

Abstract
The aim of this study was to evaluate the influence of 3-[4-(3-trifluoromethyl-phenyl)-piperazin-1-yl]-dihydrofuran-2-one (LPP1) on nociceptive thresholds in mouse models of persistent pain. Influence of LPP1 on motor coordination and its antioxidant capacity in mouse brain tissue homogenates were also assessed. Pain sensitivity thresholds in animals treated with LPP1 were established using 5 % formalin solution in normoglycemic mice and in streptozotocin (STZ)-treated diabetic mice in the von Frey, hot plate, innocuous, and noxious cold water tests (water at 10 °C and 4 °C, respectively). Motor deficits were assessed in the rotarod test, whereas antioxidant capacities were evaluated using ferric reducing ability of plasma (FRAP) assay, catalase (CAT), and superoxide dismutase (SOD) activities. LPP1was antinociceptive in both phases of the formalin test, in particular, in the late phase (at doses 0.9–30 mg/kg for 66–99 % vs. control normoglycemic mice) and in a statistically significant manner increased nociceptive thresholds in response to mechanical, heat, and noxious cold stimulation in neuropathic mice (at 30 mg/kg for 274, 192, and 316 %, respectively vs. diabetic control). LPP1 did not impair motor coordination of mice in the rotarod revolving at 6 or 18 rpm. In brain tissue homogenates, it demonstrated antioxidant capacity in FRAP assay and increased SOD activity for 63 % (acute administration) and 28 % (chronic administration) vs. control. No influence on CAT activity was observed. LPP1 has significant antinociceptive properties in the formalin model and elevates pain thresholds in neuropathic mice. It has antioxidant capacity and is devoid of negative influence on animals' motor coordination.
Keywords: Dihydrofuran-2-one, Enzymatic antioxidant defense, Formalin-induced tonic pain, Streptozotocin, Total antioxidant status
Introduction
Neuropathic pain is a debilitating form of chronic pain that results from dysfunction or damage to the peripheral or central nervous system (CNS). This type of pain is considered as a drug-resistant complication that still remains a serious medical problem worldwide. The term “neuropathic pain” comprises a variety of painful conditions, including postamputation pain, painful neuropathies (e.g., painful diabetic neuropathy and postherpetic neuralgia), posttraumatic neuralgia, and others. So far, multiple factors responsible for development of neuropathic pain have been identified: metabolic diseases (e.g., diabetes), neuronal tissue injuries caused by ischemia, toxicological factors or mechanical damage to the spinal cord, and others (Nickel et al. 2012; Woolf and Mannion 1999). Hence, pharmacotherapy used to relieve neuropathic pain comprises several pharmacological classes, of which antiepileptic drugs (AEDs), antidepressant drugs, opioid analgesics, and local anesthetic agents play a pivotal role (Christoph et al. 2011; Davis 2007; Davis 2010; Gilron et al. 2009; Miranda et al. 2012; Takeuchi et al. 2007; Yamama et al. 2010). Despite this, approximately 10–30 % of patients suffering from neuropathic pain syndromes are drug resistant (Blackburn-Munro and Erichsen 2005), so still, there is a great need for seeking new analgesic compounds able to attenuate neuropathic pain episodes.
Many lines of evidence indicate that oxidative stress is implicated in a variety of disorders, including degenerative diseases (Kasznicki et al. 2012; Reynolds et al. 2007; Trushina and McMurray 2007; Uttara et al. 2009), atherosclerosis, inflammation (Barton et al. 2007; Reuter et al. 2010; Salvemini et al. 2011), and chronic pain (Janes et al. 2012; Salvemini et al. 2011). Painful diabetic neuropathy is one of the most serious complications of diabetes in which the role of oxidative stress has been postulated. Imbalance between enhanced generation of reactive oxygen and nitrogen species and diminished activity of enzymatic and nonenzymatic antioxidant defenses as a key factor underlying diabetic neuropathy in mammals has been demonstrated (Di Naso et al. 2011; Pacher et al. 2005).
In our previous studies, we demonstrated significant antinociceptive, antiinflammatory, and local anesthetic activities (Salat et al. 2009; Salat et al. 2012a; Salat et al. 2012b; Więckowski et al. 2012) as well as antioxidant properties (Salat et al. 2012a; Salat et al. 2012b) of several dihydrofuran-2-one derivatives, including the 3-[4-(3-trifluoromethyl-phenyl)-piperazin-1-yl]-dihydrofuran-2-one (LPP1). Observed cell membrane-stabilizing properties of these derivatives, together with their antioxidant capacity, suggest that they might be effective as antiallodynic and antihyperalgesic agents in diabetes-induced neuropathic pain models.
In the present study, we focus on antinociceptive activity of the compound LPP1. We evaluate its efficacy in tonic (formalin) and neuropathic pain models in normoglycemic and diabetic mice, respectively. In view of the observed significant antioxidant capacity in 2,2'-azino-bis-3-ethylbenzothiazoline-6-sulfonic acid (ABTS) radical cation scavenging assay, we assess influence of LPP1 on selected markers of oxidative stress in mouse brain tissues (total antioxidant status in ferric reducing ability of plasma (FRAP) assay, activity of superoxide dismutase (SOD), and catalase (CAT)). The influence of LPP1 on motor coordination of diabetic animals in the rotarod test is also presented below.
Materials and methods
Animals and housing conditions
Adult male Albino Swiss (CD-1) mice weighing 18–24 g were used in behavioral experiments. The animals were kept in groups of 15 mice in cages at room temperature of 22 ± 2 °C under light/dark (12:12) cycle and had free access to food and water before experiments. Ambient temperature of the room and humidity were kept consistent throughout all tests. For the experiments, the animals were randomly selected. Each group consisted of eight to 18 animals per dose and each mouse was used only once. The mice were allowed to acclimate to holding cages prior to the test for a minimum of 30 min. The experiments were performed between 8 a.m. and 3 p.m. Behavioral measures were scored by trained observers blind to experimental conditions. The animals were killed by cervical dislocation immediately after the assay. All the procedures were approved by the local ethics committee of the Jagiellonian University in Cracow (ZI/595/2011).
Chemicals used in pharmacological tests
Synthesis of the investigated compound, LPP1, was described previously (Salat et al. 2009). For behavioral experiments, LPP1, pregabalin (a reference drug in the neuropathic pain model), and morphine (a reference compound in the formalin test) were suspended in a 0.5 % methylcellulose solution (Loba Chemie, Germany) and administered by the intraperitoneal (i.p.) route 30 min before the test. Control animals were given an appropriate amount of vehicle (0.5 % methylcellulose suspension; i.p.) 30 min before the test. To evaluate antioxidant capacity, LPP1 and pregabalin were administered in two protocols: acutely (a single i.p. injection of each compound) and chronically (a 10-day i.p. administration of LPP1 or pregabalin).
Formalin (37 % formaldehyde solution), acetic acid, hydrochloric acid, toluidine blue, glutaraldehyde (solution 3 % pure), and potassium phosphate were purchased from Polskie Odczynniki Chemiczne (Poland). Streptozotocin (STZ), 2,4,6-tripyridyl-S-triazine (TPTZ), and iron (III) chloride water solution (FeCl3) were purchased from Sigma-Aldrich (Poland). Morphine hydrochloride and pregabalin were provided by Polfa Kutno (Poland) and Tocris Bioscience (Germany), respectively. Hydrogen peroxide (H2O2) was provided by Stanlab (Poland) and 0.1 % adrenaline solution was purchased from Warszawskie Zakłady Farmaceutyczne Polfa (Poland).
Behavioral testing paradigm
Evaluation of antinociceptive activity and influence on paw edema formation in the formalin model in normoglycemic mice
In mice, i.p. injection of diluted formalin produces a biphasic nocifensive behavioral response (i.e., licking or biting the injected paw). The acute nociceptive (neurogenic) phase lasts for the first 5 min, whereas the second (inflammatory) phase occurs between 15 and 30 min after formalin injection. The formalin test in mice was performed according to Laughlin et al. (2002). The mice were pretreated with the test compound or the vehicle and were allowed to acclimate in Plexiglas observation chambers (20 × 30 × 15 cm) for 30 min before the test. Then, 20 μl of a 5 % formalin solution was injected intraplantarly into the right hind paw using a 26-gauge needle. Immediately after formalin injection, the animals were placed individually into glass beakers and were observed during the next 30 min. Time (in seconds) spent on licking or biting the injected paw in selected intervals, 0–5, 15–20, 20–25, and 25–30 min, was measured in each experimental group and was an indicator of nociceptive behavior.
