

Abstract
Researchers often decide whether to average multiple results in order to produce more precise data, and clinicians often decide whether to repeat a laboratory test in order to confirm its validity or to follow a trend. Some of the major sources of variation in laboratory tests (analytical imprecision, within-subject biological variation and between-subject variation) and the effects of averaging multiple results from the same sample or from the same person over time are discussed quantitatively in this article. This analysis leads to the surprising conclusion that the strategy of averaging multiple results is only necessary and effective in a limited range of research studies. In clinical practice, it may be important to repeat a test in order to eliminate the possibility of a rare type of error that has nothing to do analytical imprecision or within-subject variation, and for this reason, paradoxically, it may be most important to repeat tests with the highest sensitivity and/or specificity (i.e., ones that are critical for clinical decision-making).
Keywords: analytical imprecision, reference change value, repeating tests, study design, within-subject biological variation
In a recent grant application to study biomarkers in vasculitis, my colleagues and I proposed assaying each sample in duplicate. We did not discuss the issue – it just seemed like the proper way to obtain high-quality data – and we received no commentary, for or against, from the reviewers, suggesting that they had made the same assumption. However, once the funds were gratefully received, we confronted the fact that making each measurement in duplicate would cut in half the number of markers or samples that we could assay and thereby limit the scope of the project. In order to make a decision on how to use our funds most effectively, I investigated the effects of averaging test results in research studies. Because I am also a clinician, I also considered the role of repeating tests in clinical practice and was struck by the differences from the research arena.
Quantitative description of the sources of variation in laboratory tests has an extensive literature in laboratory medicine [1] that is, I suspect, little known to clinicians or clinical researchers. The improvement produced by averaging data obtained from replicate measurements has been described by Callum Fraser in publications aimed primarily at educating laboratory medicine professionals and the panels of experts who produce guidelines for the proper use of such tests [1,2]. This topic is also highly relevant, but in different ways, to those designing research studies and to practicing physicians, hence my impetus to introduce it to a more general medical audience. I will turn to C-reactive protein (CRP) repeatedly as an example, since it is being measured in my research project and is used in multiple ways in clinical medicine. I hope that my conclusions will be useful both to researchers faced with decisions regarding distribution of resources and to clinicians who seek to optimize their use of laboratory tests to make decisions about their patients.
Sources of variation
Variation in the measured concentrations of analytes for a given subject derives from three sources [1].
Pre-analytical variation encompasses numerous factors related to the status of the individual (e.g., posture, time of day, food intake, exercise and medications) and the collection and handling of the sample (e.g., tourniquet time, transport time and storage). Identifying relevant sources of pre-analytical variation is the responsibility of anyone hoping to bring a new assay into clinical use, and minimizing pre-analytical variation once an assay is in clinical use is one of the many quality-control tasks of the clinical laboratory.
Analytical variation is comprised of analytical imprecision (random error) and analytical bias, which is a systematic error defined as “the difference between the expectation of measurement results and the true value of the measured quantity” [1]. Analytical imprecision resulting from random errors (which have also been referred to as measurement error, within-subject standard deviation [SD] [3], analytic error [4], technical error of measurement, method error and Dahlberg’s d [5]) is a feature of any testing modality. These errors are expected to fall in a normal distribution around a mean value. Analytical imprecision must be determined before bringing an assay to clinical use, and it is recommended that individual laboratories determine analytical imprecision individually for commonly used assays [1]. Analytical bias should also be assessed during assay development in two ways: comparison to a gold-standard methodology if one is available, and evaluation for variation in analytical bias related to factors such as reagent lots and recalibration. Analytical bias can contribute to variation in results obtained from the same subject over time or when results obtained with different methodologies are compared. Detection and minimization of analytical bias is, thus, also an important ongoing task in the clinical laboratory for any assay in clinical use.
