

CRISPR (Clustered Regularly Interspaced Palindromic Repeats)-CAS (CRISPR-associated) systems are a bacterial defense against invading foreign nucleic acids derived from bacteriophages or exogenous plasmids1–4. These systems utilize an array of small CRISPR RNAs (crRNAs) consisting of repetitive sequences flanking unique spacers to recognize their targets, and conserved CAS proteins to mediate target degradation5–8. Recent studies have suggested that these systems may have broader functions in bacterial physiology, and it is unknown if they regulate expression of endogenous genes9,10. Here, we demonstrate that the CAS protein Cas9 of Francisella novicida utilizes a unique, small, CRISPR-CAS-associated RNA (scaRNA) to repress an endogenous transcript encoding a bacterial lipoprotein (BLP). As BLPs trigger a proinflammatory innate immune response aimed at combating pathogens11,12, CRISPR-CAS mediated repression of BLP is critical for F. novicida to dampen this host response and promote virulence. Since Cas9 proteins are highly enriched in pathogenic and commensal bacteria, our work suggests that CRISPR-CAS-mediated gene regulation may broadly contribute to the regulation of endogenous genes, particularly during the interaction of such bacteria with eukaryotic hosts.
F. novicida is a model intracellular pathogen that evades host defenses as it traffics through the phagosome of eukaryotic cells to replicate to high numbers within the cytosol. Specifically, it has developed mechanisms to prevent recognition by a variety of pattern recognition receptors (PRR) that detect bacteria and localize to the surface and phagosomes of host phagocytic cells13. One PRR, Toll-like Receptor 2 (TLR2), recognizes BLP and is critical for defense against F. novicida11–15. By dampening TLR2 activation, F. novicida reaches its replicative niche in the cytosol without inducing significant inflammatory signaling, promoting its pathogenesis13. We demonstrated that F. novicida gene FTN_0757 is involved in the repression of a BLP, FTN_1103, although its mechanism of action was unclear16. Unexpectedly, bioinformatic analysis revealed that FTN_0757 has significant sequence similarity to the CRISPR-CAS system protein Cas9 (15–65% across conserved regions)(Supplementary Fig. 1), typically known to degrade foreign DNA6,7, and not currently known to regulate endogenous gene expression. Furthermore, FTN_0757 is present in a complete Type II CRISPR-CAS locus, similar to those in the genomes of pathogens and commensals such as Streptococcus spp., Neisseria spp., Campylobacter spp., and Lactobacillus spp. (Supplementary Fig. 2 and Table S1). The locus contains Cas1, Cas2, and Cas4, all predicted to be involved in acquisition of new targeting crRNAs1,8,17, as well as a predicted trans-activating RNA (tracrRNA), an accessory small RNA (sRNA) necessary for crRNA activity18. It also contains a unique sRNA19 previously undescribed in a CRISPR-CAS locus, distinct from the crRNAs and tracrRNA, which we term small CRISPR-CAS-associated RNA (scaRNA)(Fig. 1a).
Figure 1. Cas9, tracrRNA, and scaRNA are necessary for FTN_1103 repression.
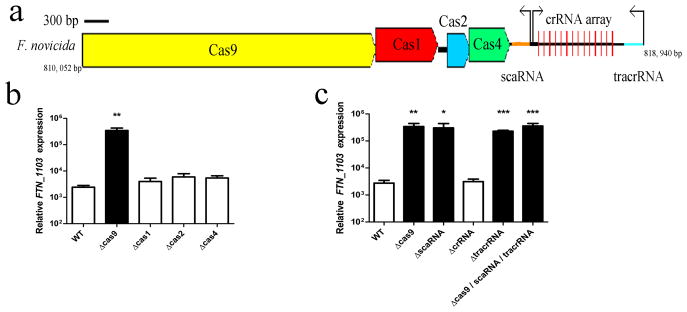
(A) Schematic of the F. novicida Type II CRISPR-CAS locus, containing cas9, cas1, cas2, and cas4, as well as the crRNA array (repeats indicated by vertical red lines), tracrRNA (blue), scaRNA (orange), and predicted promoters (black arrows). Relative expression of FTN_1103 in (B) wild-type (WT), Δcas9, Δcas1, Δcas2, and Δcas4 strains and (C) WT, Δcas9, ΔscaRNA, ΔcrRNA, and ΔtracrRNA strains (n = 4, bars represent s.d.).
