

Abstract
The nociceptin receptor (NOP) and its endogenous agonist, nociceptin/Orphanin FQ (N/OFQ), members of the opioid receptor and peptide families respectively, modulate the pharmacological effects of classical opioids, particularly opioid-induced reward and nociception. We hypothesized that compounds containing both NOP and opioid receptor activity in a single molecule may have useful pharmacological profiles as non-addicting analgesics or as drug abuse medications. We report here our forays into the structure-activity relationships for discovering `bifunctional' NOP-mu opioid receptor (MOP) ligands, starting from our NOP-selective scaffolds. This initial SAR suggests pharmacophoric elements that may be modified to modulate/increase opioid affinity, while maintaining high affinity for the NOP receptor, to result in potent bifunctional small-molecule NOP/MOP ligands.
Keywords: N/OFQ, Nociceptin/orphaninFQ, Nociceptin receptor, Bifunctional ligands, NOP/opioid, multi-target ligand
The nociceptin opioid receptor (NOPr, previously known as the opioid receptor-like receptor ORL1) is now rather well-known as the `fourth' opioid receptor, is also a G-protein receptor and shares a significant homology with the other three opioid receptors, mu, delta and kappa.1–3 It is distributed in the central nervous system and periphery similarly to the other opioid receptors. Its endogenous peptide ligand, nociceptin/orphaninFQ (N/OFQ), is a heptadecapeptide, which also shares significant similarities particularly with the kappa opioid peptide dynorphin, but does not have affinity for any of the three classical opioid receptors.4,5 Intense interest in the pharmacology of the nociceptin receptor and N/OFQ have revealed that this receptor-peptide system plays a significant role in pathways related to pain, drug reward, anxiety, feeding and learning/memory.6–8 Indeed, there has been a significant effort by the pharmaceutical industry to harness this pharmacology for medication development, and several pharma programs have reported the discovery and development of `highly selective' NOP receptor agonists9–13(refs) and antagonists14–16, which have been explored for applications as anxiolytics, anti-tussives and for chronic pain respectively,17,18 among others.
The NOP-N/OFQ system has a `modulatory' effect on opioid receptor-mediated pharmacology and it is co-localized with opioid receptors in most neuronal circuitry.6,19 Activation of the NOP receptor with endogenous ligand N/OFQ has been shown to reverse opioid analgesia supraspinally, but potentiate it spinally.20,21 N/OFQ has also been shown to decrease opioid-induced dopamine release in reward pathways,22,23 and to have anxiolytic activity via an anti-corticotropin releasing factor (CRF) function in the amygdala.24 These functional interactions have led us to the hypothesize that bifunctional compounds possessing NOP agonist activity and mu agonist activity may have a useful pharmacological profile, especially as non-addicting analgesics or medications to treat opioid dependence.25 The concept of bifunctional or even multitargeted ligands is clinically validated among opioid-based therapies, e.g. buprenorphine is a mu opioid receptor (MOP) agonist and a kappa opioid receptor (KOP) antagonist.26
Bifunctional or multifunctional pharmacological effects may be obtained by using a drug cocktail; however, there are obvious pharmacokinetic advantages to developing single chemical entities possessing bifunctional activity.27 Different strategies may be employed to obtain bifunctional activity in single chemical entities.28 Two distinct pharmacophores for the two target receptors may linked by a spacer, as has been explored previously for opioid receptor targets.29–31 However, to obtain suitable drug-like bifunctional molecules, an ideal strategy would be to develop `integrated pharmacophores',28 in a single chemical scaffold. From a drug design perspective, it is a challenge to obtain and maintain the desired spectrum of activities at dual or multiple targets within a single chemical scaffold. However, with careful SAR and optimization, it is possible to rationally design potent bifunctional ligands with the desired pharmacological profile and drug-like suitability. We report here the design of bifunctional NOPr/MOPr agonists and their structure-activity relationships, leading to varying profiles of bifunctional activity, starting with NOP receptor-selective scaffolds.