The importance of within-subject biological variation around a homeostatic set-point unique to each individual has been recognized by the laboratory medicine community [6]. This variation has been quantified for many common analytes [101] and is felt to be applicable across laboratories [7] and, thus, in contrast to analytical imprecision, does not require calculation in each clinical laboratory. Such variation is often similar in health and disease but sometimes differs [8,9].
Between-subject biological variation is, of course, a fourth source of variation in any situation in which data from different individuals are compared. Such variation is important in clinical medicine when an individual is being compared with the reference interval for the population, and in research studies in which groups of individuals are compared, but it is not relevant to clinical settings or studies in which only the change in results for each individual is evaluated.
Having acknowledged the importance of pre-analytical variation and analytical bias, I will focus on the inter-relationships between analytical imprecision, within-subject biological variation and between-subject biological variation. The effect of analytical imprecision on interpretation of data depends not only upon the magnitude of imprecision for a given test procedure, but also upon how that magnitude compares to within-subject biological variation and often between-subject variation, and on how the test is being applied in a research study or in the care of an individual patient. In a research study, the impact of analytical imprecision depends upon whether that study aims only to measure differences between populations or to determine the ability of the test to classify subjects as being in one population or another, and on whether the study involves interpretation of changes over time. In clinical care, these processes of classification (i.e., diagnosis) and longitudinal follow-up are paramount, but in addition, the decision to repeat a test depends on the clinical context.
Quantifying variation
If a distribution of data is normal (Gaussian or bell-shaped), then it is appropriate to describe the variation using the SD. Taken out of context, the SD as a number has limited meaning. A better sense of the spread in a set of data is gained by calculating the coefficient of variation (CV), which is the SD divided by the mean, usually multiplied by 100 and expressed as a percentage. Analytical imprecision and within-subject biological variation (assuming a stable state of health) are normally distributed and are typically described using the CV [1]. Analytical imprecision can be calculated in slightly different ways using replicate measurements carried out on the same samples, and within-subject biological variation can be calculated using measurements taken from multiple samples from the same subjects over time.
When multiple sources of variation contribute to the total variation in a set of data, then the mathematical relationship between those sources is expressed through variances (i.e., SD2). The many equations I will show may appear daunting but are conceptually simple, all being derived from the following relationship between total variation and component sources of variation:
Therefore,
and
Strictly speaking, these equations are true only for variances characterizing infinite numbers of results in normal distributions (σ2), but use of empirical SD2 is a reasonable and practical approximation, and in most cases, the equations are equally valid using CV rather than SD. These equations also require the reasonable assumption that there is no correlation (covariance) between the sources of variation.
If the sources of variation are broadly considered as true variation and variation due to error, then the variation observed (obs) in a group of measurements can be expressed as:
The ratio of SD2true to SD2obs is a widely used definition for test reliability, and reliability is commonly estimated from data using the principles of analysis of variance to calculate an intraclass correlation coefficient [5,10,11]. Having noted these terms, I will proceed to discuss the specific relationship between analytical imprecision, within-subject biological variation and between-subject biological variation.
For a set of laboratory data collected from one person, and if pre-analytical variation is minimized, then the total variation observed (SDT or CVT) is a function of analytical imprecision (SDA or CVA) and within-subject biological variation (SDI or CVI) [1]:
or
Plugging numbers into these equations is informative. If the variances SDA 2 and SDI 2 are identical, then the total SD observed in the data is 21/2 (~1.41) times the SD of either individual component. Otherwise, the nature of the calculation (square root of a sum of squares) emphasizes the larger of the two components, as shown in Table 1 (see column showing results for one measurement). For example, if the within-subject biological variation is twice that of analytical imprecision (CVA/CVI = 0.5 in Table 1), then the total observed CV will only be 12% greater than the true within-subject variation CVI [4,12]. For this reason, goals for analytical imprecision based on how an assay’s analytical CV compares to within-subject CV have been proposed and widely adopted: CVA/CVI ≤0.75 is regarded as a reasonable minimum standard for precision, with CVA/CVI ≤0.5 considered desirable and CVA/CVI ≤0.25 optimal [2,6,12]. Standards for analytical imprecision based purely on the CV of the assay (e.g., considering a CV of 10–15% to be the minimum standard [13,14]) are a poor substitute for this type of analysis and could result in standards being either too lax or too strict.