To investigate whether or not nociception caused by formalin is associated with development of edema formation and to assess influence of LPP1 on edema formation in formalin-treated mice, paw edema was measured after intraplantar formalin injection in control and LPP1-treated animals. For this purpose, immediately after the formalin test, the mice were sacrificed by cervical dislocation and both paws were cut at the ankle joint and weighted on an analytical balance. The difference in weight of the formalin-treated paw and the weight of the nontreated (control) paw was compared (Beirith et al. 2002).
Influence on pain sensitivity thresholds in STZ-induced neuropathic pain model in diabetic mice Induction and assessment of diabetes
STZ kills insulin-secreting islet cells. To induce type I diabetes, mice were intraperitoneally injected with STZ (a single injection of STZ—200 mg/kg) dissolved in 0.1 N citrate buffer. Age-matched control mice received an equal volume of citrate buffer. Body weight and blood glucose level were measured 1 day before (referred to as “day 0”) and repeatedly 1, 2, and 3 weeks after STZ injection using a blood glucose monitoring system (Accu-Chek Active, Roche, France). Blood samples for measurement of glucose concentration were obtained from the tail vein of the mice. The animals were considered as diabetic when their blood glucose concentration exceeded 300 mg/dl (Tanabe et al. 2008) and only these mice (diabetic mice) were used in subsequent pain tests. In order to follow development of diabetic neuropathy in STZ-treated mice, time courses of mechanical and thermal nociceptive thresholds were evaluated for naïve and diabetic animals in the von Frey and hot plate tests along with blood glucose and body weight monitoring on day 0 as well as 1, 2, and 3 weeks later.
Evaluation of mechanical nociceptive thresholds in diabetic mice
Mechanical hypersensitivity (tactile allodynia) in mice was assessed using an electronic von Frey unit (Panlab, Spain) supplied with a single flexible filament applying increasing force (from 0 to 10 g) against the plantar surface of the hind paw of the mouse. The nocifensive paw withdrawal response automatically turned off the stimulus and the mechanical pressure that evoked the response was recorded.
On the day of the experiment, the mice were placed individually in test compartments with a wire mesh bottom and were allowed to habituate for 1 h. After the habituation period, in order to obtain baseline values, each mouse was tested three times alternately in each hind paw, allowing at least 30 s between each measurement. Then, the mice were pretreated with the test compound or vehicle. Thirty minutes later, the animals were tested again and mean values for each mouse were obtained (Tanabe et al. 2008).
Evaluation of pain sensitivity thresholds for cold stimuli in diabetic mice
Cold allodynia and cold hyperalgesia were assessed as paw withdrawal latencies in response to temperature, either non-noxious or noxious cold stimulation of hind paws when dipped in water bath maintained at 10 °C or 4 °C, respectively (Barriere et al. 2012; Pabreja et al. 2011). After establishment of baseline values of latency time for each mouse, the animals were pretreated with the test compound or vehicle. Thirty minutes later, they were observed until paw withdrawal or struggle signs with a cutoff time of 30 s were established to avoid paw tissue damage. Mice not responding within 30 s were removed from the apparatus and assigned a score of 30 s. The reaction time was measured two to three times, with an interval of at least 15 min between the two measurements to obtain two consecutive values that differed by no more than 10 %. The hind paws were immediately dried with cellulose paper to avoid paw cooling between the two measurements. Final results were expressed as a percentage according to the following formula:
Cold threshold (%) = 100 % − [(mean value in drug-treated group × 100 %) / mean value of the vehicle-treated group)].
Evaluation of pain sensitivity thresholds for heat stimuli in diabetic mice
Thermal hyperalgesia was assessed in the hot plate test as described by Eddy and Leimbach (1953) with some minor modification. Briefly, mice were treated i.p. either with the test compound or vehicle 30 min before placing the animal on the hot plate apparatus (Hot Plate 2A Type Omega, Poland). This apparatus has an electrically heated surface and is supplied with a temperature controller that maintains the temperature at 55–56 °C. The time until the animal licked its hind paws or jumped was recorded by means of a stopwatch. In this assay, the cutoff time was established (30 s) to avoid tissue damage, and mice not responding within 30 s were removed from the apparatus and assigned a score of 30 s.
Formation of degenerative changes (e.g., demyelination and axonal degeneration), i.e., toxic effects of STZ within nerves, could explain the differences in pain sensitivity in experimental mice, including hypoalgesia due to STZ-induced fiber degeneration. In order to confirm correctness of results obtained in pain tests, we investigated whether STZ induced abnormalities within the nerve structure. For this purpose, we used the sciatic nerve that is relatively easy to isolate. We investigated whether these changes (if any) could be seen in a light microscope. Immediately after the pain tests, the sciatic nerves were isolated from control (nondiabetic) mice and STZ-treated mice. The nerves were fixed in 3 % glutaraldehyde in 0.1 % phosphate buffer (pH 7.4). After dehydration in a graded series of ethanol followed by acetone, the nerves were embedded in Epon. Blocks were cut on an ultramicrotome. Semithin sections were stained with toluidine blue and examined with a light microscope.
Evaluation of motor impairing properties in diabetic mice (the rotarod test)
The test was performed according to the method described by Talarek et al. (2010) with some minor modifications. Mice were trained daily for 3 days on the rotarod apparatus (rotarod apparatus, May Commat RR0711, Turkey; rod diameter: 2 cm) rotating at a constant speed of 18 rpm. During each training session, the animals were placed on a rotating rod for 3 min with an unlimited number of trials. Proper experimentation was conducted at least 24 h after the final training trial. On the test day, 30 min before the rotarod test, the mice were intraperitoneally pretreated with the test compound or vehicle. Then, the animals were tested on the rotarod, revolving at 6 or 18 rpm. Motor impairments, defined as the inability to remain on the rotating rod for 1 min, were measured at each speed and were expressed as the mean time to fall off the rotating rod.
Evaluation of antioxidant capacity
For evaluation of antioxidant capacity of LPP1 and pregabalin, whole-brain tissues of normoglycemic mice receiving these compounds at a dose of 30 mg/kg (i.p.) acutely or chronically (for details, see “Chemicals used in pharmacological tests”) were homogenized in 0.05 M potassium phosphate buffer (pH 7.0) to obtain 20 % homogenates. The homogenates were centrifuged (800 × g, 20 min, 4 °C), and the supernatants were used in all further assays. Protein concentrations were measured by means of a biochemical analyzer MaxMat PL (Maxmat, France) using ready-made reagents and applications from AllMed (Poland).
Determination of total antioxidant status in the FRAP assay
The FRAP assay was used to measure the total antioxidant effect of LPP1 and pregabalin in mouse brain tissues. The FRAP method is based on reduction of ferric tripyridyltriazine (Fe3+–TPTZ) complex to the ferrous (Fe2+) form at low pH by the low molecular weight plasma antioxidants. Reduced Fe2+–TPTZ forms an intense blue color, with an absorption maximum at 593 nm. Absorption changes proportionally to antioxidant concentration (Benzie and Strain 1996). FRAP measurement was performed using the following reagents: 0.2 mol/l acetate buffer (pH = 3.6), 0.01 mol/l TPTZ solution in 0.04 mol/l hydrochloric acid, and 0.02 mol/l iron (III) chloride water solution (FeCl3). Briefly, 5 μl of plasma along with 15 μl deionized water was added to 150 μl freshly prepared FRAP reagent containing acetate buffer, TPTZ, and FeCl3 at 10:1:1 ratio. Blank samples consisted of 20 μl deionized water against plasma. Absorbance reading was taken after 6-min incubation period using a biochemical analyzer MaxMat PL. Change in absorption (∆A) between the sample reading (A) and the reagent blank reading (A 1) was calculated for each sample and related to ∆A of a Fe2+ standard solutions tested in parallel. The calibration curve was prepared with the use of five Fe2+ standard solutions: 0.2, 0.4, 0.8, 1.2, and 1.6 mmol/l for each set of sample measurements.
