. Courtesy Ljudmila Kolker, NISBC and Imperial College.
Young stem cell-derived cardiomyocytes demonstrate a positive inotropic and chronotropic effect in response to the non-selective β-agonist isoprenaline, but show a poor lusitropic response. However, stem cell-derived cardiomyocytes can be matured through extended time in culture, and further maturation is being developed using such techniques as mechanical stretching, pharmacological or neurohormonal treatment, and chemical or conformational modulation of the growth substrate. Matured cells also seem to adopt a more adult phenotype in terms of arrhythmic propensity (Abdul Kadir et al., 2009).
Stem cell-derived cardiomyocytes show increased automacity compared with human ventricular cardiomyocytes due to under-expression of the inward rectifier potassium channel, therefore resulting in a large inward sodium current and increasing the spontaneity of contraction (Satin et al., 2004). Forced expression of the gene encoding the Iki channel reduces the observed automacity and results in an electrophysiological phenotype more akin to mature adult cardiomyocytes (Kong et al., 2010).
The concentration–response curves of stem cell–cardiomyocytes to cardioactive drugs such as isoprenaline match those of the embryonic and also of the failing human heart (Brito-Martins et al., 2008). This suggests the possibility that cardiomyocytes in heart failure regress towards the ‘protective’ fetal phenotype. If this is the case, hiPSC-CMs may paradoxically be a better model for diseased mature human cardiomyocytes than for healthy adult cardiomyocytes. Cardiotoxicity modelling using both adult and fetal phenotypes may give better predictive abilities since they will span normal responses and those from patients with sub-clinical or clinical heart disease.
Conclusion
We are currently in the transition phase from validation to discovery. A powerful testament to the utility of hiPSC-CMs in drug screening would be cardiotoxicity detection in compounds that did not show cardiotoxicity in animal screens but which subsequently failed in phase I trials due to cardiac effects. This is an extremely important set of compounds to validate the predictive power of any assay, and will expedite the widespread adoption of the model to pharmaceutical use. More extensive testing will need to be done to ensure that the model does not generate too many false negatives. This would be problematic, as many drugs with great therapeutic potential may be prematurely withdrawn. Difficulties could arise with drugs that not only have clear toxic effects but also have great therapeutic benefit in certain scenarios, the use of thalidomide in multiple myeloma being a prime example.
Once we know the sensitivity, specificity, reproducibility and predictive power of hiPSC-CMs, they can be confidently used in drug screening. The initial results are promising, but further work still needs to be done. A limitation of using cell models for drug screening is the difficulty in mimicking the drug pharmacokinetics and durations of drug exposure that occur in whole organisms. Future areas of research could involve injecting hiPSC-CMs into animal models (e.g. nude mice) to form chimeras (Friese et al., 2006; Behringer, 2007). This may allow testing toxicity on human-like cardiomyocytes in an in vivo environment that replicates the complex pathophysiology of multiple tissue interactions and also allows toxic effects with long latency periods to be assessed. Along with drug screening, an exciting new field of disease modelling has emerged with hiPSC, with greater understanding of the developmental process of cardiac disease phenotypes aiding drug discovery. We have also moved a step closer to individualised therapies, where drugs can be tested for patient organ-specific effects and toxicities. This would be a significant advance in the field of targeted health care and have great potential benefit in the clinic.
Glossary
- ADA
adenosine deaminase
- ANP
atrial natriuretic peptide
- AP
action potential
- BMP
bone morphogentic protein
- BNP
brain natriuretic peptide
- CK
creatine kinase
- EADs
early after depolarizations
- EB
embryoid body
- ESC
embryonic stem cells
- FGF
fibroblast growth factor
- FP
field potential
- hERG
human ether-a-go-go
- hESC
human embryonic stem cells
- hESC-CM
human embryonic stem cell-derived cardiomyocytes
- hiPSC
human induced pluripotent stem cells
- hiPSC-CM
human pluripotent stem cell-derived cardiomyocytes
- iPSC
induced pluripotent stem cells
- LQT
long-QT syndrome
- MEA
multi-electrode array
- NYHA
New York Heart Association
- PVA
polyvinyl alcohol
- RyR
ryanodine receptor
- SCID
severe combined immunodeficiency
- siRNA
short interfering RNA
- SR
sarcoplasmic reticulum
- TdP
Torsade de pointes
Conflict of interest
The authors have no conflicts of interest.