Since FTN_0757 (cas9) is in a CRISPR-CAS locus (Fig. 1a), we tested whether its ability to repress FTN_1103 required the canonical CRISPR-CAS system or if an alternative mechanism was involved. Deletion of cas9, but not other CAS genes, led to 100-fold increased levels of FTN_1103 transcript (Fig. 1b). Since Cas9 degrades DNA targeted by crRNAs, we tested whether the crRNA array or the tracrRNA were necessary for FTN_1103 repression. Deletion of the crRNA array did not alter FTN_1103 transcript levels (Fig. 1c); however, deletion of the tracrRNA resulted in increased FTN_1103 transcript, similar to the cas9 mutant (Fig. 1c). Additionally, deletion of the scaRNA resulted in increased FTN_1103 transcript, indicating that it is also critical for FTN_1103 repression. Complementation of the cas9, tracrRNA, and scaRNA mutants restored FTN_1103 expression to near wild-type levels, and levels of FTN_1103 transcript in the mutants correlated with an increase in protein production (Supplementary Figs 3 and 4). Furthermore, a triple mutant lacking cas9, tracrRNA, and scaRNA expressed similar levels of FTN_1103 as the single mutants, providing genetic evidence that these components may act together within the same regulatory pathway.
CRISPR-CAS systems mediate degradation of their nucleic acid targets, so we tested whether Cas9, tracrRNA, and scaRNA mediated repression of FTN_1103 via degradation. Following treatment with rifampin to prevent mRNA production, FTN_1103 transcript was rapidly depleted in wild-type cells (Fig. 2a). In contrast, FTN_1103 transcript was not degraded in cas9, tracrRNA, or scaRNA mutants (Fig. 2a), indicating that each of these components is required for its degradation. Cas9 proteins contain four RuvC endonuclease domains (RuvC-I through RuvC-IV) and an HNH endonuclease domain (Fig 2b, Supplementary Fig. 1)17. RuvC-I and the HNH are necessary for degradation of target DNA6,7. We constructed point mutant strains lacking conserved residues17 in each endonuclease domain to determine if they were necessary for repression of FTN_1103 (Fig. 2b). These strains maintained wild-type levels of FTN_1103 (as well as cas9), indicating that none of these domains were required for this activity (Fig. 2c, Supplementary Fig. 5). Additionally, we found no role for known RNases in FTN_1103 repression (Supplementary Fig. 6). Cas9 proteins also contain a previously uncharacterized, conserved, arginine-rich motif (ARM)17(Fig. 2b, Supplementary Fig. 1), motifs that are known to mediate protein-RNA interactions20. A point mutation in the ARM (R59A) completely abrogated the ability of Cas9 to repress FTN_1103 (Fig. 2c), implicating the potential importance of Cas9:RNA interactions.
Figure 2. Cas9, tracrRNA, and scaRNA associate and mediate FTN_1103 degradation.
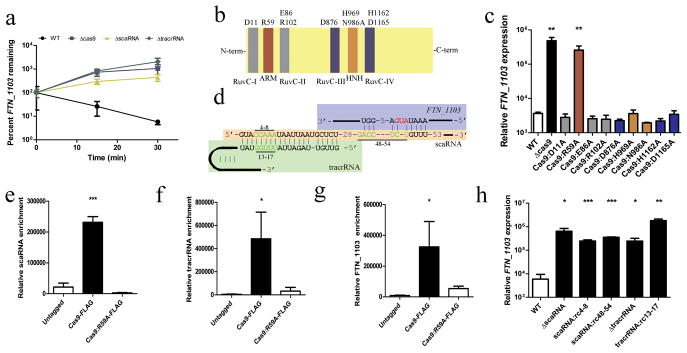
(A) FTN_1103 stability in the indicated strains (n = 3). (B) Schematic of Cas9 indicating five endonuclease domains (RuvC-I - RuvC-IV, HNH) and the ARM. (C) Relative expression of FTN_1103 in the indicated strains (n = 4). (D) Schematic of predicted hybridization between tracrRNA, scaRNA, and FTN_1103. Bars highlight mutated bases (green), red represents the FTN_1103 start codon. (E–G) Immunoprecipitation from WT, Cas9-FLAG, or Cas9:R59A-FLAG, and qRT-PCR for (E) scaRNA, (F) tracrRNA or (G) FTN_1103 (n = 4). (H) Relative expression of FTN_1103 in WT, ΔscaRNA, scaRNA:rc4-8 (reverse complement of bases 4–8), scaRNA:rc48–54, ΔtracrRNA, and tracrRNA:rc13-17 strains (n = 4, bars represent s.d.).
