

Abstract
Sporadic Creutzfeldt-Jakob disease (sCJD) is a rapidly progressive dementia (RPD) that can be difficult to identify ante-mortem, with definitive diagnosis requiring tissue confirmation. We describe the clinical, magnetic resonance imaging (MRI), cerebrospinal fluid (CSF), and electroencephalogram (EEG) measures of a small cohort of 30 patients evaluated for RPD. Clinical and diagnostic measures were cross-sectionally obtained from 17 sCJD patients (15 definite, 2 probable), 13 non-prion rapidly progressive dementia patients (npRPD), and 18 unimpaired controls. In a subset of patients (9 sCJD and 9 npRPD) diffusion tensor imaging (DTI) measures [fractional anisotropy (FA), mean diffusivity (MD), axial diffusivity (AD), and radial diffusivity (RD)] were also obtained for the caudate, corpus callosum, posterior limb of the internal capsule, pulvinar, precuneus, and frontal lobe. Differences among groups were assessed by an analysis of variance. Compared to npRPD individuals, sCJD patients had cerebellar dysfunction, significantly higher CSF tau, “positive” CSF 14-3-3, and hyperintensities on diffusion weighted imaging (DWI) that met previously established imaging criteria for sCJD. EEG changes were similar for the two groups. In addition, sCJD patients had significant decreases in DTI measures (MD, AD, RD but not FA) within the caudate and pulvinar compared to either npRPD patients or unimpaired controls. Our results confirm that CSF abnormalities and MRI (especially DWI) can assist in distinguishing sCJD patients from npRPD patients. Future longitudinal studies using multiple measures (including CSF and MRI) are needed for evaluating pathological changes seen in sCJD patients.
Keywords: Creutzfeldt-Jakob disease, diffusion magnetic resonance imaging, cerebrospinal fluid
INTRODUCTION
Creutzfeldt-Jakob Disease (CJD) is a fatal rapidly progressive neurodegenerative disease (RPD) [1]. Most CJD cases are sporadic (sCJD) in nature [2]. The pathogenesis of sCJD remains poorly characterized but is believed to be due to a conformational change in abnormal prion protein (PrPSc) that causes widespread neuronal degeneration [3].
Definitive diagnosis of sCJD requires tissue confirmation via biopsy or autopsy. However, these methods are often impractical and, in some instances, problematic due to the collection of suboptimal specimens and increased exposure risk to healthcare personnel [4]. Non-invasive diagnostic tests currently recommended for differentiating sCJD from non-prion rapidly progressive dementia (npRPD) include magnetic resonance imaging (MRI), electroencephalogram (EEG) and cerebrospinal fluid (CSF) measurements of 14-3-3 and tau. The sensitivity and specificity of each of these tests varies and changes depending on when they are obtained during the duration of the illness [5,3,6-8]. Additional non-invasive pre-mortem diagnostic methods may assist in identification of sCJD [9].
Characteristic changes within grey and white matter using MRI [i.e. fluid level attenuated inversion recovery (FLAIR) and diffusion weighted imaging (DWI)] can assist in the diagnosis of sCJD [6,8]. In addition, DTI measures the anisotropic diffusion of water molecules in brain [10]. Movement of water is usually greater along the length of the fiber (longitudinal direction) than perpendicular to it (radial or transverse direction), as myelin may restrict flow. An ellipsoid-shaped tensor is commonly used to model diffusion within any given voxel. Diffusion along the major axis of this ellipsoid is assumed to reflect diffusivity parallel to the white matter (WM) tract and is termed axial diffusivity (AD). The two minor axes can be averaged to determine radial diffusivity (RD), which reflects diffusion perpendicular to the major axis of the WM tracts. Mean diffusivity (MD) reflects the average diffusion from all three directions. Finally, fractional anisotropy (FA) represents the ratio of axial to radial diffusivity and provides a measure of the general shape of the ellipsoid [11].
We describe the clinical signs, MRI (including FLAIR, DWI, and DTI) characteristics, CSF values (including 14-3-3 and tau), and EEG measurements from 30 patients who presented to a single tertiary care medical center for RPD. In our cohort, we observed that CSF and MRI (both DWI and DTI) measures assisted in distinguishing sCJD from npRPD and unimpaired controls.
METHODS
Patient Collection
Clinical history and diagnostic studies (CSF, EEG, MRI, and laboratory values) were collected from RPD patients evaluated between the years 2005-2010. During this interval a total of 30 patients were admitted with the diagnosis of RPD at our institution. Analysis of all patient records was approved by the Institutional Review Board at our institution.
All RPD patients had a detailed neurological exam and met the following criteria for entry: 1) rapid decline in cognition (less than 2 years in duration), 2) additional evaluations excluding alternative diagnoses, and 3) at least one additional clinical symptom (myoclonus, pyramidal/extrapyramidal signs, visual disturbances, cerebellar abnormalities, higher cortical dysfunction, or akinetic mutism). Cerebellar signs included central nystagmus, truncal or appendicular ataxia, or dysdiadokinesia. Higher cortical abnormalities included aphasia, apraxia, acalculia, or neglect. A diagnosis of probable sCJD was made using combined previously established guidelines from the MRI-CJD Consortium [8] and UCSF [12,6]. A diagnosis of definite sCJD required the classical histopathologic triad of spongiform change, neuronal loss, and astrogliosis on tissue examination.
