

Abstract
Background and Purpose
Nuclear factor erythroid 2-related factor (Nrf2) is a transcription factor that up-regulates a diverse array of antioxidant genes and protects cells from oxidative damage. This study is designed to determine whether D-L-sulforaphane (SFN) can protect neural crest cells (NCCs), an ethanol-sensitive cell population implicated in fetal alcohol spectrum disorders, against ethanol-induced apoptosis and whether protective effects of SFN are mediated by the induction of Nrf2-mediated antioxidant response.
Experimental Approach
Control, SFN-treated or Nrf2-siRNA transfected NCCs were exposed to ethanol. Nrf2 activation, the expression and activities of Nrf2 downstream antioxidant proteins, reactive oxygen species generation and apoptosis were determined in control and ethanol-exposed NCCs.
Key Results
Exposure of NCCs to SFN alone significantly increased Nrf2 activation and the expression of Nrf2 downstream antioxidants as well as the activities of the antioxidant enzymes. Treatment of NCCs with SFN along with ethanol significantly decreased ethanol-induced oxidative stress and apoptosis. In contrast, knockdown of Nrf2 by siRNA significantly increased the sensitivity of NCCs to ethanol-induced oxidative stress and apoptosis. Suppression of Nrf2 signalling in NCCs also significantly diminished SFN-mediated antioxidant response and abolished the protective effects of SFN on ethanol-induced oxidative stress and apoptosis.
Conclusions and Implications
These results demonstrated that Nrf2-mediated antioxidant response plays an important role in the susceptibility of NCCs to ethanol-induced oxidative stress and apoptosis and that the protection of SFN against ethanol-induced oxidative stress and apoptosis in NCCs is mediated by the induction of Nrf2 signalling.
Keywords: Nrf2, oxidative stress, ethanol, sulforaphane, apoptosis, neural crest cell
Introduction
Fetal alcohol spectrum disorder (FASD) is one of the most common permanent birth defects caused by maternal consumption of alcohol during pregnancy. The effects of prenatal ethanol exposure include physical, mental, behavioural and/or learning disabilities with possible lifelong implications. Prenatal alcohol exposure is considered to be the leading known non-genetic cause of mental retardation (Burd, 2004).
The pathogenesis of FASD is a complex, multifactorial process that includes the vulnerability of selected cell populations to ethanol-induced apoptosis (Kotch and Sulik, 1992; Cartwright and Smith, 1995; Smith, 1997; Dunty et al., 2001). Among the vulnerable cell populations are neural crest cells (NCCs) (Sulik et al., 1981; Kotch and Sulik, 1992; Cartwright and Smith, 1995; Chen and Sulik, 1996; Chen and Sulik, 2000). The NCC is a multipotent progenitor cell population that can give rise to a diversity of neural and non-neural cell types, such as melanocytes, neurons, glial cells, endocrine cells, as well as mesenchymal cells that form craniofacial cartilages, bones, dermis, adipose tissue and vascular smooth muscle cells (Teng and Labosky, 2006; Hall, 2008). Studies have demonstrated that NCCs are vulnerable to ethanol-induced cytotoxicity. Ethanol can diminish NCC population by the induction of apoptosis, which appears to contribute heavily to subsequent abnormalities (Sulik et al., 1981; Kotch and Sulik, 1992; Cartwright and Smith, 1995; Chen and Sulik, 1996; Smith, 1997).
Previous studies have revealed that oxidative stress plays an important role in ethanol-induced apoptosis and dysmorphology (Kotch et al., 1995; Chen and Sulik, 1996; Henderson et al., 1999; Chen and Sulik, 2000; Dong et al., 2008). Reactive oxygen species (ROS) generation has been observed in ethanol-exposed cultured cells, including NCCs (Chen and Sulik, 1996; Chen and Sulik, 2000; Yan et al., 2010), as well as in mouse embryos exposed to ethanol both in vitro and in vivo (Kotch et al., 1995; Dong et al., 2008). In addition, superoxide dismutase (SOD) has been shown to diminish ethanol-induced superoxide anion generation, cell death and the incidence of neural tube defects in cultured mouse embryos (Kotch et al., 1995). Studies have also shown that maternal treatment with an SOD and catalase mimetic, EUK-134, reduced ethanol-induced apoptosis in selected cell populations in the developing limb buds and diminished subsequent limb defects in mouse embryos (Chen et al., 2004). More recently, it has been demonstrated that nuclear factor erythroid 2-related factor 2 (Nrf2) signalling is involved in ethanol-induced apoptosis in NCCs and in mouse embryos (Dong et al., 2008; Yan et al., 2010; Chen, 2012).
Nrf2 is a CNC (cap ‘n’ collar) bZIP (basic region leucine zipper) group of transcription factors which is broadly expressed in a variety of tissues (Kensler et al., 2007). Quiescent Nrf2 localizes in the cytoplasm and is rapidly turned over through a specific ubiquitin-26S proteasome pathway controlled by the Keap1/Cul3-independent ubiquitin ligase (E3) (Li and Kong, 2009). Nrf2 is activated in response to a range of oxidative and electrophilic stimuli including ROS, heavy metals and certain disease processes (Nguyen et al., 2009). Upon activation, Nrf2 mediates antioxidant response by the induction of a broad range of genes including phase 2 enzymes, such as NAD(P)H:quinone oxidoreductase 1 (NQO1) and heme oxygenase-1, and antioxidant proteins, such as SOD and catalase (Kensler et al., 2007; Nguyen et al., 2009). Both genetic and biochemical studies have implicated the Nrf2 signalling pathway in the defence against a wide range of chemical toxicity, cancer and chronic diseases in which oxidative stress is involved (Kensler et al., 2007). Recent studies have also shown that maternal ethanol treatment increased Nrf2 protein levels and Nrf2-antioxidant response element (ARE) binding in mouse embryos. Up-regulation of Nrf2 signalling by the Nrf2 inducer, 3H-1,2-dithiole-3-thione (D3T), significantly decreased ethanol-induced ROS generation and apoptosis in mouse embryos. These results demonstrate that Nrf2 signalling is involved in the induction of an antioxidant response in ethanol-exposed mouse embryos (Dong et al., 2008). This premise has also been supported by studies that have shown that Nrf2-dependent maintenance of GSH homeostasis in cerebral cortical neurons is critical for preventing ethanol-induced oxidative stress and apoptosis (Narasimhan et al., 2011), and that resveratrol can protect the cerebellar granule neurons against ethanol-induced cell death in a rat model of FASD by the induction of Nrf2 (Kumar et al., 2011).
D,L-sulforaphane (SFN) is a chemical that is abundant in broccoli sprouts (Keum et al., 2005). Compelling evidence indicates that SFN-rich broccoli sprouts and other sources of dietary SFN trigger the induction of phase 2 detoxifying genes and antioxidant enzymes through activation of Nrf2 signalling and can aid in preventing cancer and other diseases (Dinkova-Kostova et al., 2002). It has been demonstrated that SFN reacts with thiol groups of Keap1 to form thionoacyl adducts and that the modification of Keap1 releases Nrf2 from its sequestration and activates Nrf2 (Hong et al., 2005). While being a well-recognized and effective strategy in cancer chemoprevention (Cheung and Kong, 2010; Wang and Shan, 2012), the use of SFN to enhance cellular defences through the up-regulation of endogenous antioxidants in embryonic cells has not been investigated.
