

Abstract
Homologous recombination (HR) and nonhomologous end-joining (NHEJ) are two distinct DNA double-strand break (DSB) repair pathways. Here we report that DNA-dependent protein kinase (DNA-PK), the core component of NHEJ, partnering with DNA-damage checkpoint kinases ataxia telangiectasia mutated (ATM) and ATM- and Rad3-related (ATR), regulates HR repair of DSBs. The regulation was accomplished through modulation of the p53 and replication protein A (RPA) interaction. We show that upon DNA damage, p53 and RPA were freed from a p53-RPA complex by simultaneous phosphorylations of RPA at the N-terminus of RPA32 subunit by DNA-PK and of p53 at Ser37 and Ser46 in a Chk1/Chk2-independent manner by ATR and ATM, respectively. Neither the phosphorylation of RPA nor of p53 alone could dissociate p53 and RPA. Furthermore, disruption of the release significantly compromised HR repair of DSBs. Our results reveal a mechanism for the crosstalk between HR repair and NHEJ through the co-regulation of p53-RPA interaction by DNA-PK, ATM and ATR.
Introduction
DNA damage is a major cause of genome instability and, thus, the development of human cancer. In cells, DNA damage is removed by DNA repair pathways in coordination with DNA damage checkpoints. The latter halts cell cycle progression to allow time for DNA repair before cell cycling can resume (1–6). DNA double-strand breaks (DSBs) are the most lethal form of DNA damage and mainly are repaired by homologous recombination (HR) and nonhomologous end-joining (NHEJ) pathways in mammalian cells. NHEJ repairs the DSBs induced by genotoxic agents such as ionizing radiation. By contrast, HR repairs DSBs induced by genotoxins such as camptothecin (CPT). CTP is a topoisomerase I inhibitor that arrests the topoisomerase I-nicked DNA intermediate complex and leads to replication fork collapse at the nicked site to form DSBs (7, 8). Although crosstalk may occur between HR and NHEJ (9, 10), the molecular mechanism remains unknown.
DNA-PK plays a key role in NHEJ by recognizing DSBs, initiating NHEJ repair and assembling the repair machinery. DNA-PK is a 615 kDa heterotrimeric complex consisting of the catalytic subunit of DNA protein kinase (DNA-PKcs), plus Ku70 and Ku80. As a member of the phosphatidylinositol 3-kinase-related kinase (PIKK) family, DNA-PK also phosphorylates proteins such as H2AX, RPA, p53, XRCC4, Ku70 (XRCC6), and Ku80 (XRCC5) involved in DNA damage responses (DDRs) (11, 12). Of those proteins, replication protein A (RPA) is the major eukaryotic single-stranded DNA (ssDNA) binding protein and is a heterotrimer containing RPA70, RPA32, and RPA14 subunits. In addition to binding ssDNA, RPA also interacts with other proteins during DDRs (5, 13–25) and is involved in almost all DNA metabolic pathways including the HR repair pathway. A mutation in RPA also is implicated in cancer (26, 27). A remarkable fact about RPA is that upon DNA damage, the N-terminus of RPA32 is hyperphosphorylated by PIKK kinases (28). We and others have presented evidence supporting a role of RPA in coordinating DDR pathways via the RPA32 hyperphosphorylation (13, 14, 29–35). We have shown that upon hyperphosphorylation RPA undergoes a structural reorganization (32).
Among RPA-protein interactions, the p53-RPA interaction (24, 36–41) is of particular interest as p53 is a tumor suppressor whose inactivation is a key step of carcinogenesis for over half of human cancers (42, 43). As “the guardian of the genome” p53 is a key regulator of genome stabilization through its roles in cell cycle checkpoints, apoptosis and facilitating DNA repair (44). It is well known that phosphorylation of p53 plays a critical role in regulating p53 activities in various DDR pathways. Almost all the post-translational modifications on p53 occur in the unstructured region of the protein formed by the transactivation domain (TAD), the linker between the DNA-binding and TET domains, and the C-terminal 30 residues (45). These same regions are involved in the p53 interaction with RPA (24, 37, 45). However, how the p53-RPA interaction is modulated and affects DDR reactions is poorly understood.
In the present study, we determined the mechanism by which the p53-RPA interaction is modulated as well as the impacts of the regulation on HR repair. We found that the p53-RPA complex was disassembled upon the phosphorylations of RPA and p53 by DNA-PK and ATM/ATR, respectively, in a synergistic manner. While phosphorylation of RPA or p53 alone showed no effect, phosphorylation deficiency of either p53 or RPA inhibited the dissociation of p53 and RPA. Also, the inhibition of phosphorylation significantly reduced the efficiency of HR repair. Our results unveil the mechanistic details of a crosstalk between HR and NHEJ repair machineries which involves highly coordinated interactions between p53, RPA, DNA-PK, ATM and ATR in DDRs.
Results
Interaction of RPA with p53 in cells
In order to address the functional implications of the p53-RPA interaction, we examined the ability of p53 to bind to the hyperphosphorylated form of RPA32 in cells by co-immunoprecipitation (co-IP). Cells expressing phosphorylation-deficient RPA32 (PD-RPA) and wild-type RPA32 (34), respectively, were treated with CPT for 3 hrs. CPT is a DNA DSB inducer and was able to induce RPA hyperphosphorylation in cells as indicated by the bands of hyperphosphorylated RPA32 (hyp-RPA32) which migrate slower than the nonphosphorylated RPA32 band on SDS-PAGE (Figure 1A). In contrast, as expected, CPT treatment resulted in no hyperphosphorylation of RPA32 in the PD-RPA cells. As shown in Figure 1A, the association of p53 with RPA predominately occurred between p53 and the unphosphorylated RPA with little or no hyp-RPA32 associating with p53. This suggests that RPA hyperphosphorylation may have disrupted the p53-RPA association. Note that a DNase I pretreatment of the cell lysate precludes a DNA linkage between RPA and p53 as an explanation for these immunoprecipitation (IP) results. To confirm that the preferential binding of p53 to unphosphorylated RPA was due to direct protein-protein interaction, we used purified recombinant RPA (17) which had been hyperphosphorylated (30). After p53 IP from cell lysates with anti-p53 antibody, the immunoprecipitates were washed with buffer containing concentrations of NaCl up to 1 M to remove possible p53-associated proteins (Figure 1B). The wash was efficient as indicated by the removal of bound endogenous RPA. Then, an equimolar mixture of purified RPA and hyp-RPA was supplied to allow for interaction with the immunoprecipitated endogenous p53. Subsequent blotting analysis of the co-immunoprecipitates confirmed that p53 directly interacted with the nonphosphorylated RPA while having little or no affinity to the hyp-RPA (Figure 1C).
Figure 1. Hyperphosphorylated RPA is unable to interact with endogenous p53.