We previously reported the 2-indolinone class of NOP receptor ligands, which were modestly selective for the NOPr over the other opioid receptors.32 We developed a 2-D pharmacophore model (comprised of three pharmacophoric features common to most NOPr ligands, Figure 1) and used this as a working guide, to determine pharmacophoric features and SAR leading to (i) selectivity versus the classical opioid receptors and (ii) functional efficacy, i.e. agonist or antagonist activity.33 We report here the SAR studies of the 2-indolinone class of NOPr-selective ligands that resulted in potent, bifunctional NOPr agonist/MOPr agonist profiles.
Figure 1.
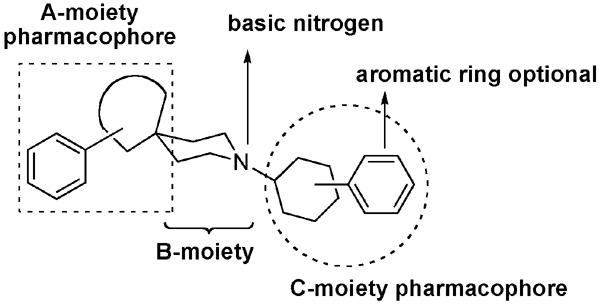
Working model of 2D pharmacophore depicting the three pharmacophoric elements present in most NOP ligands
Given that the 2-indolinone-based NOPr ligands were modestly selective (15–30-fold over MOPr), we used this scaffold to introduce modifications that would improve the binding affinity at MOPr, but retain the high affinity for the NOPr. Since most NOP ligands contain the piperidine ring as the B-moiety pharmacophore, we systematically explored the A-moiety pharmacophore (the piperidine 4-position heterocyclic ring) and the C-moiety pharmacophore (the piperidine N-substituent), to modulate NOP and mu selectivity. The structures of the various SAR modifications are shown in Table 1.
Table 1.
Binding affinities and functional activities of bifunctional NOP/opioid ligands
| Structure | Receptor Binding Ki (nM) | [35S]GTPγS NOP | [35S]GTPγS μ | [35S]GTPγS κ | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NOP | μ | κ | EC50 nM | % Stim | EC50 nM | % Stim | EC50 nM | % Stim | ||
| C-moietypharmaeophore (pipetidine N-substituent) modifications | ||||||||||
| 1 |
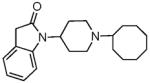
|
1.39 ± 0.42 | 29.9 ± 2.09 | 42.7 ± 1.02 | 20.8 ± 3.1 | 54 ± 11 | 99 ± 12 | 23.4 ± 3 | 276 ± 75 | 38 ± 3 |
| 2 |
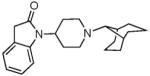
|
7.49 ± 0.78 | 2.70 ± 0.05 | 31.74 ± 4.8 | 28.7 ± 0.6 | 45 ± 5 | 29 ± 10 | 30 ± 0.2 | <10K | |
| 3 |

|
3.96 ± 1.55 | 8.0 ± 0.97 | 148.7 ± 8.7 | 26.5 ± 4.3 | 100± 15 | 73.5 ± 5 | 49 ± 0.45 | <10K | |
| 4 |

|
7.63 ± 1.87 | 5.39 ± 0.16 | 190.2 ± 45 | 107 ± 8 | 62 ± 6 | 595±493 | 28 ± 8 | NTa | NT |
| 5 |
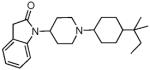
|
15.26 ± 0.29 | 16.13 ± 1.2 | 159.7 ± 4.2 | 83 ± 2 | 18.5 ± 2 | 189 ± 78 | 39 ± 6 | NT | NT |
| 6 |

|
212.8 ± 0.95 | 533 ± 42.3 | NTa | NT | NT | NT | NT | NT | NT |
| 7 |
|
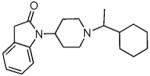