With a great deal of evidence indicating that NCCs are particularly vulnerable to ethanol-induced apoptosis and that ROS plays a major role in alcohol's teratogenesis, along with recognition that antioxidant capabilities are minimal in embryos, strategies directed towards up-regulation of endogenous antioxidant activity in this particular cell population provide a novel and promising approach for reducing alcohol's teratogenic impact. To this end, using an NCC cell line, JoMa 1.3 cells, the current study was designed to test the hypothesis that SFN, a naturally occurring isothiocyanate, can provide protection against ethanol-induced oxidative stress and apoptosis in NCCs by the induction of Nrf2-mediated antioxidant response. This is, to our knowledge, the first study attempting to prevent ethanol-induced apoptosis in embryonic cells through the use of bioactive compounds naturally present in vegetables. The results from this study demonstrated that Nrf2-mediated antioxidant response plays an important role in the susceptibility of NCCs to ethanol-induced oxidative stress and apoptosis and that the protection of SFN against ethanol-induced oxidative stress and apoptosis in NCCs is mediated by the induction of Nrf2 signalling. These findings support the premise that Nrf2 is a key molecular target for diminishing alcohol's teratogenesis and provide a note of optimism for the development of clinically applicable antioxidant strategies for the treatment or prevention of FASD.
Methods
Cell culture, treatments and siRNA transfection
NCCs (JoMa1.3 cells) were provided by Dr. Schorle and cultured as described previously by Maurer et al. (2007). Briefly, cells were grown on cell culture dishes coated with fibronectin and maintained in DMEM: Ham's F12 (1:1) at 37°C in 5% CO2/95% air. For dose–response analysis, NCCs were treated with 0.25, 0.5, 1, 2 or 4 μM SFN (LKT Laboratories, St. Paul, MN, USA) alone for 24 h, followed by 24 h of concurrent exposure to SFN and 50, 100 or 200 mM ethanol. For time–response study, NCCs were treated with 1 μM SFN alone for 24 h, followed by 24, 48 or 72 h of concurrent exposure to 1 μM SFN and 100 mM ethanol. For the majority of this study, NCCs were treated with 1 μM SFN alone for 24 h, followed by 24 h of concurrent exposure to SFN and 100 mM ethanol. Stable ethanol levels were maintained by placing the cell culture plates in a plastic desiccator containing 50, 100 or 200 mM ethanol, respectively, in distilled water as described previously (Yan et al., 2010). For siRNA inhibition studies, NCCs were transfected with MISSION® Nrf2-siRNA or negative control siRNA (Sigma-Aldrich, St Louis, MO, USA) at a final concentration of 40 nM in the presence of N-TER™ Nanoparticle siRNA Transfection System (Sigma-Aldrich), following the manufacturer's instructions. The cells were harvested 48 h after transfection for mRNA and protein extraction and additional analysis.
DNA transfection and reporter gene assays
For luciferase reporter gene assays, NCCs were transfected with pNQO1 ARE-luciferase plasmid (a generous gift from Dr. John D. Hayes) (Nioi et al., 2003) and the pRL-TK Renilla control plasmid (Promega, Madison, WI, USA) using Lipofectamine 2000 reagent (Life Technologies Inc., Gaithersburg, MD, USA), according to the manufacturer's instructions. Transfected cells treated with ethanol or/and SFN were lysed, and Renilla and firefly luciferase activities were measured using the Dual-Lucifease Reporter Assay System (Promega) with a lumat LB 9507 ultra-sensitive tube luminometer (Berthold Technologies, Oak Ridge, TN, USA). The firefly luciferase activities were normalized to Renilla luciferase enzyme activity.
Quantitative real-time PCR
To determine the mRNA expression of Nrf2 downstream target genes, total RNAs were isolated from NCCs using the QIAGEN RNeasy mini kit (QIAGEN, Valencia, CA, USA), according to the manufacturer's instructions. The reverse transcription reaction was carried out by incubation of 500 ng of total RNA with a reaction mixture containing 4 μL of transcriptase reaction buffer (5×), 2 μL of dNTPs (contains 10 mM each dNTP), 10 U of reverse transcriptase, 20 U of RNase inhibitor and 1200 pmol random hexamer primer. Quantitative RT-PCR was carried out using a standard protocol on a Rotor-Gene 6000 Real-Time system (Corbett Life Science, Sydney, Australia) with the FastStart Universal SYBR Green Master qPCR kit (Roche Diagnostics, Mannheim, Germany). The following primer pairs were used for this analysis: SOD1: forward: 5′-TAACTGAAGGCCAGCATGGG-3′; reverse: 5′-CATGGACCACCATTGTACGG-3′; catalase: forward: 5′-CACTCAGGTGCGGACATTCT-3′; reverse: 5′-TCCGGAGTGGGAGAATCCAT-3′; β-actin: forward: 5′-AGCCTTCCTTCTTGGGTATGGAATC-3′; reverse: 5′-GGAGCAATGATCTTGATCTTCATGG-3′. These primers were designed using Primer3 and synthesized by Integrated DNA Technologies, Inc. (IDT, Coralville, IA, USA). All real-time PCR assays were performed in triplicate. Relative quantitative analysis was carried out by comparing threshold cycle number for target genes and a reference β-actin mRNA.
Western blotting
Western blotting was performed as described previously (Dong et al., 2011). Briefly, NCCs were washed with PBS and then lysed in pre-cold RIPA (Cell Signaling, Beverly, MA, USA) with 1 mM freshly prepared phenylmethylsulfonyl fluoride (Sigma-Aldrich) and protease cocktail inhibitors (Roche Applied, Indianapolis, IN, USA). Cell lysates were then centrifuged at 12 000× g for 10 min at 4°C and the supernatants were used for Western blot. The protein concentration in each sample was determined using bicinchoninic acid protein assay kit (Pierce, Rockford, IL, USA) following the manufacturer's instructions. Western blots were performed by standard protocols. The levels of Nrf2, SOD1, catalase and caspase-3 were analysed with the following antibodies respectively: rabbit polyclonal anti-Nrf2 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit polyclonal anti-superoxide dismutase (SOD) antibody (Abcam, Cambridge, MA, USA), rabbit polyclonal anti-catalase antibody (Abcam) and rabbit monoclonal anti-cleaved caspase-3 antibody (Cell Signaling). The membranes were then developed on a Kodak X-OMAT 2000A imaging system (Kodak, Rochester, NY, USA) and the intensity of the protein band was analysed using the Adobe Photoshop CS software (Adobe Systems, San Jose, CA, USA). All Western blot analyses were performed in triplicate.
Determination of antioxidant enzyme activities
The activities of two antioxidant enzymes, SOD and catalase, were determined as described previously (Yan et al., 2010). Total SOD activity was determined using an SOD Assay Kit (Dojindo Molecular Technologies, Inc., Gaithersburg, MD, USA), according to the manufacturer's protocol, and was measured using a SpectraMax M5 Microplate Reader (Molecular Devices, Sunnyvale, CA, USA). The activity of catalase was determined using an Amplex Red Catalase Assay Kit (Molecular Probes, Eugene, OR, USA) according to the manufacturer's instruction and was measured using a SpectraMax M5 Microplate Reader.
Detection of intracellular ROS generation
Intracellular ROS generation was evaluated using a 2′,7′-dichlorodihydrofluoroscein diacetate (DCHFDA) assay as described previously (Yan et al., 2010). Briefly, control and treated NCCs were washed with HBSS without phenol red and lysed in pre-cold RIPA. Cell lysates were centrifuged at 12 000× g for 10 min at 4°C. The supernatant, which was equivalent to 40 μg of protein, was transferred to a 96-well microplate and incubated with 20 μM DCHFDA (Molecular Probes) at 37°C for 30 min. The fluorescence intensity was determined using a SpectraMax M5 Microplate Reader with a fluorescence excitation at 485 nm and emission at 538 nm. The ROS levels in experimental groups were expressed as fold change over control.