Determination of SOD activity
SOD activity was determined by the method described by Misra and Fridovich (1972) that is based on inhibition of autooxidation of adrenaline to adrenochrome at alkaline pH. Briefly, the assay mixture consisted of 240 μl 50 mM carbonate buffer (pH 10.2), 40 μl of the brain homogenate supernatant, 10 μl of 0.1 % adrenaline, and 10 μl of 20 mM FeCl3 solution (fivefold diluted in 0.05 M potassium phosphate buffer pH 7.0). The initial absorbance was recorded after FeCl3 addition and the final absorbance after 2 min. The reaction was followed at 480 nm on the biochemical analyzer, MaxMat PL. The control sample contained 40 μl 0.05 M potassium phosphate buffer (pH 7.0) against the sample homogenate. One unit of SOD activity was defined as the amount of the enzyme that caused 50 % reduction in the autooxidation of adrenaline. The final results were expressed as units per gram of protein.
Determination of CAT activity
CAT activity was evaluated by Aebi method (Aebi 1983) that is based on reduction of H2O2 to generate H2O and O2. Activity of the enzyme was determined by measuring the decrease in absorbance at 240 nm. The reagent mixture was composed of 30 mM H2O2 in 0.05 M potassium phosphate buffer (pH 7.0). The sample (50 μl) was added to 1 ml of the reagent mixture. Initial absorbance was recorded after addition of the sample and final absorbance after 1 min. The reaction was followed at 240 nm with the aid of the U-2800A UV/Vis spectrophotometer (Hitachi, Japan). Results were expressed as units per gram of protein. One unit of CAT activity is defined as the amount of enzyme decomposing 1 μmol of H2O2 per minute.
Data analysis
Data analysis of the results was provided by GraphPad Prism Software (v.5). Numerical results from behavioral tests are expressed as mean ± standard error of the mean (SEM). Results were statistically evaluated using Student's t test or one-way analysis of variance (ANOVA), followed by Tukey's or Dunnett's post hoc comparisons to compare the results obtained in drug-treated and control groups. Two-way repeated measures ANOVA, followed by Bonferroni's comparison were applied for statistical evaluation of time courses of effects obtained in pharmacological tests. In every case, p < 0.05 was considered significant.
The log-probit method (Litchfield and Wilcoxon 1949) was applied to establish median effective doses (ED50) for LPP1 and morphine in the first phase of the formalin test. Here, the ED50 value is defined as the dose of the investigated compound that reduces the duration of formalin-induced licking or biting response (i.e., the dose that diminishes pain reaction) for 50 % as compared to vehicle-treated animals.
Results
Antinociceptive activity in the formalin test in normoglycemic mice
In both phases of the formalin test, LPP1 demonstrated a highly significant antinociceptive activity (Table 1), reducing duration of nocifensive responses as compared to vehicle-treated mice. The ED50 value obtained for this compound in the first phase of the assay (2.1 mg/kg) was comparable to that of morphine (3.0 mg/kg). A very pronounced antinociceptive activity of LPP1 was also observed in the second (inflammatory) phase of the test, in which this compound reduced duration of paw licking response from 66 % at 0.9 mg/kg to 99.9 % at 30 mg/kg (significant at p < 0.001).
Table 1.
Antinociceptive activity of LPP1 and morphine in the formalin test
| Compound | Dose (mg/kg) | Duration of licking response (s) ± SEM (phase I) | Antinociceptive activity (%) | ED50 (mg/kg) (phase I) | Duration of licking response (s) ± SEM (phase II) | Antinociceptive activity (%) |
|---|---|---|---|---|---|---|
| Vehicle (0.5 % MC) | – | 91.2 ± 11.1 | – | – | 133.8 ± 22.6 | – |
| LPP1 | 0.9 | 59.2 ± 2.8* | 35.1 | 2.1 (0.8–5.3) | 44.9 ± 27.7** | 66.4 |
| 1.8 | 55.9 ± 7.5** | 38.6 | 4.5 ± 4.2*** | 96.6 | ||
| 7.5 | 18.4 ± 3.6*** | 79.9 | 1.2 ± 0.8*** | 99.1 | ||
| 30.0 | 15.8 ± 4.8*** | 82.7 | 0.1 ± 0.1*** | 99.9 | ||
| Vehicle (0.5 % MC) | – | 91.2 ± 11.1 | – | – | 133.8 ± 22.6 | – |
| Morphine | 2.1 | 64.7 ± 12.4* | 29.1 | 3.0 (1.9–4.7) | 3.4 ± 1.7*** | 97.5 |
| 3.0 | 41.2 ± 5.7** | 54.8 | 18.5 ± 6.6*** | 86.1 | ||
| 5.0 | 25.1 ± 3.9*** | 72.5 | 17.9 ± 8.0*** | 86.6 |
Results are shown as mean time of licking response in phase I (0–5 min after intraplantar injection of 5 % formalin) and in phase II (15–30 min after formalin injection). Statistical analysis: one-way ANOVA followed by post hoc Dunnett's test: phase I: F[4,26] = 18.01; p < 0.0001 (LPP1) and F[3,28] = 9.007; p < 0.0001 (morphine). Phase II: F[4,26] = 14.09; p < 0.0001 (LPP1) and F[3,28] = 26.17; p < 0.0001 (morphine). Statistical significance compared to MC-treated animals: * p < 0.05, ** p < 0.01, *** p < 0.001
MC methylcellulose
Evaluation of the time course of the antinociceptive activity of LPP1 at selected intervals revealed that this compound effectively attenuated the nocifensive response during the first 5 min of the test. This effect was highly significant (p < 0.001; Fig. 1). Antinociceptive effects observed between 15 and 20 min of the test were not statistically significant (p > 0.05 for each dose tested). A significant reduction of the licking response was observed in LPP1-treated mice between 20–25 min (p < 0.05) and 25–30 min (p < 0.001 for each tested dose; Fig. 1).
Fig. 1.
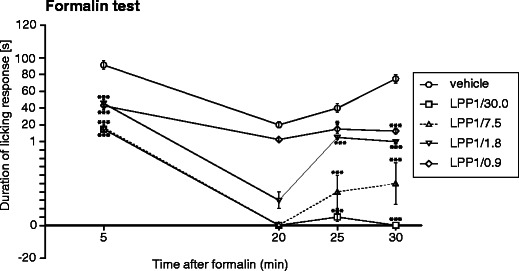
Time course of the antinociceptive activity of LPP1 in the formalin test. Data are shown as mean duration of the licking response (±SEM) measured at selected intervals: 0–5 min, 15–20 min, 20–25 min, and 25–30 min. Statistical analysis of the results was conducted using two-way repeated measures ANOVA, followed by Bonferroni's multiple comparison. Drug effect: F[4,105] = 32.57; p < 0.0001; time effect: F[3,105] = 38.55; p < 0.0001; interaction: F[12,105] = 4.53; p < 0.0001. Results compared to vehicle-treated mice at the same time points: *p < 0.05; ***p < 0.001
The pilot study that aimed at the evaluation of antiedematous activity of LPP1 in formalin-treated mice showed that this compound can attenuate some signs of peripheral inflammation, being able to reduce edema formation induced by intraplantar formalin. A main overall effect of treatment was observed (F[3,28] = 73.41; p < 0.0001). The injection of 5 % formalin caused a statistically significant increase in the paw weight in control animals (formalin-treated paw weight: 256.3 ± 8.8 mg vs. nontreated paw weight: 154.7 ± 2.5 mg; p < 0.001). Pretreatment with LPP1 (30 mg/kg; i.p.) had no effect on the nontreated paw weight but it caused an 11 % and statistically significant (p < 0.05) decrease of the formalin-treated paw weight, reducing the paw edema induced by intraplantar administration of formalin.