Using these criteria we identified 17 patients with sCJD (15 patients with definite sCJD and 2 patients with probable sCJD) (see Figure 1). Thirteen patients were defined as npRPD (4 patients having autopsies confirming alternative diagnoses). Patients with npRPD were excluded from probable sCJD diagnosis because either their survival was outside the typical range of sCJD subjects (> 3 years), they had definitive (e.g. pathology, serology, or genetic) evidence of alternative diagnosis, and/or they improved with (or without) treatment [6]. This npRPD group included the following diagnoses: Alzheimer’s disease (n=4), malignancy (i.e. paraneoplastic) (n=3), dementia with Lewy bodies (n=3), vascular dementia (n=1), vasculitis (n=1), and psychiatric (n=1). All patients with neurodegenerative disease met established diagnostic clinical criteria [13-15]. For the subset of RPD patients that had DTI, an additional unimpaired control group was recruited. This group was matched by age and sex to sCJD and npRPD patients and had similar MRI studies (see imaging protocol below) but not CSF analyses.
Figure 1. Classification of patients.
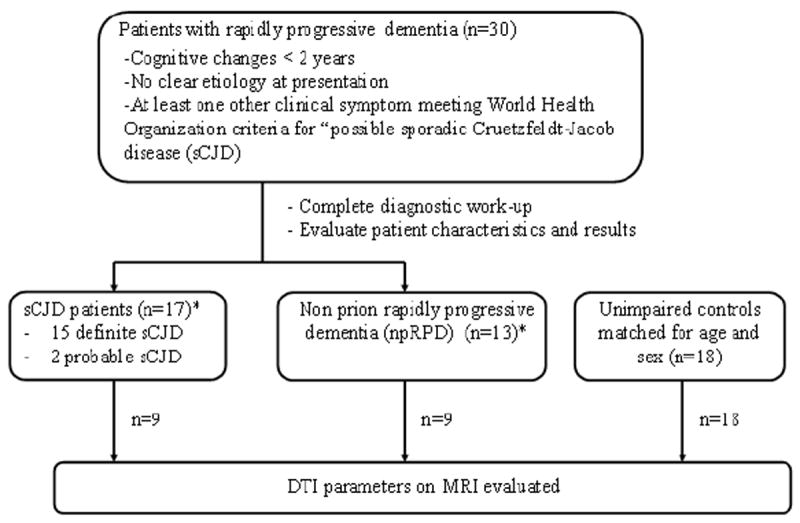
Diagram for classification of subjects as sporadic Creutzfeldt-Jakob disease (sCJD), non-prion rapidly progressive dementia (npRPD), or unimpaired control.
* = 15 of 17 sCJD patients and 4 of 13 RPD controls had tissue confirmation of their diagnoses
Brain autopsies of all definite sCJD patients were performed by the National Prion Disease Pathology Surveillance Center (NPDPSC) at Case Western Reserve University. At the NPDPSC, immunohistological and Western blot analysis was assessed for PrPSc (PrP 27-30) from brain autopsy tissue using a monoclonal antibody 3F4 (to residues 109–112) [16,17,1]. Molecular classification was performed to determine the codon 129 polymorphism of the prion gene (MM, MV, or VV) and pathologic prion protein isotype (PrPSc 1 or 2).
CSF and EEG Studies
All CSF samples were also sent to the NPDPSC for evaluation. CSF tau and presence of CSF 14-3-3 were assessed using previously described methods [18]. EEG records were scored as positive or characteristic of sCJD if they fulfilled a validated criteria including: sustained periodic sharp wave complexes (PSWC) with a variability of <500 ms or periodic generalized complexes (lasting 100–600 ms) that were bi- or tri-phasic in morphology [19].
Imaging Protocol
Standard MRI Studies
All participants (npRPD, sCJD, and unimpaired controls) were imaged on 1.5 T Siemens scanners. For all subjects, sequences performed included a standard clinical T1, T2, fluid attenuated inversion recovery (FLAIR), and DWI. All images were reviewed to ensure that excessive movement did not occur during scanning. DWI and FLAIR images were independently evaluated by a board-certified neuroradiologist (D.R.) blinded to the diagnosis. This rater used previously defined criteria [12,6,8] to evaluate if DWI changes were absent or present within cortical and/or subcortical areas. For the purposes of comparing criteria analyses via sensitivity/specificity analyses, scores of “positive” and “negative” were classified according using the Zerr and colleagues criteria [8] and scores of “probably sCJD” or “definitely sCJD” were considered “positive” while “probably not sCJD” and “definitely not sCJD” were considered “negative” when using the Vitali and colleagues criteria[6].
DTI studies
A subgroup of RPD patients (9 CJD, 9 npRPD) and 18 unimpaired controls had DTI performed using 60 contiguous 4-mm axial sections with the following parameters [12 directions, 3 b values, bmax = 1400 msec, field of view (FOV)= 230, time to repeat (TR) = 3,200 milliseconds, time to echo (TE) =107 milliseconds, parallel imaging factor = 2, and 2.5 mm × 2.5 mm × 2.7mm isotropic voxels] [20]. Imaging time was approximately 6 minutes in duration. DTI data were then transferred to a workstation (Leonardo, Siemens) for further analysis. Using the Neuro3D package (Siemens) diffusion scans were co-registered to the T1 and FLAIR sequences. Regions of interest (ROI, 10 mm3 in diameter) were manually drawn by a blinded reviewer (E.P.) on the anatomical scans and subsequently verified on the diffusion scans in order to ensure adequate placement (Figure 2). ROIs that included both cortical and subcortical areas were selected based on previous possible involvement by CJD [6]. These regions included: caudate, corpus callosum, posterior limb of the internal capsule (PLIC), pulvinar, precuneus, and frontal lobe. The left and right hemispheres of each of the ROIs were averaged for each patient to ensure adequate sampling.