We therefore analyzed the sequences of the tracrRNA and scaRNA and predicted that the tracrRNA could hybridize to a degenerate repeat region in the scaRNA (Fig. 2d, Supplementary Figure 7), similar to the interaction between the tracrRNA and the repeat region of a crRNA, which is necessary for targeting DNA18. We also predicted that a region of the scaRNA could hybridize to a portion of the FTN_1103 transcript encompassing the start codon and ribosomal binding site (RBS)(Fig. 2d, Supplementary Figure 7). To determine whether Cas9 and the RNAs associated, we immunoprecipitated Cas9 from a strain expressing a FLAG-tagged version of this protein. tracrRNA, scaRNA, and FTN_1103 mRNA were significantly enriched in association with Cas9 (Fig. 2e–g). However, these associations were abrogated in the Cas9 ARM mutant (R59A), suggesting this motif is necessary for Cas9 interaction with these RNAs. We next generated reverse complement mutations in the tracrRNA region (bases 13–17) predicted to interact with the scaRNA, as well as the scaRNA regions predicted to interact with the tracrRNA (bases 4–8) or FTN_1103 mRNA (bases 48–54). All three mutations resulted in the inability to repress FTN_1103 (Fig. 2h), while a strain that expressed the altered versions of both the tracrRNA and scaRNA significantly restored FTN_1103 repression (Supplementary Fig. 8a). Additionally, the mutations predicted to disrupt the interaction between scaRNA and tracrRNA significantly dampened the ability of either small RNA to associate with Cas9, which was immunoprecipitated with equal efficiency in all strains (Supplementary Figs. 8b,c, 9). Thus, the sequence-specific association of Cas9, tracrRNA, and scaRNA is necessary for the repression of FTN_1103.
Since Cas9, tracrRNA, and scaRNA repress the expression of the FTN_1103 BLP, and BLPs are ligands for host TLR2, we tested if these CRISPR-CAS components were involved in evasion of TLR2 recognition. Membrane protein fractions of the tracrRNA and scaRNA mutants stimulated increased TLR2-dependent secretion of the proinflammatory cytokine IL-6, similar to those from the cas9 mutant as shown previously16(Fig. 3a). This response was rescued in double mutants lacking FTN_1103, indicating that overexpression of FTN_1103 in these strains was largely responsible for the increased TLR2 signaling (Fig. 3a). Mutants lacking cas9, tracrRNA, or the scaRNA also elicited enhanced TLR2-dependent IL-6 secretion during macrophage infection compared to wild-type F. novicida, which was dependent on FTN_1103 (Fig. 3b). This is in contrast to mutants in other CAS genes or the crRNA array, which did not alter TLR2 signaling (Supplementary Fig. 10). As a control, a mutant lacking only FTN_1103 did not have significantly altered membrane protein content nor did it induce altered host signaling (Supplementary Fig. 11). Together these data indicate that CRISPR-CAS component-mediated suppression of BLP facilitates evasion of TLR2.
Figure 3. Cas9, tracrRNA, and scaRNA facilitate evasion of TLR2 signaling by temporal repression of FTN_1103.