Analysis of cell viability and apoptosis
Cell viability was measured using an MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt) assay kit (Promega), following the manufacturer's protocol. Apoptosis was determined by the analysis of caspase-3 cleavage and activity, and phosphatidylserine externalization. Caspase-3 cleavage was determined by Western blot as described previously (Dong et al., 2008). Caspase-3 activity was determined by using homogeneous luminescent Caspase-Glo™ 3/7 Assay (Promega), following the manufacturer's instructions. The phosphatidylserine externalization was determined by flow cytometry using an Annexin V FITC Apoptosis Detection Kit (BD Bioscience, San Jose, CA, USA) as described previously (Yan et al., 2010).
Statistical analysis
Statistical analyses were performed using GraphPad Prism software (GraphPad Software, San Diego, CA, USA). All data are expressed as means ± SEM of three separate experiments. Comparisons between groups were analysed by a one-way anova. Multiple comparison post-tests between groups were conducted by using Bonferroni's test. Differences between groups were considered significant at P < 0.05.
Results
SFN treatment significantly reduced ethanol-induced cell death in NCCs
To determine whether SFN can reduce ethanol-induced cell death in NCCs, JoMa 1.3 cells were treated with ethanol or/and SFN at different concentrations for 24 h or treated with 100 mM ethanol or/and 1 μM SFN for 24, 48 or 72 h. JoMa 1.3 cells were chosen as a model for the proposed studies because (i) they are NCCs derived from mouse embryos; (ii) this cell line expresses early NCC markers and can be instructed to differentiate into neurons, glia, smooth muscle cells, melanocyte and chondrocytes (Maurer et al., 2007); and (iii) this cell line represents a powerful tool for studying the mechanisms of NCC development (Murphy et al., 2011) and has been used to elucidate the role of miRNAs in NCC development (Cordes et al., 2009; Sheehy et al., 2010). As shown in Figure 1A–C, while SFN at 0.5, 1 or 2 μM and at 0.25, 0.5, 1 or 2 μM significantly reduced cell death in NCCs exposed to 50 and 100 mM ethanol, respectively, SFN at the concentrations up to 4 μM did not significantly diminish cell death in NCCs treated with 200 mM ethanol. The maximum reduction in ethanol-induced cell death was observed in the cells treated with 1 μM SFN. In addition, time–response analyses illustrated that while 1 μM SFN can significantly reduce cell death in NCCs exposed to 100 mM ethanol for up to 72 h, the maximum protection was observed in NCCs exposed to ethanol for 24 h (Figure 1D–F). These results indicate that SFN can potently prevent ethanol-induced cell death in NCCs. Based on these observations, 1 μM SFN, 100 mM ethanol and the 24 h culture period were utilized for the subsequent experiments.
Figure 1.

SFN treatment significantly reduced ethanol-induced cell death in NCCs. (A–C) NCCs were treated with 50, 100 or 200 mM ethanol alone or co-treated with SFN at indicated concentrations for 24 h. (D–F) NCCs were treated with 100 mM ethanol alone or in combination with 1 μM SFN for 24, 48 or 72 h. Ethanol-induced cell death was determined by MTS assay as described in the Method section. Data are expressed as percentage of control and represent the mean ± SEM of three separate experiments. *P < 0.05 versus control. #P < 0.05 versus EtOH.
Ethanol exposure and SFN treatment significantly increased Nrf2 protein expression in NCCs
To determine whether treatment with ethanol or SFN can increase Nrf2 protein expression in NCCs, the levels of Nrf2 protein were examined in NCCs exposed to ethanol or SFN alone or in combination by Western blot analysis. As shown in Figure 2A, exposure to 50, 100 or 200 mM ethanol for 24 h resulted in a moderate increase in Nrf2 expression. Treatment with 1 μM SFN alone significantly increased the levels of Nrf2 protein in NCCs. The combination of the SFN treatment and 100 mM ethanol exposure resulted in a significantly greater increase in Nrf2 protein expression as compared to the group treated with ethanol alone. However, induction of Nrf2 expression by ethanol or SFN alone or in combination was dramatically decreased in NCCs in which Nrf2 expression was inhibited by siRNA, indicating that Nrf2 signalling in NCCs can be efficiently knocked down by siRNA (Figure 2B).
Figure 2.
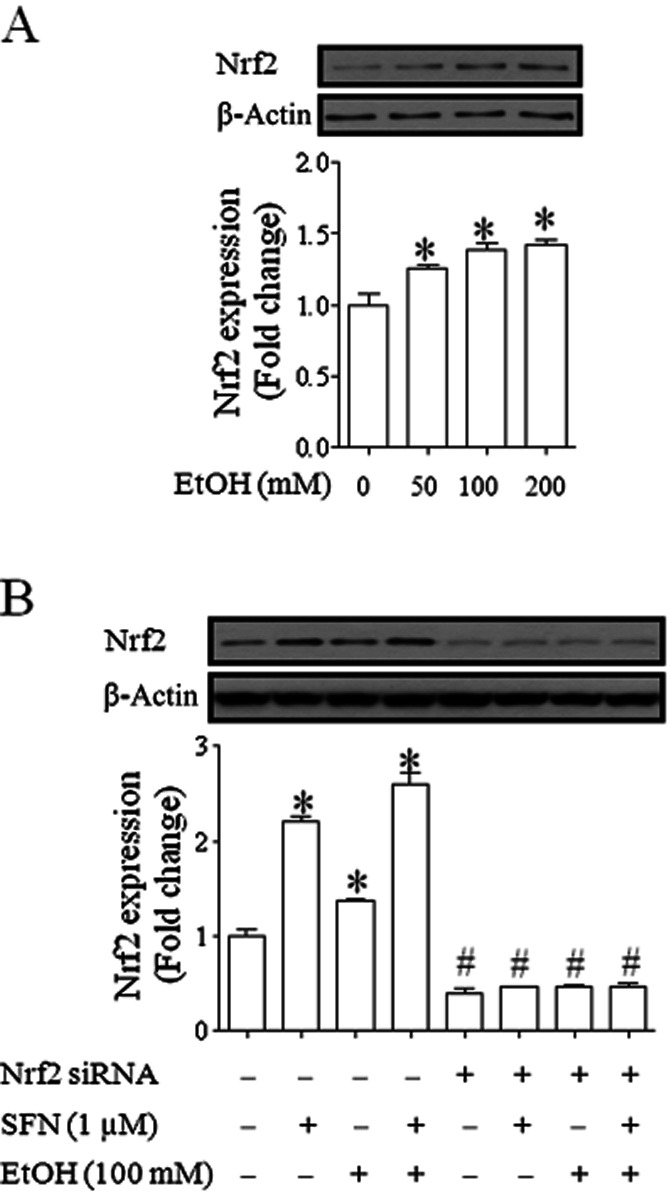
Ethanol exposure and SFN treatment significantly increased Nrf2 protein expression in NCCs. (A) NCCs were treated with 50, 100 or 200 mM ethanol for 24 h. (B) NCCs transfected with control or Nrf2-siRNA were treated with 100 mM ethanol or 1 μM SFN alone or co-treated with ethanol and SFN. Protein expression of Nrf2 was determined by Western blot. Data are expressed as fold change over control and represent the mean ± SEM of three separate experiments. (A) *P < 0.05 significant difference between control and treatment groups. (B) *P < 0.05 significant difference between control and treatment groups in NCCs transfected with control siRNA. #P < 0.05 compared with corresponding treatment in control siRNA group.