References
- Abdul Kadir SH, Ali NN, Mioulane M, Brito-Martins M, Abu-Hayyeh S, Foldes G, et al. Embryonic stem cell-derived cardiomyocytes as a model to study fetal arrhythmia related to maternal disease. J Cell Mol Med. 2009;13:3730–3741. doi: 10.1111/j.1582-4934.2009.00741.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 2003;17:126–140. doi: 10.1101/gad.224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer RR. Human-animal chimeras in biomedical research. Cell Stem Cell. 2007;1:259–262. doi: 10.1016/j.stem.2007.07.021. [DOI] [PubMed] [Google Scholar]
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Binah O, Dolnikov K, Sadan O, Shilkrut M, Zeevi-Levin N, Amit M, et al. Functional and developmental properties of human embryonic stem cells-derived cardiomyocytes. J Electrocardiol. 2007;40(Suppl.):S192–S196. doi: 10.1016/j.jelectrocard.2007.05.035. [DOI] [PubMed] [Google Scholar]
- Boland MJ, Hazen JL, Nazor KL, Rodriguez AR, Gifford W, Martin G, et al. Adult mice generated from induced pluripotent stem cells. Nature. 2009;461:91–94. doi: 10.1038/nature08310. [DOI] [PubMed] [Google Scholar]
- Braam SR, Denning C, Matsa E, Young LE, Passier R, Mummery CL. Feeder-free culture of human embryonic stem cells in conditioned medium for efficient genetic modification. Nat Protoc. 2008;3:1435–1443. doi: 10.1038/nprot.2008.140. [DOI] [PubMed] [Google Scholar]
- Braam SR, Tertoolen L, van de Stolpe A, Meyer T, Passier R, Mummery CL. Prediction of drug-induced cardiotoxicity using human embryonic stem cell-derived cardiomyocytes. Stem Cell Res. 2010;4:107–116. doi: 10.1016/j.scr.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Brito-Martins M, Harding SE, Ali NN. beta(1)- and beta(2)-adrenoceptor responses in cardiomyocytes derived from human embryonic stem cells: comparison with failing and non-failing adult human heart. Br J Pharmacol. 2008;153:751–759. doi: 10.1038/sj.bjp.0707619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge PW, Thompson S, Millrod MA, Weinberg S, Yuan X, Peters A, et al. A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PLoS ONE. 2011;6:e18293. doi: 10.1371/journal.pone.0018293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell MB, Cheng H, Lederer WJ. The control of calcium release in heart muscle. Science. 1995;268:1045–1049. doi: 10.1126/science.7754384. [DOI] [PubMed] [Google Scholar]
- Cao F, Wagner RA, Wilson KD, Xie X, Fu JD, Drukker M, et al. Transcriptional and functional profiling of human embryonic stem cell-derived cardiomyocytes. PLoS ONE. 2008;3:e3474. doi: 10.1371/journal.pone.0003474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson L. In vitro and in vivo models for testing arrhythmogenesis in drugs. J Intern Med. 2006;259:70–80. doi: 10.1111/j.1365-2796.2005.01590.x. [DOI] [PubMed] [Google Scholar]
- Carpenter L, Carr C, Yang CT, Stuckey DJ, Clarke K, Watt SM. Efficient differentiation of human induced pluripotent stem cells generates cardiac cells that provide protection following myocardial infarction in the rat. Stem Cells Dev. 2012;21:977–986. doi: 10.1089/scd.2011.0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright P, McLean C, Sheppard A, Rivett D, Jones K, Dalton S. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development. 2005;132:885–896. doi: 10.1242/dev.01670. [DOI] [PubMed] [Google Scholar]
