

Abstract
Objective
Semaphorin (Sema) 7a regulates TGF- β1 induced fibrosis. Using a murine model of pulmonary fibrosis in which an inducible, bioactive form of the human TGF- β1 gene is overexpressed in the lung, we tested the hypothesis that Sema-7a exerts its pro-fibrotic effects in part by promoting the tissue accumulation of CD45+ fibrocytes.
Methods
Fibrosis and fibrocytes were evaluated in TGF- β1 transgenic mice in which the Sema-7a locus had been disrupted. The effect of replacement or deletion of Sema-7a on bone marrow derived cells was ascertained using bone marrow transplantation. The role of the Sema-7a receptor β1 integrin was assessed using neutralizing antibodies. The applicability of these findings to TGF-β1-driven fibrosis in humans was examined in patients with scleroderma-related interstitial lung disease.
Results
The appearance of fibrocytes in the lungs in TGF- β1 transgenic mice requires Sema-7a. Replacement of Sema-7a in bone marrow derived cells restores lung fibrosis and fibrocytes. Immunoneutralization of β1 integrin reduces pulmonary fibrocytes and fibrosis. Peripheral blood mononuclear cells from patients with scleroderma-related interstitial lung disease show increased mRNA for Sema-7a and the β1 integrin, with Sema-7a located on collagen producing fibrocytes and CD19+ lymphocytes. Peripheral blood fibrocyte outgrowth is enhanced in these patients. Stimulation of normal human peripheral blood mononuclear cells with recombinant Sema-7a enhances fibrocyte differentiation; these effects are attenuated by β1 integrin neutralization.
Conclusion
Interventions that reduce Sema-7a expression or prevent the Sema-7a - β1 integrin interaction may be ameliorative in TGF- β1-driven or fibrocyte-associated autoimmune fibroses.
The Semaphorins (Semas) are a family of highly conserved, secreted or membrane-bound proteins that are divided into eight classes based on primary sequence similarity and distinct structural features (1, 2). Semas are expressed on nerve, myeloid, and lymphoid cells, and they regulate immune responses as well as developmental processes related to organogenesis, angiogenesis, apoptosis, and neoplasia (3–5). Semaphorin 7a (Sema-7a), also called CDw108, is a GPI-anchored membrane protein that signals through at least two receptors: the β1-integrin subunit and Plexin C1 (1, 3). Sema-7a-mediated activation of β1-integrin enhances central and peripheral axonal growth and is requiredfor proper axonal tracking during embryonic development (4, 5), while Plexin C1 appears to inhibit some of these β1 integrin-mediated effects (3). Interactions between Sema-7a and its receptors also contribute to inflammation and immunity by stimulation of macrophage chemotaxis and cytokine production (6), regulation of dendritic cell migration and chemokine expression (4), modulation of T cell function (7), and regulation of melanocyte spreading and melanoma invasion (3, 8). Our recent studies advance the understanding of Sema-7a by demonstrating that it also plays an important role in the pathogenesis of transforming growth factor (TGF)-β1 induced inflammation and fibrosis (9). However, the mechanism(s) by which Sema-7a promotes these outcomes remains obscure.
The CD14+ fraction of peripheral blood contains a heterogeneous group of monocyte progenitors with important roles in tissue injury and repair. A CD34+CD45+ subpopulation of CD14+ monocytes differentiates into fibrocytes by acquiring a fibroblast-like morphology and expressing collagens I and III (10). These events occur in a TGF-β1-dependent, PI3 kinase-dependent manner (11, 12), and over time, CD14 and CD34 expression may be down-regulated. Fibrocytes traffic into and accumulate in injured tissue in response to chemokines (13, 14), and their presence is associated with various fibrosing disorders including asthma, pulmonary fibrosis, and scleroderma (15–17). Interestingly, while Sema-7a is known to affect monocyte activation in vitro via β1 integrin mediated effects (6) the role of Sema-7a in the development of fibrocytes has not been assessed.
Systemic sclerosis (SSc), or scleroderma, is a multisystem autoimmune disease characterized by progressive cutaneous and visceral fibrosis and over-activation of TGF-β1 signaling pathways (18, 19). Advances in the treatment of SSc-related renal disease have led to the emergence of pulmonary involvement as the greatest cause of mortality in SSc (20). The majority of patients with SSc demonstrate pathologic findings of interstitial lung disease (SSc-ILD) and show replacement of the normal lung parenchyma with inflamed and fibrotic tissue that is ineffective for gas exchange (21, 22). Up to 42% of patients with SSc-ILD will die of disease progression within ten years of diagnosis (20). Treatment with cyclophosphamide (23) or lymphocyte modulating agents (24, 25) show a modest benefit in delaying disease progression, but patients often relapse. The prevalence of gastroesophageal reflux disease (GERD) and ongoing autoimmunity in these patients frequently leads to poor outcomes following lung transplantation (26, 27). Thus, a better understanding of the pathogenesis of SSc-ILD may ameliorate the most frequent cause of death in these patients.
We hypothesized that Sema-7a promotes TGF-β1-induced pulmonary fibrosis by stimulating the accumulation of intrapulmonary fibrocytes and that the β1 integrin subunit plays an important role in these responses. Fibrocytes have emerged as an important area of research in SSc-ILD (28). However immunologic factors regulating their accumulation remain obscure, and a role for Sema-7a has not been considered. We characterized the role of Sema-7a in fibrocyte accumulation in lung-targeted, TGF-β1 transgenic mice and defined the tissue compartment and molecular pathway through which Sema-7a exerts these effects. Our studies demonstrate that TGF-β1 induces fibrocyte accumulation in a Sema-7a dependent manner, that Sema-7a expression on bone marrow derived cells is sufficient for the induction of fibrosis and fibrocyte appearance, and that β1 integrin blockade ameliorates these responses. This signaling axis and pathway for fibrocyte activation exists in normal human cells and appears enhanced in patients with SSc-ILD.