Assessment of pain sensitivity thresholds in diabetes-induced neuropathic pain model
Assessment of diabetes
Measurements of the body weight and blood glucose level
Mean body weight of mice before STZ administration (day 0) was 23.8 ± 0.4 g, whereas 21 days later, the mean body weight in diabetic mice was 30.1 ± 0.4 g. This difference was statistically significant (p < 0.001). In control (normoglycemic) mice, the mean body weight at the same time point was 38.9 ± 0.6 (p < 0.001 vs. day 0 body weight).
A statistically significant increase in mean blood glucose concentration was observed the first 7 days after STZ administration. At this time point, the mean blood glucose concentration in diabetic mice increased from 130.4 ± 5.5 mg/dl (day 0—before STZ administration) to 475.5 ± 39.2 mg/dl (7 days after STZ). In STZ-treated mice, this high glucose level was maintained almost constant as measured 14 and 21 days after STZ (474.0 ± 41.3 mg/dl and 462.5 ± 24.6 mg/dl, respectively). In normoglycemic control mice, the blood glucose level remained almost unaltered as compared to day 0 level (day 0: 131.5 ± 5.4 mg/dl; 1 week later: 133.4 ± 6.9 mg/dl; 2 weeks later: 127.0 ± 6.1 mg/dl; 3 weeks later: 119.3 ± 5.9 mg/dl) (Fig. 2a).
Fig. 2.
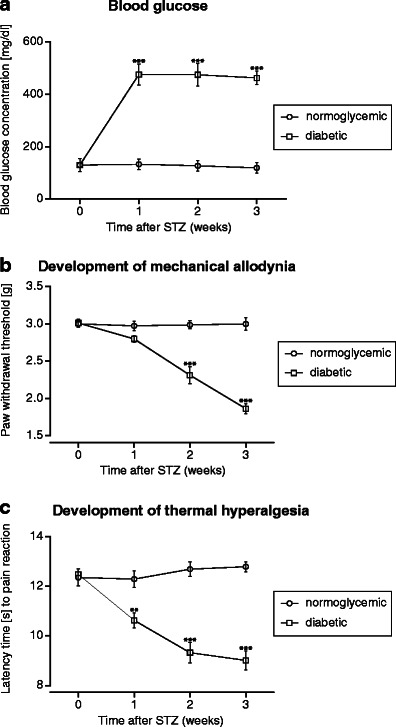
Time courses of blood glucose levels (a) and the development of mechanical allodynia (b) and thermal hyperalgesia (c) in normoglycemic mice and in STZ-treated animals. Statistical analysis of the results was conducted using two-way repeated measures ANOVA, followed by Bonferroni's multiple comparison. Blood glucose concentration: drug effect: F[1,42] = 392.57; p < 0.0001; time effect: F[3,42] = 25.3; p < 0.0001; interaction: F[3,42] = 26.7; p < 0.0001. ***p < 0.001 compared to nondiabetic mice. Mechanical allodynia was evaluated using von Frey test and thermal hyperalgesia was assessed by means of hot plate test (56 °C). Mechanical nociceptive thresholds: drug effect: F[1,42] = 96.33; p < 0.0001; time effect: F[3,42] = 28.64; p < 0.0001; interaction: F[3,42] = 29.41; p < 0.0001. Thermal nociceptive thresholds: drug effect: F[1,42] = 80.51; p < 0.0001; time effect: F[3,42] = 10.68 p < 0.0001; interaction: F[3,42] = 17.84; p < 0.0001. Significance: **p < 0.01; ***p < 0.001 compared to normoglycemic mice
No clear differences in the general structure of the sciatic nerves were observed in STZ-treated control (Fig. 3b) compared to control nondiabetic mice (Fig. 3a) and LPP1-treated diabetic mice (Fig. 3c). It can, therefore, be assumed that in diabetic mice, the nerves were not damaged 3 weeks after diabetes induction so the pharmacological effects obtained in our study were not due to STZ-induced nerve degeneration with the subsequent development of hypoalgesia.
Fig. 3.
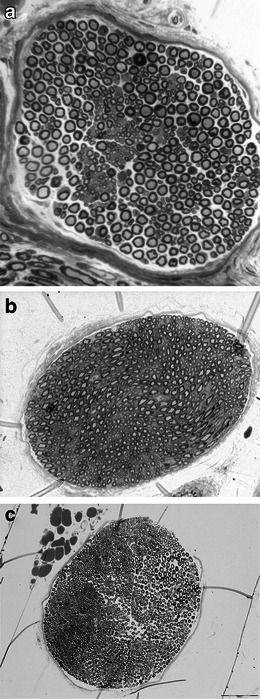
Representative semithin cross section of the sciatic nerve of male normoglycemic mice (a), STZ-treated control (b), and LPP1-treated diabetic mice (c). Toluidine blue stained
Evaluation of mechanical and thermal nociceptive thresholds after STZ injection
Pain sensitivity thresholds (i.e., development of tactile allodynia and thermal hyperalgesia) in normoglycemic and diabetic mice were assessed in course of time using von Frey and hot plate tests. In diabetic animals, a significant decrease of mechanical nociceptive thresholds was observed the first 14 days after STZ injection (p < 0.001; Fig. 2b). Thermal hyperalgesia developed earlier, i.e., 7 days after STZ (p < 0.01; Fig. 2c). A very significant (p < 0.001) reduction of pain thresholds for both tactile and thermal stimuli was observed 21 days after STZ (Fig. 2b, c), so at this time point, the antinociceptive activities of LPP1 and the reference pregabalin were evaluated.
Influence of LPP1 on mechanical nociceptive thresholds
On the day of measurement in nondiabetic control mice, mean pain sensitivity threshold for mechanical stimulation was 3.04 ± 0.2 g (Fig. 2b). STZ injection significantly increased pain sensitivity of experimental animals resulting in reduction of mechanical nociceptive threshold to 1.98 ± 0.14 g (baseline value; p < 0.001 vs. normoglycemic animals—Fig. 2b). Both LPP1 and pregabalin induced significant elevation of pain thresholds in STZ-treated mice (Fig. 4a) and LPP1 was more efficacious than pregabalin in this respect.
Fig. 4.
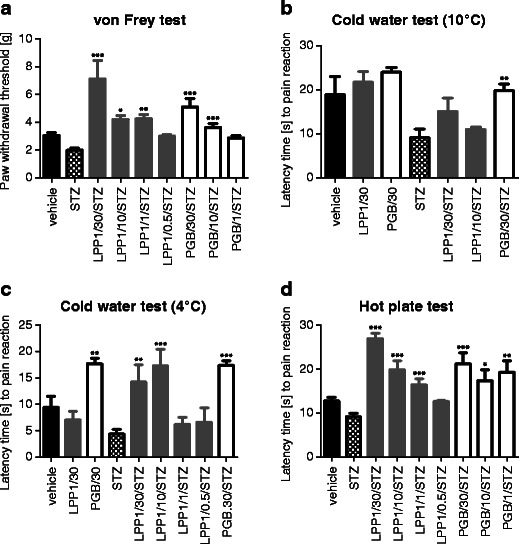
Influence of LPP1 and pregabalin (PGB) on nociceptive thresholds measured as paw withdrawal thresholds in the von Frey test (a), paw withdrawal latencies in cold water tests (b, c), and the latency time to pain reaction in the hot plate assay (d). Von Frey test: statistical analysis: one-way ANOVA followed by post hoc Dunnett's multiple comparison: F[4,52] = 13.48; p < 0.0001 (LPP1) and F[3,44] = 19.90; p < 0.0001 (pregabalin). Significance compared to STZ-baseline: *p < 0.05; **p < 0.01; ***p < 0.001. Cold water (10 °C) test: influence of LPP1 and pregabalin on latency time to pain reaction in normoglycemic mice (LPP1/30; PGB/30) and in STZ-treated mice (LPP1/30/STZ; LPP1/10/STZ; PGB/30/STZ). Statistical analysis: one-way ANOVA followed by Dunnett's multiple comparison: F[3,30] = 4.568; p < 0.01. Significance compared to STZ baseline latency value: **p < 0.01. Cold water (4 °C) test: influence of LPP1 and pregabalin on latency time to pain reaction in normoglycemic mice (LPP1/30; PGB/30) and in STZ-treated mice (LPP1/30/STZ; LPP1/10/STZ; LPP1/1/STZ; LPP1/0.5/STZ; PGB/30/STZ). Statistical analysis: one-way ANOVA followed by Dunnett's multiple comparison test: F[5,63] = 8.456; p < 0.0001. Significance vs. STZ baseline latency: **p < 0.01; ***p < 0.001. Hot plate test: antinociceptive activity of LPP1 and pregabalin in diabetic mice. Statistical analysis: one way ANOVA followed by post hoc Dunnett's multiple comparison: F[4,50] = 33.50; p < 0.0001 (LPP1) and F[3,41] = 8.078; p < 0.001 (pregabalin). Statistical significance compared to STZ baseline latency: *p < 0.05; **p < 0.01; ***p < 0.001. The antinociceptive activity of LPP1 in normoglycemic mice in the hot plate test has already been reported. (Salat et al. 2009)
Influence on cold sensitivity thresholds
Neither LPP1 nor pregabalin affected cold sensitivity thresholds in normoglycemic mice in water maintained at 10 °C (F[2,22] = 0.7747; NS). In STZ-treated animals, LPP1-induced prolongation of latency time to paw withdrawal (at 30 mg/kg: 65 % vs. STZ baseline latency and at 10 mg/kg: 20 % vs. STZ baseline latency) was not statistically significant. Pregabalin at 30 mg/kg (i.p.) prolonged the latency time to cold-induced nocifensive response in diabetic animals (112 % vs. STZ baseline value; p < 0.01) (Fig. 4b).