- Carvajal-Vergara X, Sevilla A, D'Souza SL, Ang YS, Schaniel C, Lee DF, et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010;465:808–812. doi: 10.1038/nature09005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi O, Itzhaki I, Kehat I, Gepstein A, Arbel G, Huber I, et al. In vitro electrophysiological drug testing using human embryonic stem cell derived cardiomyocytes. Stem Cells Dev. 2009;18:161–172. doi: 10.1089/scd.2007.0280. [DOI] [PubMed] [Google Scholar]
- Chan TY. Aconite poisoning. Clin Toxicol (Phila) 2009;47:279–285. doi: 10.1080/15563650902904407. [DOI] [PubMed] [Google Scholar]
- Chin MH, Mason MJ, Xie W, Volinia S, Singer M, Peterson C, et al. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell. 2009;5:111–123. doi: 10.1016/j.stem.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ponti F, Poluzzi E, Cavalli A, Recanatini M, Montanaro N. Safety of non-antiarrhythmic drugs that prolong the QT interval or induce torsade de pointes: an overview. Drug Saf. 2002;25:263–286. doi: 10.2165/00002018-200225040-00004. [DOI] [PubMed] [Google Scholar]
- Demiryürek AT, Demiryürek S. Cardiotoxicity of digitalis glycosides: roles of autonomic pathways, autacoids and ion channels. Auton Autacoid Pharmacol. 2005;25:35–52. doi: 10.1111/j.1474-8673.2004.00334.x. [DOI] [PubMed] [Google Scholar]
- Deng J, Shoemaker R, Xie B, Gore A, LeProust EM, Antosiewicz-Bourget J, et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat Biotechnol. 2009;27:353–360. doi: 10.1038/nbt.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick E, Rajamohan D, Ronksley J, Denning C. Evaluating the utility of cardiomyocytes from human pluripotent stem cells for drug screening. Biochem Soc Trans. 2010;38:1037–1045. doi: 10.1042/BST0381037. [DOI] [PubMed] [Google Scholar]
- Dolnikov K, Shilkrut M, Zeevi-Levin N, Gerecht-Nir S, Amit M, Danon A, et al. Functional properties of human embryonic stem cell-derived cardiomyocytes: intracellular Ca2+ handling and the role of sarcoplasmic reticulum in the contraction. Stem Cells. 2006;24:236–245. doi: 10.1634/stemcells.2005-0036. [DOI] [PubMed] [Google Scholar]
- Drysdale CM, McGraw DW, Stack CB, Stephens JC, Judson RS, Nandabalan K, et al. Complex promoter and coding region beta 2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. Proc Natl Acad Sci U S A. 2000;97:10483–10488. doi: 10.1073/pnas.97.19.10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Extramiana F, Maison-Blanche P, Cabanis MJ, Ortemann-Renon C, Beaufils P, Leenhardt A. Clinical assessment of drug-induced QT prolongation in association with heart rate changes. Clin Pharmacol Ther. 2005;77:247–258. doi: 10.1016/j.clpt.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Feng J, Li GR, Fermini B, Nattel S. Properties of sodium and potassium currents of cultured adult human atrial myocytes. Am J Physiol. 1996;270:H1676–H1686. doi: 10.1152/ajpheart.1996.270.5.H1676. [DOI] [PubMed] [Google Scholar]
- Fermini B, Fossa AA. The impact of drug-induced QT interval prolongation on drug discovery and development. Nat Rev Drug Discov. 2003;2:439–447. doi: 10.1038/nrd1108. [DOI] [PubMed] [Google Scholar]
- Földes G, Mioulane M, Wright JS, Liu AQ, Novak P, Merkely B, et al. Modulation of human embryonic stem cell-derived cardiomyocyte growth: a testbed for studying human cardiac hypertrophy? J Mol Cell Cardiol. 2011;50:367–376. doi: 10.1016/j.yjmcc.2010.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friese MA, Jensen LT, Willcox N, Fugger L. Humanized mouse models for organ-specific autoimmune diseases. Curr Opin Immunol. 2006;18:704–709. doi: 10.1016/j.coi.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Frothingham R. Rates of torsades de pointes associated with ciprofloxacin, ofloxacin, levofloxacin, gatifloxacin, and moxifloxacin. Pharmacotherapy. 2001;21:1468–1472. doi: 10.1592/phco.21.20.1468.34482. [DOI] [PubMed] [Google Scholar]