MATERIALS AND METHODS
TGFβ1 transgenic mice
All mouse experiments were approved by the Yale School of Medicine Institutional Animal Care and Use Committee. The CC10-tTS-rtTA- TGF-β1 transgenic mice used in this study have been described (29). These mice use the Clara cell 10-kD protein (CC10) promoter to specifically express bioactive human TGF-β1 to the lung, and were backcrossed for >10 generations onto a C57BL/6 background (29). The Sema-7a null mice were provided by Dr. Alex Kolodkin (Johns Hopkins) and have been described previously (5)
Doxycycline Administration
Eight-to-10 week old CC10-tTS-rtTA- TGF-β1 transgene positive (Tg+) or their wild-type littermate controls (transgene negative, Tg−) with the Sema-7a locus null or intact were given doxycycline 0.5mg/ml in their drinking water for up to two weeks.
Bone marrow transplantation
Mice were prepared for bone marrow transplantation using 400 cGy total body irradiation. Bone marrow harvest, preparation, and injection were performed as previously described (30).
β1 integrin blocking antibodies
TGF-β1 Tg+ and Tg− mice with an intact Sema-7a locus were injected with 125 μg of a neutralizing anti-β1 integrin antibody or isotype control (both from Biolegend) as previously described (31).
Lung inflammation
Euthanasia and bronchoalveolar lavage were performed as previously described (29). Lung inflammation was assessed via bronchoalveolar lavage (BAL) samples as described previously (29).
Collagen assessment
Total left lung collagen was measured using the Sircol Assay following the manufacturer’s protocol (Biocolour, Ireland).
Flow cytometry for fibrocytes
Lungs were digested for flow cytometry and total viable cells were quantified using Trypan blue staining as previously described (30) and flow cytometry for fibrocytes was performed according to our published method (32). Percentages of live cells co-expressing CD45 and Col-Iα were multiplied by total viable cell count of digested lung to determine the absolute number of CD45+ Col-Iα+ cells per left lung.
For flow cytometry of human cells, antibodies to Sema-7a or isotype control were purchased from R&D systems. Antibodies against human CD45, CD34, CD14, β1 integrin, and appropriate isotype controls were obtained from BD Pharmingen. Flow cytometry and cell sorting was performed using a BD FACSCalibur. Data were analyzed using Flow Jo v. 7.5 software (TreeStar, Inc). For all analyses, isotype control staining was subtracted from true antibody staining to determine the percentage of positive cells.
Histologic analysis
Formalin-fixed and paraffin-embedded lung sections were stained with hematoxylin and eosin to assess gross morphology or Mallory’s trichrome stains to visualize collagen deposition.
mRNA analyses
Total RNA was obtained using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. Primers specific for human Sema-7a, β1 integrin subunit, Plexin C1 and GAPDH, and murine β1 integrin subunit, Plexin C1, and β-actin were purchased from Superarray Bioscience. Gene expression levels were quantified using real time RT-PCR (Applied Biosystems), according to the manufacturer’s protocols and normalized to GAPDH or β-actin mRNA.
Human cell isolation and culture
All studies were performed with HIC approval and written informed consent at Yale University School of Medicine. Individuals who self identified as having no known medical conditions were included as controls. Patients with SSc according to current American College of Rheumatology criteria with or without ILD were used as our study group. Following informed consent, 30 ml of peripheral blood was drawn and peripheral blood mononuclear cells (PBMCs) isolated as previously described (33). Cells were cultured in 96 well plates that had been pre-coated with 80 μg of recombinant Sema-7a (Abnova, Taiwan) or human serum albumin in the presence or absence of β1 integrin blocking antibody or isotype control. Experiments examining the effect of Plexin C1 inhibition utilized lentiviral particles encoding specific shRNAs against Plexin C1 or negative control “scramble” sequences (Santa Cruz Biotechnology, Santa Cruz, CA) according to the manufacturer’s instructions. Confirmation of Plexin C1 inhibition was assessed using immunofluorescence. Cultured cells were assessed visually for fibrocytes based on spindle shaped morphology and by FACS as we have previously described (28).
Statistics
Normally distributed data were expressed as means ± SEM and assessed for significance by Student’s t test or ANOVA asappropriate. Data that were not normally distributed were assessed for significance using the Mann-Whitney U test.
RESULTS
Role of Sema-7a in TGF-β1-induced fibrocyte accumulation
We and others have previously shown that profibrotic stimuli or transgenic TGF-β1 overexpression leads within 14 days to the robust accumulation of fibrocytes in the murine lung (13, 32). To further evaluate these findings, studies were undertaken to determine if Sema-7a plays a role in this response. TGF-β1 Tg+ mice were interbred with Sema-7a null (−/−) mice. The pulmonary phenotype and accumulation of fibrocytes in Tg+ mice with WT and null Sema-7a loci was enumerated after 14 days of doxycyline. Consistent with prior findings (13), Sema-7a deficiency did not significantly affect BAL cell counts at this time-point (1.11 ± 0.17 × 106 cells vs. 0.88 ± 0.1 × 106 cells, p = 0.3, Figure 1A). Sircol assay revealed that total left lung collagen was significantly reduced (574.6 ± 30 μg vs. 151.8 ± 17.3 μg, p<0.0001, Fig 1B). Studies of fibrocytes revealed a 59.6% decrease in the number of CD45+Col-Iα+ cells in the Sema-7a null mice (1.29 ± 0.23 × 105 vs. 0.525 ± 0.10 × 105, p<0.001, Figure 1C, D). These data demonstrate that in addition to its well-described effects on fibrosis, Sema-7a mediates fibrocyte accumulation in the TGF-β1 exposed murine lung.
Figure 1.