In the paw immersion test in water at 4 °C in normoglycemic mice, a main overall effect of treatment was observed (F[2,29] = 10.93; p < 0.001). Post hoc analysis revealed that only pregabalin at 30 mg/kg prolonged the latency time to nociceptive reaction (88 % vs. control; p < 0.01). Strong antihyperalgesic effects of both LPP1 and pregabalin were observed in STZ-treated mice in water maintained at 4 °C (noxious cold stimulus). A main overall effect of treatment was observed (F[5,63] = 8.456; p < 0.0001). Post hoc analysis showed that LPP1 prolonged the latency time to nocifensive reaction in diabetic animals (224 % and 294 % for 30 mg/kg and 10 mg/kg, respectively vs. STZ baseline latency time) in a statistically significant manner (p < 0.01 and p < 0.001, respectively). In diabetic animals, pregabalin (30 mg/kg) was also able to prolong the latency time to pain reaction (258 % vs. STZ; p < 0.001) (Fig. 4c).
Influence on heat nociceptive thresholds
In normoglycemic animals, mean baseline latency time to nociceptive response was 12.75 ± 0.8 s. The baseline latency time of STZ-treated animals' response to heat stimulus was shortened for 28 % (p < 0.001 vs. nondiabetic mice) and was an indicative of hyperalgesia. In diabetic mice, both LPP1 and pregabalin at doses 1–30 mg/kg prolonged the latency time to jump or lick the hind paw (Fig. 4d).
Influence on motor coordination (rotarod test)
Effect of LPP1 on motor coordination in normoglycemic mice was reported previously (Salat et al. 2012a). In the present study, to evaluate whether LPP1 and pregabalin can cause motor impairments in diabetic animals, the rotarod test was performed. Neither LPP1 nor pregabalin impaired animals' motor coordination as demonstrated using rotarod apparatus revolving at 6 and 18 rpm (Fig. 5a, b).
Fig. 5.
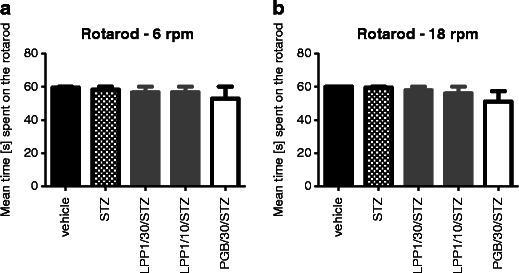
Influence of LPP1 and pregabalin (PGB) on motor coordination in diabetic mice evaluated using rotarod revolving at 6 rpm (a) or 18 rpm (b). Statistical analysis: one-way ANOVA followed by post hoc Tukey's multiple comparison test: 6 rpm: F[3,28] = 0.3216; NS (LPP1) and F[2,21] = 0.7179; NS (pregabalin); 18 rpm: F[3,28] = 0.6636; NS (LPP1) and F[2,21] = 1.914; NS (pregabalin)
Antioxidant capacity
In the FRAP assay, a statistically significant antioxidant capacity of both LPP1 and pregabalin administered as a single i.p. dose of 30 mg/kg was demonstrated (50.7 % and 28.4 % vs. vehicle-treated mice, respectively; F[4,44] = 9.057; p < 0.0001). Chronic administration of these compounds revealed no effect (p > 0.05) (Fig. 6a).
Fig. 6.
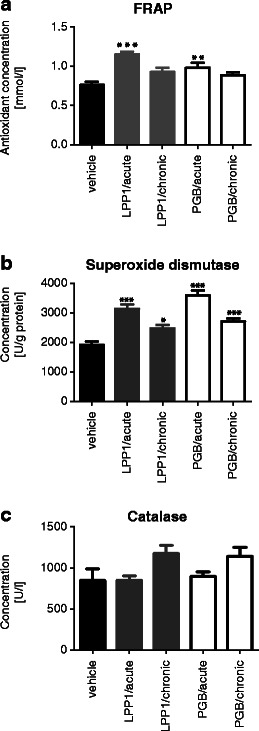
Antioxidant effects of LPP1 and pregabalin (PGB) measured as total antioxidant capacity in the FRAP assay (a), superoxide dismutase activity (b), and catalase activity (c) in mouse brains. Statistical analysis: one-way ANOVA followed by Dunnett's multiple comparison test. Significance vs. methylcellulose-treated mice: *p < 0.05; **p < 0.01; ***p < 0.001
Both LPP1 and pregabalin administered acutely and chronically had a very significant influence on SOD activity in mouse brain tissues (Fig. 6b). A main overall effect of the treatment was observed (F[4,44] = 25.66; p < 0.0001). Influence of LPP1 and pregabalin on SOD activity was particularly enhanced in acute administration (63.4 % and 87.2 % vs. vehicle-treated mice).
Acute and chronic treatments with LPP1 or pregabalin had no effect on CAT activity in mouse brain tissues (F[4,44] = 2.63; p > 0.05). In LPP1-treated mice (acute administration), the activity of CAT remained similar to that of control animals (850.2 ± 55.3 U/g of protein vs. 846.6 ± 142.9 U/g of protein, respectively). Chronic administration of LPP1 increased the activity of CAT (1,175 ± 100.9 U/g of protein); however, these results compared to control values were not statistically significant (Fig. 6c).
Discussion
In this paper, an extended investigation regarding the antinociceptive activity of LPP1 in tonic (formalin) and neuropathic pain models as well as its antioxidant capacity are demonstrated. Here, we show that LPP1 significantly influences pain sensitivity thresholds in persistent pain models and has antioxidant properties in mouse brain homogenates.
In our previous studies (Salat et al. 2009; Salat et al. 2012a), we demonstrated that LPP1 is a very effective antinociceptive and local anesthetic compound in rodent models of acute pain (hot plate and writhing tests, capsaicin and glutamate pain models in mice) and local anesthesia in mice and guinea pigs. Both central and peripheral antinociceptive mechanisms seem to contribute to LPP1-evoked antinociception, as indicated by the results obtained in the hot plate test (a pain model of spinal/supraspinal origin) and in the writhing test (a model of chemically induced peripheral pain), respectively.