Table 1.
Effect of analytical imprecision and replication on the variation observed in a distribution.†
| CVA/CVtrue‡ | CVT/CVtrue with listed number of measurements§ | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 5 | 10 | |
| 0.2 | 1.02 | 1.01 | 1.01 | 1.00 | 1.00 |
| 0.5 | 1.12 | 1.06 | 1.04 | 1.02 | 1.01 |
| 1.0 | 1.41 | 1.22 | 1.15 | 1.10 | 1.05 |
| 2.0 | 2.24 | 1.73 | 1.53 | 1.34 | 1.18 |
| 5.0 | 5.10 | 3.67 | 3.06 | 2.45 | 1.87 |
The relative magnitudes of analytical imprecision and the true population variation.
Entries express the relative magnitudes of observed total CV to the true population CV.
CV: Coefficient of variation.
For a set of data collected from multiple persons, between-subject biological variation (SDG or CVG) also contributes:
or
As above, the largest sources of variation contribute disproportionately to the total observed variation.
Reported CVs for CRP assays are typically <10% and often <5%. Within-subject and between-subject variation are much higher [15-18]. In one of several studies on this topic, mean CRP was 1.96 mg/l, analytical imprecision SD was approximately 0.1 mg/l, within-subject SD was 1.19 mg/l and between-subject SD was 1.66 mg/l. Within-subject variation contributed 34% to total variation, between-subject variation contributed 66% and the contribution of analytical imprecision was tiny [18]. Similar results have been reported for many common clinical chemistry tests [19,102]. However, since within- and between-subject variation are only 1–5% for some electrolytes [12,19], analytical imprecision can contribute greatly to observed variability in these cases.
Effects of averaging repeated measurements
The (only) other statistical principle that will play a role in the following equations is that the effect of averaging data (i.e., determining the mean and using that number) is expressed by dividing the variance (SD2) by the number of data points being averaged. Although this concept may seem unfamiliar to many readers, it is the basis of very familiar determination of the standard error (SE) from the standard deviation:
Averaging replicate results from the same samples reduces the impact of analytical imprecision. Averaging repeated test results from independent samples from the same subjects over time reduces the effect of within-subject biological variation. Neither of these types of replication has any impact on the systematic errors that can result from use of a particular assay method (analytical bias) or technical failures; these are detected and addressed differently [20-22,103].
Replicate measurements on the same sample
Averaging multiple results from the same specimen reduces the effective analytical imprecision as follows:
where nA is the number of replicates [2].
For example, when using an assay with a CV of 21%, one would have to measure each sample nine times in order to match the precision of an assay with a CV of 7%. However, as above, the contribution of analytical imprecision to total variation depends on the relative magnitudes of other sources of variation. The effect on total observed variation of performing replicate measurements and averaging them is:
if samples are from one person, or
if samples are from multiple persons, where nA is the number of replicates [2].
As can be seen in Table 1, averaging replicates always reduces the total CV (or SD), but the degree of improvement depends heavily on the context. If the analytical CV is much lower than the true CV (within-subject or the combination of within- and between-subject), then the total CV closely resembles the true CV to begin with, and the effect of replication is minuscule. If the analytical CV is higher than the true CV, then replication has a sizable impact, but large numbers of replicates may be needed to bring the observed CV close to the true CV.
Repeated measurements on the same person
By contrast, averaging repeated measurements on multiple specimens taken from the same person over time mitigates the effects of both within-subject biological variation and analytical imprecision:
where nI is the number of repeated samples.