(A) Stable U2-OS cells expressing WT- or PD-RPA32 were treated with 10 uM CPT for 3 hrs to induce RPA hyperphosphorylation. Cells were harvested and chromatin-bound proteins were isolated. Chromatin was subjected to DNase I digestion and 10% of the sample was loaded onto the gel (INPUT). The remaining lysate was immunoprecipitated using anti-p53 antibody. Samples were analyzed by Western blotting. (B) Immunoprecipitation was performed with A549 cell lysate using anti-p53 antibody. Immunoprecipitates were washed with buffer of increasing salt concentrations (0 – 1.0 M) to remove proteins bound to p53, including RPA and Mdm2. Samples were then analyzed by Western blotting. (C) p53 from A549 cell lysates treated with 10 uM CPT for 2 hrs or 2 mM HU for 24 hrs was isolated by IP with anti-p53 antibody, followed by a 1 M salt buffer wash. Equimolar amounts of purified RPA and hyp-RPA were added and the proteins were allowed to interact for 6 hrs. Then, the p53 complexes were pulled down, washed and analyzed by Western blotting.
In vitro interaction of p53 with native and hyperphosphorylated RPA in the presence or absence of ssDNA
To further describe the p53-RPA interaction, co-IP assays with purified RPA/hyp-RPA and p53 proteins were performed. Surprisingly, the binding of recombinant p53 to the hyp-RPA is greater than that to native RPA (Figure 2A), contradicting the cellular results shown in Figure 1. The same experiment also was performed with RPA hyperphosphorylated by purified DNA-PK (Promega Corp., Madison, WI) (32) and a similar preference for hyp-RPA was obtained (Figure 2B). To investigate this discrepancy, similar in vitro immunoprecipitation was conducted in the presence of ssDNA as the binding to ssDNA is a major function of RPA in cells. After RPA pre-incubation with 5’-biotinylated dT30mer or dT90mer ssDNA, the ssDNA-bound RPA was pulled down with streptavidin-agarose beads, and then the RPA-ssDNA complex was incubated with purified recombinant p53. Recombinant p53 still bound more efficiently to hyp-RPA than native RPA in the presence of dT30mer or dT90mer (Figures 2A, 2B and 2C). To further determine the possible effect of ssDNA on the p53 interaction with native RPA and hyp-RPA, the immunoprecipitated p53-RPA complex of purified proteins was titrated with increasing concentrations of dT30mer ssDNA (Figure 2D). The ssDNA had little or no effect on the p53-RPA binding when the ssDNA had a 1:1 molar ratio to the proteins, but did competed with p53 for hyp-RPA at significantly higher ssDNA-to-protein ratios. Alternatively, RPA also was pre-incubated with various concentrations of ssDNA and then p53 was supplied. Similar results were obtained although native RPA binding to p53 also was affected at high ssDNA-to-protein ratios (Figure 2E). These data indicate that 1) hyperphosphorylation of RPA does not disrupt the RPA interaction with recombinant p53 in vitro; and 2) ssDNA does not play a significant role in mediating the phosphorylation-induced disruption of cellular p53-RPA interaction observed in Figure 1. Thus, the hyperphosphorylation of RPA alone may not be sufficient to substantially impact the p53-RPA interaction; the post-translational modifications on p53 also may be important.
Figure 2. In vitro p53-RPA interaction with and without ssDNA.

(A) Recombinant GST-tagged p53 protein was incubated with either recombinant RPA or hyp-RPA. Except for the 10% of sample volume loaded for input, samples were incubated with GST-agarose beads, collected by centrifugation, washed and analyzed by Western blotting. (B) Recombinant RPA protein was phosphorylated in vitro using DNA-PK kinase, and then incubated with GST-p53, followed by IP as in (A). (C) Recombinant RPA or hyp-RPA was incubated with either dT90 or dT30 ssDNA. Recombinant p53 was supplied in excess to the RPA-ssDNA complex and incubated overnight. Biotinylated DNA complexes were collected by centrifugation, washed and analyzed by Western blotting. (D) Recombinant RPA and hyp-RPA were mixed followed by incubation with p53 for 6 hrs. The ssDNA (dT30) was added in increasing molar ratios of DNA-to-RPA. The samples were subjected to immunoprecipitation with GST beads and analyzed by Western blotting. (E) Recombinant RPA and hyp-RPA were mixed and incubated with increasing molar ratios of ssDNA (dT30). Purified p53 protein then was supplied at an equimolar RPA amount for binding for 6 hrs. The samples were subjected to p53 immunoprecipitation with GST beads and analyzed by Western blotting.
Effect of p53 phosphorylation on p53-RPA interaction
To determine whether post-translational modifications of p53 are involved in the modulation of p53-RPA interactions, cells were treated with CPT followed by immunoprecipitation of p53 from the nuclear lysates. The p53 immunoprecipitates were washed with the 1M NaCl buffer to remove p53-associated proteins (Figure 1B). A portion of the endogenous p53 was treated with Calf Intestinal Alkaline Phosphatase (CIAP) to remove the endogenous phosphorylations. Then, recombinant RPA and hyp-RPA were supplied as an equimolar mix to allow for the interaction with p53. Western blot analysis of the samples is shown in Figure 3 where the endogenous p53 predominately bound to the unphosphorylated form of RPA (lane 5). However the binding preference was reversed after the same endogenous p53 was de-phosphorylated with CIAP, then the p53-hypRPA interaction is favored (lane 4). The results indicated that phosphorylation of p53 also is involved in the modulation of the p53-RPA interaction.
Figure 3. p53 phosphorylation is required for regulation of p53-RPA binding.
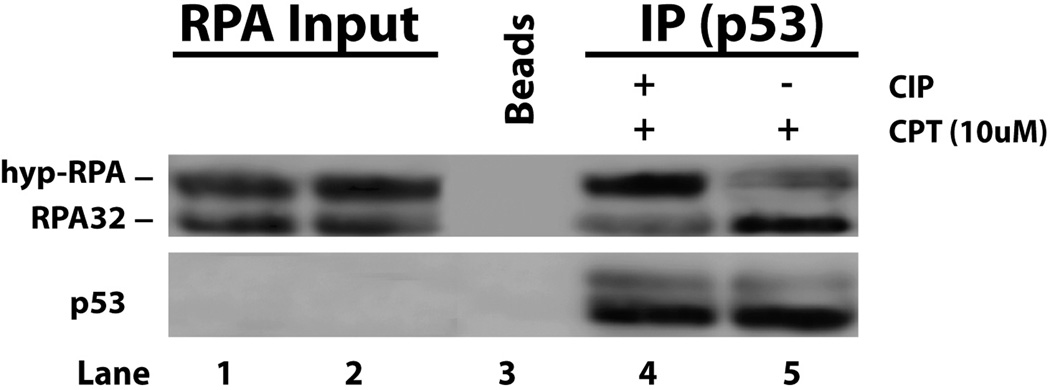
p53 is immunoprecipitated from A549 cell lysates using anti-p53 antibody. Samples were washed with 1 M salt buffer and incubated with Calf Intestinal Alkaline Phosphatase (CIAP). After washing, recombinant RPA and hyp-RPA were added in equal molar amounts and incubated with the endogenous p53 overnight. Samples were then spun down, washed and analyze by Western blotting.