Figure 2. Regions of interest for diffusion imaging analysis.
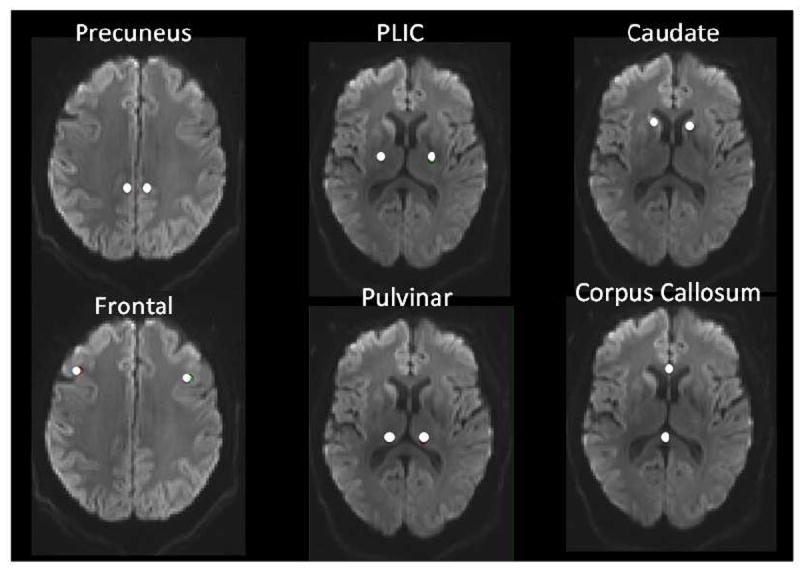
Representative placement of six ROIs on diffusion images of a definite sCJD patient.
Statistical Analysis
Diagnostic odds ratios with 95% confidence intervals were determined for each of the variables (including clinical symptoms, laboratory data, and MRI values) using two-tailed Fisher’s exact tests. Both sensitivity and specificity values were subsequently calculated for each variable. A two-tailed Student’s t-test was performed for comparing CSF tau values between sCJD and npRPD patients as CSF was not collected from unimpaired controls. For the DTI analysis, the p-values for each of the ROIs from the analysis of variance (ANOVA) for the three group comparisons were adjusted to account for the testing of the multiple regions using Bonferroni correction. For all analyses standard error was calculated. GraphPad Prism software (Version 3.0, La Jolla, CA) was used for all analyses.
RESULTS
Patient characteristics and clinical findings
All 17 sCJD patients resided within a 200-mile radius. The estimated population of this entire catchment area is ~ 3–3.5 million people (2010 U.S. Census Data). While formal modeling (similar to that performed by Klug et al [21]) was not pursued, the calculated incidence of sCJD is approximately 0.81 to 0.94 per million per year for the catchment area based on current population values.
No significant differences in demographic variables existed between sCJD and npRPD patients (Table 1). Of note, one of the definitive sCJD patients was diagnosed with a new form of sporadic prion disease, variable protease-sensitive prionopathy (VPSPr) [22], but did not have an MRI performed. On clinical exam, sCJD patients were more likely to have cerebellar signs and myoclonus than npRPD patients. The most commonly observed cerebellar abnormality in sCJD patients was truncal ataxia causing gait instability.
Table 1.
| sCJD (n=17) | RPD (n=13)1 | OR | 95% CI | p | ||
|---|---|---|---|---|---|---|
| Male, n (%) | 8 (47) | 7 (54) | - | - | 0.99 | |
| Age at diagnosis, years (SEM) | 63.5 (1.8) | 68.2 (3.6) | - | - | 0.23 | |
| Age Range, years | 52.3-76 | 40-90.5 | - | - | - | |
| Onset to diagnosis, days (SEM) | 255 (83) | 144 (24) | - | - | 0.25 | |
| Onset to DTI scan, days (SEM) | 115 (21) | 128 (27) | - | - | 0.70 | |
| Clinical Data n (%) | ||||||
| Dementia | 17 (100) | 13 (100) | - | - | ||
| Cerebellar | 15 (88) | 2 (15) | 41 | 5.5 -311 | ||
| Pyr/EPyr | 14 (82) | 9 (69) | 2.1 | 0.4 – 11 | ||
| Akinetic Mutism | 9 (53) | 3 (23) | 3.8 | 0.8 – 17 | ||
| Visual | 3 (18) | 2 (15) | 1.2 | 0.2 -7 | ||
| Myoclonus | 13 (76) | 5 (38) | 5.2 | 1.1 - 24 | ||
| High Cortical | 8 (47) | 3 (23) | 3.0 | 0.6 -14 | ||
| Diagnostic Testing | Sensitivity (95% CI) | Specificity (95% CI) | ||||
| EEG PSWC (%) | 7/17 (41) | 6/13 (46) | 0.8 | 0.2 – 3.4 | 41 (27 - 56) | 54 (35 - 73) |
| CSF 14-3-3 “+” (%) | 10/15 (67) | 3/12 (25) | 6 | 1.2 - 31 | 67 (50 -79) | 75 (54 -90) |
| CSF T-tau “+” (%) | 12/15 (80) | 0/12 (0) | Inf | 9.2 - Inf | 80 (66 -80) | 100 (83 -100) |
| MRI according to Vitali and colleagues (2011) and Zerr and colleagues (2009) (%) | 12/14 (86) | 1/13 (8) | 72 | 4 - 2903 | 86 (65 – 93) | 92 (70 -100) |
Please see the Methods section for diagnoses
Abbreviations – SEM = standard error of the mean, Pyr/EPyr = pyramidal/extrapyramidal, T-tau = total tau, “+” = positive value per National Prion Disease Pathology Surveillance Center Report, 95 % CI = 95% Confidence Interval, Inf = Infinity, PSWC = periodic sharp wave complexes