Incorporating the averaging of replicate measurements from the same specimen and the averaging of repeated measurements from the same person gives the following:
if samples are from one person, or
if samples are from multiple persons.
Thus, averaging of test results taken over time is a more efficient way to reduce total variation than replicate testing of individual samples, although only replicate testing can allow calculation of analytical imprecision (CVA) and estimation of within-subject biological variation using:
The practice of averaging values separated in time also carries the assumptions that the person is in the same state of health on all occasions and that no change in analytical bias has occurred.
Averaging replicate measurements of CRP on individual samples would not improve the precision of estimating a patient’s homeostatic concentration, since analytical imprecision is much smaller than within-person biological variation. Averaging results obtained on multiple occasions would reduce the impact of within-subject variation (e.g., averaging three results would reduce the contribution of within-subject variation to 15% of the total variation in a population).
Effects of analytical imprecision, within-subject biological variation & averaging of repeated measurements in research studies
In many research studies, it is the comparison of two populations that is of interest. Analytical imprecision and within-subject variation will contribute to the observed distributions in both populations, but their impact depends strongly on how those distributions relate to each other and on the way in which the data are used.
Pre-analytical variation and analytical bias may have a large or small effect on results, depending on the analyte and on how assays are performed. If an analyte varies greatly with posture and time of day, for example, and blood is not collected in a standardized manner, then pre-analytical variation may contribute significantly to total variation. If samples are collected over time but are all tested at the same time, then analytical bias can only influence comparison to results outside the study. If test results are obtained over time and/or in different laboratories, then attention to the possibility of varying analytical bias may be needed in order to properly interpret the data [23].
The comparison of two distributions is affected by analytical imprecision and/or within-subject variation if, and only if, the degree of overlap between the distributions is increased, as shown in Figure 1. If the spread is driven by true variation among individuals’ homeostatic set-points within each population, then analytical imprecision and/or within-subject variation are of very little consequence (Figure 1a). If their contribution to total variation is high but the test shows excellent discrimination between groups (Figure 1b), then these sources of variation still have no effect. This latter situation is probably uncommon, but to give a trivial example, a test for blood testosterone could be very imprecise and still distinguish healthy men from healthy women, since the lower limit of the reference interval in men is three times the upper limit of the reference interval in women.
Figure 1. Effect of analytical imprecision on the distribution of data.
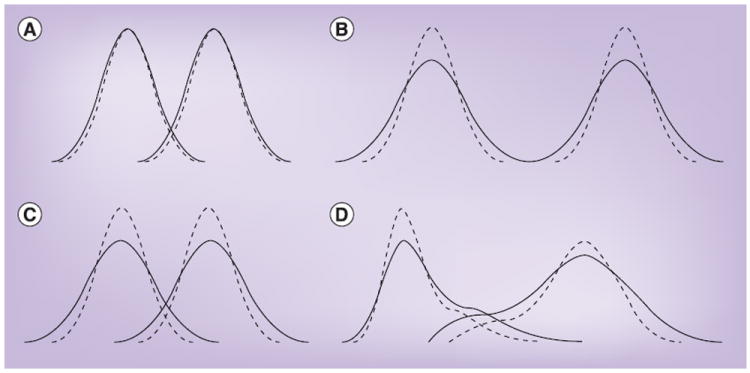
In each diagram of two populations, the dotted lines represent the true distributions of concentrations, and the solid lines represent the distributions observed as a consequence of the combination of true variation and analytical imprecision. (A) Analytical imprecision is very small relative to true variation and therefore has little adverse effect on the observed distribution. (B) Analytical imprecision contributes meaningfully to observed variation, but the distributions being compared still do not overlap, so there is no impact on analyses comparing these distributions. (C) Analytical imprecision contributes to the observed variation and results in greater overlap between the populations, which will affect analysis. (D) Analogous effects in non-normally distributed populations.