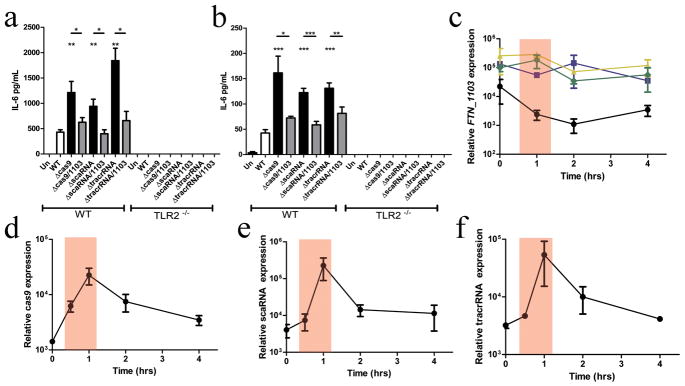
IL-6 secretion from wild-type (WT) and TLR2−/ − bone marrow-derived macrophages (BMDM) (A) unstimulated (Un) or stimulated with membrane proteins (relative MOI of 20:1 for 5 hours) from wild-type (WT), the indicated single mutants, or double deletion strains also lacking FTN_1103 (n = 3), or (B) infected with the same strains at an MOI of 20:1 for 5 hours (n =6). Relative expression of (C) FTN_1103, (D) cas9, (E) scaRNA, and (F) tracrRNA during infection of BMDM with the indicated strains (n = 3, bars represent s.d.).
To determine if repression of FTN_1103 was an active evasion process, we analyzed the temporal expression of CRISPR-CAS components during intracellular infection. We found that FTN_1103 expression decreased when the bacteria were in the phagosome, as shown previously16 (Fig. 3c), directly correlating with the ~100-fold induction of cas9, tracrRNA, and scaRNA (Fig. 3d–f). In the absence of Cas9, tracrRNA, or scaRNA, the temporal repression of FTN_1103 was completely abrogated (Fig. 3c). These data indicate that cas9, tracrRNA, and scaRNA are induced during intracellular infection, allowing temporal repression of FTN_1103 when the bacteria are in the proximity of TLR2 in the phagosome, thus facilitating evasion of this innate immune pathway. Interestingly, although cas1, cas4, and the crRNA array are not required for FTN_1103 repression, they were similarly expressed during infection (Supplementary Fig. 12). However, their expression differed during in vitro growth (Supplementary Fig. 13), suggesting specific co-regulation of these CRISPR-CAS components during intracellular infection.
We next tested the impact of the inability to repress FTN_1103 on fitness during murine infection. We performed competitive infections with wild-type F. novicida, and either the cas9, tracrRNA or scaRNA deletion mutants, and measured bacterial burden in the spleen 48 hours post-infection. All three mutants were highly attenuated (1,000 to 10,000 fold) compared to wild-type (Fig. 4a), demonstrating that all three components are critical for F. novicida virulence. This attenuation was significantly rescued by deletion of FTN_1103 from the mutants. Notably, mutants lacking the crRNA array or other CAS genes were not attenuated (Supplementary Fig. 14). The cas9, tracrRNA, and scaRNA mutants were also highly attenuated when inoculated individually; they were non-lethal even at 100x LD50 doses, while mice infected with wild-type or cis-complemented strains (which restored repression of FTN_1103, Supplementary Fig. 15) rapidly succumbed to disease (Fig. 4b). We hypothesized that the mice surviving an initial infection might be protected against subsequent lethal challenge with F. novicida. While naïve mice rapidly succumbed to a challenge, mice immunized with cas9, tracrRNA or scaRNA mutants were completely protected (Fig. 4c), demonstrating that mutants lacking these CRISPR-CAS components can efficiently vaccinate mice. Given that CRISPR-CAS systems of other bacteria may also contribute to virulence, mutants of these genes may represent attractive vaccine strains for other pathogens.
Figure 4. Cas9, tracrRNA, and scaRNA are necessary for virulence.
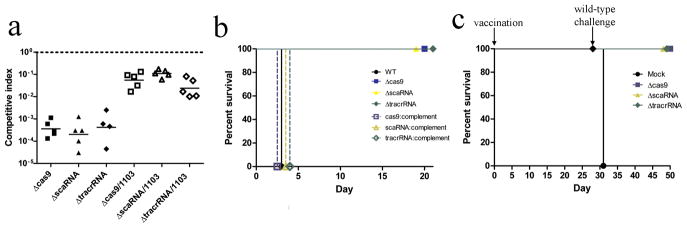
(A) Competitive indices of wild-type and the indicated mutant or double mutant strains from murine spleens, 48 hours post-infection. Bars represent the geometric mean. (B) Mice were infected with 107 cfu of either wild-type (black circle), Δcas9 (blue square), ΔscaRNA (yellow triangle), ΔtracrRNA (green diamond), or the corresponding cis-complemented strains (open symbols), and survival monitored over time. (C) Mice were vaccinated with 104 cfu of either Δcas9, ΔscaRNA, or ΔtracrRNA strains, or PBS. Twenty-eight days later, mice were challenged with 107 cfu wild-type.