- Fusaki N, Ban H, Nishiyama A, Saeki K, Hasegawa M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:348–362. doi: 10.2183/pjab.85.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gai H, Leung EL, Costantino PD, Aguila JR, Nguyen DM, Fink LM, et al. Generation and characterization of functional cardiomyocytes using induced pluripotent stem cells derived from human fibroblasts. Cell Biol Int. 2009;33:1184–1193. doi: 10.1016/j.cellbi.2009.08.008. [DOI] [PubMed] [Google Scholar]
- Germanguz I, Sedan O, Zeevi-Levin N, Shtrichman R, Barak E, Ziskind A, et al. Molecular characterization and functional properties of cardiomyocytes derived from human inducible pluripotent stem cells. J Cell Mol Med. 2011;15:38–51. doi: 10.1111/j.1582-4934.2009.00996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi MA, Bolaños R, Gonzalez CD, Di Girolamo G. QT interval prolongation: preclinical and clinical testing arrhythmogenesis in drugs and regulatory implications. Curr Drug Saf. 2010;5:54–57. doi: 10.2174/157488610789869148. [DOI] [PubMed] [Google Scholar]
- Gonzalez F, Barragan Monasterio M, Tiscornia G, Montserrat Pulido N, Vassena R, Batlle Morera L, et al. Generation of mouse-induced pluripotent stem cells by transient expression of a single nonviral polycistronic vector. Proc Natl Acad Sci U S A. 2009;106:8918–8922. doi: 10.1073/pnas.0901471106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Abrams RM, Babiarz JE, Cohen JD, Kameoka S, Sanders MJ, et al. Estimating the risk of drug-induced proarrhythmia using human induced pluripotent stem cell-derived cardiomyocytes. Toxicol Sci. 2011a;123:281–289. doi: 10.1093/toxsci/kfr158. [DOI] [PubMed] [Google Scholar]
- Guo L, Qian JY, Abrams R, Tang HM, Weiser T, Sanders MJ, et al. The electrophysiological effects of cardiac glycosides in human iPSC-derived cardiomyocytes and in guinea pig isolated hearts. Cell Physiol Biochem. 2011b;27:453–462. doi: 10.1159/000329966. [DOI] [PubMed] [Google Scholar]
- Gupta MK, Illich DJ, Gaarz A, Matzkies M, Nguemo F, Pfannkuche K, et al. Global transcriptional profiles of beating clusters derived from human induced pluripotent stem cells and embryonic stem cells are highly similar. BMC Dev Biol. 2010;10:98. doi: 10.1186/1471-213X-10-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib M, Caspi O, Gepstein L. Human embryonic stem cells for cardiomyogenesis. J Mol Cell Cardiol. 2008;45:462–474. doi: 10.1016/j.yjmcc.2008.08.008. [DOI] [PubMed] [Google Scholar]
- Halbach M, Egert U, Hescheler J, Banach K. Estimation of action potential changes from field potential recordings in multicellular mouse cardiac myocyte cultures. Cell Physiol Biochem. 2003;13:271–284. doi: 10.1159/000074542. [DOI] [PubMed] [Google Scholar]
- Harding SE, Ali NN, Brito-Martins M, Gorelik J. The human embryonic stem cell-derived cardiomyocyte as a pharmacological model. Pharmacol Ther. 2007;113:341–353. doi: 10.1016/j.pharmthera.2006.08.008. [DOI] [PubMed] [Google Scholar]
- He JQ, Ma Y, Lee Y, Thomson JA, Kamp TJ. Human embryonic stem cells develop into multiple types of cardiac myocytes: action potential characterization. Circ Res. 2003;93:32–39. doi: 10.1161/01.RES.0000080317.92718.99. [DOI] [PubMed] [Google Scholar]
- ICH. 2000. Safety Pharmacology Studies for Human Pharmaceuticals. ICH Safety Guideline S7A. The International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use.
- Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011a;471:225–229. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- Itzhaki I, Rapoport S, Huber I, Mizrahi I, Zwi-Dantsis L, Arbel G, et al. Calcium handling in human induced pluripotent stem cell derived cardiomyocytes. PLoS ONE. 2011b;6:e18037. doi: 10.1371/journal.pone.0018037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- January CT, Riddle JM, Salata JJ. A model for early afterdepolarizations: induction with the Ca2+ channel agonist Bay K 8644. Circ Res. 1988;62:563–571. doi: 10.1161/01.res.62.3.563. [DOI] [PubMed] [Google Scholar]
- Jonsson MK, Duker G, Tropp C, Andersson B, Sartipy P, Vos MA, et al. Quantified proarrhythmic potential of selected human embryonic stem cell-derived cardiomyocytes. Stem Cell Res. 2010;4:189–200. doi: 10.1016/j.scr.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Jost N, Acsai K, Horváth B, Bányász T, Baczkó I, Bitay M, et al. Contribution of I Kr and I K1 to ventricular repolarization in canine and human myocytes: is there any influence of action potential duration? Basic Res Cardiol. 2009;104:33–41. doi: 10.1007/s00395-008-0730-3. [DOI] [PubMed] [Google Scholar]
- Kaji K, Norrby K, Paca A, Mileikovsky M, Mohseni P, Woltjen K. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458:771–775. doi: 10.1038/nature07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang L, Wang J, Zhang Y, Kou Z, Gao S. iPS cells can support full-term development of tetraploid blastocyst-complemented embryos. Cell Stem Cell. 2009;5:135–138. doi: 10.1016/j.stem.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Kannankeril PJ, Roden DM. Drug-induced long QT and torsade de pointes: recent advances. Curr Opin Cardiol. 2007;22:39–43. doi: 10.1097/HCO.0b013e32801129eb. [DOI] [PubMed] [Google Scholar]
- Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A, et al. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell. 2011;8:228–240. doi: 10.1016/j.stem.2010.12.008. [DOI] [PubMed] [Google Scholar]
- Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A, et al. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest. 2001;108:407–414. doi: 10.1172/JCI12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Jeon K, Park HY, Han YJ, Yang BC, Park SB, et al. Differences between cellular and molecular profiles of induced pluripotent stem cells generated from mouse embryonic fibroblasts. Cell Reprogram. 2010;12:627–639. doi: 10.1089/cell.2010.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- Kong CW, Akar FG, Li RA. Translational potential of human embryonic and induced pluripotent stem cells for myocardial repair: insights from experimental models. Thromb Haemost. 2010;104:30–38. doi: 10.1160/TH10-03-0189. [DOI] [PubMed] [Google Scholar]
- Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25:1015–1024. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- Li Y, McClintick J, Zhong L, Edenberg HJ, Yoder MC, Chan RJ. Murine embryonic stem cell differentiation is promoted by SOCS-3 and inhibited by the zinc finger transcription factor Klf4. Blood. 2005;105:635–637. doi: 10.1182/blood-2004-07-2681. [DOI] [PubMed] [Google Scholar]
- Lian Q, Chow Y, Esteban MA, Pei D, Tse HF. Future perspective of induced pluripotent stem cells for diagnosis, drug screening and treatment of human diseases. Thromb Haemost. 2010;104:39–44. doi: 10.1160/TH10-05-0269. [DOI] [PubMed] [Google Scholar]
- Liu J, Fu JD, Siu CW, Li RA. Functional sarcoplasmic reticulum for calcium handling of human embryonic stem cell-derived cardiomyocytes: insights for driven maturation. Stem Cells. 2007;25:3038–3044. doi: 10.1634/stemcells.2007-0549. [DOI] [PubMed] [Google Scholar]
- Lowry WE, Richter L, Yachechko R, Pyle AD, Tchieu J, Sridharan R, et al. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc Natl Acad Sci U S A. 2008;105:2883–2888. doi: 10.1073/pnas.0711983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HR, Mariën R, Saels A, De Clerck F. Species plays an important role in drug-induced prolongation of action potential duration and early afterdepolarizations in isolated Purkinje fibers. J Cardiovasc Electrophysiol. 2001;12:93–102. doi: 10.1046/j.1540-8167.2001.00093.x. [DOI] [PubMed] [Google Scholar]