Diagnostic testing
The presence of periodic sharp wave complexes on EEG was similar for both sCJD and npRPD participants. The presence of a “positive” CSF 14-3-3 was observed with a greater frequency in sCJD subjects compared to npRPD. However, this test was neither sensitive nor specific for distinguishing sCJD from npRPD when compared to CSF tau. Mean CSF tau (abnormal defined as >1,200 pg/ml) was significantly elevated in sCJD patients (5,239 ± 1,801 pg/ml (range 440 – 28,342)) compared to npRPD subjects (486 ± 89 pg/ml (range 70 – 1,135)). No correlation existed between CSF tau values and the onset of symptoms for sCJD patients (p=0.28) (See Supplementary Figure 1).
Genetic studies were performed in eighty-eight percent (15/17) of sCJD subjects (Supplemental Table 1). Most definite sCJD patients were MM 1 (n=7) with VV 1-2 (n=3) being the second most common form. The diagnostic characteristics of our group were similar to a previous report [23].
Three definite sCJD patients did not have MRI scans due to either contraindications (presence of a pacemaker (n=2)) or lack of availability (scan performed outside our institution and not available for review (n=1)). Of the remaining sCJD cohort (n=14), both previously defined MRI scoring criteria [6,8], yielded identical results. The sensitivity and specificity of DWI were 86 and 92, respectively (Table 1). Observed results are consistent with values established for DWI by previous studies with larger cohorts of sCJD patients [6,8]. In particular, a single patient in the npRPD group was scored as “positive” using either MRI criteria [6,8]. This individual did not undergo DTI analysis and did not have CSF findings consistent with sCJD. The patient was diagnosed with a paraneoplastic epileptic encephalopathy. DWI signal abnormalities were observed only within the cortex and were not simply a result of “T2 shine through.”
DTI analysis
DTI imaging was performed in only a subset of RPD patients. The sCJD patients that had DTI (n=9) were representative of the rest of the group. MD parameters were reduced for sCJD patients compared to either npRPD or matched unimpaired controls (p=0.01). Even after correction for multiple comparisons, MDs were significantly decreased within the caudate and pulvinar regions for sCJD subjects (576 ± 29 (range 89-736) and 649 ± 34 (range 67-805), respectively) compared to the npRPD patients (657±27 (range 532 – 867) and 802±41 (range 663-1,254), respectively) and unimpaired controls (727±19 (range 642-1,048) and 749 ± 11 (range 687 – 843), see Figure 3). Observed decreases in MD were primarily due to a reduction in both AD and RD within these two regions. MD parameters were not significantly different in other regions after correcting for multiple comparisons. FA measures were similar among the three groups for each of the ROIs (Figure 3).
Figure 3. Diffusion weighted imaging (DWI) measurements with ROIs for patients.
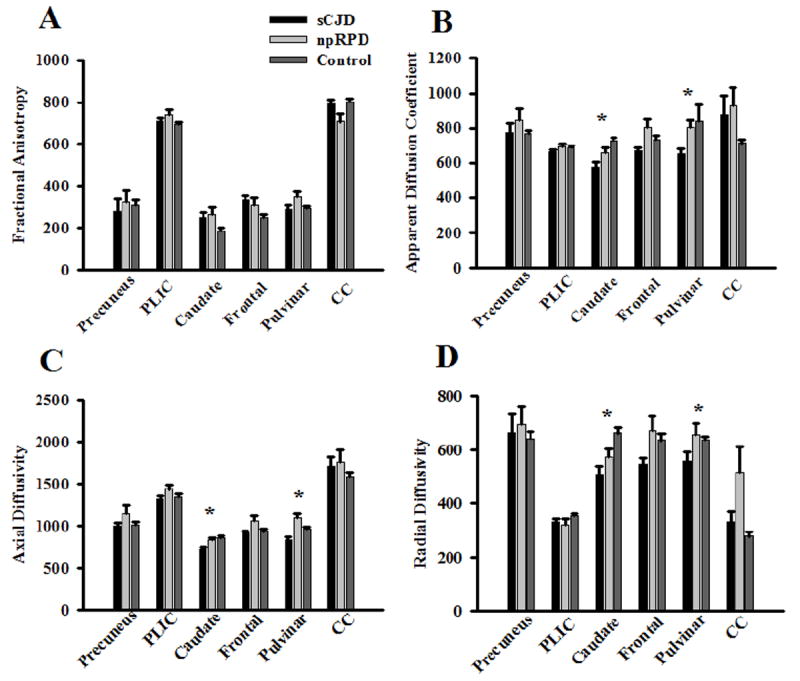
Fractional anisotropy (FA; panel A), mean diffusivity (MD; panel B), axial diffusivity (AD; panel C), and radial diffusivity (RD; panel D) values for the 6 ROIs in sCJD, npRPD, and unimpaired control subjects. sCJD patients had significant reductions in MD (primarily due to changes in both AD and RD) in the caudate and pulvinar compared to npRPD and unimpaired controls. No significant changes in FA were seen for the ROIs among the three groups. *= p < 0.05.