Ethanol exposure and SFN treatment significantly increased the ARE promoter activity in NCCs
Nrf2 activates transcription of its target genes through binding specifically to ARE found in the gene promoters (Ohtsuji et al., 2008; Nguyen et al., 2009). To determine whether treatment with ethanol and SFN can activate Nrf2, we examined the ARE promoter activity in NCCs exposed to ethanol and SFN. ARE-luciferase reporter assay showed an increased ARE promoter activity in NCCs exposed to ethanol alone. Treatment with SFN alone or in combination with ethanol resulted in a significantly greater increase in the ARE promoter activity as compared to the group treated with ethanol alone. However, ethanol- or SFN-induced increase in ARE promoter activity was significantly decreased in NCCs transfected with Nrf2-siRNA (Figure 3). These data demonstrate that, in addition to increasing Nrf2 protein expression, ethanol and SFN can activate ARE activity in NCCs.
Figure 3.
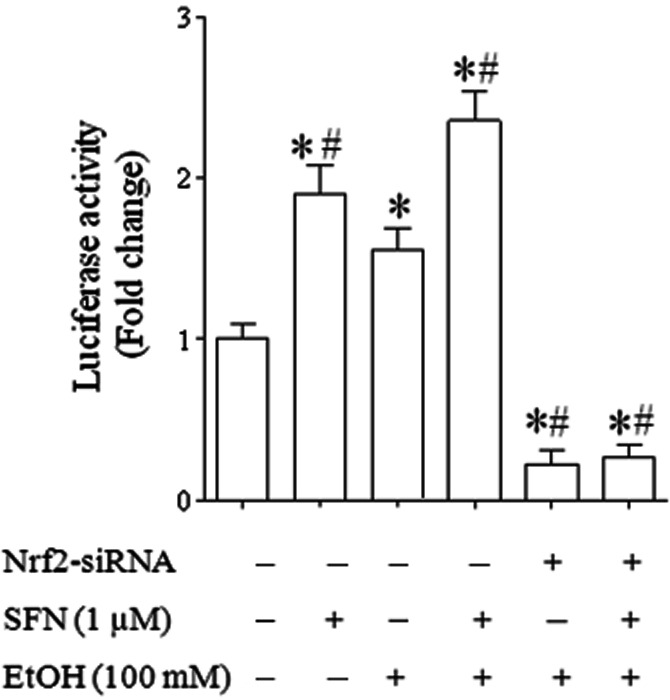
Ethanol exposure and SFN treatment significantly increased the ARE promoter activity in NCCs. NCCs transfected with control or Nrf2-siRNA and pNQO1 ARE-luciferase plasmid were treated with 100 mM ethanol or 1 μM SFN alone or co-treated with ethanol and SFN. The firefly luciferase activities were measured as described in the Methods section. Data are expressed as fold change over control and represent the mean ± SEM of three separate experiments. *P < 0.05 versus control. #P < 0.05 versus EtOH.
SFN treatment significantly increased Nrf2 transcriptional activity in control and ethanol-exposed NCCs
To further confirm that ethanol and SFN can increase Nrf2 transcriptional activity in NCCs, the effects of ethanol and SFN on mRNA expression of Nrf2 target genes, SOD1 and catalase, were determined in NCCs. We found that ethanol exposure resulted in a moderate increase in mRNA expression of SOD1 and catalase. mRNA expression of these two genes was significantly increased in NCCs treated with SFN alone or in combination with ethanol. However, these effects were not observed in Nrf2-siRNA transfected NCCs exposed to ethanol and SFN (Figure 4).
Figure 4.
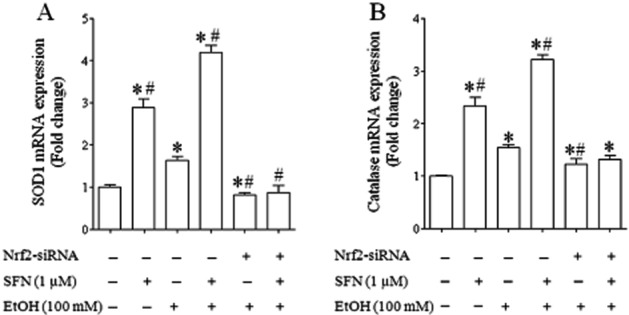
SFN treatment significantly increased Nrf2 transcriptional activity in control and ethanol-exposed NCCs. NCCs transfected with control or Nrf2-siRNA were treated with 100 mM ethanol or 1 μM SFN alone or co-treated with ethanol and SFN. The mRNA expression of SOD (A) and catalase (B) were determined as described in the Methods section. Data are expressed as fold change over control and represent the mean ± SEM of three separate experiments. *P < 0.05 versus control. #P < 0.05 versus EtOH.
Treatment with SFN significantly increased the expression of antioxidant proteins and their catalytic activity in control and ethanol-exposed NCCs
To examine whether increases in mRNA expression of Nrf2 target genes in NCCs exposed to ethanol and SFN are accompanied by over-expression of Nrf2 downstream antioxidant proteins, the expression of antioxidant proteins, SOD1 and catalase, was determined by Western blot. As shown in Figure 5, exposure of NCCs to ethanol alone resulted in a moderate increase in the protein expression of SOD1 and catalase. Treatment with SFN alone or in combination with ethanol resulted in a significantly greater increase in the protein expression of SOD1 and catalase, confirming the corresponding changes in major antioxidant proteins that accompany mRNA induction. In contrast, ethanol- or SFN-induced up-regulation of these two antioxidant proteins was significantly decreased in NCCs transfected with Nrf2-siRNA, demonstrating that SFN-induced up-regulation of antioxidant proteins in ethanol-exposed NCCs is Nrf2-dependent.
Figure 5.
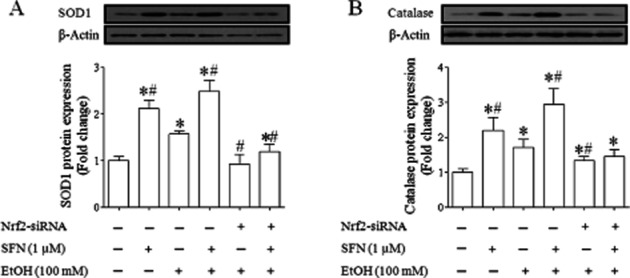
SFN treatment significantly increased the expression of antioxidant proteins in control and ethanol-exposed NCCs. NCCs transfected with control or Nrf2-siRNA were treated with 100 mM ethanol or 1 μM SFN alone or co-treated with ethanol and SFN. The protein expression of SOD (A) and catalase (B) were determined as described in the Methods section. Data are expressed as fold change over control and represent the mean ± SEM of three separate experiments. *P < 0.05 versus control. #P < 0.05 versus EtOH.
To determine the corresponding changes in the activities of major antioxidant enzymes that accompany protein induction in NCCs, the activities of SOD and catalase were determined in the NCCs exposed to ethanol and SFN. While treatment with ethanol or SFN alone significantly increased the activities of SOD and catalase, treatment with SFN in combination with ethanol resulted in significantly greater increases in the activities of these antioxidant enzymes. However, knockdown of Nrf2 expression by siRNA significantly reduced ethanol- and SFN-induced increases in the activities of SOD and catalase in NCCs (Figure 6), indicating that SFN-induced antioxidant response in ethanol-exposed NCCs is mediated by Nrf2 signalling.