Modulation of p53-RPA binding upon CPT treatment is DNA-PK, ATR and ATM dependent
Hyperphosphorylation of RPA in response to DNA damage is carried out by members of the phosphoinositide-3-kinase-related protein kinase (PIKK) family which includes ATM, ATR and DNA-PK (5, 39, 46). To identify the protein kinases involved in the phosphorylation-mediated regulation of the cellular p53-RPA interaction in response to CPT treatment, RPA hyperphosphorylation was evaluated in the cells treated with protein kinase inhibitors (Figures 4A and 4B), or depleted of ATR, ATM or DNA-PK by siRNAs (Figure 4C). The kinase activities of ATR and ATM were efficiently inhibited by caffeine, an inhibitor of ATR and ATM, as demonstrated by the inhibition of p53 phosphorylation at Ser15, a downstream DNA damage signaling event in the ATR and ATM checkpoint pathways (Figure 4A, left). The caffeine treatment inhibited the release of hyp-RPA from p53 since the hyp-RPA remained bound efficiently to p53 as compared with native RPA following DNA damage (Figure 4A, right). The results were further confirmed by the more specific ATM and ATR inhibitors Ku55933 and Nu6027, respectively (Figure 4B). Consistent results were also obtained with ATR-deficient cells (Figure S1). To further assess the effect of individual PIKK proteins on modulation of p53-RPA interaction, siRNAs were used to knockdown ATR, ATM, or DNA-PK (Figure 4C). Subsequent co-immunoprecipitation assays of cell lysates indicated that in agreement with the results of inhibitor treatments, depletion of ATR or ATM significantly increased the level of hyp-RPA binding to p53 versus control siRNA (Figure 4C). In addition, we found that DNA-PK was required for the CPT-induced RPA hyperphosphorylation while ATM and ATR are not, which is consistent with the previous reports (39, 46–48). As expected, knockdown of DNA-PK kept RPA bound to p53 (Figure 4C).
Figure 4. Modulation of p53-RPA binding is dependent on DNA-PK as well as ATM and ATR.

(A) A549 cells were treated with caffeine to inhibit ATM and ATR activities prior to the CPT treatment. Whole cell lysates were loaded in a 10% SDS page (left). Nuclear lysates then were isolated and subjected to DNase I digestion and 10% of sample was loaded as input. IP was subsequently performed using anti-p53 antibody and co-immunoprecipitated proteins were analyzed by Western blotting with the indicated antibodies. (B) A549 cells were synchronized in S-phase and treated with 10 µM ATM and/or ATR inhibitors (Ku55933 and Nu6027) for 1 hour prior to CPT treatment (10 µM for 2.5 hrs.) Whole cell lysates were subjected to DNase I digestion and 5% of the sample was loaded as input. IP was subsequently performed using anti-p53 antibody and the co-immunoprecipitated proteins were analyzed by Western blotting with the indicated antibodies. (C) A549 cells were transfected with ATM siRNA, ATR siRNA, DNA-PK siRNA or their combinations. Cells were treated with CPT. Whole cell lysates were collected and analyzed by Western blotting with indicated antibodies. Nuclear lysates then were isolated and subjected to DNase I digestion. Complexes with p53 were isolated by IP using anti-p53 antibody and the co-immunoprecipitated proteins were analyzed by Western blotting.
Phosphorylation of p53 at Ser37 and Ser46 is important for regulation of p53-RPA binding
Since phosphorylation of p53 at serine 15 is involved in DNA damage checkpoint signaling, it is of interest to determine if phosphorylation of this site is involved in modulating the p53-RPA interaction. We therefore transfected constructs for expressing wild-type and mutant p53 in which the serine was replaced with an alanine (S15A), respectively, into H1299 cells (p53−/−). After transfection cells were treated with CPT, nuclear lysates were prepared, and co-immunoprecipitation performed using anti-p53 antibody. In agreement with our in vivo data described above, we found that only non-phosphorylated RPA32 was able to be co-immunoprecipitated with p53 and that the S15A mutation did not affect the p53 binding to RPA (Figure 5A). To confirm the results, the same immunoprecipitates were washed with 1 M NaCl buffer to remove p53-associated. Then, an equimolar amount of recombinant RPA and hyp-RPA proteins were added. As shown in Figure 5B, the mutation at Ser15 in p53 did not affect p53-RPA binding.
Figure 5. Phosphorylations of Ser37 and Ser46 of p53 are important for regulation of p53-RPA binding.

(A) H1299 cells (p53−/−) were transfected with p53 wild type (WT) and p53 S15A constructs for 72 hrs. The cells were synchronized with APH before treatment with 10 uM CPT for 2 hrs. Nuclear fractions were isolated, treated with DNase I, followed by immunoprecipitation with anti-p53 antibody. The co-immunoprecipitated proteins were analyzed by Western blotting using indicated antibodies. (B) The immunoprecipitates generated as in (A) were washed with 1 M salt buffer to remove co-immunoprecipitated proteins. Then equimolar quantities of RPA and hyp-RPA were supplied to allow interaction with the immunoprecipitated p53. The p53-RPA interaction was analyzed by collecting the p53 immunoprecipitates for analysis by Western blotting. (C) H1299 cells were transfected with four different p53 constructs in which one single serine was mutated to alanine on the N terminus of p53 (WT, S15A, S20A, S37A, S46A). Transfections were done for 72 hrs, followed by treatment with CPT to induce p53 phosphorylation. (D) Whole cell lysates were prepared and subjected to IP with anti-p53 antibody. The immunoprecipitates were washed with 1 M salt buffer, and then supplied with equimolar mixture of purified RPA and hyp-RPA. The p53-RPA complex formation was analyzed by Western blotting. (E) Cells were transfected with ATM siRNA, ATR siRNA, or a combination of both, followed by CPT treatment. Prepared whole cell lysates with or without lambda phosphatase treatment were analyzed by Western blotting.
To identify the phosphorylation site(s) of p53 important for regulation of the p53-RPA interaction, we transfected H1299 cells with a series of p53 mutant expression constructs in which one single serine had been mutated to alanine. The mutations were all localized in the N-terminus of p53 (S15A, S20A, S37A, S46A). The transfected H1299 cells were treated with CPT to induce phosphorylation of p53 (Figure 5C). Anti-p53 antibody then was used to pull-down the p53. After washing with 1 M salt buffer, the immunoprecipitates were mixed with equimolar amounts of recombinant RPA and hyp-RPA to test their interactions with the p53 proteins. The S37A and S46A mutations prevented p53 dissociation from hyp-RPA relative to WT-p53, indicating that phosphorylations at Ser37 and Ser46 of p53 are required for release of RPA upon phosphorylation of RPA32 (Figure 5D). These observations suggest that the two particular serines are involved in regulating p53-RPA complex formation and stability in the CPT-induced DDR. Furthermore, individual knockdown of ATR and ATM identify the checkpoint kinases responsible for specific serine phosphorylation: the CPT-induced phosphorylation of p53 at Ser37 is primarily dependent on ATR while the phosphorylation at Ser46 depends on ATM.