9.98 ± 2.8 | 3.44 ± 0.46 | 43.9 ± 9.2 | 82.3 ± 16 | 60± 10 | 28 ± 3.4 | 80.5 ± 11 | 873±293 | 48 ± 12 |
| 8 |

|
201.3 ± 51.7 | 91.1 ± 16.6 | 84.5 ± 0.76 | 174 ± 51 | 18± 2 | 1981±606 | 36 ± 5 | <10K | |
| 9 |
|
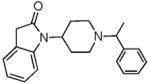
26.9 ± 0.25 | 2.34 ± 0.45 | 29.2 ± 0.06 | 138 ± 34 | 76± 14 | 50.6 ± 7 | 91 ± 4.9 | 422.60 | 37.5 |
| 10 |
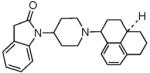
|
11.43 ± 0.91 | 79.9 ± 3.86 | 681.3 ± 61 | 46.1 ± 20 | 107± 7.4 | 129 ± 48 | 18 ± 1.6 | <10K | |
| A-moietypharmaeophore (thepipetidine 4-position heterocyclic ring) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 11 |
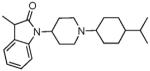
|
6.53 ± 1.48 | 3.62 ± 0.38 | 41.77 ± 2.4 | 54.9 ± 41 | 100± 40 | 119.6 | 84.3 | <10K |
| 12 |
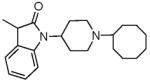
|
12.65 ± 1.93 | 10.49 ± 0.4 | 39.2 ± 15.3 | 30.2 ± 0.3 | 40± 10 | 94 ± 1.4 | 53.2± 14 | <10K |
| 13 |
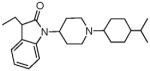
|
5.22 ± 0.65 | 1.07 ± 0.17 | 82.4 ± 16.4 | 8.5 ± 0.8 | 95± 12 | 4.7 ± 1.2 | 44 ± 5.2 | <10K |
| 14 |
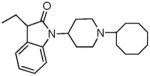
|
8.67 ± 0.89 | 16.26 ± 5.2 | 43.4 ± 17.8 | 6.8 ± 0.96 | 62 ± 12 | 37.3 ± 5 | 63 ± 18 | <10K |
| 15 |

|
20.4 ± 5.55 | 7.62 ± 1.01 | 70.04 ± 8.4 | 127 ± 5.3 | 75± 5 | 68 ± 5.9 | 50 ± 0.55 | <10K |
| 16 |

|
18.66 ± 5.05 | 46.97 ± 5.4 | 37.92 ± 32 | 101 ± 20 | 32 ± 2.2 | NT | NT | <10K |
| 17 |
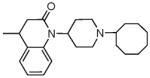
|
40.28 ± 1.8 | 23.27 ± 0.7 | 27.69 ± 3.6 | 94.7 ± 12 | 38 ± 2 | 58 ± 18 | 59 ± 5.1 | <10K |
NT=not tested. In some cases where the binding affinity was above 40 nM, the functional assays were not done.
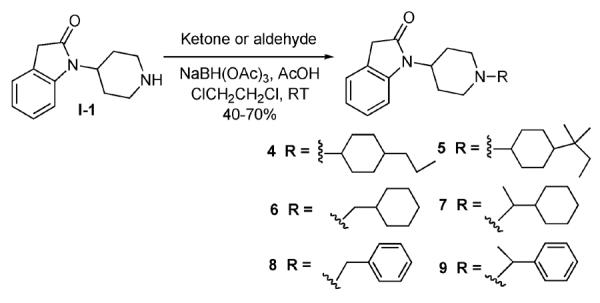
The synthesis of these bifunctional ligands in the SAR campaign was designed to be modular, to enable the use of common intermediates as much as possible. We have previously reported these approaches for the 2-indolinone class of compounds.32 Most piperidine N-substituents (for compounds 4–9) were introduced via reductive alkylation of a common intermediate I-1, an indolinyl-piperidine which was synthesized as reported previously (Scheme I).32 All ketones used for synthesizing compounds 4–9 were commercially available. For 4-alkyl substituted cyclohexanones, the reductive alkylations typically yielded a mixture of cis and trans isomers, with the cis isomers being the predominant products. These mixtures were readily separable by chromatography, and compounds 4 and 5 were obtained and tested as their cis isomers.
Scheme I.