In the present study, we used the formalin model of chemogenic pain to further evaluate the antinociceptive efficacy of LPP1. In rodents, the formalin model reflects persistent pain that is regarded to be dependent on sensory C-fiber activation, sensitization within the spinal cord dorsal horn, and the brain; however, peripheral inflammation is also an important contributor to this type of pain (Hunskaar and Hole 1987; Tjølsen et al. 1992; Yashpal and Coderre, 1998). The compound LPP1 demonstrated a very strong and statistically significant antinociceptive activity in both phases of this test. The ED50 value obtained for LPP1 in the first (neurogenic) phase of the test was similar to that of morphine (2.1 mg/kg vs. 3.0 mg/kg, respectively). LPP1 was also highly active in the second (late) phase of the formalin test that is thought to be a pain reaction dependent on combination of inflammation in peripheral tissues and functional changes in the dorsal horn of the spinal cord (central sensitization of pain and neuroplasticity of the CNS) (Hunskaar and Hole 1987; Pabreja et al. 2011; Tjølsen et al. 1992; Wang et al. 2004).
Several lines of evidence indicate that there is a strong concentration dependence of nociceptive responses induced by formalin both in mice and rats. Also, there is evidence that distinct nociceptive mechanisms might underlie animals' pain reaction in response to low or high concentrations of formalin (Yashpal and Coderre 1998). In our study, we used a 5 % formalin solution. At this concentration of formalin in the late phase of the test, a significant dependence of the nociceptive response on peripheral inflammatory changes and, to a lesser degree, on central sensitization is observed (Munro 2009; Yashpal and Coderre 1998). In our research, this fact was confirmed by the observed significant degree of edema formation when 5 % formalin solution was injected into control animals' hind paws and these changes in paw thickness correlated well with behavioral pain responses of these animals. Our observation remains in agreement with results reported by other authors (Yashpal and Coderre 1998) who demonstrated that 5 %, but not 1 %, formalin produced significant inflammation and plasma extravasation in peripheral tissues that were attenuated by high doses of antiinflammatory drugs (Munro 2009; Yashpal and Coderre 1998). Regarding the antinociceptive activity of LPP1 in the second phase of the formalin test, it can be concluded that this compound is not only able to attenuate pain symptoms accompanying formalin-evoked peripheral inflammation (Taylor et al. 2000) but it can also affect other signs of inflammation having a weak, albeit statistically significant, antiedematous activity shown as the decrease of the formalin-treated paw weight. The antiedematous effect demonstrated previously for some other gamma-butyrolactone (GBL) derivatives as well (Salat et al. 2012b) is not due to the influence of LPP1 on the inhibition of proinflammatory and hyperalgesic prostaglandin E2 synthesis (Sałat et al. 2012c), so the underlying mechanism for this activity seems to be distinct from that of nonsteroidal antiinflammatory drugs and requires further studies.
Diabetic mice with symptoms of mechanical allodynia and thermal hyperalgesia were used as a model of chronic pain that may reflect painful diabetic neuropathy in humans (Anjaneyulu and Chopra 2004; Obrosova 2009). In the present study, mice injected with STZ exhibited significantly increased plasma glucose levels, urine output, and decreased body weight gain compared to their normoglycemic littermates and these effects were observed as early as the first week after STZ administration. In our research, a significant decrease of pain thresholds with the subsequent development of thermal hyperalgesia and tactile allodynia were observed in the first 7 and 14 days after STZ, respectively. Conflicting data have been accumulated regarding the nociceptive thresholds, in particular, for thermal stimuli in STZ diabetic model of pain. These differences can be explained, in part, by the kind or intensity of thermal stimuli as well as the duration of diabetes (Obrosova 2009; Ulugol et al. 2012). For the above-mentioned reasons, in our study, pain sensitivity tests were performed 21 days after STZ administration to avoid development of hypoalgesia that often occurs during the advanced phase of diabetic process (Ulugol et al. 2012) in STZ-diabetic animals with longer term (≥12 weeks) diabetes (Obrosova 2009). Lack of visible degenerative changes in the general structure of the sciatic nerve of diabetic animals at the time point at which we carried out behavioral tests was proven under a light microscope, so the effects observed in behavioral tests (lowering of nociceptive thresholds in baseline measurement and elevation of pain thresholds induced by LPP1) cannot be attributed to STZ-induced hypoalgesia.
We showed that in diabetic mice, LPP1 significantly elevated pain reactivity thresholds for mechanical and thermal (noxious cold and heat) stimuli. However, it is worth noting that increases in paw withdrawal thresholds and prolonged latency times to pain reactions are only apparently beneficial effects. These high efficacies of LPP1 and, to a lesser degree, also pregabalin may be potentially harmful. Nociception as a part of mechanical, thermal, or chemical perception protects the body against potential harm. This means that maintenance or restoration (e.g., by use of analgesic drugs) of physiological pain sensitivity thresholds is pivotal to avoid a potential damage to the body. In our research, the nociceptive thresholds were elevated by LPP1 far beyond baseline values of untreated control mice and this effect might be responsible for a diminished protection against harmful stimuli, either mechanical or thermal. In other words, this effect, mainly observed at doses 10 and 30 mg/kg of LPP1, should rather be interpreted as an undesirable adverse effect and not a measure of its antinociceptive efficacy.
An interesting observation from the present study was that the effect of LPP1 varied depending on the type of thermal stimulus. No influence of LPP1 on pain thresholds was observed either in normoglycemic or diabetic mice subjected to innocuous cold (10 °C). LPP1 induced the elevation of nociceptive thresholds in diabetic animals subjected to noxious cold (water at 4 °C) and noxious heat (hot plate maintained at 56 °C) and under these circumstances, this activity may be potentially harmful. The observed temperature dependence of LPP1 influence on nociceptive thresholds is hard to explain at present. In view of the well-known fact that different biologically active molecules, in particular, thermosensitive channels belonging to the Transient Receptor Potential family, are involved in the sensation of heat (mainly TRPV1 channels), innocuous cold (TRPM8 channels), and noxious cold (TRPA1 channels) (Salat et al. 2013b), this fact is an interesting matter for further studies.
The LPP1-induced elevation of pain thresholds observed in the present experiment can be confidently attributed to its action within nociceptive circuits and these effects are not due to sedation or motor dysfunction. These results support our earlier findings that showed that LPP1 produces antinociception in several pain models (Salat et al. 2009; Salat et al. 2012a). The mechanisms explaining this activity in diabetic mice are not fully understood. In our previous studies, we showed that LPP1 had no affinity for GABAA, opioid μ, and serotonergic 5-HT1A receptors (Salat et al. 2012a), but it possessed potent membrane-stabilizing properties demonstrated in rodent models of local anesthesia (Salat et al. 2009). Here, the observed LPP1-induced increases in pain thresholds considerably over the thresholds of healthy controls might be due to its potent membrane-stabilizing activity shown previously that might also be an unwanted side effect, as it can enhance, typical for diabetes, subjective symptoms of the disease, such as paw numbness, sensation of rigidity, paresthesia, or heaviness in the sole upon walking. Although the rotarod test did not reveal any impairment of motor functions in diabetic mice, the incidence of the above-mentioned disorders cannot be excluded.
To gain further insight into mechanisms that could potentially contribute to the observed pharmacological effects of LPP1, in the present study, we have estimated this compound's influence on the activity of selected enzymatic markers of the oxidative stress: SOD and CAT in mouse brain tissues. Accumulating data indicate that reactive oxygen species are implicated in nociceptive responses and central sensitization of pain (Janes et al. 2012; Salvemini et al. 2011), in particular, chronic inflammatory (Wang et al. 2004) and neuropathic pain types (Kasznicki et al. 2012; Little et al. 2012) both in animals and humans. Available data from numerous studies show that targeting nitrosative and oxidative stress can be beneficial as a way to treat neuropathic pain syndromes and diminish central sensitization of pain (Janes et al. 2012; Pacher et al. 2005; Salat et al. 2013a; Salvemini et al. 2011; Salvemini and Neumann 2010). Sharma et al. (2006) showed that superoxide and nitric oxide are key mediators of glucose-induced oxidative injuries and they observed a marked increase in the whole brain nitrite levels in diabetic animals.