- Ma J, Guo L, Fiene SJ, Anson BD, Thomson JA, Kamp TJ, et al. High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am J Physiol Heart Circ Physiol. 2011;301:H2006–H2017. doi: 10.1152/ajpheart.00694.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JS, Robertson RT. Toxicity testing in the 21st century: a view from the pharmaceutical industry. Toxicol Sci. 2009;110:40–46. doi: 10.1093/toxsci/kfp088. [DOI] [PubMed] [Google Scholar]
- Maherali N, Sridharan R, Xie W, Utikal J, Eminli S, Arnold K, et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1:55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Mandenius CF, Steel D, Noor F, Meyer T, Heinzle E, Asp J, et al. Cardiotoxicity testing using pluripotent stem cell-derived human cardiomyocytes and state-of-the-art bioanalytics: a review. J Appl Toxicol. 2011;31:191–205. doi: 10.1002/jat.1663. [DOI] [PubMed] [Google Scholar]
- Marbán E. Cardiac channelopathies. Nature. 2002;415:213–218. doi: 10.1038/415213a. [DOI] [PubMed] [Google Scholar]
- Matsa E, Rajamohan D, Dick E, Young L, Mellor I, Staniforth A, et al. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur Heart J. 2011;32:952–962. doi: 10.1093/eurheartj/ehr073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauritz C, Schwanke K, Reppel M, Neef S, Katsirntaki K, Maier LS, et al. Generation of functional murine cardiac myocytes from induced pluripotent stem cells. Circulation. 2008;118:507–517. doi: 10.1161/CIRCULATIONAHA.108.778795. [DOI] [PubMed] [Google Scholar]
- Meiry G, Reisner Y, Feld Y, Goldberg S, Rosen M, Ziv N, et al. Evolution of action potential propagation and repolarization in cultured neonatal rat ventricular myocytes. J Cardiovasc Electrophysiol. 2001;12:1269–1277. doi: 10.1046/j.1540-8167.2001.01269.x. [DOI] [PubMed] [Google Scholar]
- Meyer T, Leisgen C, Gonser B, Günther E. QT-screen: high-throughput cardiac safety pharmacology by extracellular electrophysiology on primary cardiac myocytes. Assay Drug Dev Technol. 2004;2:507–514. doi: 10.1089/adt.2004.2.507. [DOI] [PubMed] [Google Scholar]
- Mitcheson JS, Hancox JC, Levi AJ. Cultured adult cardiac myocytes: future applications, culture methods, morphological and electrophysiological properties. Cardiovasc Res. 1998;39:280–300. doi: 10.1016/s0008-6363(98)00128-x. [DOI] [PubMed] [Google Scholar]
- Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flügel L, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363:1397–1409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- Mummery C, Ward D, van den Brink CE, Bird SD, Doevendans PA, Opthof T, et al. Cardiomyocyte differentiation of mouse and human embryonic stem cells. J Anat. 2002;200:233–242. doi: 10.1046/j.1469-7580.2002.00031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummery C, Ward-van Oostwaard D, Doevendans P, Spijker R, van den Brink S, Hassink R, et al. Differentiation of human embryonic stem cells to cardiomyocytes: role of coculture with visceral endoderm-like cells. Circulation. 2003;107:2733–2740. doi: 10.1161/01.CIR.0000068356.38592.68. [DOI] [PubMed] [Google Scholar]
- Narazaki G, Uosaki H, Teranishi M, Okita K, Kim B, Matsuoka S, et al. Directed and systematic differentiation of cardiovascular cells from mouse induced pluripotent stem cells. Circulation. 2008;118:498–506. doi: 10.1161/CIRCULATIONAHA.108.769562. [DOI] [PubMed] [Google Scholar]
- Narsinh KH, Sun N, Sanchez-Freire V, Lee AS, Almeida P, Hu S, et al. Single cell transcriptional profiling reveals heterogeneity of human induced pluripotent stem cells. J Clin Invest. 2011;121:1217–1221. doi: 10.1172/JCI44635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95:379–391. doi: 10.1016/s0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- Niwa H, Miyazaki J, Smith AG. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet. 2000;24:372–376. doi: 10.1038/74199. [DOI] [PubMed] [Google Scholar]
- Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- Okita K, Nakagawa M, Hyenjong H, Ichisaka T, Yamanaka S. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322:949–953. doi: 10.1126/science.1164270. [DOI] [PubMed] [Google Scholar]