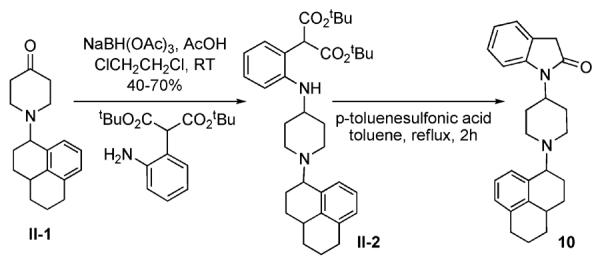
In cases where the reductive alkylation of the piperidine nitrogen was very low-yielding, an alternate approach was used, in which the piperidine nitrogen substituent (C-moiety) was introduced prior to the construction of the indolinyl A-moiety, as shown in Scheme II for compound 10. Piperidone II-1 containing the C-moiety was synthesized as a mixture of diastereomers according to the procedures described in Wichman et al34 and was used as such, to obtain 10, which also contained a equal proportion of the diastereomers, as seen by NMR. Compound 10 was tested as a diastereomeric mixture.
Scheme II.
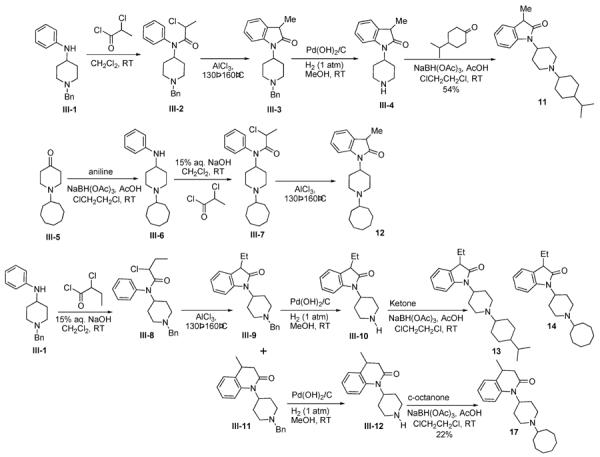
Analogs with alkyl modifications (3-Me and 3-Et) in the indolinyl A-moiety pharmacophore (11–14) were synthesized as shown in Scheme III, using a Friedel-Crafts alkylation-mediated cyclization,35 as for the parent indolinones 1 and 3.32 In most cases, the piperidine nitrogen substituent could be introduced by reductive alkylation, after construction of the alkyl-indolinone heterocycle and deprotection of the common N-benzyl substituent (III-4 and III-10). The reductive alkylation of III-4 and III-10 with isopropylcyclohexanone (Scheme III) typically results in a mixture of cis and trans isomers of the N-isoproylcyclohexyl group, containing the cis isomer as the major product. This isomeric mixture is readily separable by chromatography. Since the cis isomer has been shown to be significantly more active than the trans isomer,32,34,36 only the cis isomer of compounds 11 and 13 were tested. However, the reductive alkylation of the piperidine nitrogen with cyclooctanone (for compound 12) was typically very sluggish and low-yielding. Therefore, for 12, the cyclooctyl group was introduced onto the piperidine nitrogen (III-5)32 prior to the construction of the indolinone heterocycle (Scheme III).
Scheme III.
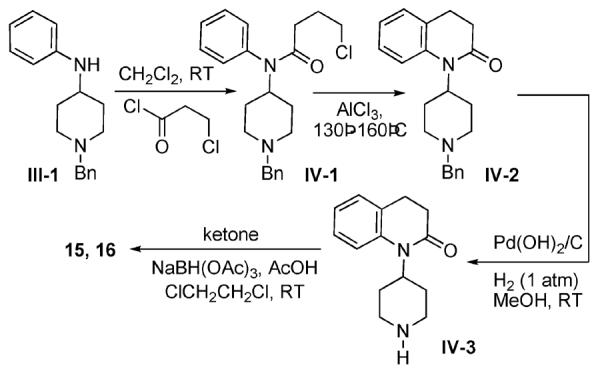
During the cyclization to the 3-Et indolinone intermediate III-9, the isomeric 4-methyl tetrahydroquinolinone heterocycle (III-11) was obtained as a byproduct in isolable yields. This was further debenzylated and N-alkylated to yield 4-Me tetrahydroquinolinone analog 17. The other tetrahydroquinolinone A-moiety analogs 15 and 16 were obtained by a Friedel-Crafts alkylation-cyclization, shown in Scheme IV.