Figure 6.
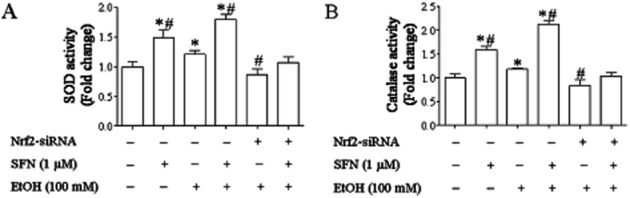
SFN treatment significantly increased the catalytic activities of antioxidant proteins in control and ethanol-exposed NCCs. NCCs transfected with control or Nrf2-siRNA were treated with 100 mM ethanol or 1 μM SFN alone or co-treated with ethanol and SFN. The catalytic activities of SOD (A) and catalase (B) were determined as described in the Methods section. Data are expressed as fold change over control and represent the mean ± SEM of three separate experiments. *P < 0.05 versus control. #P < 0.05 versus EtOH.
ROS generation was significantly decreased in ethanol-exposed NCCs treated with SFN but increased in ethanol-exposed NCCs transfected with Nrf2-siRNA
To determine whether SFN-induced activation of Nrf2 and induction of Nrf2 downstream antioxidant proteins can reduce ethanol-induced ROS generation in NCCs, a DCHFDA assay was used to analyse the ROS generation in NCCs transfected with control or Nrf2-siRNA. Consistent with our previous studies, ethanol exposure resulted in a significant increase in ROS generation. Exposure of NCCs transfected with Nrf2-siRNA to ethanol resulted in a significantly greater increase in ROS generation, indicating that Nrf2 signalling is important in preventing ethanol-induced oxidative stress in NCCs. Treatment with SFN significantly diminished ethanol-induced ROS in NCCs transfected with control siRNA. However, SFN was not able to reduce ethanol-induced ROS generation in NCCs transfected with Nrf2-siRNA (Figure 7).
Figure 7.
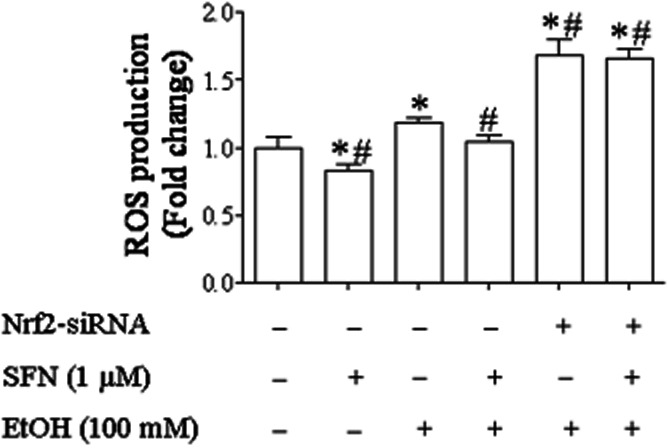
ROS generation in ethanol-exposed NCCs treated with SFN or transfected with Nrf2- siRNA. NCCs transfected with control or Nrf2-siRNA were treated with 100 mM ethanol or 1 μM SFN alone or in combination. ROS generation was determined as described in the Methods section. Data are expressed as fold change over control and represent the mean ± SEM of three separate experiments. *P < 0.05 versus control. #P < 0.05 versus EtOH.
Ethanol-induced apoptosis was significantly decreased in NCCs treated with SFN but increased in NCCs transfected with Nrf2-siRNA
To examine the potential of SFN in preventing ethanol-induced apoptosis in NCCs, ethanol-induced apoptosis was determined in control and SFN-treated NCCs. As shown in Figure 8A, MTS analysis showed a significant increase in cell death in ethanol-exposed NCCs. While transfection of NCCs with siRNA did not increase cell death in NCCs, suppression of Nrf2 signalling by siRNA increased the sensitivity of NCCs to ethanol-induced cell death. Flow cytometric analysis of FITC Annexin V showed a very similar result (Figure 8B). Ethanol exposure also resulted in a significant increase in caspase-3 cleavage and activity (Figure 8C,D). Suppression of Nrf2 signalling by siRNA resulted in significantly greater increases in apoptosis as well as caspase-3 cleavage and activity, indicating that Nrf2 signalling is an important determinant for the vulnerability of NCCs to ethanol-induced apoptosis. In contrast, treatment with SFN significantly reduced ethanol-induced apoptosis in NCCs. However, as expected, the protection of SFN against ethanol-induced apoptosis was lost in NCCs transfected with Nrf2-siRNA, demonstrating that SFN protects against ethanol-induced apoptosis by the induction of Nrf2-mediated antioxidant response.
Figure 8.
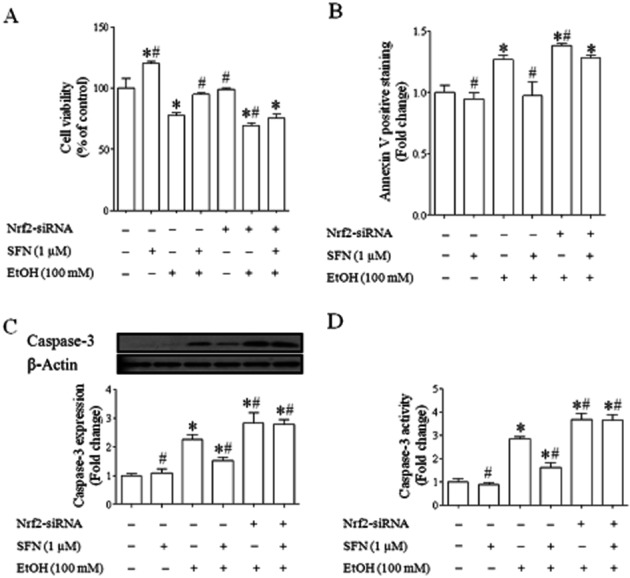
Ethanol-induced apoptosis in NCCs treated with SFN or transfected with Nrf2-siRNA. NCCs transfected with control or Nrf2-siRNA were treated with 100 mM ethanol or 1 μM SFN alone or in combination. Ethanol-induced apoptosis was determined by MTS assay (A), flow cytometry analysis of FITC Annexin V (B) and analysis of the cleavage and activity of caspase-3 (C, D). Data are expressed as percentage of control (A) or fold change over control (B, C, D) and represent the mean ± SEM of three separate experiments. *P < 0.05 versus control. #P < 0.05 versus EtOH.