A. BAL cell counts in (white bar) wild type mice and (black bar) TGF-β1 Tg+ mice. Left: Sema-7a locus intact. Right: Sema-7a locus deleted. B. Total left lung collagen, same comparisons. C. Flow cytometry for fibrocytes. Gates for collagen staining were determined based on CD45 and intracellular isotype staining). Top left: Anti-CD45-PerCP (Y axis) vs. FITC-stained isotype control (X axis) on lung suspension from TGF-β1 mouse with intact Sema 7a locus. Top right: Anti-Col-Iα-FITC (X axis) vs anti-CD45-PerCP (Y axis) on same sample. Bottom left: Anti-CD45-PerCP (Y axis) vs. FITCstained isotype control (X axis) on lung suspension from TGF-β1 mouse with disrupted Sema 7a locus. Bottom right: Anti-Col-Iα-FITC (X axis) vs anti-CD45-PercCP (Y axis) on lung cell suspensions obtained from TGF-β1 x Sema-7a null mouse. Compared to the TGF-β1 x Sema 7a +/+A large decrease in CD45+Col-Iα+ cells is evident in the right upper quadrant. D. Compared to TGF-β1 x WT mice, lungs from the TGF-β1 x Sema-7a null mice contain significantly decreased quantities of CD45+Col-Iα+ cells. White bar: WT. Black bar: TGF-β1 Tg+ mouse. ***p<0.001.
Site of expression of Sema-7a
Sema-7a is expressed on stromal cells and bone marrow derived inflammatory cells in the TGF-β1-exposed lung (9). In order to determine the tissue compartment through which Sema-7a exerts its effects on fibrosis and fibrocyte accumulation, bone marrow transplantation was used to create chimeras in which Sema-7a expression was restricted to the stroma (Sema-7a null donor marrow transplanted into TGF-β1 x WT recipient, “Sema-7a null → TGF-β1 x WT mice”) or bone marrow derived cells (BMDCs) (WT donor marrow transplanted into TGF-β1 x Sema-7a null recipient, “WT → TGF-β1 x WT mice”). A set of transplanted mice in which Sema-7a null marrow was transferred into TGF-β1 x Sema-7a null mice (Sema-7a null → TGF-β1 x Sema null mice), and WT bone marrow was transferred into TGF-β1 x WT mice (WT → TGF-β1 x WT mice), were also studied as controls. After one month, mice were assessed for engraftment, exposed to doxycycline for two weeks, and assessed for TGF-β1-relevant endpoints including lung inflammation, fibrosis, and fibrocytes. Compared to the WT → TGF-β1 x WT mice, Sema-7a null → TGF-β1 x WT mice did not exhibit significantly altered lung inflammation (Figure 2A and data not shown), collagen content (529.3 ± 56.80 μg vs 371.0 ± 46.06 μg, p = 0.06, Figure 2B), or intrapulmonary fibrocytes (9.60 ±1.20 × 104 vs 7.50 ± 1.19 × 104 p = 0.2, Figure 2C). However, when compared to the Sema-7a null → TGF-β1 x Sema null mice, WT → TGF-β1 x Sema-7a null mice demonstrated unchanged lung inflammation (Figure 2A and data not shown), increased lung fibrosis (181.5 ± 14.11 μg vs 479.1 ± 30.05 μg, p < 0.001 Figure 2B) and fibrocytes (4.40 ± 0.51 × 104 vs 9.60 ±0.75 × 104, p =0.0004, Figure 2C). These data indicate that while Sema-7a-expressing BMDCs may not be required for pulmonary fibrosis and fibrocyte accumulation, they are sufficient for the development of these pathologies.
Figure 2.

Sema-7a expression by bone marrow derived cells is sufficient for fibrosis and fibrocyte accumulation in the TGF-β1 exposed lung. A. BAL cell counts in TGF-β1 Tg+ mice with the Sema-7a locus intact (left comparisons) or deleted (right comparisons). B. Total left lung collagen content, same comparisons. Transplantation with Sema-7a null donor marrow did not significantly reduce collagen accumulation while transplantation with WT bone marrow was sufficient to restore collagen accumulation in the TGF-β1 x Sema-7a null mice. C. CD45+Col-Iα+ cells, same comparisons. As with the collagen content, transplantation of TGF-β1 x WT mice with Sema-7a null mice did not reduce fibrocytes. However, transplantation of TGF-β1 x Sema-7a null mice with WT marrow restored fibrocyte accumulation to wild type levels. D. Expression of Plexin C1 (left) and β1 integrin (right) in TGF-β1 x Sema-7a recipients of marrow with the Sema-7a locus disrupted or intact. Replacement of Sema-7a on BM-derived cells is accompanied by a trend towards increased expression of β1 integrin. *p<0.05. **p<0.01.
Expression of Receptors for Sema-7a in recipients of bone marrow transplantation
Sema-7a signals through two known receptors, the β1 integrin subunit and Plexin C1. In order to assess which of these receptors was responsible for the increased fibrosis seen in the WT → TGF-β1 x Sema-7a null animals, whole lung RNA was obtained from the TGF-β1 x Sema-7a null mice that had received WT or Sema-7a null BM and assessed for expression of β1 integrin and Plexin C1. Here we found that in contrast to the Sema-7a → TGF-β1 x Sema-7a null mice, the lungs of WT → TGF-β1 x Sema-7a null mice contained a 55.5% increase in β1 integrin subunit expression that approached statistical significance (Figure 2D). Plexin C1 expression was not enhanced in these mice (Figure 2D). These data demonstrate that the lung fibrosis caused by replacement of Sema-7a on BMDCs is accompanied by a trend towards increasedβ1 integrin expression, suggesting that the profibrotic effects of Sema-7a may be mediated through this receptor.