Scheme IV.
The compounds were tested for binding affinity at human NOP, μ-, δ-, and κ-opioid receptors transfected into CHO cells. Binding to NOP was conducted with [3H] N/OFQ, as described previously.37 Binding to the opioid receptors utilized the selective agonists [3H] DAMGO, [3H] Cl-DPDPE, and [3H] U69593 for the μ, δ, and κ receptors respectively.38 Functional activity of these compounds was determined by stimulation of [35S] GTPγS binding to cell membranes, as we have described previously.38–40 Binding affinities and functional activity are shown in Table 1.
The SAR seen in Table 1 shows several interesting trends that suggest approaches for obtaining bifunctional mu opioid receptor affinity in NOP-selective ligands. The goal of our SAR studies was to obtain a bifunctional NOP/mu opioid ligand with equipotent binding affinity at both targets, and an agonist profile at both targets in functional assays. Our systematic exploration of the piperidine nitrogen substituent (C-moiety) showed that while the N-cyclooctyl substituent (lead compound 1) afforded high affinity and modest selectivity for the NOP receptor, a conformationally restricted, bicyclic N-substituent (as in 2) and the 4-isopropylcyclohexyl substituent (as in 3) increased mu opioid receptor affinity. A n-propylcyclohexyl substituent (as in 4) afforded a further increase in mu opioid receptor affinity. However, increasing bulk in the N-cyclohexyl substituent appeared detrimental to the binding affinity (compound 5). Interestingly, the N-cyclohexylmethyl substituent (compound 6) has poor affinity for the NOP and MOP receptor, compared to its homologous cyclooctylmethyl substituent, which we showed has potent NOP binding affinity and is a NOP antagonist.32 Surprisingly, introd34ucing a methyl substituent on the methylene bridge restored the high binding affinity at NOP as well as the MOP receptor, resulting in 7, which has a reasonable bifunctional profile and agonist activity at both receptors in the GTPγS functional assays.
We also explored the effect of increasing the aromatic character of the piperidine N-substituent. However, the N-benzyl susbstituent (as in 8) had low binding affinity at both receptors but introducing an α-methyl substituent in the methylene linker (compound 9) significantly increased affinity as well as agonist potency at the MOP receptor. The hexahydro-phenalen-1-yl substituent (as in 10) however, resulted in a NOP-selective full agonist ligand, which has low agonist efficacy at the mu opioid receptor. Although 10 was tested as a diastereomeric mixture of the piperidine N-substituent, this N-substituent is chemically similar to that on the selective NOP agonist Ro 64-6198, a triazaspirodecanone, which is about 100-fold selective for the NOP receptor over other opioid receptors.34
For SAR exploration of the indolinyl pharmacophore (A-moiety) for bifunctional activity, we introduced small alkyl substituents on the 3-indolinyl position, to modulate its NOP/MOP bifunctional profile. As seen in Table 1, a 3-methyl substituent on the indolinyl heterocyclic moiety of the NOP-selective lead compound 1, resulted in a 3-fold increase in MOP receptor affinity and agonist activity compared to the lead compound 1, but a slight drop in NOP receptor affinity as well as potency. However, compound 12 has equipotent binding affinity at both the NOP and MOP receptor. A 3-ethyl analog of the lead compound 1 (compound 14) also showed a modest increase in MOP receptor affinity and potency, and a small drop in affinity and potency at the NOP receptor. However, similar modifications made to the lead compound 3, containing the N-isopropylcyclohexyl substituent, resulted in significant enhancements to MOP receptor affinity and importantly to MOP receptor agonist efficacy. The 3-methyl-indolinyl analog 11 has equipotent binding affinity at the NOP and MOP receptor, and has full agonist activity at both NOP and MOP. The 3-ethyl-indolinyl analog 13 was even more potent than 11, at both receptors and has full agonist activity at NOP.