DISCUSSION
In our case-control study, we analyzed the sensitivity and specificity of clinical symptoms, laboratory measures, and MRI values for discriminating sCJD from npRPD patients. Compared to npRPD patients, sCJD patients were more likely to have myoclonus and cerebellar signs on exam, significantly higher CSF tau, positive CSF 14-3-3, and restricted diffusion using DWI. Typical EEG findings, a diagnostic feature often used in many CJD diagnostic criteria, failed to distinguish sCJD from npRPD. Our results are consistent with recent reports questioning the diagnostic utility of EEG [3,19]. Additionally, a smaller cohort of sCJD patients had significant decreases in MD but not FA within the caudate and pulvinar compared to RPD patients and unimpaired controls. Our results confirm that specific clinical signs, CSF studies, and MRI could assist differentiating sCJD patients from npRPD patients.
The overall prevalence observed at our site closely approximates the widely accepted incidence of approximately 1 per million per year [21]. Our catchment area was quite diverse and incorporated both primary and tertiary hospitals that have referred patients to our institution for further evaluation.
In regards to clinical signs, cerebellar ataxia and myoclonus were sensitive and specific in distinguishing sCJD from npRPD [24]. These results are in good agreement with a recent CJD neurological status scale developed to assist in distinguishing sCJD patients [25]. While sCJD may sometimes have a predilection for the cerebellum, MRI methods such as DWI may be insensitive to abnormalities in these regions [26].
A thorough analysis of the diagnostic test results of our cohort yielded findings that are consistent with previous larger studies [27,12,16,8]. In particular, controversy remains concerning the role of CSF 14-3-3. The sensitivity and specificity of this test ranges for various studies from 48 – 86% and 66 - 74% respectively [18,27,12]. In our small cohort, the presence of a “positive” CSF 14-3-3 was less robust than CSF tau in distinguishing sCJD from npRPD patients. Results from our cohort also highlight an increasing role for MRI based imaging methods, specifically DWI in distinguishing sCJD from npRPD. We observed that DWI hyperintensities using previously defined criteria [6,8] were both sensitive and specific in differentiating sCJD from npRPD or unimpaired controls. DWI had higher accuracy than clinical symptoms, EEG, or certain CSF measures (14-3-3) [6,8].
Significant advances have been made in DTI over the past decade. This technique has been previously used to study pathological changes seen in other neurodegenerative diseases such as Alzheimer’s disease and dementia with Lewy bodies [28,29]. This method has also been utilized in a cohort of sCJD patients that had cerebellar signs due to the E200K mutation [26]. Family members with no dementia served as controls. An elevation in apparent diffusion coefficient was seen in the cerebellum of sCJD patients compared to controls. However, these authors did not report findings using other DTI parameters. This may in part be due to the fact that DTI results were primarily used for generating DWI measures. In contrast, our study evaluated a more representative cohort of sCJD patients and included not only healthy controls but also npRPD patients. In a subgroup of subjects we assessed changes in multiple DTI parameters. While we do not propose for DTI to replace DWI for assisting in the diagnosis of sCJD, our results suggest DTI may provide additional information concerning pathological changes seen in sCJD.
Observed decreases in MD in matter tracts within the caudate and pulvinar of sCJD patients are similar to previous findings using DWI [30-33]. These previous studies have observed a correlation between prion protein deposits and areas of diffusion restriction on DWI. The exact etiology as to why differences in MD were observed within the caudate and pulvinar remains unknown. One possible mechanism may be the presence of inflammatory cells or cytotoxic edema due to neuronal cell loss [33,3]. Reductions in AD could represent axonal compromise, while alterations in RD may indicate myelin degradation with sCJD. We hypothesize the presence of spongiform changes due to membrane-bound intra-neuronal vacuoles [34] could impair water diffusion and lead to decreased diffusion of water molecules in all directions and a relative preservation of FA [35,36].
The differential for rapidly progressive dementia can be extensive with MRI imaging assisting in the diagnosis. In particular, observed DTI changes for sCJD were different than those seen for stroke, which is sometimes considered in the radiological differential. Stroke is often characterized by a reduction in MD and an increase in FA. Pathological changes due to stroke cause neuronal damage that eliminates directional diffusion [37]. In contrast, we observed a decrease in MD with preservation in FA for sCJD.