Role of β1 integrin in the Effects of Sema-7a
These data led us to explore the role of β1 integrin in our model. In addition to epithelial cells and fibroblasts, the β1 integrin subunit is expressed strongly on monocyte derived cells, and signals through PI3 kinase to promote monocyte recruitment, adherence, and activation (30). The β1 integrin also has been shown previously to modulate fibrosis in this TGF-β1 model (9) and shows induction in the lungs of mice in which Sema-7a was replaced via bone marrow transplantation. In contrast, Plexin C1 is not known to influence fibrosis directly, and its expression was not affected in the transplanted mice. Accordingly, we chose to examine the role of the β1 integrin subunit in our experimental system. TGF-β1 Tg+ or Tg− mice with the Sema-7a locus intact thus were exposed to doxycycline to induce transgene expression and then randomized to receive intraperitoneal injection of 125 μg of a neutralizing anti-β1 integrin antibody or isotype control every other day. Mice were sacrificed at day 14 and inflammation, fibrosis, and fibrocyte content were assessed. Treatment with anti-β1 integrin antibody caused a 61.8% decrease in BAL cell counts (55.7±14.0 × 106 vs. 21.0±6.4 × 106, p<0.01, Figure 3A), a 34.4% reduction in right upper lobe fibrosis and collagen accumulation (125 ± 2.5 μg vs 82 ± 5.5μg, p< 0.01, Figure 3B), and an 86.5% decrease in fibrocyte content (5.2 ± 0.80 × 104 vs 0.70 ± 0.34 × 104, p<0.001, Figure 3C). These data demonstrate that interventions that reduce Sema-7a and/or β1 integrin have similar effects on TGF-β1 induced fibrosis and fibrocyte accumulation in the lung, thus supporting the contention that the effects of Sema-7a are mediated in part via β1 integrin.
Figure 3.
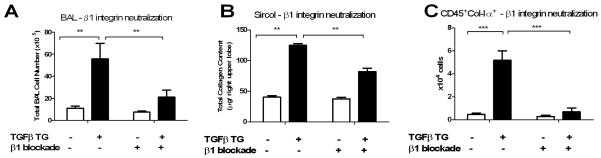
A, B. Effect of β1 integrin blockade on collagen accumulation and fibrocyte content. Treatment of TGF- β1 x WT mice with a neutralizing anti-β1 integrin blocking antibody reduced A) BAL cell counts, B) total right upper lobe collagen content and C) CD45+Col-Iα+ cells to WT levels. White bar: WT. Black bar: TGF- β1 Tg+. Left: isotype control injected. Right: injections with 125 μg of a β1 integrin blocking antibody every other day. *p<0.05, **p<0.01, ***p<0.001.
Expression of Semaphorin 7a and β1 integrin are increased in the circulation of patients with Scleroderma-related ILD
In order to better understand the mechanisms behind these findings and to support a role for the Sema-7a -β1 integrin-axis in human lung disease, we investigated the expression and action of these proteins in primary cells from patients with SSc that did or did not have ILD. SSc-ILD is strongly associated with TGF-β1 over-activation (34). PBMCs obtained from patients with SSc-ILD (n=9), SSc only (n=4), and normal controls (n=11) were assessed for the expression of Sema-7a and its receptors β1 integrin and Plexin C1 using qRT-PCR. We observed that the relative expression of Sema-7a was significantly increased in the PBMCs of SSc-ILD patients when compared to normal controls or SSc patients that did not have ILD (p<0.05 both comparisons, Figure 4A). β1 integrin expression was increased in the SSc-ILD patients compared to normal; however, there was no difference in expression of this gene in patients with SSc with or without ILD (Figure 4B). The relative expression of Plexin C1 demonstrated great variability and thus did not meet statistical significance for enhanced expression in the SSc-ILD group (Figures 4A–C). These data indicate that PBMCs from SSc patients that have ILD demonstrate robust expression of Sema-7a and its receptor the β1 integrin, suggesting that the Sema-7a -β1 integrin axis might contribute to the development or maintenance of this disease.
Figure 4.
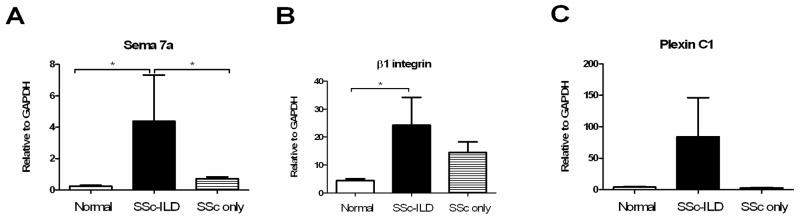
Expression of Sema-7a (A) is increased in the blood of patients with SSc-ILD compared to normal controls and patients with SSc that lack ILD. β1 integrin is increased in SSc-ILD patients compared to control (B). Expression of Plexin C1 (C) is not significantly increased. White bar: normal subjects. Grey striped bar: SSc without ILD. Black bar: SSc-ILD. *p<0.05. **p<0.01.
Expression of Semaphorin 7a and β1 integrin in the circulating cells of patients with SSc-ILD
Given the observation that Sema-7a gene expression is increased in the blood of SSc-ILD patients, we further interrogated these findings using flow cytometry. PBMCs from SSc-ILD (n = 8) and normal controls (n = 8) were obtained and characterized for co-expression of hematopoietic lineage markers and Sema-7a. The clinical characteristics of participants are shown in Figure 1. Because our murine data suggest that Sema-7a participates in the transition from CD14+ monocyte to collagen-producing fibrocyte, we initially considered that Sema-7a would be expressed on collagen-expressing fibrocytes or CD14+ fibrocyte precursors. Evaluation of collagen-producing cells revealed that low level Sema-7a expression was evident on collagen producing cells in normal subjects. This expression was significantly increased in the patients with SSc-ILD (1.530 ± 1.018% vs. 37.18 ± 12.08%, p<0.02, Figure 5A–D). In contrast, no significant differences were observed in Sema-7a expression in CD14+ monocytes from normal subjects when compared to SSC-ILD (8.213 ± 1.143% vs. 5.284 ± 1.689%, p = 0.16, Figure 5E–H).
Figure 5.