While the alkylation of the A-moiety indolinyl pharmacophore afforded improved bifunctional activity, homologation of the indolinyl moiety to a tetrahydroquinolinyl heterocycle, resulted in a decrease in binding affinity at both the NOP and MOP receptor (compounds 15 and 16), with the decrease being more pronounced when the N-piperidine substituent was cyclooctyl (15). In this series however, a small 4-alkyl substituent in the tetrahydroquinolinyl A-moiety (compound 17) resulted in a decrease in binding affinity at NOP but a small increase in binding affinity and potency at MOP. This SAR trend, i.e. increase in MOP receptor binding affinity upon alkylation of the A-moiety pharmacophore, is consistent with that observed in the indolinyl series of compounds discussed above.
The SAR presented clearly suggests that it is possible to increase MOP receptor affinity and potency, starting with NOP-selective scaffolds. While most NOP ligands contain the central piperidinyl scaffold, it appears that modifications to the piperidinyl N-substituent and/or the A-moiety heterocyclic pharmacophore can lead to an increase in MOP receptor activity, maintaining the NOP activity, to give a bifunctional profile.
Bifunctional NOP/MOP agonists are hypothesized to be useful as non-addicting analgesics or drug abuse medications.25 However, the ratio of the NOP agonist activity to that of the MOP agonist activity that provides a non-addicting analgesic profile remains to be determined. The series of bifunctional NOP/MOP agonists designed and reported in this study are very useful in this regard because several bifunctional compounds in this series have varying ratios of NOP/MOP agonist potency. For instance, compound 11 has equipotent binding affinity at both receptors, and is a full agonist at NOP and MOP. Compound 13 on the other hand, has full agonist activity at NOP, but about 44% agonist potency in the GTPγS functional assay. Compound 10 has full agonist activity at NOP and has low, but measurable agonist efficacy at MOP. We have previously reported the in vivo characterization of 10 (SR16835) and 13 (SR16507) for their antinociceptive and rewarding properties.25 Compound 10, which has low agonist efficacy at MOP has no acute antinociceptive activity and is not rewarding but blocks morphine conditioned place preference. Compound 13, which has partial agonist efficacy at MOP shows significant antinociceptive activity but is also rewarding on its own.
Modulation of the ratio of NOP agonist versus MOP agonist potency using SAR approaches described here will be useful for developing bifunctional NOP/MOP ligands for medication development.
Acknowledgments
Research funding by grants R01DA14026 and R01DA027811 to N.Z., is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Mollereau C, Parmentier M, Mailleux P, Butour JL, Moisand C, Chalon P, Caput D, Vassart G, Meunier JC. FEBS Lett. 1994;341:33. doi: 10.1016/0014-5793(94)80235-1. [DOI] [PubMed] [Google Scholar]
- 2.Wang JB, Johnson PS, Imai Y, Persico AM, Ozenberger BA, Eppler CM, Uhl GR. FEBS Lett. 1994;348:75. doi: 10.1016/0014-5793(94)00557-5. [DOI] [PubMed] [Google Scholar]