The current study has several limitations. First, our results are primarily confirmatory. Observed differences in some measures were seen at the group level and it remains unknown if each of the methods can be reliably and practically applied to an individual patient. Future longitudinal MRI-based imaging studies of sCJD patients are needed to determine temporal changes associated with disease progression. Second, a relatively small number of patients were evaluated at a single institution. Larger studies involving multiple centers are needed to compare CSF and MRI measures. The small sample size also precluded our ability to determine whether changes in DTI values were specific for particular codon 129 genotypes or PrPSc protein type. Third, our study focused on a limited number of regions. Future studies that include additional white matter regions would allow us to determine if observed changes are specific to certain subcortical gray matter regions or if they occur throughout the brain. MD values within the caudate and pulvinar can be more variable due to partial volume effects and eddy current distortion [38]. Fourth, recently developed blood and CSF tests have been proposed to assist in the diagnosis of sCJD [39,40]. However, both of these methods are not commercially available and require additional confirmatory analyses. Additionally if these tests (similar to the CSF 14-3-3 and tau analyses) are to be performed at commercial laboratories, a significant lag time exists between collection and reporting of results. In contrast, the MRI modalities utilized in this study are relatively easy to perform with results almost immediately available. It is important to recognize that we do not suggest that any one test is sufficient for the diagnosis of sCJD and that the best approach is most likely multimodal.
In summary, a combination of clinical and diagnostic tests (including CSF and MRI) is useful in diagnosing sCJD. DWI and DTI may be helpful for assessing pathological changes associated with sCJD.
Supplementary Material
Acknowledgments
The authors would like to thank current and former residents at our institution for their assistance in patient identification and collection of data. We would also like to thank many of the faculty at our institution for their assistance and guidance. Finally, we would also like to thank all the families of patients for their assistance and efforts.
FUNDING SOURCE
Funding support came from NIMH (1K23MH081786) (BMA), NINR (1R01NR012907) (BMA), Dana Foundation (DF10052) (BMA), Institute of Clinical and Translational Sciences at Washington University in St. Louis (BMA), and Missouri Just-In-Time Core Usage Funding Program (BMA).
Footnotes
AUTHOR CONTRIBUTION:
Drs. Wang, Bucelli, Patrick, Alvarez, Lim, DeBruin, Sharma, Ward, and Ances were involved in the recruitment of participants. Drs. Wang, Bucelli, Patrick, Rajderkar, Benzinger, and Ances were involved in the analysis of data. Drs. Wang, Bucelli, Patrick, Alvarez, Lim, DeBruin, Sharma, Dahiya, Benzinger, Ward, and Ances were involved in manuscript preparation.
FINANCIAL DISCLOSURE INFORMATION:
Drs. Wang, Patrick, Bucelli, Alvarez, Lim, DeBruin, Sharma, Dahiya, Benzinger, and Ward report no disclosures. Dr. Ances is currently participating in a clinical trial of anti-dementia drugs sponsored by Pfizer. He also serves on the scientific advisory board for Lilly Pharmaceuticals and Medscape.
References
- 1.Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti B, Gambetti P, Kretzschmar H. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46(2):224–233. [PubMed] [Google Scholar]
- 2.Kovacs GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, van Duijn C, Collins SJ, Boyd A, Giulivi A, Coulthart M, Delasnerie-Laupretre N, Brandel JP, Zerr I, Kretzschmar HA, de Pedro-Cuesta J, Calero-Lara M, Glatzel M, Aguzzi A, Bishop M, Knight R, Belay G, Will R, Mitrova E. Genetic prion disease: the EUROCJD experience. Human genetics. 2005;118(2):166–174. doi: 10.1007/s00439-005-0020-1. [DOI] [PubMed] [Google Scholar]
- 3.Geschwind MD, Shu H, Haman A, Sejvar JJ, Miller BL. Rapidly progressive dementia. Ann Neurol. 2008;64(1):97–108. doi: 10.1002/ana.21430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World OH. Global surveillance, diagnosis and therapy of human transmissible spongiform encephalopathies: report of a WHO consultation. September 1, 2010 1998 [Google Scholar]
- 5.Collins SJ, Sanchez-Juan P, Masters CL, Klug GM, van Duijn C, Poleggi A, Pocchiari M, Almonti S, Cuadrado-Corrales N, de Pedro-Cuesta J, Budka H, Gelpi E, Glatzel M, Tolnay M, Hewer E, Zerr I, Heinemann U, Kretszchmar HA, Jansen GH, Olsen E, Mitrova E, Alperovitch A, Brandel JP, Mackenzie J, Murray K, Will RG. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain. 2006;129(Pt 9):2278–2287. doi: 10.1093/brain/awl159. [DOI] [PubMed] [Google Scholar]