Loss of hyperphosphorylation of RPA compromises DSB repair
DNA damage-induced hyperphosphorylation of RPA stimulates RPA localization to DSB repair and checkpoint complexes (13, 14), thus likely enhancing DSB repair. Also, the interaction of p53 with RPA mediates suppression of HR (24). Therefore, it is of interest to determine if phosphorylation-mediated regulation of the p53-RPA interaction plays a role in modulating DSB repair. Neutral comet assays were performed to assess the HR repair of CPT-induced DSBs in cells expressing PD-RPA versus cells expressing WT-RPA32. As shown in Figures 6A and 6B, repair of CPT-induced DSBs was significantly compromised in cells with PD-RPA in comparison to cells with WT-RPA. Consistently, in parallel experiments unphosphorylated RPA was efficiently co-immunoprecipitated with p53 in the cells expressing PD-RPA, while most hyp-RPA in the cells expressing wt-RPA was incapable of co-immunoprecipitation with p53 (Figure 6C, compare hyp-RPA to RPA ratios in lanes 6– 8 with lanes 14–16, respectively). These data suggest that RPA was unphosphorylated and, thus, sequestered in a p53-RPA complex in PD-RPA cells, inhibiting HR repair of CTP-induced DSBs. By contrast, RPA was extensively hyperphosphorylated and mostly free of binding to p53 in WT-RPA cells, making them available for HR repair.
Figure 6. Phosphorylation-mediated regulation of p53-RPA binding is required for DSB repair.

(A) Stable U2OS cells expressing WT- or PD-RPA32 were treated with CPT in a dose-dependent manner for 2 hrs. Comet assay under neutral conditions was performed to assess the efficiency of DSB repair. (B) Tail moment was measured using the Comet Assay IV software (Perceptive). At least 50 cells were assessed per treatment (* represents a p-value less than 0.001). (C) Co-immunoprecipitation assay was performed simultaneously using duplicate cell cultures. Nuclear lysates were isolated and anti-p53 antibody was used for immunoprecipitation; samples were then analyzed by Western blotting using the indicated antibodies. (D) A549 cells were treated with CPT or mock treated, followed by nuclear fractionation and incubation with DNase I. Soluble fractions were incubated with anti-p53 antibodies for co-immunoprecipitation (lanes 3–4). The supernatant after p53 IP then was immunoprecipitated again using Rad51 antibodies (lanes 7–8). (E) Cells expressing WT- or PD-RPA32 were treated with CPT and subjected to immunofluorescence microscopic determination of nuclear focus formation of Rad52. (F) Quantitative analysis of the data from (E). 100 cells were randomly selected in three separate experiments. Cells with at least one focus were counted (* represents a p-value less than 0.001).
We reasoned that RPA released from p53 sequestration by RPA32 phosphorylation would remain in the supernatant after IP pull-down of p53 and show association with DSB repair proteins. To test this, lysates from CPT-treated A549 cells were subjected to two consecutive immunoprecipitation steps in which p53 was immunoprecipitated first and then Rad51 was immunoprecipitated from the remaining supernatant. Although native RPA was efficiently sequestered by p53, little hyp-RPA was bound to the p53 in CPT-treated or -untreated cells (Figure 6D, lanes 3 and 4). Subsequently, anti-Rad51 antibody co-immunoprecipitated Rad51 and hyp-RPA from the remaining supernatant (lane 7) while little non-phosphorylated RPA was co-immunoprecipitated with Rad51. Similar results were obtained with U2OS cells expressing PD-RPA32 as compared with WT-RPA (Figure S2). Furthermore, CPT-induced nuclear focus formation of Rad52 was significantly reduced in cells expressing PD-RPA32 than those expressing wild-type RPA32 (Figures 6E and 6F). Rad51 interaction with ssDNA-bound RPA plays an important role in promoting Rad51 presynaptic filament assembling at DSBs (49–51), Thus, a significant amount of cellular RPA is sequestered in a p53-RPA complex under normal conditions and upon DNA damage, phosphorylation releases RPA or prevents hyp-RPA from binding to p53, promoting DSB repair.
Phosphorylation of Ser37 and Ser46 of p53 are important for homologous recombination repair
To further confirm the above results, constructs for expression of p53 with S37A or S46A mutation were generated. Then, we performed the pDR-GFP-based HR assays (52, 53) in H1299 cells transfected with the S37A or S46A p53 constructs in the presence or absence of CPT. As shown in Figures 7A and 7B, homologous recombination repair of the CPT-induced DSBs, as indicated by the cells emitting green fluorescence, was significantly compromised in cells expressing the S37A or the S46A p53 constructs in comparison to the cells expressing WT p53.
Figure 7. ATM- and ATR-dependent phosphorylation of Ser37 and Ser46 of p53 is important for efficient homologous recombination repair of DSBs.

(A) H1299 cells (p53−/−) were transfected simultaneously with the HR reporter pDR-GFP and a p53 construct (WT, S37A or S46A) for 48 hrs. The cells then were either treated with 5 uM CPT for 24 hrs to induce phosphorylation of p53 and DNA double-strand breaks or transfected with an I-SceI endonuclease expression vector for 36 hrs as a positive control. Phase contrast microscopy was used to visualize cells and GFP expressions was scored by fluorescence microscopy. The cells with green fluorescence indicated functional HR in these cells. (B) Percentage of GFP-positive cells was measured from a random selection of 100 cells in three separate experiments. (C) A549 cells were transfected with the HR reporter pDR-GFP for 48 hrs. Cells then were treated with 10 uM ATM and/or ATR inhibitors for 1 hr prior to CPT treatment (5 µM for 24 hrs). (D) Percentage of GFP positive cells was measured and analyzed as above.
ATM and ATM inhibition impairs homologous recombination repair
The same pDR-GFP-based HR assays also were performed with cells treated with ATM and ATR inhibitors KU55933 and NU6027, respectively. Figures 7C and 7B show that the inhibition of ATR kinase significantly reduced HR efficiency in cells treated with CPT. Furthermore, in the cells treated with the ATM inhibitor, the HR activity was also reduced, though not statistically significant (p = 0.08), as compared to the mock-treated cells. Consistently, when both inhibitors were used, the HR rate was significantly reduced in the inhibitor-treated versus mock-treated cells. Together, these results support a role of ATM and ATR kinases in regulation of HR, at least partially through their regulation of the p53-RPA interaction.
Discussion
Cellular DDRs are a complex defense system against genome instability which involves multiple biochemical pathways. In particular, HR and NHEJ repair pathways and ATM and ATR checkpoints play pivotal roles in cellular response to DSB damage. This study addresses important questions concerning how these pathways are regulated and coordinated with one another, important information for our understanding of the mechanisms of DDRs. We provide evidence that DNA-PK, the hallmark protein of NHEJ, together with ATR and ATM plays a regulatory role in the repair of CPT-induced DSBs, and this regulation is mediated by synergistic phosphorylations of both p53 and RPA. This finding reveals a novel crosstalk mechanism between HR and NHEJ pathways and coordination between ATM/ATR/p53 checkpoints and DNA-PK.