Thus, analytical imprecision affects comparisons of populations only under certain circumstances (Figure 1C & 1D), but those circumstances occur frequently. The different ways in which such overlapping distributions can be analyzed show striking differences in the impact of analytical imprecision.
Comparison of average values
If the goal of a study is to compare means of populations, then increasing the number of subjects reduces the SE for estimating the mean of a population regardless of whether the variation is driven by between- or within-person variation, analytical imprecision, or some combination of these:
where n is the number of subjects.
Averaging results of repeated measurements on individual samples and/or results of repeated measurements from multiple samples from individual subjects has the following effect:
where nI is the number of repeated results averaged from the same subject over time, nA the number of replicate results averaged on each sample and n the number of subjects.
Thus, although averaging test results (increasing nA or especially nI above 1) may reduce the SE meaningfully if analytical imprecision and/or within-subject biological variation contribute importantly to total variation, increasing the number of subjects always reduces SE more efficiently.
The early studies by Paul Ridker and colleagues that showed an association between CRP levels and risk of future cardiovascular disease included comparisons of average CRP levels between groups that did or did not have subsequent cardiovascular events [24-26]. Based on the SD reported in another study [18] and the use of groups with >100 subjects, the SE of each estimated mean was approximately 0.20 mg/l. Averaging three independent samples from each subject would have reduced the SE further, but only to 0.18 mg/l.
Classification, sensitivity & specificity
When analytical imprecision is large enough to increase the overlap of two distributions, then analyses that discriminate between populations, such as calculation of sensitivity and specificity, are also affected [4]. Within-subject biological variation has the same effect. Figure 2 shows how such an increase in variation produces an increase in false negatives (resulting in lower sensitivity) and false positives (resulting in lower specificity) simultaneously, and adversely shifts the test’s receiver operating characteristic curve, which is a useful visualization of the trade-off between the true-positive rate (sensitivity) and the false-positive rate (1 – specificity) over a range of cut-off values.
Figure 2. Effect of analytical imprecision on the calculation of sensitivity and specificity.
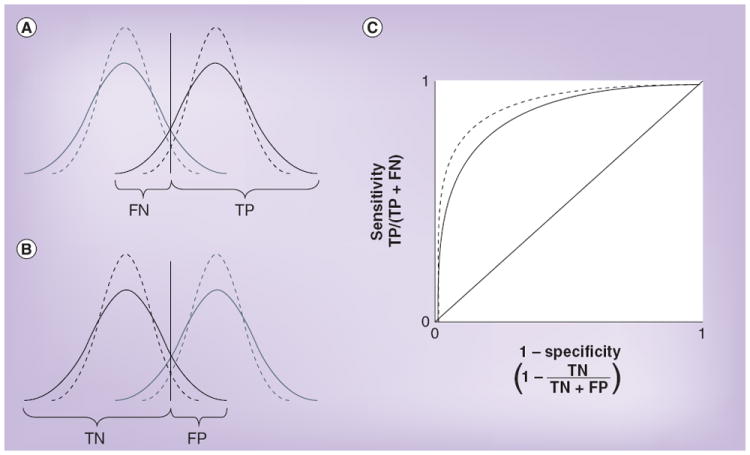
(A & B) The dotted lines represent the true distributions of concentrations, the solid lines represent the distributions that are observed as a consequence of both true variation and analytical imprecision, and the vertical lines show the cut-off points used to distinguish a positive from a negative test result. (A) Analytical imprecision increases the FN rate and correspondingly reduces the TP rate, thereby reducing sensitivity. (B) Analytical imprecision increases the FP rate and correspondingly reduces the TN rate, thereby reducing specificity. (C) The effect on a receiver operating characteristic curve. FN: False negative; FP: False positive; TN: True negative; TP: True positive.
In Figure 3, the effect of a range of increases in variation (caused by analytical imprecision, within-subject biological variation or both) upon a range of sensitivities (or specificities) is calculated using ratios of the sum of the effect of these two sources of variation to the true between-subject variation in the population, also known as the index of individuality [1,27-29]:
Figure 3. Effect of the index of individuality on sensitivity or specificity.