- 6.Vitali P, Maccagnano E, Caverzasi E, Henry RG, Haman A, Torres-Chae C, Johnson DY, Miller BL, Geschwind MD. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology. 2011;76(20):1711–1719. doi: 10.1212/WNL.0b013e31821a4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Young GS, Geschwind MD, Fischbein NJ, Martindale JL, Henry RG, Liu S, Lu Y, Wong S, Liu H, Miller BL, Dillon WP. Diffusion-weighted and fluid-attenuated inversion recovery imaging in Creutzfeldt-Jakob disease: high sensitivity and specificity for diagnosis. AJNR Am J Neuroradiol. 2005;26(6):1551–1562. [PMC free article] [PubMed] [Google Scholar]
- 8.Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, Breithaupt M, Varges D, Meissner B, Ladogana A, Schuur M, Haik S, Collins SJ, Jansen GH, Stokin GB, Pimentel J, Hewer E, Collie D, Smith P, Roberts H, Brandel JP, van Duijn C, Pocchiari M, Begue C, Cras P, Will RG, Sanchez-Juan P. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. 2009;132(Pt 10):2659–2668. doi: 10.1093/brain/awp191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perry DC, Geschwind MD. Prion disease: Thorough work-up and new diagnostic criteria needed for CJD. Nat Rev Neurol. 2011;7(9):479–480. doi: 10.1038/nrneurol.2011.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamada K, Sakai K, Akazawa K, Yuen S, Nishimura T. MR tractography: a review of its clinical applications. Magn Reson Med Sci. 2009;8(4):165–174. doi: 10.2463/mrms.8.165. [DOI] [PubMed] [Google Scholar]
- 11.Naismith RT, Xu J, Tutlam NT, Scully PT, Trinkaus K, Snyder AZ, Song SK, Cross AH. Increased diffusivity in acute multiple sclerosis lesions predicts risk of black hole. Neurology. 2010;74(21):1694–1701. doi: 10.1212/WNL.0b013e3181e042c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geschwind MD, Haman A, Miller BL. Rapidly progressive dementia. Neurol Clin. 2007;25(3):783–807. doi: 10.1016/j.ncl.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Litvan I, MacIntyre A, Goetz CG, Wenning GK, Jellinger K, Verny M, Bartko JJ, Jankovic J, McKee A, Brandel JP, Chaudhuri KR, Lai EC, D’Olhaberriague L, Pearce RK, Agid Y. Accuracy of the clinical diagnoses of Lewy body disease, Parkinson disease, and dementia with Lewy bodies: a clinicopathologic study. Arch Neurol. 1998;55(7):969–978. doi: 10.1001/archneur.55.7.969. [DOI] [PubMed] [Google Scholar]
- 14.McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 15.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34(7):939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 16.Otto M, Wiltfang J, Cepek L, Neumann M, Mollenhauer B, Steinacker P, Ciesielczyk B, Schulz-Schaeffer W, Kretzschmar HA, Poser S. Tau protein and 14-3-3 protein in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology. 2002;58(2):192–197. doi: 10.1212/wnl.58.2.192. [DOI] [PubMed] [Google Scholar]
- 17.Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson DW, Sima AA, Trojanowski JQ, Petersen RB, Gambetti P. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39(6):767–778. doi: 10.1002/ana.410390613. [DOI] [PubMed] [Google Scholar]
- 18.Castellani RJ, Colucci M, Xie Z, Zou W, Li C, Parchi P, Capellari S, Pastore M, Rahbar MH, Chen SG, Gambetti P. Sensitivity of 14-3-3 protein test varies in subtypes of sporadic Creutzfeldt-Jakob disease. Neurology. 2004;63(3):436–442. doi: 10.1212/01.wnl.0000135153.96325.3b. [DOI] [PubMed] [Google Scholar]
- 19.Steinhoff BJ, Zerr I, Glatting M, Schulz-Schaeffer W, Poser S, Kretzschmar HA. Diagnostic value of periodic complexes in Creutzfeldt-Jakob disease. Ann Neurol. 2004;56(5):702–708. doi: 10.1002/ana.20261. [DOI] [PubMed] [Google Scholar]
- 20.Shimony JS, Sheline YI, D’Angelo G, Epstein AA, Benzinger TL, Mintun MA, McKinstry RC, Snyder AZ. Diffuse microstructural abnormalities of normal-appearing white matter in late life depression: a diffusion tensor imaging study. Biol Psychiatry. 2009;66(3):245–252. doi: 10.1016/j.biopsych.2009.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klug GM, Wand H, Boyd A, Law M, Whyte S, Kaldor J, Masters CL, Collins S. Enhanced geographically restricted surveillance simulates sporadic Creutzfeldt-Jakob disease cluster. Brain. 2009;132(Pt 2):493–501. doi: 10.1093/brain/awn303. [DOI] [PubMed] [Google Scholar]
- 22.Gambetti P, Dong Z, Yuan J, Xiao X, Zheng M, Alshekhlee A, Castellani R, Cohen M, Barria MA, Gonzalez-Romero D, Belay ED, Schonberger LB, Marder K, Harris C, Burke JR, Montine T, Wisniewski T, Dickson DW, Soto C, Hulette CM, Mastrianni JA, Kong Q, Zou WQ. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol. 2008;63(6):697–708. doi: 10.1002/ana.21420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003;66:213–239. doi: 10.1093/bmb/66.1.213. [DOI] [PubMed] [Google Scholar]
- 24.Binelli S, Agazzi P, Canafoglia L, Scaioli V, Panzica F, Visani E, Di Fede G, Giaccone G, Bizzi A, Bugiani O, Avanzini G, Tagliavini F, Franceschetti S. Myoclonus in Creutzfeldt-Jakob disease: polygraphic and video-electroencephalography assessment of 109 patients. Movement disorders : official journal of the Movement Disorder Society. 2010;25(16):2818–2827. doi: 10.1002/mds.23397. [DOI] [PubMed] [Google Scholar]