- Papkovsky DB. Methods in optical oxygen sensing: protocols and critical analyses. Methods Enzymol. 2004;381:715–735. doi: 10.1016/S0076-6879(04)81046-2. [DOI] [PubMed] [Google Scholar]
- Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, et al. Disease-specific induced pluripotent stem cells. Cell. 2008a;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008b;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- Pekkanen-Mattila M, Chapman H, Kerkelä E, Suuronen R, Skottman H, Koivisto AP, et al. Human embryonic stem cell-derived cardiomyocytes: demonstration of a portion of cardiac cells with fairly mature electrical phenotype. Exp Biol Med. 2010;235:522–530. doi: 10.1258/ebm.2010.009345. [DOI] [PubMed] [Google Scholar]
- Pelzmann B, Schaffer P, Bernhart E, Lang P, Mächler H, Rigler B, et al. L-type calcium current in human ventricular myocytes at a physiological temperature from children with tetralogy of Fallot. Cardiovasc Res. 1998;38:424–432. doi: 10.1016/s0008-6363(98)00002-9. [DOI] [PubMed] [Google Scholar]
- Ren Y, Lee MY, Schliffke S, Paavola J, Amos PJ, Ge X, et al. Small molecule Wnt inhibitors enhance the efficiency of BMP-4-directed cardiac differentiation of human pluripotent stem cells. J Mol Cell Cardiol. 2011;51:280–287. doi: 10.1016/j.yjmcc.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reppel M, Boettinger C, Hescheler J. Beta-adrenergic and muscarinic modulation of human embryonic stem cell-derived cardiomyocytes. Cell Physiol Biochem. 2004;14:187–196. doi: 10.1159/000080326. [DOI] [PubMed] [Google Scholar]
- Reppel M, Igelmund P, Egert U, Juchelka F, Hescheler J, Drobinskaya I. Effect of cardioactive drugs on action potential generation and propagation in embryonic stem cell-derived cardiomyocytes. Cell Physiol Biochem. 2007;19:213–224. doi: 10.1159/000100628. [DOI] [PubMed] [Google Scholar]
- Rosen MR. Consequences of the Sicilian Gambit. Eur Heart J. 1995;16(Suppl. G):32–36. doi: 10.1093/eurheartj/16.suppl_g.32. [DOI] [PubMed] [Google Scholar]
- Saha K, Jaenisch R. Technical challenges in using human induced pluripotent stem cells to model disease. Cell Stem Cell. 2009;5:584–595. doi: 10.1016/j.stem.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara Y, Furukawa T, Singer DH, Jia H, Backer CL, Arentzen CE, et al. Sodium current in isolated human ventricular myocytes. Am J Physiol. 1993;265:H1301–H1309. doi: 10.1152/ajpheart.1993.265.4.H1301. [DOI] [PubMed] [Google Scholar]
- Sartiani L, Bettiol E, Stillitano F, Mugelli A, Cerbai E, Jaconi ME. Developmental changes in cardiomyocytes differentiated from human embryonic stem cells: a molecular and electrophysiological approach. Stem Cells. 2007;25:1136–1144. doi: 10.1634/stemcells.2006-0466. [DOI] [PubMed] [Google Scholar]
- Satin J, Kehat I, Caspi O, Huber I, Arbel G, Itzhaki I, et al. Mechanism of spontaneous excitability in human embryonic stem cell derived cardiomyocytes. J Physiol. 2004;559:479–496. doi: 10.1113/jphysiol.2004.068213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satin J, Itzhaki I, Rapoport S, Schroder EA, Izu L, Arbel G, et al. Calcium handling in human embryonic stem cell-derived cardiomyocytes. Stem Cells. 2008;26:1961–1972. doi: 10.1634/stemcells.2007-0591. [DOI] [PubMed] [Google Scholar]
- Sedan O, Dolnikov K, Zeevi-Levin N, Leibovich N, Amit M, Itskovitz-Eldor J, et al. 1,4,5-Inositol trisphosphate-operated intracellular Ca(2+) stores and angiotensin-II/endothelin-1 signaling pathway are functional in human embryonic stem cell-derived cardiomyocytes. Stem Cells. 2008;26:3130–3138. doi: 10.1634/stemcells.2008-0777. [DOI] [PubMed] [Google Scholar]
- Shinozawa T, Furukawa H, Sato E, Takami K. A novel purification method of murine embryonic stem cell- and human-induced pluripotent stem cell-derived cardiomyocytes by simple manual dissociation. J Biomol Screen. 2012;14:239–245. doi: 10.1177/1087057111434145. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- Snir M, Kehat I, Gepstein A, Coleman R, Itskovitz-Eldor J, Livne E, et al. Assessment of the ultrastructural and proliferative properties of human embryonic stem cell-derived cardiomyocytes. Am J Physiol Heart Circ Physiol. 2003;285:H2355–H2363. doi: 10.1152/ajpheart.00020.2003. [DOI] [PubMed] [Google Scholar]