A–D Sema-7a on collagen-producing cells from a SSc-ILD subject. A. FITC isotype control (X Axis) vs anti-CD45-PerCP (Y axis). B. Anti-pro-collagen-Iα-FITC (X axis) vs anti-CD45-PerCP (Y axis) in SSc-ILD PBMCs. C. Sema-7a on CD45+/Pro-Col-Iα+ cells. Green: PE-isotype control. Red: Anti-CD45-PE. Blue: Sema-7a on the CD45+/Pro-Col-Iα+cells indicated in B. D. Sema-7a on CD45+/Pro-Col-Iα+ cells from normal and SSc-ILD. E–H. Sema-7a + CD14+ monocytes obtained from a subject with SSc-ILD. E. Isotype-APC (X axis) vs anti-CD45-PerCP (Y axis). F. Anti-CD14-APC (X axis) vs anti-CD45-PerCP (Y axis) in SSC-ILD monocytes. G. Sema-7a expression (blue) on the CD14+ population indicated in F. Red: Isotype control. Green: Anti-CD45-PE. H. Sema-7a expression in controls vs SSc-ILD. I–L. Sema-7a expression on lymphocytes obtained from a patient with SSc-ILD. I. Lymphocytes identified by FSC (X axis) and SSC (Y axis) characteristics. J. PE isotype control (X axis) vs anti-CD45-PerCP (Y axis). K. Anti-Sema-7a -PE (X Axis) and anti-CD45-PerCP (Y axis). L Sema-7a + lymphocytes in control vs SSc-ILD. M–P. Sema-7a on CD19+ cells (M) CD8+ (N) or CD4+ (O) cells in SSc-ILD patients. P. β1 integrin expression on CD45+Pro-Col Iα+ cells from normal subjects and SSc-ILD patients. White: control. Black: SSc-ILD. *p<0.05.
Because collagen-producing cells comprise only a small component of total PBMCs in patients with SSc-ILD (17), we thought it unlikely that this rare population accounts fully for the observed increase in Sema-7a mRNA. Thus, we examined other cell populations, such as lymphocyte subtypes, which are abundant in circulation and are known to express Sema-7a. Using forward scatter (FSC) vs. side scatter (SSC) criteria, total lymphocytes were gated and Sema-7a expression was quantified as shown in Figure 5I–K. This approach revealed a robust increase in the percentage of Sema-7a+ lymphocytes in the blood of patients with SSc-ILD (3.946 ± 0.9602% vs 9.223 ± 2.401%, p<0.025, Figure 5L). Further analysis using lymphocyte specific markers revealed that this increase in Sema-7a expression localized to CD19+ B cells (10.79 ± 4.429% vs. 23.80 ± 5.880%, p<0.03, Figure 5M). There was no demonstrable change in Sema-7a expression on CD4+ or CD8+ T cells between controls and SSc-ILD subjects (Figure 5N–O and data not shown).
Having found that Sema-7a+ lymphocytes were increased in the circulation of patients with SSc-ILD, we next analyzed the cell population where the β1 integrin subunit was expressed. We reasoned that if the Sema-7a -β1 integrin axis was involved in fibrocyte appearance, then the β1 integrin also would be expressed on collagen-producing fibrocytes. This indeed was the case, as, consistent with prior reports (35), nearly all of the collagen producing cells demonstrated surface expression of the β1 integrin subunit (Figure 5P). These data indicate that both components of the Sema-7a –β1 integrin axis are expressed in higher levels on multiple cell populations in patients with SSc-ILD than in normal controls, with increased Sema-7a located on collagen-expressing fibrocytes and CD19+ lymphocytes, and β1 integrin expression present on collagen producing cells.
Role of Sema-7a and its receptors on fibrocyte differentiation in humans
Lastly, to better understand the functional relationship between Sema-7a, the β1 integrin, and fibrocyte differentiation, PBMCs were obtained from normal individuals (n = 8) and patients with SSc-ILD (n=9), cultured under conditions that favor fibrocyte outgrowth, and the impact of recombinant Sema-7a and/or anti-β1 integrin antibody assessed (8). Given that Plexin C1 was so highly expressed in the circulating cells from SSc-ILD patients, we also explored the effect of Plexin C1 inhibition in this system. Fibrocyte phenotype was assessed visually by morphology (Figure 6A) and confirmed by flow cytometry for co-expression of CD45, pro-collagen-Iα, CD14, and CD34 (Figure 6B–F). In these experiments, incubation with Sema-7a led to a nearly threefold increase in fibrocytes in the normal controls (13.25 ± 1.548 × 104 vs. 35.25 ± 2.056 × 104, p = 0.0001, Figure 6G). When compared to normal individuals, baseline fibrocyte outgrowth was enhanced in the subjects with SSc-ILD (23.46 ± 2.252 × 104 vs 13.25 ± 1.548 × 104, p<0.02, Figure 6G) and did not respond to stimulation with Sema-7a (Figure 6G and data not shown). Importantly, these events were in all cases inhibited by β1 integrin blockade (Figure 6G, p<0.001 all comparisons), while Plexin C1 inhibition enhanced collagen production in Sema 7a stimulated monocytes from both control and SSc-ILD subjects, as well as in unstimulated monocytes obtained from SSc-ILD patients (Figure 6H). These studies demonstrate that exogenous Sema-7a is sufficient to induce fibrocyte outgrowth in normal controls and that this axis is abnormal in patients with SSc-ILD. These data also support the conclusion that a balance between the β1 integrin subunit and Plexin C1 plays a critical role in these Sema-7a -induced inductive events.
Figure 6.

A. Fibrocytes identified by spindle shaped morphology in cultures of SSc-ILD PBMCs. B–F. FACS for CD45, Pro-Col-Iα, CD14, and CD34 on cultured PBMCs from a normal control. B. FITC detected intracellular isotype control (X axis) and anti-CD45-PE (Y axis). This negative control was used to set the negative gate for Pro-Col-Iα. C. Anti-Pro-Col-Iα-FITC (X axis) and anti-CD45-PE (Y axis) on normal PBMCs cultured in the presence of HSA. D. Anti-Pro-Col-Iα-FITC (X axis) and anti-CD45-PE (Y axis) on normal PBMCs cultured in the presence of Sema-7a. Increased collagen-producing cells are seen in the right upper quadrant. E. Staining for anti-CD14-APC (X axis) and anti-CD34-PerCP (Y axis) reveals that these collagen-producing cells demonstrate high level CD34 expression and variable CD14 expression. F. Anti-Pro-Col-Iα-FITC (X axis) and anti-CD45-PE (Y axis) on normal PBMCs cultured in the presence of human serum albumin. G. Sema-7a stimulated PBMCs show increased fibrocyte differentiation (white, left comparisons). In contrast, SSc-ILD subjects show increased fibrocyte outgrowth at baseline (black, left comparisons) with no response to exogenous Sema stimulation but pronounced reduction via immunoneutralization of the β1 integrin subunit (right comparisons). H. Inhibition of Plexin C1promotes fibrocyte differentiation in Sema-7a stimulated monocytes from control and SSc-ILD subjects as well as in unstimulated SSc-ILD monocytes. Overall quantities may be lower in these studies due to the exposure to lentiviral particles. Same comparisons as in (G). *p<0.05. **p<0.01. ***p<0.001.