In recent years, much attention has been paid to the role of SOD, an enzyme catalyzing dismutation of superoxide radicals (McCord and Fridovich 1969), in suppression of inflammatory and neuropathic pain (Janes et al. 2012; Little et al. 2012; Wang et al. 2004). It is well appreciated that the superoxide anion formed during hyperalgesia states plays a pivotal role in development of pain through direct peripheral sensitization and is also regarded as a newly identified mediator of pain (Wang et al. 2004). The centrally released superoxide anions can play a crucial role in maintenance of nociception (Wang et al. 2004), whereas inactivation or decreased activity of mitochondrial manganese SOD is a critical event in the hyperalgesic response. As a result, a massive increase of superoxide anions and an indirect increase of peroxynitrite anions occur (Salvemini et al. 2011; Wang et al. 2004).
The observed strong antinociceptive activity in the second phase of the formalin test and the elevation of pain sensitivity thresholds in the STZ model of neuropathic pain implicate that antioxidant capacity of LPP1 might, at least in part, contribute to its pharmacological properties, including efficacy in some rodent pain models. Our earlier studies regarding the antioxidant capacity of LPP1 proved its high ABTS radical cation-scavenging properties (Salat et al. 2012a). The results from the present research confirm that LPP1 has antioxidant activity in tissues.
In conclusion, the significant antinociceptive efficacy of LPP1 in acute and tonic pain models in rodents as well as its ability to elevate nociceptive thresholds in the diabetic neuropathic pain model indicate an interesting lead structure in the search for new analgesic agents active in a wide range of painful conditions, provided carefully selecting dose ranges that maintain or restore physiological nociceptive thresholds. The pharmacological activity of LPP1 in mice, together with the antioxidant effect of this compound, in particular, its influence on SOD activity, is interesting for further studies.
Acknowledgments
This study was financially supported by the Jagiellonian University grant K/ZDS/003329.
Conflict of interest
None.
Abbreviations
- AEDs
Antiepileptic drugs
- CAT
Catalase
- CNS
Central nervous system
- FRAP
Ferric reducing ability of plasma
- H2O2
Hydrogen peroxide
- i.p.
Intraperitoneal
- MC
Methylcellulose
- PGB
Pregabalin
- ROS
Reactive oxygen species
- SOD
Superoxide dismutase
- STZ
Streptozotocin
- TPTZ
2,4,6-Tripyridyl-S-triazine
References
- Aebi HE (1983) Catalase. In: “Method of enzymatic analysis”, VCH, Weinheim.(3):273-286
- Anjaneyulu M, Chopra K. Fluoxetine attenuates thermal hyperalgesia through 5-HT1/2 receptors in streptozotocin-induced diabetic mice. Eur J Pharmacol. 2004;497:285–292. doi: 10.1016/j.ejphar.2004.06.063. [DOI] [PubMed] [Google Scholar]
- Barrière DA, Rieusset J, Chanteranne D, Busserolles J, Chauvin MA, Chapuis L, Salles J, Dubray C, Morio B. Paclitaxel therapy potentiates cold hyperalgesia in streptozotocin-induced diabetic rats through enhanced mitochondrial reactive oxygen species production and TRPA1 sensitization. Pain. 2012;153(3):553–561. doi: 10.1016/j.pain.2011.11.019. [DOI] [PubMed] [Google Scholar]
- Barton M, Minotti R, Haas E. Inflammation and atherosclerosis. Circ Res. 2007;101:750–751. doi: 10.1161/CIRCRESAHA.107.162487. [DOI] [PubMed] [Google Scholar]
- Beirith A, Santos AR, Calixto JB. Mechanisms underlying the nociception and paw oedema caused by injection of glutamate into the mouse paw. Brain Res. 2002;924(2):219–228. doi: 10.1016/S0006-8993(01)03240-1. [DOI] [PubMed] [Google Scholar]
- Benzie IF, Strain JJ. The ferric reducing ability of plasma (FRAP) as measure of antioxidant power: the FRAP assay. Anal Biochem. 1996;239:70–76. doi: 10.1006/abio.1996.0292. [DOI] [PubMed] [Google Scholar]
- Blackburn-Munro G, Erichsen HK. Antiepileptics and the treatment of neuropathic pain: evidence from animal models. Curr Pharm Des. 2005;11(23):2961–2976. doi: 10.2174/1381612054865000. [DOI] [PubMed] [Google Scholar]
- Christoph T, De Vry J, Schiene K, Tallarida RJ, Tzschentke TM. Synergistic antihypersensitive effects of pregabalin and tapentadol in a rat model of neuropathic pain. Eur J Pharmacol. 2011;666:72–79. doi: 10.1016/j.ejphar.2011.05.029. [DOI] [PubMed] [Google Scholar]
- Davis MP. What is new in neuropathic pain? Suppor Care Cancer. 2007;15(4):363–372. doi: 10.1007/s00520-006-0156-0. [DOI] [PubMed] [Google Scholar]
- Davis MP (2010) Recent advances in the treatment of pain, F1000 Med Rep 2 63. doi: 10.3410/M2-63 [DOI] [PMC free article] [PubMed]
- Di Naso FC, Simões Dias A, Porawski M, Marroni NA. Exogenous superoxide dismutase: action on liver oxidative stress in animals with streptozotocin-induced diabetes. Exp Diabetes Res. 2011;2011:754132. doi: 10.1155/2011/754132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy N, Leimbach D. Synthetic analgesics. II. Dithienylbutenyl- and dithienylbutylamines. J Pharmacol Exp Ther. 1953;107:385–393. [PubMed] [Google Scholar]
- Gilron I, Bailey JM, Tu D, Holden RR, Jackson AC, Houlden RL. Nortriptyline and gabapentin, alone and in combination for neuropathic pain: a double-blind, randomised controlled crossover trial. Lancet. 2009;374(9697):1252–1261. doi: 10.1016/S0140-6736(09)61081-3. [DOI] [PubMed] [Google Scholar]
- Hunskaar S, Hole K. The formalin test in mice: dissociation between inflammatory and non-inflammatory pain. Pain. 1987;30:103–114. doi: 10.1016/0304-3959(87)90088-1. [DOI] [PubMed] [Google Scholar]
- Janes K, Neumann WL, Salvemini D. Anti-superoxide and anti-peroxynitrite strategies in pain suppression. Biochim Biophys Acta. 2012;1822(5):815–821. doi: 10.1016/j.bbadis.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasznicki J, Kosmalski M, Sliwinska A, Mrowicka M, Stanczyk M, Majsterek I, Drzewoski J. Evaluation of oxidative stress markers in pathogenesis of diabetic neuropathy. Mol Biol Rep. 2012;39(9):8669–8678. doi: 10.1007/s11033-012-1722-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughlin TM, Tram KV, Wilcox GL, Birnbaum AK. Comparison of antiepileptic drugs tiagabine, lamotrigine, and gabapentin in mouse models of acute, prolonged, and chronic nociception. J Pharmacol Exp Ther. 2002;302(3):1168–1175. doi: 10.1124/jpet.302.3.1168. [DOI] [PubMed] [Google Scholar]
- Litchfield JT, Wilcoxon E. A simplified method of evaluating dose–effect experiments. J Pharmacol Exp Ther. 1949;96:99–113. [PubMed] [Google Scholar]
- Little JW, Doyle T, Salvemini D. Reactive nitroxidative species and nociceptive processing: determining the roles for nitric oxide, superoxide, and peroxynitrite in pain. Amino Acids. 2012;42(1):75–94. doi: 10.1007/s00726-010-0633-0. [DOI] [PubMed] [Google Scholar]
- McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244(22):6049–6055. [PubMed] [Google Scholar]
- Miranda HF, Noriega V, Prieto JC. Previous administration of naltrexone did not change synergism between paracetamol and tramadol in mice. Pharmacol Biochem Behav. 2012;102(1):72–76. doi: 10.1016/j.pbb.2012.03.008. [DOI] [PubMed] [Google Scholar]