Our results demonstrate that the Cas9 system has a non-canonical function in acting with a non-crRNA (scaRNA) to regulate gene expression via the degradation of an endogenous mRNA, leading to innate immune evasion and promoting virulence (Supplementary Fig. 16). This surprising observation shows that CRISPR-CAS components have been co-opted to perform functions distinct from foreign nucleic acid defense. Interestingly, they have also been implicated in DNA repair10 and biofilm formation9. Our work further suggests that predicted self-targeting crRNAs21 may have natural roles in endogenous gene regulation, functioning with the Cas9 machinery.
Eighty-five of the 109 bacteria we and others22 show encode Cas9 are known pathogens or commensals, making it interesting to speculate that the regulatory mechanism we describe may function in numerous other organisms that interact with eukaryotic cells (Supplementary Table 1). To further explore the breadth of this phenomenon, we generated a cas9 deletion mutant in Neisseria meningitidis str. 92045 and assayed virulence traits. We observed a significant decrease in the ability of the cas9 mutant to adhere to, invade, and replicate in human epithelial cells, leading to an overall defect in survival (Supplementary Fig. 17), indicating that Cas9 plays an important role in N. meningitidis pathogenesis. Additionally, a recent study identified Campylobacter jejuni Cas9 as critical for interactions with host cells 23. Bioinformatic analysis predicted that N. meningitidis, C. jejuni, and other pathogens may encode a scaRNA, which is critical for Cas9 targeting of endogenous mRNA (Supplementary Table 2). Together, these results clearly show that Cas9 controls virulence traits of several bacteria. It is interesting to note, however, that the CRISPR-CAS locus in highly virulent Francisella tularensis is likely nonfunctional, as it lacks the tracrRNA and contains an internal deletion within Cas9. F. tularensis is known to potently inhibit TLR signaling13 and may therefore not limit, or use distinct mechanisms to limit BLP expression. While its role in different species may therefore vary, the enrichment of Cas9 within the genomes of pathogens and commensals and its demonstrated role in controlling virulence traits in F. novicida, N. meningitidis, and C. jejuni, strongly suggest that it is involved in regulating the interaction of bacteria with eukaryotic hosts. Our data support a model whereby Type II CRISPR-CAS systems can function in endogenous gene regulation, ultimately promoting both pathogenesis and commensalism.
Methods
Bacterial Growth
Francisella novicida U112 was a kind gift from Dr. Denise Monack, Stanford University. Cultures were grown overnight at 37°C with aeration in tryptic soy broth (TSB) supplemented with 0.2% L-cysteine (BD Biosciences, Sparks, MD, USA) or on tryptic soy agar (TSA; BD Biosciences). When necessary, media was supplemented with kanamycin (30 μg/mL) or tetracycline (20 μg/mL). Meningococcal strains were grown with 5% CO2 at 37°C on GC base (GCB; Difco) agar containing supplements of 0.4% glucose and 0.68 mM Fe(NO3)3, or GC broth with the same supplements and 0.043% NaHCO3. Brain heart infusion (BHI) medium with 1.25% fetal bovine serum was used when kanamycin selection was required. N. meningitidis was transformed by the procedure of Janik et al. 30. To measure growth rate, overnight cultures of wild-type and the indicated F. novicida mutant strains were diluted to an OD600 of 0.03, incubated at 37°C with aeration and OD600 was measured hourly in a BioTek Synergy MX plate reader (BioTek, Winooski, VT) for 20 hours.