- Soldner F, Hockemeyer D, Beard C, Gao Q, Bell GW, Cook EG, et al. Parkinson's disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964–977. doi: 10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008;322:945–949. doi: 10.1126/science.1162494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Tohyama S, Murata M, Nomura F, Kaneko T, Chen H, et al. In vitro pharmacologic testing using human induced pluripotent stem cell-derived cardiomyocytes. Biochem Biophys Res Commun. 2009;385:497–502. doi: 10.1016/j.bbrc.2009.05.073. [DOI] [PubMed] [Google Scholar]
- Thomsen MB, Beekman JD, Attevelt NJ, Takahara A, Sugiyama A, Chiba K, et al. No proarrhythmic properties of the antibiotics Moxifloxacin or Azithromycin in anaesthetized dogs with chronic-AV block. Br J Pharmacol. 2006a;149:1039–1048. doi: 10.1038/sj.bjp.0706900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen MB, Matz J, Volders PG, Vos MA. Assessing the proarrhythmic potential of drugs: current status of models and surrogate parameters of torsades de pointes arrhythmias. Pharmacol Ther. 2006b;112:150–170. doi: 10.1016/j.pharmthera.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Wadhwa D, Fallah-Rad N, Grenier D, Krahn M, Fang T, Ahmadie R, et al. Trastuzumab mediated cardiotoxicity in the setting of adjuvant chemotherapy for breast cancer: a retrospective study. Breast Cancer Res Treat. 2009;117:357–364. doi: 10.1007/s10549-008-0260-6. [DOI] [PubMed] [Google Scholar]
- Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- Woltjen K, Michael IP, Mohseni P, Desai R, Mileikovsky M, Hämäläinen R, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458:766–770. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Police S, Rao N, Carpenter MK. Characterization and enrichment of cardiomyocytes derived from human embryonic stem cells. Circ Res. 2002;91:501–508. doi: 10.1161/01.res.0000035254.80718.91. [DOI] [PubMed] [Google Scholar]
- Xu H, Yi BA, Wu H, Bock C, Gu H, Lui KO, et al. Highly efficient derivation of ventricular cardiomyocytes from induced pluripotent stem cells with a distinct epigenetic signature. Cell Res. 2012;22:142–154. doi: 10.1038/cr.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XQ, Soo SY, Sun W, Zweigerdt R. Global expression profile of highly enriched cardiomyocytes derived from human embryonic stem cells. Stem Cells. 2009;27:2163–2174. doi: 10.1002/stem.166. [DOI] [PubMed] [Google Scholar]
- Yang L, Soonpaa MH, Adler ED, Roepke TK, Kattman SJ, Kennedy M, et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature. 2008;453:524–528. doi: 10.1038/nature06894. [DOI] [PubMed] [Google Scholar]
- Yazawa M, Hsueh B, Jia X, Pasca AM, Bernstein JA, Hallmayer J, et al. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011;471:230–234. doi: 10.1038/nature09855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoo N, Baba S, Kaichi S, Niwa A, Mima T, Doi H, et al. The effects of cardioactive drugs on cardiomyocytes derived from human induced pluripotent stem cells. Biochem Biophys Res Commun. 2009;387:482–488. doi: 10.1016/j.bbrc.2009.07.052. [DOI] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaza A. Control of the cardiac action potential: The role of repolarization dynamics. J Mol Cell Cardiol. 2010;48:106–111. doi: 10.1016/j.yjmcc.2009.07.027. [DOI] [PubMed] [Google Scholar]
- Zhang J, Wilson GF, Soerens AG, Koonce CH, Yu J, Palecek SP, et al. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 2009;104:e30–e41. doi: 10.1161/CIRCRESAHA.108.192237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XY, Li W, Lv Z, Liu L, Tong M, Hai T, et al. iPS cells produce viable mice through tetraploid complementation. Nature. 2009;461:86–90. doi: 10.1038/nature08267. [DOI] [PubMed] [Google Scholar]
- Zwi L, Caspi O, Arbel G, Huber I, Gepstein A, Park IH, et al. Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation. 2009;120:1513–1523. doi: 10.1161/CIRCULATIONAHA.109.868885. [DOI] [PubMed] [Google Scholar]