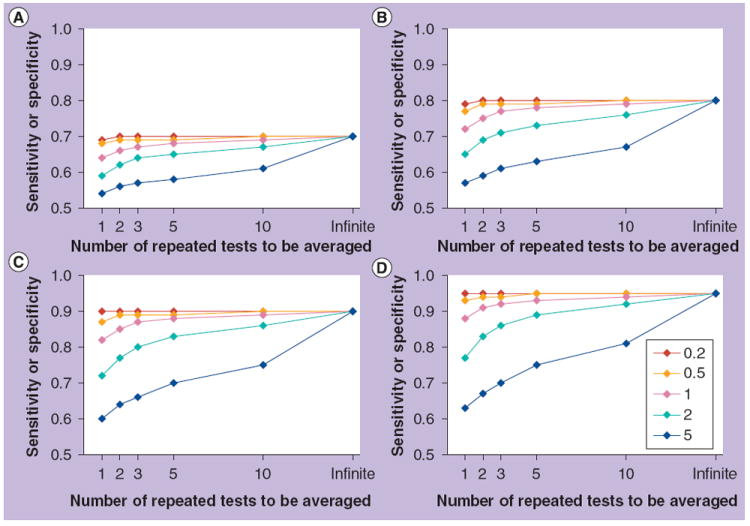
In each panel, five lines show the observed reduction in sensitivity or specificity at different indices of individuality, which is the ratio of analytical imprecision and within-subject variation to between-subject variation. Improvements obtained by averaging different numbers of results are shown. (A–D) indicate true sensitivities/specificities of (A) 0.7, (B) 0.8, (C) 0.9 and (D) 0.95. As discussed in the text, for laboratory tests in common use, indices of individuality are usually 0.5–1.0 and rarely greater than 1.4.
As a caveat, these calculations assume a normal distribution of data in the population as well as precise estimation of analytical imprecision, within-subject biological variation and the observed total CV. However, the principle is valid for any situation in which the distribution is unimodal.
In a study in which sensitivity and specificity are calculated, each subject is classified individually on the basis of a test result. Thus, increasing the numbers of subjects does not overcome the effect of either analytical imprecision or within-subject biological variation as it does with comparison of means. As above, averaging replicate results from the same samples will reduce the effect of analytical imprecision, and averaging repeated results from the same subjects over time (caveats as above) will reduce the effect of both within-person variation and analytical imprecision. Hypothetical examples of the effects of averaging two to ten results are included in Figure 3. Note that if the impact of analytical imprecision and within-subject biological variation is small (index of individuality ≤0.5), averaging of repeated results is unnecessary, but if the index is large, then a large number of results may need to be averaged to neutralize the adverse effect.
For clinical chemistry tests in common use, the index of individuality is usually 0.5–1.0 and rarely greater than 1.4 [1,7,19]. This index is usually driven by within-subject variation, but occasionally by analytical imprecision for analytes in which within-subject variation is low [19]. Experimental biomarkers, which are often the subject of research studies, are likely to behave similarly, but this cannot be assumed.
Ridker and colleagues also divided subjects into categories based on quartiles of CRP level [24]. With this type of analysis, within-subject variation makes misclassification into the wrong quartile a very real possibility, since the index of individuality for CRP is approximately 0.78. Indeed, in a separate study, a second measurement fell into the same quartile as the first measurement only 64% of the time [18]. However, the conclusion that there is a ‘dose–response’ relationship between CRP and cardiovascular risk remains valid – misclassification would bias against finding an association that is truly present.