DISCUSSION
TGF–β1 is essential for wound healing, stimulates matrix molecule deposition, and is implicated in the pathogenesis of fibrotic disorders including SSc-ILD. Because prior studies from our laboratory demonstrated that Sema-7a promotes TGF–β1 induced injury and fibrosis (13), and that TGF-β1 induces the accumulation of fibrocytes (32), experiments were undertaken to better define the mechanism by which Sema-7a influences fibrocyte biology. Our results demonstrate a previously unrecognized relationship between Sema-7a and fibrocytes. Specifically, they demonstrate that Sema-7a expression on BMDCs is sufficient for the appearance of fibrosis and fibrocytes in TGF–β1 exposed murine lung and that these effects are mediated in part through β1-integrin. We also demonstrate that the expression of Sema-7a and its receptors are increased in the circulation of patients with SSc-ILD, that exposure to exogenous Sema-7a induces fibrocyte differentiation in PBMCs obtained from normal controls, and that immunoneutralization of the β1 integrin which functions as a receptor for Sema-7a abrogates fibrocyte outgrowth, while inhibition of Plexin C1 promotes fibrocyte accumulation in these subjects. Our data do not rule out a role for other β1 integrin-dependent mechanisms that may involve such as epithelial to mesenchymal transition and/or fibroblast activation. The increase in fibrocytes that develop in response to Plexin C1 blockade suggests that the increased expression of this protein in the circulation of patients with SSc-ILD reflects a counter-regulatory response of either Sema-7a signaling (3) or other Plexin C1 targets such as cofilin-1, an actin-modifying protein known to be activated in myofibroblasts (3). Murine modeling using Plexin C1 null and/or in vivo gene knockdown approaches may be useful strategies to test this hypothesis. These data nevertheless identify a novel role for Sema-7a in the regulation of fibrocyte accumulation in mice and humans.
Fibrocytes were first described as blood-borne, fibroblast-like cells that appeared in exudative fluid at the earliest phases of wound repair (10). They now are considered to originate from CD14+ myeloid cells that co-express collagen, CD45, and the progenitor marker CD34; this later marker is downregulated as these cells mature in situ (36). Fibrocytes contribute to normal wound healing and pathologic fibrosis (37), and they are present in increased quantities in the circulation and tissues of patients with fibrotic disorders of the lung and other organs (38, 39). It should be noted that while CD45 and Col-Iα expression has classically been sufficient for the identification of fibrocytes in the murine lung (39), the unambiguous identification of human fibrocytes in certain contexts may additionally require the presence of CD34 and/or confirmation of spindle shaped morphology (28) (as performed in the present study). The difference we detected in Sema-7a expression on circulating cells in normal control versus SSc-ILD subjects may relate to phenotypic differences in the collagen-producing fibrocytes and their precursors. The differentiation and accumulation of fibrocytes are regulated by a complex interaction of cytokines, chemokines and plasma proteins. The present studies add to our understanding of fibrocyte homeostasis by demonstrating, for the first time, that TGF–β1 stimulates fibrocyte accumulation via a Sema-7a -dependent, β1-integrin-dependent pathway and that Sema-7a expression on BMDCs is sufficient for these outcomes. The molecular mechanism of these effects is unclear. Because Sema-7a is required for TGF-β1 induced epithelial apoptosis (9) it is possible that Sema-7a+ BMDCs have an important role in regulating cell death responses. This notion may be especially relevant given the recent finding that SSc phenotypes are associated with polymorphisms in the promoter for the pro-apoptotic molecule, Fas (40). An alternate hypothesis is suggested in other models of inflammation (7, 41), where the Sema-7a-β1 integrin interaction affects T cell and monocyte responses. It is notable that while we found that Sema-7a was increased in the circulation of SSc-ILD patients, stimulation of monocytes with this protein failed to augment fibrocyte outgrowth. This finding could relate to the enhanced expression of Plexin C1 or an inherent propensity for collagen production in monocytes from the SSc-ILD subjects. Because we have recently shown that monocytes from aged but healthy individuals are enriched for collagen production (28), it is also possible that the increased fibrocytes in these patients represents a form of immunosenescence.
While the role of lymphocytes in lung fibrosis remains controversial (43), the response of SSc-ILD to lymphocyte modulating agents (24, 25) suggests that these cells also influence the development and maintenance of SSc-ILD. Because CD4+ cells are reported to induce fibrocyte outgrowth in a murine model of renal fibrosis (43), we were surprised to find that CD19+ cells expressed Sema-7a. It is possible that had we investigated cells from a larger number of patients, or focused investigation on certain disease subtypes, these results may have differed. Mechanism(s) through which B lymphocytes may affect fibrocytes include contact mediated effects, antigen presentation, or secretion of soluble mediators and humoral factors. Notably, CD19 depletion ameliorates fibrosis, and B cell autoantibody production is a characteristic but pathogenically poorly understood feature of SSc. The role of B cells in fibrogenesis may be clarified in studies using genetic or pharmacologic techniques to modulate B cell populations in murine models of disease. To our knowledge these are the first studies to suggest a relationship between B lymphocytes and fibrocyte biology in fibrogenic disorders.