The complex mechanism unveiled in this study is centered on the regulation of p53-RPA interaction via site-specific post-translational modifications of p53 and RPA. Remarkably, the regulation requires participation of all three major PIKK family members involved in DDRs, DNA-PK, ATM and ATR. Upon DNA damage, each kinase phosphorylates specific sites of p53 or RPA to make a synergistic contribution to inducing p53-RPA dissociation. Specifically, DNA-PK hyperphosphorylates RPA at multiple sites in the N-terminal domain of RPA32, while ATR and ATM phosphorylate p53 at Ser37 and Ser46, respectively (Figure 5). Surprisingly, phosphorylation of p53 at Ser15, well known for its role in ATR/ATM-dependent checkpoint activation and DDRs (54–57) is not required (Figure 5). In addition, phosphorylation of p53 at Ser20 by Chk2 (58) does not participate either (Figure 5) consistent with the lack of effect of Chk2 or Chk1 on p53-RPA interaction (Figure S3). These data suggest that p53 phosphorylations involved in modulating p53-RPA interactions are carried out directly by ATR and ATM in a Chk1/Chk2-independent manner. Although excess ssDNA interfered with RPA-p53 complex formation (38), we found that equimolar ssDNA did not substantially inhibit the p53-RPA interaction (Figures 2C and 2D).
The impact of p53-RPA association/dissociation on their cellular functions could occur at multiple levels. Normally, RPA expression is constant at a relatively abundant level during cell cycle transit (59). It is known that p53 interacts with RPA via p53’s N-terminal domain containing the transactivation and trans-repression functions of the protein (60). Since a basal level of p53 is required for antioxidant activities in normal cell growth (61), the p53-RPA complex formation may serve to mask this p53 domain and prevent the above-basal levels of free p53 from interrupting normal cellular functions, complementing the MDM2 function of sequestering and inactivating p53. With significant DNA damage, however, cellular p53 is greatly elevated while expression of RPA remains unaffected (62). Here, disruption of the p53-RPA complex may be necessary to free RPA for functioning in DDRs as RPA plays indispensable roles in DNA damage checkpoint and repair pathways. Indeed, our results indicate that a deficiency in RPA phosphorylation and release from the p53-RPA complex significantly reduces repair efficiency of DSBs induced by CPT (Figures 6 and 7). The released hyp-RPA binds much more efficiently to Rad51 than does native RPA (Figure 6D) (13). These observations suggest that the phosphorylations of RPA and p53 not only frees RPA during DDR, but also allows RPA to more efficiently recruit Rad51 to the DSB sites during an early step of HR, thus promoting the repair process (13, 33). In addition, the phosphorylations may serve to prevent RPA sequestration by increasing amounts of p53. Furthermore, although p53 is highly expressed in cells following DNA damage, it is also possible that released phosphorylated p53 could enhance the DNA damage checkpoints and transcriptional activation of genes involved in DDRs. In this RPA might be a regulatory element ensuring that p53 would be available only after DNA damage.
The multiple diverse functions for both RPA and p53 imply that the DNA-PK/ATM/ATR modulation of the p53-RPA interaction may have multiple, varied impacts on the DDRs beyond HR repair. Activation of tumor suppressor protein p53 orchestrates multiple cellular responses involved in cell cycle control and apoptosis (42, 43). Also, RPA is involved in almost every, if not all, DDR pathways, from damage signaling, checkpoint activation through DNA repair (5). Also, hyp-RPA is more efficient in recruiting the checkpoint complex Rad9/Rad1/Hus1 (14), preventing its association with replication centers (29), facilitating mitotic exit in response to mitotic DNA damage (63), and regulating mismatch repair (31). These potential hyp-RPA activities form a complex and interacting DDR network dependent on the stability of the p53-RPA interaction regulated by the PIKK members.
Given that p53 interacts with RPA via its N-terminal domain (60) and that the phosphorylation at S37 and S46 in the N-terminus of p53 by ATR/ATM disrupted p53-RPA interactions (Figure 5), these phosphorylations may interfere with RPA binding to the N-terminus of p53. This disruption of the p53-RPA complex requires the concomitant hyperphosphorylation of RPA32. As reported previously, hyperphosphorylation alters RPA conformation (32). Thus, this may structurally change the p53-binding domain/motif of RPA although this change alone may not be sufficient to disrupt the formation of the p53-RPA complex. On the other hand, the phosphorylation at S37 and S46 in the N-terminal domain of p53 changes both the chemistry and structure of the domain. It is likely that combination of these changes with those in RPA due to hyperphosphorylation prevents RPA from binding to p53. However, revealing the details of the phosphorylation-induced structural changes is beyond the scope of the current study but deserves further investigation.
Taken together, we propose that under unstressed conditions, the low level of ‘free’ p53 is sequestered by the abundant RPA in cells. The sequestration not only prevents relatively high levels of p53 from interfering with normal cellular functions and cell cycle progression, but also may help to maintain a basal level of p53 for upregulation of a few genes for activities against DNA damage induced by endogenous reactive oxygen species in cells under normal growth conditions. Upon severe DNA damage, however, phosphorylation of p53 and RPA by ATM/ATR and DNA-PK, respectively, prevents RPA sequestration by the damage-induced high level accumulation of p53, freeing phosphorylated forms of both p53 and RPA for DDR functions.
Materials and methods
Cells, cell culture, proteins and antibodies
A549 cells were maintained at 37°C under a humidified atmosphere of 5% CO2 in Dulbecco’s modified Eagle medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS; HyClone), 1% penicillin/streptomycin. U2OS cells expressing RPA32 wild-type (WT-RPA) or a hyperphosphorylation-deficient mutant (PD-RPA) (kindly provided by Dr. Xiaohua Wu; Scripps Research Institute, La Jolla, California, USA) were maintained in DMEM supplemented with 10% FBS and antibiotics as described above. These U2OS cells were grown in hygromycin (200 µg/mL) and puromycin (1 µg/mL) to maintain plasmid expression. These are stable cell lines in which the endogenous RPA32 was stably knocked down while recombinant WT-RPA or PD-RPA were stably produced (34). The HCT-116 ATR−/− cells, also known as ATRflox/− cells (kindly provided by Dr. Stephen Elledge, Harvard University), were grown as described above using McCoy’s 5A medium (ATCC).
Recombinant human RPA was expressed and purified as described (Yang et al., 2002). Hyp-RPA was purified using previous procedures (Patrick et al., 2005). In addition, hyperphosphorylation of RPA by purified DNA-PK (Promega, Madison, WI, U.S.A.) was carried out as described (Liu et al., 2005).
Antibodies used in this study include anti-RPA32 (Sigma R3280), anti-p53 (Invitrogen AHO0142 or Santa Cruz sc-6243), anti-phospho-p53(pSer15) (R&D AF1043 and Cell Signaling 9286), anti-phospho-p53(pSer20) (AnaSpec 54428), anti-phospho-p53(pSer37) (Santa Cruz sc-135633), anti-phospho-p53(Ser46) (Cell Signaling 2521), anti-DNA-PK (Santa Cruz sc-9051), anti-ATM (Bethyl Lab A300-299A), anti-ATR (Bethyl Lab A300-138A) and anti-RAD51 (Santa Cruz sc-8349).