Mutagenesis
Francisella deletion mutant and point mutant strains were constructed by allelic replacement as described previously24 using primers in Supplemental Table S3. Double deletion strains were created using Flp-recombinase, as previously described31, and transforming unmarked strains with the second targeting construct. scaRNA and tracrRNA were complemented in trans by ligation into the broad host range vector, pBAV1K-T5-gfp32 at the EcoRI and BamHI sites, and transformed into unmarked ΔscaRNA or ΔtracrRNA strains. The N. meningitidis cas9 mutant was generated using a targeting construct generated by overlapping PCR, using primers in Supplemental Table S3, that created a 2615 bp deletion in the cas9 (3246 bp) coding sequence. The final PCR product with the expected size was gel-purified and used directly for transformation of a meningococcal serogroup W135 strain Nm92045. Colonies were selected on BHI agar plates with 80 μg/ml of kanamycin. Removal of the cas9 internal sequence was confirmed by PCR.
Membrane Protein Fractionation and SDS-PAGE Analysis
Membrane protein fractions were prepared as previously described16. Membrane proteins were normalized by colony forming units (cfu) to 108 cfu, separated via 12–20% SDS-PAGE (Bio-Rad, Hercules, CA, USA) and stained with Coomassie Blue G-250 (Teknova, Hollister, CA, USA).
RNA Isolation and qRT-PCR
RNA was isolated from bacterial cultures or macrophage infections at the given time points using TRI Reagent (Molecular Research Center, Cincinnati, OH, USA) and purified using the RNeasy Mini Kit (Qiagen, Germantown, MD, USA) and on-column DNase treatment (Qiagen) according to the manufacturers’ instructions. Quantitative real-time RT-PCR (qRT-PCR) was performed with 40ng total RNA using the Power Sybr Green RNA to CT 1-Step Kit (Applied Biosystems, Carlsbad, CA, USA) and gene-specific primers (Supplemental Table S3) using an Applied Biosystems StepOne Cycler. Relative transcript levels were calculated by normalizing CT values to DNA helicase II (uvrD, FTN_1594) and plotted as 2−ΔΔCT.
RNA Degradation Assay
RNA degradation assays were performed as previously described29. Overnight cultures of bacteria were subcultured 1:10 into 10 mL of TSB with 0.2% cysteine and grown to an OD600 of ~0.4. Rifampin (USB Corporation, Cleveland, OH) was added to a final concentration of 500 μg/mL, cultures were incubated at 37°C with aeration, and aliquots were taken at the indicated time points for RNA isolation.
Immunoprecipitation
Immunoprecipitation was performed on bacterial lysates using the FLAG Immunoprecipitation Kit (Sigma) according to the manufacturer’s instructions and the addition of 0.05% NP-40 during wash steps. Total RNA was isolated from the precipitate and qRT-PCR performed, normalizing to uvrD.
Macrophage Infections and Stimulations
Murine bone marrow-derived macrophages were prepared from 6 to 8 week-old wild-type and TLR2−/ − C57BL/6 mice and cultured as described 28. Macrophages were seeded into 96-well plates (~5×104 cells per well) for cytokine analysis, or 24-well plates (~3.2×105 cells per well) for RNA isolation, in high glucose Dulbecco’s Modified Eagle’s Medium (DMEM) (Lonza) supplemented with 10% heat-inactivated fetal bovine serum (HyClone, Logan, UT, USA) and 10% L929-conditioned media (conditioned DMEM) containing macrophage colony stimulating factor (M-CSF) overnight. Bacteria were added at a multiplicity of infection (MOI) of 20:1 bacteria per macrophage and centrifuged for 15 min at 335 × g at room temperature to facilitate bacterial uptake. Infected macrophages were incubated for 30 min at 37°C and washed two times before adding warm conditioned DMEM. The concentrations of IL-6 in culture supernatants at the indicated time points after infection were quantified by ELISA (BD Biosciences, Sparks, MD, USA). For stimulation with bacterial membrane protein fractions, cells were washed gently and media containing membrane fractions at a relative MOI of 20:1 were added. Macrophages were stimulated for the indicated duration of time, before the cell culture supernatant was collected and assayed for IL-6 by ELISA.