In summary, these studies demonstrate that TGF–β1-induced Sema-7a stimulates tissue accumulation of fibrocytes via its expression on BMDCs. They also reveal the importance of β1 integrin activation in these fibrocyte responses and that this axis functions in humans. This pathway is abnormal in patients with SSc-ILD but may be manipulated for therapeutic benefit.
Table 1.
Clinical characteristics of subjects.
| SSc-ILD | Control | P value | |
|---|---|---|---|
| Age (years) | 50.37±14.49 | 64.25±18.0 | NS |
| Sex (female) | 6/8 | 6/8 | NS |
| Race (white vs nonwhite) | 7/8 | 6/8 | NS |
| Diffuse | 7/8 | N/A | N/A |
| Length of disease, years | 5.18 ± 5.72 | N/A | N/A |
| Known Pulmonary hypertension | 0/8 | N/A | N/A |
| FVC, % predicted | 65.4 ± 12.4 | N/A | N/A |
| DLCO, % predicted | 49.2 ± 15.6 | N/A | N/A |
Acknowledgments
Sources of Support: P30AR053495-01A1, K08 HL079066, UL1RR024139, an Edward Mallinckrodt, Jr., Scholar Award and Established Investigator Award from the Scleroderma Foundation (all to ELH).
References
- 1.Eickholt BJ. Functional diversity and mechanisms of action of the semaphorins. Development. 2008;135(16):2689–94. doi: 10.1242/dev.019968. [DOI] [PubMed] [Google Scholar]
- 2.Kumanogoh A, Kikutani H. Immune semaphorins: a new area of semaphorin research. J Cell Sci. 2003;116(Pt 17):3463–70. doi: 10.1242/jcs.00674. [DOI] [PubMed] [Google Scholar]
- 3.Lazova R, Gould Rothberg BE, Rimm D, Scott G. The semaphorin 7A receptor Plexin C1 is lost during melanoma metastasis. Am J Dermatopathol. 2009;31(2):177–81. doi: 10.1097/DAD.0b013e318196672d. [DOI] [PubMed] [Google Scholar]
- 4.Maruyama T, Matsuura M, Suzuki K, Yamamoto N. Cooperative activity of multiple upper layer proteins for thalamocortical axon growth. Dev Neurobiol. 2008;68(3):317–31. doi: 10.1002/dneu.20592. [DOI] [PubMed] [Google Scholar]
- 5.Pasterkamp RJ, Peschon JJ, Spriggs MK, Kolodkin AL. Semaphorin 7A promotes axon outgrowth through integrins and MAPKs. Nature. 2003;424(6947):398–405. doi: 10.1038/nature01790. [DOI] [PubMed] [Google Scholar]
- 6.Holmes S, Downs AM, Fosberry A, Hayes PD, Michalovich D, Murdoch P, et al. Sema7A is a potent monocyte stimulator. Scand J Immunol. 2002;56(3):270–5. doi: 10.1046/j.1365-3083.2002.01129.x. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki K, Okuno T, Yamamoto M, Pasterkamp RJ, Takegahara N, Takamatsu H, et al. Semaphorin 7A initiates T-cell-mediated inflammatory responses through alpha1beta1 integrin. Nature. 2007;446(7136):680–4. doi: 10.1038/nature05652. [DOI] [PubMed] [Google Scholar]
- 8.Scott GA, McClelland LA, Fricke AF. Semaphorin 7a promotes spreading and dendricity in human melanocytes through beta1-integrins. J Invest Dermatol. 2008;128(1):151–61. doi: 10.1038/sj.jid.5700974. [DOI] [PubMed] [Google Scholar]
- 9.Kang HR, Lee CG, Homer RJ, Elias JA. Semaphorin 7A plays a critical role in TGF-beta1-induced pulmonary fibrosis. J Exp Med. 2007;204(5):1083–93. doi: 10.1084/jem.20061273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1(1):71–81. [PMC free article] [PubMed] [Google Scholar]
- 11.Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol. 2001;166(12):7556–62. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- 12.Mehrad B, Burdick MD, Strieter RM. Fibrocyte CXCR4 regulation as a therapeutic target in pulmonary fibrosis. Int J Biochem Cell Biol. 2009;41(8–9):1708–18. doi: 10.1016/j.biocel.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114(3):438–46. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moore BB, Murray L, Das A, Wilke CA, Herrygers AB, Toews GB. The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am J Respir Cell Mol Biol. 2006;35(2):175–81. doi: 10.1165/rcmb.2005-0239OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mehrad B, Burdick MD, Zisman DA, Keane MP, Belperio JA, Strieter RM. Circulating peripheral blood fibrocytes in human fibrotic interstitial lung disease. Biochem Biophys Res Commun. 2007;353(1):104–8. doi: 10.1016/j.bbrc.2006.11.149. [DOI] [PubMed] [Google Scholar]
- 16.Wang CH, Huang CD, Lin HC, Lee KY, Lin SM, Liu CY, et al. Increased circulating fibrocytes in asthma with chronic airflow obstruction. Am J Respir Crit Care Med. 2008;178(6):583–91. doi: 10.1164/rccm.200710-1557OC. [DOI] [PubMed] [Google Scholar]
- 17.Mathai SK, Gulati M, Peng X, Russell TR, Shaw AC, Rubinowitz AN, et al. Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab Invest. 90(6):812–23. doi: 10.1038/labinvest.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong C, Zhu S, Wang T, Yoon W, Li Z, Alvarez RJ, et al. Deficient Smad7 expression: a putative molecular defect in scleroderma. Proc Natl Acad Sci U S A. 2002;99(6):3908–13. doi: 10.1073/pnas.062010399. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Varga J, Pasche B. Transforming growth factor beta as a therapeutic target in systemic sclerosis. Nat Rev Rheumatol. 2009;5(4):200–6. doi: 10.1038/nrrheum.2009.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis. 2007;66(7):940–4. doi: 10.1136/ard.2006.066068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harrison NK, McAnulty RJ, Haslam PL, Black CM, Laurent GJ. Evidence for protein oedema, neutrophil influx, and enhanced collagen production in lungs of patients with systemic sclerosis. Thorax. 1990;45(8):606–10. doi: 10.1136/thx.45.8.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harrison NK, Myers AR, Corrin B, Soosay G, Dewar A, Black CM, et al. Structural features of interstitial lung disease in systemic sclerosis. Am Rev Respir Dis. 1991;144(3 Pt 1):706–13. doi: 10.1164/ajrccm/144.3_Pt_1.706. [DOI] [PubMed] [Google Scholar]