- 3.Bunzow JR, Saez C, Mortrud M, Bouvier C, Williams JT, Low M, Grandy DK. FEBS Lett. 1994;347:284. doi: 10.1016/0014-5793(94)00561-3. [DOI] [PubMed] [Google Scholar]
- 4.Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, Butour JL, Guillemot JC, Ferrara P, Monsarrat B, et al. Nature. 1995;377:532. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- 5.Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, Grandy DK, Langen H, Monsma FJ, Jr., Civelli O. Science. 1995;270:792. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- 6.Mogil JS, Pasternak GW. Pharmacol Rev. 2001;53:381. [PubMed] [Google Scholar]
- 7.Lambert DG. Nat Rev Drug Discov. 2008;7:694. doi: 10.1038/nrd2572. [DOI] [PubMed] [Google Scholar]
- 8.Reinscheid RK. CNS & Neurological Disorders - Drug Targets. 2006;5:219. doi: 10.2174/187152706776359628. [DOI] [PubMed] [Google Scholar]
- 9.Rover S, Adam G, Cesura AM, Galley G, Jenck F, Monsma FJ, Jr., Wichmann J, Dautzenberg FM. J Med Chem. 2000;43:1329. doi: 10.1021/jm991129q. [DOI] [PubMed] [Google Scholar]
- 10.Palin R, Bom A, Clark JK, Evans L, Feilden H, Houghton AK, Jones PS, Montgomery B, Weston MA, Wishart G. Bioorganic & Medicinal Chemistry. 2007;15:1828. doi: 10.1016/j.bmc.2006.11.030. [DOI] [PubMed] [Google Scholar]
- 11.Ho GD, Bercovici A, Tulshian D, Greenlee WJ, Fawzi A, Fernandez X, McLeod RL, Smith Torhan A, Zhang H. Bioorganic & Medicinal Chemistry Letters. 2007;17:3028. doi: 10.1016/j.bmcl.2007.03.062. [DOI] [PubMed] [Google Scholar]
- 12.Ho GD, Bercovici A, Tulshian D, Greenlee WJ, Fawzi A, Smith Torhan A, Zhang H. Bioorganic & Medicinal Chemistry Letters. 2007;17:3023. doi: 10.1016/j.bmcl.2007.03.061. [DOI] [PubMed] [Google Scholar]
- 13.Ho GD, Anthes J, Bercovici A, Caldwell JP, Cheng KC, Cui X, Fawzi A, Fernandez X, Greenlee WJ, Hey J, Korfmacher W, Lu SX, McLeod RL, Ng F, Torhan AS, Tan Z, Tulshian D, Varty GB, Xu X, Zhang H. Bioorg Med Chem Lett. 2009;19:2519. doi: 10.1016/j.bmcl.2009.03.031. [DOI] [PubMed] [Google Scholar]
- 14.Kawamoto H, Ozaki S, Itoh Y, Miyaji M, Arai S, Nakashima H, Kato T, Ohta H, Iwasawa Y. J Med Chem. 1999;42:5061. doi: 10.1021/jm990517p. [DOI] [PubMed] [Google Scholar]
- 15.Shinkai H, Ito T, Iida T, Kitao Y, Yamada H, Uchida I. J Med Chem. 2000;43:4667. doi: 10.1021/jm0002073. [DOI] [PubMed] [Google Scholar]
- 16.Yoshizumi T, Takahashi H, Miyazoe H, Sugimoto Y, Tsujita T, Kato T, Ito H, Kawamoto H, Hirayama M, Ichikawa D, Azuma-Kanoh T, Ozaki S, Shibata Y, Tani T, Chiba M, Ishii Y, Okuda S, Tadano K, Fukuroda T, Okamoto O, Ohta H. J Med Chem. 2008;51:4021. doi: 10.1021/jm701590h. [DOI] [PubMed] [Google Scholar]
- 17.Tamai H, Sawamura S, Takeda K, Orii R, Hanaoka K. European Journal of Pharmacology. 2005;510:223. doi: 10.1016/j.ejphar.2005.01.033. [DOI] [PubMed] [Google Scholar]
- 18.Zaratin PF, Petrone G, Sbacchi M, Garnier M, Fossati C, Petrillo P, Ronzoni S, Giardina GA, Scheideler MA. J Pharmacol Exp Ther. 2004;308:454. doi: 10.1124/jpet.103.055848. [DOI] [PubMed] [Google Scholar]
- 19.Mollereau C, Mouledous L. Peptides. 2000;21:907. 20. doi: 10.1016/s0196-9781(00)00227-8. [DOI] [PubMed] [Google Scholar]
- 20.Mogil JS, Grisel JE, Reinscheid RK, Civelli O, Belknap JK, Grandy DK. Neuroscience. 1996;75:333. doi: 10.1016/0306-4522(96)00338-7. [DOI] [PubMed] [Google Scholar]