Analytical imprecision & within-subject biological variation in interpreting data collected longitudinally
In a study in which the change in a test result over time is the outcome of interest, analytical imprecision and within-subject variation at both time points contribute to the variation in the difference between the two results:
If all that is sought in the study is the average change in value associated with a change in disease status (i.e., with comparison to a null distribution), then between-subject variation in the difference (CVGΔ or SDGΔ) is the other factor that contributes to the total variation (CVTΔ or SDTΔ) in that distribution:
or
The effects on the SE of the difference between results of averaging repeated results and increasing the number of subjects are:
where nI is the number of repeated results averaged from the same subject over time, nA the number of replicate results averaged on each sample and n the number of subjects.
Thus, increasing the number of subjects will reduce the SE of the estimated mean difference regardless of the origins of variation.
On the other hand, if each individual subject is being classified according to magnitude of change, then between-subject variation ceases to be relevant, and the variation due to within-subject biological variation and/or analytical imprecision is likely to become highly relevant. Analogous to what is shown in Figure 1, if the difference sought (e.g., pre- and post-treatment) is large relative to these sources of variation, then analytical imprecision and within-subject biological variation may not play an important role; an example might be definition of responders and nonresponders on the basis of a viral load changing over several orders of magnitude. If, however, the outcome sought is a more subtle change (e.g., a 10% reduction in low density lipoprotein), then analytical imprecision and within-subject variation may reduce the sensitivity and specificity with which a change in disease status is associated with a change in the laboratory test result, analogous to what is shown in Figures 2 & 3.
The determinants of total variation are affected by averaging of repeated results in the same way as above – that is, dividing the variance due to within-subject biological variation by the number of repeated tests on different samples (SDI2/nI or CVI2/nI) and dividing the variance due to analytical imprecision by the number of repeated tests on the same or different samples (SDA2/[nI × nA] or CVA2/[nI × nA]). Increasing the number of subjects will not mitigate the effects of either analytical imprecision or within-subject biological variation in a study in which change is interpreted on a per-subject basis. Furthermore, if each transition to a new disease state is treated individually, then there is no prospect of reducing the effect of within-subject variation through averaging of test results in each state. If the numbers of results that can be averaged differ during two clinical states (e.g., good health and disease), then the effect on the CV attributable to within-subject variation and analytical imprecision is:
where n1 and n2 are the numbers of results averaged in the two clinical states and nA is the number of results averaged per sample.
Thus, the smaller of n1 and n2 drives the multiplier, which has a maximum of 21/2 = ~1.41. A balanced design (e.g., n1 = 3, n2 = 3, multiplier = 0.82) reduces the multiplier more than an unbalanced design (e.g., n1 = 5, n2 = 1, multiplier = 1.1), but that goal may not be achievable.
In our research study, approximately 200 patients with vasculitis had ten blood samples taken over 18 months. On average, each patient had active vasculitis during three of those sample collections and was in remission during the other seven. To look for associations between CRP level and disease state on average, the contribution of within-subject biological variation and analytical imprecision to total variation in the difference between the two disease states for such a patient will be reduced by approximately 50%: (1/3 + 1/7)1/2 = 0.69, rather than 21/2 = ~1.41.
If we want to look at the change in CRP associated with a change in disease state on an individual basis, then within-subject variation (in stable health) may put severe limits on the power of the study to detect small changes. If we determine the average change in CRP associated with changes in disease state, then the SE of that estimate is reduced based on the number of such changes in disease state in the entire study, and within-subject variation may still be effectively neutralized [30,31]. However, if we try to determine how often a change in clinical status is associated with a significant change in CRP (sensitivity), then the minimum change we can propose to detect is limited by this marker’s normal within-subject variation of approximately 60–80% [18,32].
Should the test be repeated in this research study?
I hope that it will come as no surprise that my answer as to whether the test should be repeated in research studies is that it depends. It depends on the study’s design and goals; the relative magnitudes of analytical imprecision, within-person biological variation and between-person variation within a population; and the differences between populations or between states of health and disease. Of course, most studies have more complex designs than the types I have analyzed above, but the principles regarding the combination of different sources of variation and the averaging of multiple results will still apply.