- 23.Tashkin DP, Elashoff R, Clements PJ, Goldin J, Roth MD, Furst DE, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354(25):2655–66. doi: 10.1056/NEJMoa055120. [DOI] [PubMed] [Google Scholar]
- 24.Zamora AC, Wolters PJ, Collard HR, Connolly MK, Elicker BM, Webb WR, et al. Use of mycophenolate mofetil to treat scleroderma-associated interstitial lung disease. Respir Med. 2008;102(1):150–5. doi: 10.1016/j.rmed.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 25.Daoussis D, Liossis SN, Tsamandas AC, Kalogeropoulou C, Kazantzi A, Sirinian C, et al. Experience with rituximab in scleroderma: results from a 1-year, proof-of-principle study. Rheumatology (Oxford) 49(2):271–80. doi: 10.1093/rheumatology/kep093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D’Ovidio F, Singer LG, Hadjiliadis D, Pierre A, Waddell TK, de Perrot M, et al. Prevalence of gastroesophageal reflux in end-stage lung disease candidates for lung transplant. Ann Thorac Surg. 2005;80(4):1254–60. doi: 10.1016/j.athoracsur.2005.03.106. [DOI] [PubMed] [Google Scholar]
- 27.D’Ovidio F, Mura M, Tsang M, Waddell TK, Hutcheon MA, Singer LG, et al. Bile acid aspiration and the development of bronchiolitis obliterans after lung transplantation. J Thorac Cardiovasc Surg. 2005;129(5):1144–52. doi: 10.1016/j.jtcvs.2004.10.035. [DOI] [PubMed] [Google Scholar]
- 28.Mathai SK, Gulati M, Peng X, Russell TR, Shaw AC, Rubinowitz AN, et al. Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab Invest. doi: 10.1038/labinvest.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee CG, Cho SJ, Kang MJ, Chapoval SP, Lee PJ, Noble PW, et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J Exp Med. 2004;200(3):377–89. doi: 10.1084/jem.20040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herzog EL, Van Arnam J, Hu B, Krause DS. Threshold of lung injury required for the appearance of marrow-derived lung epithelia. Stem Cells. 2006;24(8):1986–92. doi: 10.1634/stemcells.2005-0579. [DOI] [PubMed] [Google Scholar]
- 31.Bungartz G, Stiller S, Bauer M, Muller W, Schippers A, Wagner N, et al. Adult murine hematopoiesis can proceed without beta1 and beta7 integrins. Blood. 2006;108(6):1857–64. doi: 10.1182/blood-2005-10-007658. [DOI] [PubMed] [Google Scholar]
- 32.Murray LA, Chen Q, Kramer MS, Hesson DP, Argentieri RL, Peng X, et al. TGF-beta driven lung fibrosis is macrophage dependent and blocked by Serum Amyloid P. Int J Biochem Cell Biol. doi: 10.1016/j.biocel.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 33.Murray LA, Rosada R, Moreira AP, Joshi A, Kramer MS, Hesson DP, et al. Serum amyloid P therapeutically attenuates murine bleomycin-induced pulmonary fibrosis via its effects on macrophages. PLoS One. 5(3):e9683. doi: 10.1371/journal.pone.0009683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sargent JL, Milano A, Bhattacharyya S, Varga J, Connolly MK, Chang HY, et al. A TGFbeta-responsive gene signature is associated with a subset of diffuse scleroderma with increased disease severity. J Invest Dermatol. 130(3):694–705. doi: 10.1038/jid.2009.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pilling D, Fan T, Huang D, Kaul B, Gomer RH. Identification of markers that distinguish monocyte-derived fibrocytes from monocytes, macrophages, and fibroblasts. PLoS One. 2009;4(10):e7475. doi: 10.1371/journal.pone.0007475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidt M, Sun G, Stacey MA, Mori L, Mattoli S. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J Immunol. 2003;171(1):380–9. doi: 10.4049/jimmunol.171.1.380. [DOI] [PubMed] [Google Scholar]
- 37.Herzog EL, Bucala R. Fibrocytes in health and disease. Exp Hematol. doi: 10.1016/j.exphem.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moeller A, Gilpin SE, Ask K, Cox G, Cook D, Gauldie J, et al. Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179(7):588–94. doi: 10.1164/rccm.200810-1534OC. [DOI] [PubMed] [Google Scholar]
- 39.Herzog EL, Bucala R. Fibrocytes in health and disease. Exp Hematol. 38(7):548–56. doi: 10.1016/j.exphem.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Broen J, Gourh P, Rueda B, Coenen M, Mayes M, Martin J, et al. The FAS -670A>G polymorphism influences susceptibility to systemic sclerosis phenotypes. Arthritis Rheum. 2009;60(12):3815–20. doi: 10.1002/art.24964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Czopik AK, Bynoe MS, Palm N, Raine CS, Medzhitov R. Semaphorin 7A is a negative regulator of T cell responses. Immunity. 2006;24(5):591–600. doi: 10.1016/j.immuni.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 42.Luzina IG, Todd NW, Iacono AT, Atamas SP. Roles of T lymphocytes in pulmonary fibrosis. J Leukoc Biol. 2008;83(2):237–44. doi: 10.1189/jlb.0707504. [DOI] [PubMed] [Google Scholar]
- 43.Niedermeier M, Reich B, Gomez MR, Denzel A, Schmidbauer K, Gobel N, et al. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci U S A. 2009;106(42):17892–7. doi: 10.1073/pnas.0906070106. [DOI] [PMC free article] [PubMed] [Google Scholar]