N. meningitidis intracellular survival assay
The A549 human lung adenocarcinoma cell line was cultured in DMEM supplemented with heat inactivated FBS (10%) at 37°C and 5% CO2. For the bacterial adherence and invasion assay, A549 cells were seeded at a density of 105 cells per ml in 24-well plates (Corning) two days prior to the experiment. To prepare the bacterial inoculum, meningococcal strains were grown in GC broth to mid log phase, collected by centrifugation and resuspended in cell culture media. Bacterial cells were added to cell cultures at a multiplicity of infection (MOI) of 100, and serial dilutions of the inoculum were plated to determine the input colony-forming unit (cfu). After a 3-hr infection, the monolayers were washed three times with sterile phosphate-buffered saline to remove free bacteria and the cfu of attached bacteria determined. Separately infected cells were washed and then incubated in cell culture media containing 100 μg/ml of gentamicin for 1 hr to kill extracellular bacteria. A549 cells were lysed by incubation with 1% saponin (Sigma) for 10 minutes to release intracellular bacteria at 4 hours and 6 hours post infection. Serial dilutions of lysates in PBS were plated on GC plates for cfu counts of invasion efficiency. Each assay was conducted with 2–3 independently infected monolayers and repeated three times.
In Vivo Experiments
Female C57BL/6 mice aged 7 to 10 weeks were kept under specific-pathogen free conditions in filter-top cages at Yerkes National Primate Center, and provided food and water ad libitum. All experimental procedures were approved by the Emory University Institutional Animal Care and Use Committee (protocol #YER-2000573-061314BN). For competitive infections, groups of five mice were infected subcutaneously with 1×105 cfu of wild-type and the indicated mutant strain of F. novicida at a 1:1 ratio in sterile PBS. At 48 hours post-infection, spleens were harvested and homogenized in PBS. Appropriate dilutions were plated with or without kanamycin for enumeration of bacterial burden. The competitive index (CI) was calculated using the following formula: CI=(mutant cfu output/wild-type cfu output)/(mutant cfu input/wild-type cfu input). For vaccination experiments, groups of five mice were infected subcutaneously with 1×105 cfu of the indicated mutant strain of F. novicida in sterile PBS, or PBS alone. Twenty-eight days following, mice were challenged subcutaneously with 1×107 cfu wild-type F. novicida in sterile PBS and sacrificed when they appeared moribund.
Supplementary Material
Acknowledgments
We would like to thank Rafi Ahmed, Graeme Conn, Christine Dunham, Charles Moran, Brooke Napier, Michelle Swanson, David S. Stephens, and the Stephens lab for helpful discussions and critical reading of this manuscript. The project described was supported by NIH grant U54-AI057157 from the Southeastern Regional Center of Excellence for Emerging Infections and Biodefense and R56-AI87673 to D.S.W., and R56-AI061031 to Y.-L. T. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. T.R.S. was supported by the NSF Graduate Research Fellowship, as well as the ARCS Foundation. T.R.S. and D.S.W. have filed a related patent.
Footnotes
Supplementary Information is linked to the online version of this paper at www.nature.com/nature.
Author Contributions T.R.S performed the experiments; S.D.S and Y.-L.T. generated the N. meningitidis cas9 deletion mutant and performed associated experiments; A.C.L. generated the Cas9-FLAG expressing strain; T.R.S. and D.S.W. conceived and designed experiments, interpreted data and wrote the manuscript.
Reprints and permissions available at www.nature.com/reprints.
The authors declare no competing financial interests.