S-phase cell synchronization
To optimize RPA32 hyperphosphorylation in response to CPT treatment cells were synchronized in S-phase by incubating with aphidicolin (APH) (1 ug/mL) for 18 hrs before release into fresh media for 2 hrs. Synchronized cells were then treated with CPT.
Co-immunoprecipitation
Immunoprecipitation (IP) in U2OS cell lysates was done after subcellular fractionation: cells were collected with a policeman and resuspended in CSK buffer (10 mM PIPES, pH 6.8, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, 0.1% Triton X-100, phosphatases and protease inhibitors) and incubated at 4 °C for 5 min. Low speed centrifugation (1,300×g/5 min) separated cytoplasmic proteins from pelleted nuclei. Isolated nuclei were lysed in solution B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, phosphatase and protease inhibitors). Chromatin-bound proteins were collected (1500×g centrifugation/5 min), and resuspended in IP buffer (20 mM Tris-HCl, pH 7.8, 137 mM NaCl, 10% glycerol, 2 mM EDTA, 1% NP-40) and subjected to DNase I digestion. Lysates were cleared (centrifugation at 13,000×g/15 min, 4 °C) and received 3 ug of anti-p53 antibodies for immunoprecipitation and incubated overnight at 4 °C, followed by incubation with protein G beads (Invitrogen 10-1242) for 2 hrs. Immune complexes were collected by centrifugation at 1,000×g.
Pull-down assays
Recombinant GST-tagged p53 protein (SignalChem P05-30BG) was incubated with purified RPA or hyp-RPA in RPA binding buffer at 4 °C overnight. 10% of the sample was loaded as “Input”. Pre-equilibrated GST-agarose beads (GE) were added to the remaining sample and incubated at 4 °C for 2 hrs. Immune complexes were collected by centrifugation at 1000×g, washed 3× with RPA binding buffer and analyzed by Western blotting.
For the p53-RPA interaction involving ssDNA, purified RPA or hyp-RPA was incubated with 5’-biotinylated ssDNA (dT30mer or dT90mer) at indicated ratios for 30 min at 25 °C in RPA binding buffer. Pre-equilibrated streptavidin beads were supplied and the samples incubated for 2 hrs at 4 °C. DNA-streptavidin complexes were collected, and then washed twice with RPA binding buffer to remove unbound RPA. Subsequently, recombinant p53 was supplied and the mixture incubated overnight at 4 °C. Complexes were collected at 1000 × g, washed 3× with RPA binding buffer and analyzed by Western blotting.
siRNA and plasmid constructs transfections
Cells were transfected with siRNA for 72 hrs using INTERFERin transfection reagent (Polyplus 409-10) following the manufacturer instructions. The siRNAs include ATM: CAUACUACUCAAAGACAUUTT, AAUGUCUUUGAGUAGUAUGTT, ATR: CCUCCGUGAUGUUGCUUGATT, UCAAGCAACAUCACGGAGGTT, DNA-PK: AGGGCCAAGCUGUCACUCUTT, AGAGUGACAGCUUGGCCCUTT. The pCB6-p53-WT and pCB6-p53-S15A expression constructs (kindly provided by Dr. Karen Vousden, Beatson Institute for Cancer, Bearsden, Glasgow, UK) were transfected into cells using JetPI transfection reagent (Polyplus 101-10) according to the manufacturer’s instructions for 72 hrs. Similar transfections were performed with pCAG3.1-p53-WT, -S15A, -S20A, -S37A and -S46A expression vectors (kindly provided by Dr. Carl W. Anderson, Biology Department, Brookhaven National Laboratory, Upton, New York).
Comet Assay
U2OS cells stably expressing RPA32-WT or PD-RPA were treated with increasing doses of CPT for 2 hrs. Then, neutral comet assays were carried out using the Comet Assay System (Trevigen) according to the manufacturer’s instructions. Fluorescence images were captured using a Nikon inverted fluorescent microscope with attached CCD camera at 100× magnification and the comet tail moment was measured using Comet Assay IV software (Perceptive). At least 50 cells were assessed per treatment. In parallel with the comet assay, cell cultures with the same treatments were harvested for co-immunoprecipitation and the proteins analyzed by Western blotting.
Homologous Recombination Assays
H1299 (p53−/−) or A549 cells were transfected with the HR reporter pDR-GFP (a gift of Maria Jasin, Addgene plasmid #26475) for 48 hrs. H1299 cells also were transfected simultaneously with the p53-expression constructs (WT, S37A and S46A), while A549 cells were treated with ATM and/or ATR inhibitors. Then, cells were either treated with 5 uM CPT for 24 hrs to induce phosphorylation of RPA and p53 and DNA double-strand breaks or transfected for 36 hrs (control) with an I-SceI expression vector (pCBASceI, a gift of Maria Jasin, Addgene plasmid #26477). Following the treatments, cells were visualized in phase contract or for green fluorescence using fluorescence microscopy. At least 100 cells were scored for GFP positive in three independent experiments.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgement
We gratefully acknowledge Dr. Xiaohua Wu for providing U2OS cells expressing RPA32-WT and PD-RPA proteins. We also gratefully acknowledge Dr. Carl W. Anderson for providing the p53 expression constructs (pCAG3.1-WT, -S15A, -S20A, -S37A and -S46A) and Dr. Karen Vousden for the pCB6 expression vectors p53-WT and p53-S15A. This work is supported by National Institutes of Health grants CA86927 and GM083307 (to Y.Z.) as well as ES017214 (to M.A.S.).