Discussion
Recent studies have demonstrated that maternal ethanol exposure resulted in a moderate increase in the expression of Nrf2 and its downstream target detoxifying and antioxidant genes. In addition, maternal D3T (an Nrf2 inducer) pretreatment significantly decreased ethanol-induced ROS generation and apoptosis in mouse embryos (Dong et al., 2008). These results demonstrate that Nrf2 signalling is involved in the induction of an antioxidant response in ethanol-exposed mouse embryos. To test whether Nrf2-mediated antioxidant response can also be induced in ethanol-exposed NCCs, in this study, Nrf2 activation and the expression of Nrf2 downstream antioxidants were determined in NCCs exposed to 100 mM ethanol for 24 h. This ethanol concentration and exposure regimen was chosen because previous studies have shown that acute ethanol administration that produces peak maternal blood ethanol concentrations of 400–500 mg/100 mL (approximately 85–105 mM) is needed to induce apoptosis and major malformations that are characteristics of fetal alcohol syndrome in early mouse embryos (Sulik et al., 1981; Kotch and Sulik, 1992; Dunty et al., 2001). While this ethanol concentration is relatively high, it is not beyond that which can be observed in chronic alcoholics (Adachi et al., 1991). Studies have also shown that susceptibility to specific ethanol-induced defects is directly related to the timing of ethanol exposure and, therefore, acute ethanol exposures during critical periods of vulnerability for specific organs or cell types are desirable and essential for the studies regarding ethanol teratogenesis (Coles, 1994; Maier et al., 1996; Goodlett and Johnson, 1999). The results from this study have shown that in vitro exposure of NCCs to ethanol increased the protein expression of Nrf2 and its downstream antioxidants, SOD and catalase, confirming that Nrf2 activation and Nrf2-mediated antioxidant response can be induced in NCCs, a cell population that is sensitive to ethanol-induced apoptosis that contributes heavily to the subsequent abnormalities (Sulik et al., 1981; Kotch and Sulik, 1992; Cartwright and Smith, 1995).
Nrf2 signalling plays a significant role in protecting cells from endogenous and exogenous stresses (Kensler et al., 2007; Jin et al., 2012; Lu et al., 2012; Wu et al., 2012). It was reported that over-expression of Nrf2 protected cells from Fas-induced apoptosis (Kotlo et al., 2003). Activation of Nrf2 also protected against cell death in an in vitro model of ischaemia/reperfusion (Danilov et al., 2009). Studies have also shown that exposure to chromium (VI) and cadmium resulted in an increased ROS production and apoptosis in mouse embryonic fibroblast cells lacking Nrf2 (He et al., 2007; 2008). Loss of Nrf2 function has also been found to be associated with increased susceptibility to chemical carcinogenesis, inhalation particles, ovarian toxicants and autoimmune disease (Ramos-Gomez et al., 2001; Li et al., 2004; Hu et al., 2006; Ma et al., 2006). Consistent with these studies, we have shown that significantly greater increase in ethanol-induced ROS generation was observed in NCCs transfected with Nrf2-siRNA. In addition, suppression of Nrf2 increased the sensitivity of NCCs to ethanol-induced apoptosis. These findings suggest that Nrf2 signalling plays an important role in the susceptibility of NCCs to ethanol-induced apoptosis. This premise is further supported by the results from current studies that have shown that up-regulation of Nrf2 by SFN can increase the resistance of NCCs to ethanol-induced oxidative stress and apoptosis.
SFN is an isothiocyanate compound naturally present in high concentrations in several varieties of cruciferous vegetables such as sprouts of broccoli, cabbage and cauliflower (Soane et al., 2010). Studies have shown that systemically administered SFN after the onset of focal ischaemia decreased the total brain infarct volume (Zhao et al., 2007). SFN also protects cultured cortical neurons from glutamate toxicity (Kraft et al., 2004). In addition, SFN can prevent 6-hydroxydopamine-induced cytotoxicity in dopaminergic neurons (Han et al., 2007). More recently, studies have found that SFN has cardioprotective effects in neonatal cardiac myocytes by reducing ROS generation and increasing cell viability (Angeloni et al., 2009). Protective effects against post-ischaemia by SFN have also been reported in kidneys (Yoon et al., 2008), astrocytes (Danilov et al., 2009) and pancreatic islet cells (Solowiej et al., 2004). The current results show that treatment with SFN resulted in significant increases in Nrf2 protein expression and ARE promoter activity, and increases in the mRNA and protein expression of Nrf2 target genes, SOD1 and catalase, and their enzyme activities in ethanol-exposed NCCs. SFN treatment also significantly reduced ethanol-induced oxidative stress and apoptosis in NCCs. These findings clearly demonstrate that SFN can prevent oxidative stress and apoptosis in NCCs exposed to ethanol by activating Nrf2 signalling. However, while we have demonstrated that SFN treatment increased the protein expression and activities of SOD1 and catalase in this study, it is noteworthy that it does not imply that the protective effects of SFN are mediated only by SOD1 and catalase. Other antioxidants induced by Nrf2, which can induce a wide spectrum of antioxidants, may also contribute to SFN's anti-apoptotic actions. It has been reported that SFN can activate multiple Nrf2-induced genes (Hu et al., 2004). Our previous studies have also shown that a number of Nrf2 downstream antioxidant genes can be induced in early mouse embryos (Dong et al., 2008).
Of greater importance for the study is the observation that suppression of Nrf2 signalling abrogated the protective activity of SFN against ethanol-induced oxidative stress and apoptosis in NCCs. Although the major mechanism by which SFN protects cells is thought to be through Nrf2-mediated induction of antioxidant and phase 2 detoxification enzymes that elevates cell defence against oxidative damage, other mechanisms of action might also account for the anti-apoptotic effects of SFN. Indeed, it has been demonstrated that there are multiple mechanisms activated in response to SFN, including suppression of NF-κB (Moon et al., 2009), down-regulation of pro-apoptotic Bid and TNF-α expression, and up-regulation of anti-apoptotic Bcl-xL (Sun et al., 2011). However, the present study has shown that suppression of Nrf2 signalling abolished SFN-induced antioxidant response in NCCs. Knockdown of Nrf2 also abrogated the anti-apoptotic activity of SFN in ethanol-exposed NCCs. These results demonstrated that the protection of SFN against ethanol-induced oxidative stress and apoptosis in NCCs is mainly mediated by the induction of Nrf2 signalling.
Oxidative stress is an important contributing factor in the pathogenesis of ethanol-induced birth defects (Chen et al., 2004; Chen and Sulik, 1996; Dong et al., 2010; Kotch et al., 1995). This suggests that therapeutic strategies directed against ROS might be particularly valuable for the prevention of the ethanol-induced damage and malformations in early embryos, in which ROS are involved and the antioxidant capabilities are minimal. Therefore, either administration of exogenous antioxidants or up-regulation of endogenous antioxidant is an important therapeutic strategy to prevent ethanol-induced cell injury and subsequent malformation. The results of this study further support that up-regulation of endogenous antioxidants through the activation of Nrf2 signalling can prevent ethanol-induced oxidative stress and apoptosis. The potency of SFN in preventing ethanol-induced oxidative stress and apoptosis in NCCs, along with the fact that SFN is a natural micronutrient found in cruciferous vegetables, including cauliflower, broccoli and cabbage (Keum et al., 2005) makes SFN a promising therapeutic agent for human FASD.
In summary, the results of this study demonstrate that Nrf2-dependent antioxidant response can be induced in NCCs, an ethanol-sensitive cell population implicated in FASD, and that the loss of Nrf2 increased the sensitivity of NCCs to ethanol-induced oxidative stress and apoptosis. We have also shown that SFN-mediated Nrf2-dependent antioxidant response can prevent ethanol-induced oxidative stress and apoptosis in NCCs. These findings provide key insights into the roles of Nrf2 signalling in the induction of antioxidant response in ethanol-exposed embryonic cells and suggest that the induction of Nrf2-mediated antioxidant response by SFN is a promising safe and effective approach for the prevention of FASD.
Acknowledgments
We thank Drs Hubert Schorle and Jochen Maurer (University of Bonn Medical School, Germany) for providing JoMa neural crest cell line. We also thank Dr John D. Hayes (University of Dundee, UK) for providing pNQO1 ARE luciferase reporter. This work was supported by the National Institute of Health grant AA017446 and AA020265 (S.-Y.C) from the National Institute on Alcohol Abuse and Alcoholism.