References
- 1.Barrangou R, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 2.Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322:1843–1845. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet. 2011;45:273–297. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- 4.Garneau JE, et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 5.Hale CR, et al. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139:945–956. doi: 10.1016/j.cell.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A. 2012;109:E2579–2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Datsenko KA, et al. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat Commun. 2012;3:945. doi: 10.1038/ncomms1937. [DOI] [PubMed] [Google Scholar]
- 9.Zegans ME, et al. Interaction between bacteriophage DMS3 and host CRISPR region inhibits group behaviors of Pseudomonas aeruginosa. J Bacteriol. 2009;191:210–219. doi: 10.1128/JB.00797-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Babu M, et al. A dual function of the CRISPR-Cas system in bacterial antivirus immunity and DNA repair. Mol Microbiol. 2011;79:484–502. doi: 10.1111/j.1365-2958.2010.07465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aliprantis AO, et al. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science. 1999;285:736–739. doi: 10.1126/science.285.5428.736. [DOI] [PubMed] [Google Scholar]
- 12.Brightbill HD, et al. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science. 1999;285:732–736. doi: 10.1126/science.285.5428.732. [DOI] [PubMed] [Google Scholar]
- 13.Jones CL, et al. Subversion of Host Recognition and Defense Systems by Francisella spp. Microbiol Mol Biol Rev. 2012;76:383–404. doi: 10.1128/MMBR.05027-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malik M, et al. Toll-like receptor 2 is required for control of pulmonary infection with Francisella tularensis. Infect Immun. 2006;74:3657–3662. doi: 10.1128/IAI.02030-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abplanalp AL, Morris IR, Parida BK, Teale JM, Berton MT. TLR-dependent control of Francisella tularensis infection and host inflammatory responses. PLoS One. 2009;4:e7920. doi: 10.1371/journal.pone.0007920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones CL, Sampson TR, Nakaya HI, Pulendran B, Weiss DS. Repression of bacterial lipoprotein production by Francisella novicida facilitates evasion of innate immune recognition. Cell Microbiol. 2012 doi: 10.1111/j.1462-5822.2012.01816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Makarova KS, Aravind L, Wolf YI, Koonin EV. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol Direct. 2011;6:38. doi: 10.1186/1745-6150-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deltcheva E, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Postic G, et al. Identification of small RNAs in Francisella tularensis. BMC Genomics. 2010;11:625. doi: 10.1186/1471-2164-11-625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bayer TS, Booth LN, Knudsen SM, Ellington AD. Arginine-rich motifs present multiple interfaces for specific binding by RNA. RNA. 2005;11:1848–1857. doi: 10.1261/rna.2167605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stern A, Keren L, Wurtzel O, Amitai G, Sorek R. Self-targeting by CRISPR: gene regulation or autoimmunity? Trends Genet. 2010;26:335–340. doi: 10.1016/j.tig.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makarova KS, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Louwen R, et al. A novel link between Campylobacter jejuni bacteriophage defence, virulence and Guillain-Barre syndrome. Eur J Clin Microbiol Infect Dis. 2012 doi: 10.1007/s10096-012-1733-4. [DOI] [PubMed] [Google Scholar]
- 24.Brotcke A, et al. Identification of MglA-regulated genes reveals novel virulence factors in Francisella tularensis. Infect Immun. 2006;74:6642–6655. doi: 10.1128/IAI.01250-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janik A, Juni E, Heym GA. Genetic Transformation as a tool for detection of Neisseria gonorrhoeae. J Clin Microbiol. 1976;4:71–81. doi: 10.1128/jcm.4.1.71-81.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Llewellyn AC, Jones CL, Napier BA, Bina JE, Weiss DS. Macrophage replication screen identifies a novel Francisella hydroperoxide resistance protein involved in virulence. PLoS One. 2011;6:e24201. doi: 10.1371/journal.pone.0024201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar P, Sannigrahi S, Scoullar J, Kahler CM, Tzeng YL. Characterization of DsbD in Neisseria meningitidis. Mol Microbiol. 2011;79:1557–1573. doi: 10.1111/j.1365-2958.2011.07546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schaible UE, Kaufmann SHE. Studying trafficking of intracellular pathogens in antigen-presenting cells. Molecular Cellular Microbiology. 2002;31:343–360. [Google Scholar]
- 29.Postic G, et al. Identification of a Novel Small RNA Modulating Francisella tularensis Pathogenicity. PLoS One. 2012;7:e41999. doi: 10.1371/journal.pone.0041999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Janik A, Juni E, Heym GA. Genetic transformation as a tool for detection of Neisseria gonorrhoeae. J Clin Microbiol. 1976;4:71–81. doi: 10.1128/jcm.4.1.71-81.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gallagher LA, McKevitt M, Ramage ER, Manoil C. Genetic dissection of the Francisella novicida restriction barrier. J Bacteriol. 2008;190:7830–7837. doi: 10.1128/JB.01188-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bryksin AV, Matsumura I. Rational design of a plasmid origin that replicates efficiently in both gram-positive and gram-negative bacteria. PLoS One. 2010;5:e13244. doi: 10.1371/journal.pone.0013244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Menard R, Sansonetti PJ, Parsot C. Nonpolar mutagenesis of the ipa genes defines IpaB, IpaC, and IpaD as effectors of Shigella flexneri entry into epithelial cells. J Bacteriol. 1993;175:5899–5906. doi: 10.1128/jb.175.18.5899-5906.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.