References
- 1.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 2.Huen MS, Chen J. Assembly of checkpoint and repair machineries at DNA damage sites. Trends Biochem Sci. 2009 Feb;35(2):101–108. doi: 10.1016/j.tibs.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007 Dec 14;28(5):739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 4.Wood JL, Chen J. DNA-damage checkpoints: location, location, location. Trends Cell Biol. 2008 Oct;18(10):451–455. doi: 10.1016/j.tcb.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 5.Zou Y, Liu Y, Wu X, Shell SM. Functions of human replication protein A (RPA): from DNA replication to DNA damage and stress responses. J Cell Physiol. 2006 Aug;208(2):267–273. doi: 10.1002/jcp.20622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shell SM, Zou Y. Ahmad S, Hanaoka F. Molecular Mechanisms of Ataxia Telangiectasia Landes Bioscience. 2009. Protein-protein interactions in Ataxia telangiectesia; pp. 42–51. [Google Scholar]
- 7.Arnaudeau C, Lundin C, Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol. 2001 Apr 13;307(5):1235–1245. doi: 10.1006/jmbi.2001.4564. [DOI] [PubMed] [Google Scholar]
- 8.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006 Oct;6(10):789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 9.Couedel C, Mills KD, Barchi M, Shen L, Olshen A, Johnson RD, et al. Collaboration of homologous recombination and nonhomologous end-joining factors for the survival and integrity of mice and cells. Genes Dev. 2004 Jun 1;18(11):1293–1304. doi: 10.1101/gad.1209204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mills KD, Ferguson DO, Essers J, Eckersdorff M, Kanaar R, Alt FW. Rad54 and DNA Ligase IV cooperate to maintain mammalian chromatid stability. Genes Dev. 2004 Jun 1;18(11):1283–1292. doi: 10.1101/gad.1204304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hill R, Lee PW. The DNA-dependent protein kinase (DNA-PK): More than just a case of making ends meet? Cell Cycle. 2010 Sep;9(17):3460–3469. doi: 10.4161/cc.9.17.13043. [DOI] [PubMed] [Google Scholar]
- 12.Meek K, Dang V, Lees-Miller SP. DNA-PK: the means to justify the ends? Adv Immunol. 2008;99:33–58. doi: 10.1016/S0065-2776(08)00602-0. [DOI] [PubMed] [Google Scholar]
- 13.Wu X, Yang Z, Liu Y, Zou Y. Preferential localization of hyperphosphorylated replication protein A to double-strand break repair and checkpoint complexes upon DNA damage. Biochem J. 2005 Nov 1;391(Pt 3):473–480. doi: 10.1042/BJ20050379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu X, Shell SM, Zou Y. Interaction and colocalization of Rad9/Rad1/Hus1 checkpoint complex with replication protein A in human cells. Oncogene. 2005 Jul 7;24(29):4728–4735. doi: 10.1038/sj.onc.1208674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zou L, Liu D, Elledge SJ. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc Natl Acad Sci U S A. 2003 Nov 25;100(24):13827–13832. doi: 10.1073/pnas.2336100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003 Jun 6;300(5625):1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 17.Yang ZG, Liu Y, Mao LY, Zhang JT, Zou Y. Dimerization of human XPA and formation of XPA2-RPA protein complex. Biochemistry. 2002 Oct 29;41(43):13012–13020. doi: 10.1021/bi026064z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robison JG, Bissler JJ, Dixon K. Replication protein A is required for etoposide-induced assembly of MRE11/RAD50/NBS1 complex repair foci. Cell Cycle. 2007 Oct 1;6(19):2408–2416. doi: 10.4161/cc.6.19.4773. [DOI] [PubMed] [Google Scholar]
- 19.Mer G, Bochkarev A, Gupta R, Bochkareva E, Frappier L, Ingles CJ, et al. Structural basis for the recognition of DNA repair proteins UNG2, XPA, and RAD52 by replication factor RPA. ell. 2000 Oct 27;103(3):449–456. doi: 10.1016/s0092-8674(00)00136-7. [DOI] [PubMed] [Google Scholar]
- 20.Sugiyama T, Kantake N. Dynamic regulatory interactions of rad51, rad52, and replication protein-a in recombination intermediates. J Mol Biol. 2009 Jul 3;390(1):45–55. doi: 10.1016/j.jmb.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 21.Stauffer ME, Chazin WJ. Physical interaction between replication protein A and Rad51 promotes exchange on single-stranded DNA. J Biol Chem. 2004 Jun 11;279(24):25638–25645. doi: 10.1074/jbc.M400029200. [DOI] [PubMed] [Google Scholar]
- 22.Jackson D, Dhar K, Wahl JK, Wold MS, Borgstahl GE. Analysis of the human replication protein A:Rad52 complex: evidence for crosstalk between RPA32, RPA70, Rad52 and DNA. J Mol Biol. 2002 Aug 2;321(1):133–148. doi: 10.1016/s0022-2836(02)00541-7. [DOI] [PubMed] [Google Scholar]
- 23.Sommers JA, Sharma S, Doherty KM, Karmakar P, Yang Q, Kenny MK, et al. p53 modulates RPA-dependent and RPA-independent WRN helicase activity. Cancer Res. 2005 Feb 15;65(4):1223–1233. doi: 10.1158/0008-5472.CAN-03-0231. [DOI] [PubMed] [Google Scholar]
- 24.Romanova LY, Willers H, Blagosklonny MV, Powell SN. The interaction of p53 with replication protein A mediates suppression of homologous recombination. Oncogene. 2004;23(56):9025–9033. doi: 10.1038/sj.onc.1207982. [DOI] [PubMed] [Google Scholar]
- 25.Derheimer FA, O'Hagan HM, Krueger HM, Hanasoge S, Paulsen MT, Ljungman M. RPA and ATR link transcriptional stress to p53. Proc Natl Acad Sci U S A. 2007 Jul 31;104(31):12778–12783. doi: 10.1073/pnas.0705317104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hass CS, Gakhar L, Wold MS. Functional characterization of a cancer causing mutation in human replication protein A. Mol Cancer Res. 2010 Jul;8(7):1017–1026. doi: 10.1158/1541-7786.MCR-10-0161. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Putnam CD, Kane MF, Zhang W, Edelmann L, Russell R, et al. Mutation in Rpa1 results in defective DNA double-strand break repair, chromosomal instability and cancer in mice. Nat Genet. 2005 Jul;37(7):750–755. doi: 10.1038/ng1587. [Research Support, N.I.H., Extramural Research Support, U.S. Gov't, P.H.S.] [DOI] [PubMed] [Google Scholar]
- 28.Binz SK, Sheehan AM, Wold MS. Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair (Amst) 2004 Aug-Sep;3(8–9):1015–1024. doi: 10.1016/j.dnarep.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 29.Vassin VM, Wold MS, Borowiec JA. Replication protein A (RPA) phosphorylation prevents RPA association with replication centers. Mol Cell Biol. 2004 Mar;24(5):1930–1943. doi: 10.1128/MCB.24.5.1930-1943.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patrick SM, Oakley GG, Dixon K, Turchi JJ. DNA damage induced hyperphosphorylation of replication protein A. 2. Characterization of DNA binding activity, protein interactions, and activity in DNA replication and repair. Biochemistry. 2005 Jun 14;44(23):8438–8448. doi: 10.1021/bi048057b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo S, Zhang Y, Yuan F, Gao Y, Gu L, Wong I, et al. Regulation of replication protein A functions in DNA mismatch repair by phosphorylation. J Biol Chem. 2006 Aug 4;281(31):21607–21616. doi: 10.1074/jbc.M603504200. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Kvaratskhelia M, Hess S, Qu Y, Zou Y. Modulation of Replication Protein A Function by Its Hyperphosphorylation-induced Conformational Change Involving DNA Binding Domain B. J Biol Chem. 2005 Sep 23;280(38):32775–32783. doi: 10.1074/jbc.M505705200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi W, Feng Z, Zhang J, Gonzalez-Suarez I, Vanderwaal RP, Wu X, et al. The role of RPA2 phosphorylation in homologous recombination in response to replication arrest. Carcinogenesis. 2010 Jun;31(6):994–1002. doi: 10.1093/carcin/bgq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olson E, Nievera CJ, Klimovich V, Fanning E, Wu X. RPA2 Is a Direct Downstream Target for ATR to Regulate the S-phase Checkpoint. Biol Chem. 2006 Dec 22;281(51):39517–39533. doi: 10.1074/jbc.M605121200. [DOI] [PubMed] [Google Scholar]