Glossary
- ARE
antioxidant response element
- D3T
3H-1,2-dithiole-3-thione
- DCHFDA
2′,7′-dichlorodihydrofluoroscein diacetate
- FASD
fetal alcohol spectrum disorder
- MTS
3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H- tetrazolium salt
- NCC
neural crest cell
- NQO1
NAD(P)H:quinone oxidoreductase 1
- Nrf2
nuclear factor erythroid 2-related factor
- ROS
reactive oxygen species
- SFN
D-L-sulforaphane
- SOD
superoxide dismutase
Conflicts of interest
None.
References
- Adachi J, Mizoi Y, Fukunaga T, Ogawa Y, Ueno Y, Imamichi H. Degrees of alcohol intoxication in 117 hospitalized cases. J Stud Alcohol. 1991;52:448–453. doi: 10.15288/jsa.1991.52.448. [DOI] [PubMed] [Google Scholar]
- Angeloni C, Leoncini E, Malaguti M, Angelini S, Hrelia P, Hrelia S. Modulation of phase II enzymes by sulforaphane: implications for its cardioprotective potential. J Agric Food Chem. 2009;57:5615–5622. doi: 10.1021/jf900549c. [DOI] [PubMed] [Google Scholar]
- Burd L. Fetal alcohol syndrome. Am J Med Genet C Semin Med Genet. 2004;127C:1–2. doi: 10.1002/ajmg.c.30015. [DOI] [PubMed] [Google Scholar]
- Cartwright MM, Smith SM. Increased cell death and reduced neural crest cell numbers in ethanol-exposed embryos: partial basis for the fetal alcohol syndrome phenotype. Alcohol Clin Exp Res. 1995;19:378–386. doi: 10.1111/j.1530-0277.1995.tb01519.x. [DOI] [PubMed] [Google Scholar]
- Chen SY. Analysis of Nrf2-mediated transcriptional induction of antioxidant response in early embryos. Methods Mol Biol. 2012;889:277–290. doi: 10.1007/978-1-61779-867-2_17. [DOI] [PubMed] [Google Scholar]
- Chen SY, Sulik KK. Free radicals and ethanol-induced cytotoxicity in neural crest cells. Alcohol Clin Exp Res. 1996;20:1071–1076. doi: 10.1111/j.1530-0277.1996.tb01948.x. [DOI] [PubMed] [Google Scholar]
- Chen SY, Sulik KK. Iron-mediated free radical injury in ethanol-exposed mouse neural crest cells. J Pharmacol Exp Ther. 2000;294:134–140. [PubMed] [Google Scholar]
- Chen SY, Dehart DB, Sulik KK. Protection from ethanol-induced limb malformations by the superoxide dismutase/catalase mimetic, EUK-134. FASEB J. 2004;18:1234–1236. doi: 10.1096/fj.03-0850fje. [DOI] [PubMed] [Google Scholar]
- Cheung KL, Kong AN. Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer chemoprevention. AAPS J. 2010;12:87–97. doi: 10.1208/s12248-009-9162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles C. Critical periods for prenatal alcohol exposure: evidence from animal and human studies. Alcohol Health Res World. 1994;18:22–29. [PMC free article] [PubMed] [Google Scholar]
- Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, et al. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705–710. doi: 10.1038/nature08195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilov CA, Chandrasekaran K, Racz J, Soane L, Zielke C, Fiskum G. Sulforaphane protects astrocytes against oxidative stress and delayed death caused by oxygen and glucose deprivation. Glia. 2009;57:645–656. doi: 10.1002/glia.20793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Sulik KK, Chen SY. Nrf2-mediated transcriptional induction of antioxidant response in mouse embryos exposed to ethanol in vivo: implications for the prevention of fetal alcohol spectrum disorders. Antioxid Redox Signal. 2008;10:2023–2033. doi: 10.1089/ars.2007.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Sulik KK, Chen SY. The role of NOX enzymes in ethanol-induced oxidative stress and apoptosis in mouse embryos. Toxicol Lett. 2010;193:94–100. doi: 10.1016/j.toxlet.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Yan D, Chen SY. Stabilization of Nrf2 protein by D3T provides protection against ethanol-induced apoptosis in PC12 cells. PLoS ONE. 2011;6:e16845. doi: 10.1371/journal.pone.0016845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunty WC, Jr, Chen SY, Zucker RM, Dehart DB, Sulik KK. Selective vulnerability of embryonic cell populations to ethanol-induced apoptosis: implications for alcohol-related birth defects and neurodevelopmental disorder. Alcohol Clin Exp Res. 2001;25:1523–1535. [PubMed] [Google Scholar]
- Goodlett C, Johnson TB. Temporal windows of vulnerability to alcohol during the third trimester equivalent: why ‘knowing when’ matters. In: Hannigan JH, Spear LP, Spear NE, Goodlett CR, editors. Alcohol and Alcoholism: Effects on Brain and Development. Hillsdale, NJ: Lawrence Erlbaum Associates; 1999. pp. 59–91. [Google Scholar]
- Hall BK. The neural crest and neural crest cells: discovery and significance for theories of embryonic organization. J Biosci. 2008;33:781–793. doi: 10.1007/s12038-008-0098-4. [DOI] [PubMed] [Google Scholar]
- Han JM, Lee YJ, Lee SY, Kim EM, Moon Y, Kim HW, et al. Protective effect of sulforaphane against dopaminergic cell death. J Pharmacol Exp Ther. 2007;321:249–256. doi: 10.1124/jpet.106.110866. [DOI] [PubMed] [Google Scholar]
- He X, Lin GX, Chen MG, Zhang JX, Ma Q. Protection against chromium (VI)-induced oxidative stress and apoptosis by Nrf2. Recruiting Nrf2 into the nucleus and disrupting the nuclear Nrf2/Keap1 association. Toxicol Sci. 2007;98:298–309. doi: 10.1093/toxsci/kfm081. [DOI] [PubMed] [Google Scholar]
- He X, Chen MG, Ma Q. Activation of Nrf2 in defense against cadmium-induced oxidative stress. Chem Res Toxicol. 2008;21:1375–1383. doi: 10.1021/tx800019a. [DOI] [PubMed] [Google Scholar]
- Henderson GI, Chen JJ, Schenker S. Ethanol, oxidative stress, reactive aldehydes, and the fetus. Front Biosci. 1999;4:D541–D550. doi: 10.2741/henderson. [DOI] [PubMed] [Google Scholar]
- Hong F, Freeman ML, Liebler DC. Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem Res Toxicol. 2005;18:1917–1926. doi: 10.1021/tx0502138. [DOI] [PubMed] [Google Scholar]
- Hu R, Hebbar V, Kim BR, Chen C, Winnik B, Buckley B, et al. In vivo pharmacokinetics and regulation of gene expression profiles by isothiocyanate sulforaphane in the rat. J Pharmacol Exp Ther. 2004;310:263–271. doi: 10.1124/jpet.103.064261. [DOI] [PubMed] [Google Scholar]
- Hu R, Xu C, Shen G, Jain MR, Khor TO, Gopalkrishnan A, et al. Gene expression profiles induced by cancer chemopreventive isothiocyanate sulforaphane in the liver of C57BL/6J mice and C57BL/6J/Nrf2 (-/-) mice. Cancer Lett. 2006;243:170–192. doi: 10.1016/j.canlet.2005.11.050. [DOI] [PubMed] [Google Scholar]
- Jin M, Kumar A, Kumar S. Ethanol-mediated regulation of cytochrome P450 2A6 expression in monocytes: role of oxidative stress-mediated PKC/MEK/Nrf2 pathway. Plos ONE. 2012;7:e35505. doi: 10.1371/journal.pone.0035505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Keum YS, Jeong WS, Kong AN. Chemopreventive functions of isothiocyanates. Drug News Perspect. 2005;18:445–451. doi: 10.1358/dnp.2005.18.7.939350. [DOI] [PubMed] [Google Scholar]