- Misra HP, Fridovich I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem. 1972;247(10):3170–3175. [PubMed] [Google Scholar]
- Munro G. Pharmacological assessment of the rat formalin test utilizing the clinically used analgesic drugs gabapentin, lamotrigine, morphine, duloxetine, tramadol and ibuprofen: influence of low and high formalin concentrations. Eur J Pharmacol. 2009;605:95–102. doi: 10.1016/j.ejphar.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Nickel FT, Seifert F, Lanz S, Maihöfner C. Mechanisms of neuropathic pain. Eur Neuropsychopharmacol. 2012;22(2):81–91. doi: 10.1016/j.euroneuro.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Obrosova IG. Diabetic painful and insensate neuropathy: pathogenesis and potential treatments. Neurotherapeutics. 2009;6(4):638–647. doi: 10.1016/j.nurt.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabreja K, Dua K, Sharma S, Padi SS, Kulkarni SK. Minocycline attenuates the development of diabetic neuropathic pain: possible anti-inflammatory and anti-oxidant mechanisms. Eur J Pharmacol. 2011;661(1–3):15–21. doi: 10.1016/j.ejphar.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Pacher P, Obrosova IG, Mabley JG, Szabó C. Role of nitrosative stress and peroxynitrite in the pathogenesis of diabetic complications. Emerging new therapeutical strategies. Curr Med Chem. 2005;12(3):267–275. doi: 10.2174/0929867053363207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds A, Laurie C, Mosley RL, Gendelman HE. Oxidative stress and the pathogenesis of neurodegenerative disorders. Int Rev Neurobiol. 2007;82:297–325. doi: 10.1016/S0074-7742(07)82016-2. [DOI] [PubMed] [Google Scholar]
- Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49:1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sałat K, Filipek B, Wieckowski K, Malawska B. Analgesic activity of 3-mono-substituted derivatives of dihydrofuran-2-one in experimental rodent models of pain. Pharmacol Rep. 2009;61(5):807–818. doi: 10.1016/s1734-1140(09)70136-7. [DOI] [PubMed] [Google Scholar]
- Salat K, Moniczewski A, Salat R, Janaszek M, Filipek B, Malawska B, Wieckowski K. Analgesic, anticonvulsant and antioxidant activities of 3-[4-(3-trifluoromethyl-phenyl)-piperazin-1-yl]dihydrofuran-2-one dihydrochloride in mice. Pharmacol Biochem Behav. 2012;101(1):138–147. doi: 10.1016/j.pbb.2011.12.014. [DOI] [PubMed] [Google Scholar]
- Salat K, Librowski T, Moniczewski A, Stanisz-Wallis K, Wieckowski K, Malawska B. Analgesic, antioedematous and antioxidant activity of γ-butyrolactone derivatives in rodents. Behav Pharmacol. 2012;23(4):407–416. doi: 10.1097/FBP.0b013e3283566042. [DOI] [PubMed] [Google Scholar]
- Sałat K, Librowski T, Gdula-Argasińska J, Tyszka-Czochara M, Jastrzębska-Więsek M, Moniczewski A, Rapacz A, Pytka K, Filipek B, Więckowski K, Malawska B. Influence of gamma-butyrolactone derivatives with analgesic properties on the prostaglandin E2 level and gastric mucosa in rodents. Acta Biologica Cracoviensia Series Zoologia. 2012;53:45–52. [Google Scholar]
- Sałat K, Moniczewski A, Librowski T. Nitrogen, oxygen or sulfur containing heterocyclic compounds as analgesic drugs used as modulators of the nitroxidative stress. Mini Rev Med Chem. 2013;13(3):335–352. [PubMed] [Google Scholar]
- Sałat K, Moniczewski A, Librowski T (2013b) Transient receptor potential channels—emerging novel drug targets for the treatment of pain. Curr Med Chem (in press) [DOI] [PubMed]
- Salvemini D, Little JW, Doyle T, Neumann WL. Roles of reactive oxygen and nitrogen species in pain. Free Radic Biol Med. 2011;51(5):951–966. doi: 10.1016/j.freeradbiomed.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvemini D, Neumann W. Targeting peroxynitrite driven nitroxidative stress with synzymes: a novel therapeutic approach in chronic pain management. Life Sci. 2010;86(15–16):604–614. doi: 10.1016/j.lfs.2009.06.011. [DOI] [PubMed] [Google Scholar]
- Sharma S, Kulkarni SK, Agrewala JN, Chopra K. Curcumin attenuates thermal hyperalgesia in a diabetic mouse model of neuropathic pain. Eur J Pharmacol. 2006;536:256–261. doi: 10.1016/j.ejphar.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Takasu K, Ono H, Tanabe M. Pregabalin, S-(+)-3-isobutylgaba, activates the descending noradrenergic system to alleviate neuropathic pain in the mouse partial sciatic nerve ligation model. Neuropharmacology. 2007;53(7):842–853. doi: 10.1016/j.neuropharm.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Talarek S, Orzelska J, Listos J, Fidecka Effect of sildenafil treatment on the development of tolerance to diazepam-induced motor impairment and sedation in mice. Pharmacol Rep. 2010;62:627–634. doi: 10.1016/s1734-1140(10)70320-0. [DOI] [PubMed] [Google Scholar]
- Tanabe M, Murakami T, Ono H. Zonisamide suppresses pain symptoms of formalin-induced inflammatory and streptozotocin-induced diabetic neuropathy. J Pharmacol Sci. 2008;107(2):213–220. doi: 10.1254/jphs.08032FP. [DOI] [PubMed] [Google Scholar]
- Taylor BK, Peterson MA, Roderick RE, Tate J, Green PG, Levine JO, Basbaum AI. Opioid inhibition of formalin-induced changes in plasma extravasation and local blood flow in rats. Pain. 2000;84(2–3):263–270. doi: 10.1016/S0304-3959(99)00212-2. [DOI] [PubMed] [Google Scholar]
- Tjølsen A, Berge OG, Hunskaar S, Rosland JH, Hole K. The formalin test: an evaluation of the method. Pain. 1992;51:5–17. doi: 10.1016/0304-3959(92)90003-T. [DOI] [PubMed] [Google Scholar]
- Trushina E, McMurray CT. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience. 2007;145:1233–1248. doi: 10.1016/j.neuroscience.2006.10.056. [DOI] [PubMed] [Google Scholar]
- Ulugol A, Oltulu C, Gunduz O, Citak C, Carrara R, Shaqaqi MR, Sanchez AM, Dogrul A. 5-HT7 receptor activation attenuates thermal hyperalgesia in streptozocin-induced diabetic mice. Pharmacol Biochem Behav. 2012;102:344–347. doi: 10.1016/j.pbb.2012.05.006. [DOI] [PubMed] [Google Scholar]
- Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZQ, Porreca F, Cuzzocrea S, Galen K, Lightfoot R, Masini E, Muscoli C, Mollace V, Ndengele M, Ischiropoulos H, Salvemini D. A newly identified role for superoxide in inflammatory pain. J Pharmacol Exp Ther. 2004;309(3):869–878. doi: 10.1124/jpet.103.064154. [DOI] [PubMed] [Google Scholar]
- Więckowski K, Sałat K, Bytnar J, Bajda M, Filipek B, Stables JP, Malawska B. Search for anticonvulsant and analgesic active derivatives of dihydrofuran-2(3H)-one. Bioorg Med Chem. 2012;20(21):6533–6544. doi: 10.1016/j.bmc.2012.08.037. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet. 1999;353(9168):1959–1964. doi: 10.1016/S0140-6736(99)01307-0. [DOI] [PubMed] [Google Scholar]
- Yamama Y, Nishikawa K, Funao T, Mori T, Asada A. Intrathecal gabapentin and clonidine synergistically inhibit allodynia in spinal nerve-ligated rats. Life Sci. 2010;87(17–18):565–571. doi: 10.1016/j.lfs.2010.09.017. [DOI] [PubMed] [Google Scholar]
- Yashpal K, Coderre TJ. Influence of formalin concentration on the mantinociceptive effects of antiinflammatory drugs in the formalin test in rats: separate mechanisms underlying the nociceptive effects of low- and high-concentration formalin. Eur J Pain. 1998;2:63–68. doi: 10.1016/S1090-3801(98)90047-7. [DOI] [PubMed] [Google Scholar]