- 35.Nuss JE, Patrick SM, Oakley GG, Alter GM, Robison JG, Dixon K, et al. DNA damage induced hyperphosphorylation of replication protein A. 1. Identification of novel sites of phosphorylation in response to DNA damage. Biochemistry. 2005 Jun 14;44(23):8428–8437. doi: 10.1021/bi0480584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vise PD, Baral B, Latos AJ, Daughdrill GW. NMR chemical shift and relaxation measurements provide evidence for the coupled folding and binding of the p53 transactivation domain. Nucleic Acids Res. 2005;33(7):2061–2077. doi: 10.1093/nar/gki336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bochkareva E, Kaustov L, Ayed A, Yi G-S, Lu Y, Pineda-Lucena A, et al. Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proceedings of the National Academy of Sciences of the United States of America. 2005 Oct 25;102(43):15412–15417. doi: 10.1073/pnas.0504614102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller S, Moses K, Jayaraman L, Prives C. Complex formation between p53 and replication protein A inhibits the sequence-specific DNA binding of p53 and is regulated by single-stranded DNA. Mol Cell Biol. 1997 Apr 1;17(4):2194–2201. doi: 10.1128/mcb.17.4.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abramova NA, Russell J, Botchan M, Li R. Interaction between replication protein A and p53 is disrupted after UV damage in a DNA repair-dependent manner. Proceedings of the National Academy of Sciences of the United States of America. 1997 Jul 8;94(14):7186–7191. doi: 10.1073/pnas.94.14.7186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuhn C, Muller F, Melle C, Nasheuer HP, Janus F, Deppert W, et al. Surface plasmon resonance measurements reveal stable complex formation between p53 and DNA polymerase alpha. Oncogene. 1999 Jan 21;18(3):769–774. doi: 10.1038/sj.onc.1202327. [DOI] [PubMed] [Google Scholar]
- 41.Dutta A, Ruppert JM, Aster JC, Winchester E. Inhibition of DNA replication factor RPA by p53. Nature. 1993 Sep 2;365(6441):79–82. doi: 10.1038/365079a0. [DOI] [PubMed] [Google Scholar]
- 42.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997 Feb 7;88(3):323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 43.Ko LJ, Prives C. p53: puzzle and paradigm. Genes & Development. 1996 May 1;10(9):1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- 44.Efeyan A, Serrano M. p53: Guardian of the Genome and Policeman of the Oncogenes. Cell cycle, Landes Bioscience. 2007 doi: 10.4161/cc.6.9.4211. [DOI] [PubMed] [Google Scholar]
- 45.Kaustov L, Yi GS, Ayed A, Bochkareva E, Bochkarev A, Arrowsmith CH. p53 transcriptional activation domain: a molecular chameleon? Cell Cycle. 2006 Mar;5(5):489–494. doi: 10.4161/cc.5.5.2489. [DOI] [PubMed] [Google Scholar]
- 46.Shao RG, Cao CX, Zhang H, Kohn KW, Wold MS, Pommier Y. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. Embo J. 1999 Mar 1;18(5):1397–1406. doi: 10.1093/emboj/18.5.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oakley GG, Patrick SM, Yao J, Carty MP, Turchi JJ, Dixon K. RPA phosphorylation in mitosis alters DNA binding and protein-protein interactions. Biochemistry. 2003 Mar 25;42(11):3255–3264. doi: 10.1021/bi026377u. [DOI] [PubMed] [Google Scholar]
- 48.Sakasai R, Teraoka H, Tibbetts RS. Proteasome inhibition suppresses DNA-dependent protein kinase activation caused by camptothecin. DNA Repair (Amst) 2009 Jan 2;9(1):76–82. doi: 10.1016/j.dnarep.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kantake N, Sugiyama T, Kolodner RD, Kowalczykowski SC. The recombination-deficient mutant RPA (rfa1-t11) is displaced slowly from single-stranded DNA by Rad51 protein. The Journal of biological chemistry. 2003 Jun 27;278(26):23410–23417. doi: 10.1074/jbc.M302995200. [Research Support, U.S. Gov't, P.H.S.] [DOI] [PubMed] [Google Scholar]
- 50.Sugiyama T, Kantake N, Wu Y, Kowalczykowski SC. Rad52-mediated DNA annealing after Rad51-mediated DNA strand exchange promotes second ssDNA capture. The EMBO journal. 2006 Nov 29;25(23):5539–5548. doi: 10.1038/sj.emboj.7601412. [Research Support, Non-U.S. Gov't] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Plate I, Hallwyl SC, Shi I, Krejci L, Muller C, Albertsen L, et al. Interaction with RPA is necessary for Rad52 repair center formation and for its mediator activity. The Journal of biological chemistry. 2008 Oct 24;284(43):29077–29085. doi: 10.1074/jbc.M804881200. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pierce AJ, Hu P, Han M, Ellis N, Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001 Dec 15;15(24):3237–3242. doi: 10.1101/gad.946401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnson RD, Liu N, Jasin M. Mammalian XRCC2 promotes the repair of DNA double-strand breaks by homologous recombination. Nature. 1999 Sep 23;401(6751):397–399. doi: 10.1038/43932. [DOI] [PubMed] [Google Scholar]
- 54.Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997 Oct 31;91(3):325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 55.Khanna KK, Keating KE, Kozlov S, Scott S, Gatei M, Hobson K, et al. ATM associates with and phosphorylates p53: mapping the region of interaction. Nat Genet. 1998 Dec;20(4):398–400. doi: 10.1038/3882. [DOI] [PubMed] [Google Scholar]
- 56.Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998 Sep 11;281(5383):1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 57.Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, et al. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999 Jan 15;13(2):152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chehab NH, Malikzay A, Appel M, Halazonetis TD. Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev. 2000 Feb 1;14(3):278–288. [PMC free article] [PubMed] [Google Scholar]
- 59.Virshup DM, Wold MS, Weinberg DH, Kauffman MG, Kelly TJ. Cellular proteins involved in SV40 DNA Replication in vitro. Molecular Mechanisms in DNA Replication and Recombination. In: Richardson CC, Lehman IR, Liss Alan R, editors. Molecular Mechanisms in DNA Replication and Recombination. New York: 1990. pp. 303–314. [Google Scholar]
- 60.Leiter LM, Chen J, Marathe T, Tanaka M, Dutta A. Loss of transactivation and transrepression function, and not RPA binding, alters growth suppression by p53. Oncogene. 1996 Jun 20;12(12):2661–2668. [PubMed] [Google Scholar]
- 61.Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005 Dec;11(12):1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun J, Oma Y, Harata M, Kono K, Shima H, Kinomura A, et al. ATM modulates the loading of recombination proteins onto a chromosomal translocation breakpoint hotspot. PLoS One. 2010;5(10):e13554. doi: 10.1371/journal.pone.0013554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anantha RW, Sokolova E, Borowiec JA. RPA phosphorylation facilitates mitotic exit in response to mitotic DNA damage. Proc Natl Acad Sci U S A. 2008 Sep 2;105(35):12903–12908. doi: 10.1073/pnas.0803001105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.