- 25.Cohen OS, Prohovnik I, Korczyn AD, Ephraty L, Nitsan Z, Tsabari R, Appel S, Rosenmann H, Kahana E, Chapman J. The Creutzfeldt-Jakob disease (CJD) neurological status scale: a new tool for evaluation of disease severity and progression. Acta Neurol Scand. 2011;124(6):368–374. doi: 10.1111/j.1600-0404.2011.01489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohen OS, Hoffmann C, Lee H, Chapman J, Fulbright RK, Prohovnik I. MRI detection of the cerebellar syndrome in Creutzfeldt-Jakob disease. Cerebellum. 2009;8(3):373–381. doi: 10.1007/s12311-009-0106-8. [DOI] [PubMed] [Google Scholar]
- 27.Chohan G, Pennington C, Mackenzie JM, Andrews M, Everington D, Will RG, Knight RS, Green AJ. The role of cerebrospinal fluid 14-3-3 and other proteins in the diagnosis of sporadic Creutzfeldt-Jakob disease in the UK: a 10-year review. J Neurol Neurosurg Psychiatry. 2010;81(11):1243–1248. doi: 10.1136/jnnp.2009.197962. [DOI] [PubMed] [Google Scholar]
- 28.Kantarci K, Avula R, Senjem ML, Samikoglu AR, Zhang B, Weigand SD, Przybelski SA, Edmonson HA, Vemuri P, Knopman DS, Ferman TJ, Boeve BF, Petersen RC, Jack CR., Jr Dementia with Lewy bodies and Alzheimer disease: neurodegenerative patterns characterized by DTI. Neurology. 2010;74(22):1814–1821. doi: 10.1212/WNL.0b013e3181e0f7cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sexton CE, Kalu UG, Filippini N, Mackay CE, Ebmeier KP. A meta-analysis of diffusion tensor imaging in mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging. 2010;32(12):2322.e5–18. doi: 10.1016/j.neurobiolaging.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 30.Haik S, Dormont D, Faucheux BA, Marsault C, Hauw JJ. Prion protein deposits match magnetic resonance imaging signal abnormalities in Creutzfeldt-Jakob disease. Ann Neurol. 2002;51(6):797–799. doi: 10.1002/ana.10195. [DOI] [PubMed] [Google Scholar]
- 31.Mittal S, Farmer P, Kalina P, Kingsley PB, Halperin J. Correlation of diffusion-weighted magnetic resonance imaging with neuropathology in Creutzfeldt-Jakob disease. Arch Neurol. 2002;59(1):128–134. doi: 10.1001/archneur.59.1.128. [DOI] [PubMed] [Google Scholar]
- 32.Manners DN, Parchi P, Tonon C, Capellari S, Strammiello R, Testa C, Tani G, Malucelli E, Spagnolo C, Cortelli P, Montagna P, Lodi R, Barbiroli B. Pathologic correlates of diffusion MRI changes in Creutzfeldt-Jakob disease. Neurology. 2009;72(16):1425–1431. doi: 10.1212/WNL.0b013e3181a18846. [DOI] [PubMed] [Google Scholar]
- 33.Geschwind MD, Potter CA, Sattavat M, Garcia PA, Rosen HJ, Miller BL, DeArmond SJ. Correlating DWI MRI with pathologic and other features of Jakob-Creutzfeldt disease. Alzheimer Dis Assoc Disord. 2009;23(1):82–87. doi: 10.1097/wad.0b013e31818323ef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Armstrong RA, Lantos PL, Cairns NJ. Spatial patterns of the vacuolation in subcortical white matter in sporadic Creutzfeldt-Jakob disease (sCJD) Clin Neuropathol. 2002;21(6):284–288. [PubMed] [Google Scholar]
- 35.Liberski PP, Sikorska B, Hauw JJ, Kopp N, Streichenberger N, Giraud P, Boellaard J, Budka H, Kovacs GG, Ironside J, Brown P. Ultrastructural characteristics (or evaluation) of Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies or prion diseases. Ultrastruct Pathol. 2010;34(6):351–361. doi: 10.3109/01913123.2010.491175. [DOI] [PubMed] [Google Scholar]
- 36.Liberski PP, Streichenberger N, Giraud P, Soutrenon M, Meyronnet D, Sikorska B, Kopp N. Ultrastructural pathology of prion diseases revisited: brain biopsy studies. Neuropathol Appl Neurobiol. 2005;31(1):88–96. doi: 10.1111/j.1365-2990.2004.00595.x. [DOI] [PubMed] [Google Scholar]
- 37.Sotak CH. The role of diffusion tensor imaging in the evaluation of ischemic brain injury - a review. NMR Biomed. 2002;15(7-8):561–569. doi: 10.1002/nbm.786. [DOI] [PubMed] [Google Scholar]
- 38.Mohammadi S, Nagy Z, Moller HE, Symms MR, Carmichael DW, Josephs O, Weiskopf N. The effect of local perturbation fields on human DTI: characterisation, measurement and correction. NeuroImage. 2012;60(1):562–570. doi: 10.1016/j.neuroimage.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Atarashi R, Satoh K, Sano K, Fuse T, Yamaguchi N, Ishibashi D, Matsubara T, Nakagaki T, Yamanaka H, Shirabe S, Yamada M, Mizusawa H, Kitamoto T, Klug G, McGlade A, Collins SJ, Nishida N. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med. 2011;17(2):175–178. doi: 10.1038/nm.2294. [DOI] [PubMed] [Google Scholar]
- 40.Edgeworth JA, Farmer M, Sicilia A, Tavares P, Beck J, Campbell T, Lowe J, Mead S, Rudge P, Collinge J, Jackson GS. Detection of prion infection in variant Creutzfeldt-Jakob disease: a blood-based assay. Lancet. 2011;377(9764):487–493. doi: 10.1016/S0140-6736(10)62308-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.