- 21.Tian JH, Xu W, Fang Y, Mogil JS, Grisel JE, Grandy DK, Han JS. Br J Pharmacol. 1997;120:676. doi: 10.1038/sj.bjp.0700942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Giannuario A, Pieretti S, Catalani A, Loizzo A. Neurosci Lett. 1999;272:183. doi: 10.1016/s0304-3940(99)00579-0. [DOI] [PubMed] [Google Scholar]
- 23.Di Giannuario A, Pieretti S. Peptides. 2000;21:1125. doi: 10.1016/s0196-9781(00)00250-3. [DOI] [PubMed] [Google Scholar]
- 24.Economidou D, Hansson AC, Weiss F, Terasmaa A, Sommer WH, Cippitelli A, Fedeli A, Martin-Fardon R, Massi M, Ciccocioppo R, Heilig M. Biol Psychiatry. 2008;64:211. doi: 10.1016/j.biopsych.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toll L, Khroyan TV, Polgar WE, Jiang F, Olsen C, Zaveri NT. J Pharmacol Exp Ther. 2009;331:954. doi: 10.1124/jpet.109.157446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cremeans CM, Gruley E, Kyle DJ, Ko MC. J Pharmacol Exp Ther. 2012;343:72. doi: 10.1124/jpet.112.194308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morphy R, Rankovic Z. J Med Chem. 2005;48:6523. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- 28.Schiller PW. Life Sci. 2010;86:598. doi: 10.1016/j.lfs.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daniels DJ, Kulkarni A, Xie Z, Bhushan RG, Portoghese PS. J Med Chem. 2005;48:1713. doi: 10.1021/jm034234f. [DOI] [PubMed] [Google Scholar]
- 30.Zhang S, Yekkirala A, Tang Y, Portoghese PS. Bioorg Med Chem Lett. 2009;19:6978. doi: 10.1016/j.bmcl.2009.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng Y, Akgun E, Harikumar KG, Hopson J, Powers MD, Lunzer MM, Miller LJ, Portoghese PS. J Med Chem. 2009;52:247. doi: 10.1021/jm800174p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zaveri NT, Jiang F, Olsen CM, Deschamps JR, Parrish D, Polgar W, Toll L. J Med Chem. 2004;47:2973. doi: 10.1021/jm034249d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zaveri N, Jiang F, Olsen C, Polgar W, Toll L. AAPS J. 2005;7:E345. doi: 10.1208/aapsj070234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wichmann J, Adam G, Rover S, Hennig M, Scalone M, Cesura AM, Dautzenberg FM, Jenck F. Eur J Med Chem. 2000;35:839. doi: 10.1016/s0223-5234(00)00171-9. [DOI] [PubMed] [Google Scholar]
- 35.Ahadi S, Moafi L, Feiz A, Bazgir A. Tetrahedron. 2011;67:3954. doi: 10.1016/j.tet.2011.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rover S, Wichmann J, Jenck F, Adam G, Cesura AM. Bioorg Med Chem Lett. 2000;10:831. doi: 10.1016/s0960-894x(00)00111-6. [DOI] [PubMed] [Google Scholar]
- 37.Adapa ID, Toll L. Neuropeptides. 1997;31:403. doi: 10.1016/s0143-4179(97)90032-9. [DOI] [PubMed] [Google Scholar]
- 38.Toll L, Berzetei-Gurske IP, Polgar WE, Brandt SR, Adapa ID, Rodriguez L, Schwartz RW, Haggart D, O'Brien A, White A, Kennedy JM, Craymer K, Farrington L, Auh JS. NIDA Res Monogr. 1998;178:440. [PubMed] [Google Scholar]
- 39.Dooley CT, Spaeth CG, Berzetei-Gurske IP, Craymer K, Adapa ID, Brandt SR, Houghten RA, Toll L. J Pharmacol Exp Ther. 1997;283:735. [PubMed] [Google Scholar]
- 40.Spagnolo B, Calo G, Polgar WE, Jiang F, Olsen CM, Berzetei-Gurske I, Khroyan TV, Husbands SM, Lewis JW, Toll L, Zaveri NT. Br J Pharmacol. 2008;153:609. doi: 10.1038/sj.bjp.0707598. [DOI] [PMC free article] [PubMed] [Google Scholar]