- Kotch LE, Sulik KK. Experimental fetal alcohol syndrome: proposed pathogenic basis for a variety of associated facial and brain anomalies. Am J Med Genet. 1992;44:168–176. doi: 10.1002/ajmg.1320440210. [DOI] [PubMed] [Google Scholar]
- Kotch LE, Chen SY, Sulik KK. Ethanol-induced teratogenesis: free radical damage as a possible mechanism. Teratology. 1995;52:128–136. doi: 10.1002/tera.1420520304. [DOI] [PubMed] [Google Scholar]
- Kotlo KU, Yehiely F, Efimova E, Harasty H, Hesabi B, Shchors K, et al. Nrf2 is an inhibitor of the Fas pathway as identified by Achilles' heel method, a new function-based approach to gene identification in human cells. Oncogene. 2003;22:797–806. doi: 10.1038/sj.onc.1206077. [DOI] [PubMed] [Google Scholar]
- Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci. 2004;24:1101–1112. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Singh CK, Lavoie HA, Dipette DJ, Singh US. Resveratrol restores Nrf2 level and prevents ethanol-induced toxic effects in the cerebellum of a rodent model of fetal alcohol spectrum disorders. Mol Pharmacol. 2011;80:446–457. doi: 10.1124/mol.111.071126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Stein TD, Johnson JA. Genetic dissection of systemic autoimmune disease in Nrf2-deficient mice. Physiol Genomics. 2004;18:261–272. doi: 10.1152/physiolgenomics.00209.2003. [DOI] [PubMed] [Google Scholar]
- Li W, Kong AN. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog. 2009;48:91–104. doi: 10.1002/mc.20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhang XH, Cederbaum AI. Ethanol induction of CYP2A5: role of CYP2E1-ROS-Nrf2 pathway. Toxicol Sci. 2012;128:427–438. doi: 10.1093/toxsci/kfs164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Battelli L, Hubbs AF. Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am J Pathol. 2006;168:1960–1974. doi: 10.2353/ajpath.2006.051113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SE, Chen W-JA, West JR. The effects of timing and duration of alcohol exposure on development of the fetal brain. In: Abel EL, editor. Fetal Alcohol Syndrome: From Mechanism to Prevention. Boca Raton, FL: CRC Press; 1996. pp. 27–50. [Google Scholar]
- Maurer J, Fuchs S, Jager R, Kurz B, Sommer L, Schorle H. Establishment and controlled differentiation of neural crest stem cell lines using conditional transgenesis. Differentiation. 2007;75:580–591. doi: 10.1111/j.1432-0436.2007.00164.x. [DOI] [PubMed] [Google Scholar]
- Moon DO, Kim MO, Kang SH, Choi YH, Kim GY. Sulforaphane suppresses TNF-alpha-mediated activation of NF-kappaB and induces apoptosis through activation of reactive oxygen species-dependent caspase-3. Cancer Lett. 2009;274:132–142. doi: 10.1016/j.canlet.2008.09.013. [DOI] [PubMed] [Google Scholar]
- Murphy DA, Diaz B, Bromann PA, Tsai JH, Kawakami Y, Maurer J, et al. A Src-Tks5 pathway is required for neural crest cell migration during embryonic development. PLoS ONE. 2011;6:e22499. doi: 10.1371/journal.pone.0022499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan M, Mahimainathan L, Rathinam ML, Riar AK, Henderson GI. Overexpression of Nrf2 protects cerebral cortical neurons from ethanol-induced apoptotic death. Mol Pharmacol. 2011;80:988–999. doi: 10.1124/mol.111.073262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nioi P, McMahon M, Itoh K, Yamamoto M, Hayes JD. Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence. Biochem J. 2003;374:337–348. doi: 10.1042/BJ20030754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuji M, Katsuoka F, Kobayashi A, Aburatani H, Hayes JD, Yamamoto M. Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J Biol Chem. 2008;283:33554–33562. doi: 10.1074/jbc.M804597200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001;98:3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehy NT, Cordes KR, White MP, Ivey KN, Srivastava D. The neural crest-enriched microRNA miR-452 regulates epithelial-mesenchymal signaling in the first pharyngeal arch. Development. 2010;137:4307–4316. doi: 10.1242/dev.052647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM. Alcohol-induced cell death in the embryo. Alcohol Health Res World. 1997;21:287–297. [PMC free article] [PubMed] [Google Scholar]
- Soane L, Li Dai W, Fiskum G, Bambrick LL. Sulforaphane protects immature hippocampal neurons against death caused by exposure to hemin or to oxygen and glucose deprivation. J Neurosci Res. 2010;88:1355–1363. doi: 10.1002/jnr.22307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solowiej E, Solowiej J, Kasprzycka-Guttman T, Rowinski W. Application of sulforaphane – does it lead to improvement of islet graft survival after warm and/or cold ischemia. Ann Transplant. 2004;9:68–71. [PubMed] [Google Scholar]
- Sulik KK, Johnston MC, Webb MA. Fetal alcohol syndrome: embryogenesis in a mouse model. Science. 1981;214:936–938. doi: 10.1126/science.6795717. [DOI] [PubMed] [Google Scholar]
- Sun X, Mi L, Liu J, Song L, Chung FL, Gan N. Sulforaphane prevents microcystin-LR-induced oxidative damage and apoptosis in BALB/c mice. Toxicol Appl Pharmacol. 2011;255:9–17. doi: 10.1016/j.taap.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng L, Labosky PA. Neural crest stem cells. Adv Exp Med Biol. 2006;589:206–212. doi: 10.1007/978-0-387-46954-6_13. [DOI] [PubMed] [Google Scholar]
- Wang F, Shan Y. Sulforaphane retards the growth of UM-UC-3 xenographs, induces apoptosis, and reduces survivin in athymic mice. Nutr Res. 2012;32:374–380. doi: 10.1016/j.nutres.2012.03.014. [DOI] [PubMed] [Google Scholar]
- Wu KC, Liu J, Klaassen CD. Role of Nrf2 in preventing ethanol-induced oxidative stress and lipid accumulation. Toxicol Appl Pharmacol. 2012;262:321–329. doi: 10.1016/j.taap.2012.05.010. [DOI] [PubMed] [Google Scholar]
- Yan D, Dong J, Sulik KK, Chen SY. Induction of the Nrf2-driven antioxidant response by tert-butylhydroquinone prevents ethanol-induced apoptosis in cranial neural crest cells. Biochem Pharmacol. 2010;80:144–149. doi: 10.1016/j.bcp.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HY, Kang NI, Lee HK, Jang KY, Park JW, Park BH. Sulforaphane protects kidneys against ischemia-reperfusion injury through induction of the Nrf2-dependent phase 2 enzyme. Biochem Pharmacol. 2008;75:2214–2223. doi: 10.1016/j.bcp.2008.02.029. [DOI] [PubMed] [Google Scholar]
- Zhao J, Moore AN, Redell JB, Dash PK. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J Neurosci. 2007;27:10240–10248. doi: 10.1523/JNEUROSCI.1683-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]