Most studies will be limited in one or more necessary resources – subjects, materials or money. Table 2 outlines pros and cons of allocating resources for 100 tests in different ways. The decision whether to perform replicate measurements on individual samples and/or to repeat measurements on the same subjects over time may also depend on whether it is desirable to calculate analytical imprecision and/or within-person variation as part of the study. Information may already be available regarding an assay’s analytical imprecision and/or the analyte’s within-person variation [8,33,34,101]. Alternatively, for some studies, it may not be essential to know this information. In a study with the very simple goal of determining whether average concentrations of molecule X are different in two populations, it may not even be important to know and report the assay’s CV. However, in a study in which individual results are interpreted, particularly with regard to change over time, then not only analytical imprecision but also within-subject biological variation should be calculated and reported. This latter situation is much closer to that of clinical medicine, as will be discussed next.
Table 2.
Pros and cons of different ways of allocating resources to perform a study using 100 tests.
| Subjects (n) | Tests per subject at different times | Tests per sample | Information on analytical imprecision (SDA and CVA)† | Information on within-subject biological variation (SDI and CVI)‡ | Equation for SE of sampled population | Reduction of SE by this study design | Reduced dispersion of test result to improve classification§ | |
|---|---|---|---|---|---|---|---|---|
| 100 | 1 | 1 | No | No |
|
Most | None | |
| 50 | 2 | 1 | No | Yes/limited‡ |
|
Less | Yes | |
| 50 | 1 | 2 | Yes† | No |
|
Less | Maybe | |
| 25 | 4 | 1 | No | Yes/limited‡ |
|
Less | Yes/best | |
| 25 | 2 | 2 | Yes† | Yes† |
|
Least | Yes |
It is assumed that 100 subjects are available and that performing duplicate measurements on individual samples and/or repeated measurements on individual subjects are options.
Analytical imprecision (SDA and CVA) can be estimated by taking the square root of the average variance of pairs of measurements, or other methods.
The sum of within-subject variation (SDI or CVI) and analytical imprecision (SDA or CVA) can be estimated by taking the square root of the average variance of pairs of measurements separated in time; CVI can only be accurately estimated if CVA is determined separately and subtracted from (CVI + CVA).
Dispersion (confidence interval) for an individual test result is a function of CVA and CVI. Averaging repeated results separated in time will always reduce dispersion; averaging replicate results from each sample will only reduce dispersion if the magnitude of CVA is similar to or greater than that of CVI. How much misclassification is reduced depends on other factors (see text).
CV: Coefficient of variation; SD: Standard deviation; SDG: Between-subject SD; SE: Standard error of the mean.
Test repetition in clinical medicine
The extensive literature on the sources of variation in laboratory test results and the role of test repetition is more relevant to clinical medicine than to the design of research studies, and the interested reader may wish to consult these articles [1,2,6,27,34] (and essays and databases at [103]). I will summarize the topic briefly, and with more anecdotes, to highlight differences from the research arena.
Interpretation of laboratory tests is only one component of clinical decision-making, but it is an important and widespread one. Use of test results in clinical practice differs from use in research for three major reasons: clinical decision-making is always an exercise in classification; the unit of analysis is always an individual, never a group; and the decisions made have implications for that individual’s well-being, which means that rare sources of error that have nothing to do with statistical distributions need to be considered.
Also, as a practical matter, the decision to perform replicate testing on the same sample is made by the clinical laboratory. As a general rule, laboratories do not do this routinely except in cases where there is potential for human error at multiple steps, such as manual immunoassays [1]. Many laboratories have ‘repeat rules’ to automatically re-test the same sample in the event of either a clinically significant or critical alert value, or a value outside the measurement range, or a value that has changed more than a given amount in a minimum period of time (‘delta checks’) [35]. By contrast, the decision to perform repeated testing of an independent sample from the same patient is made by the clinician, and may be performed to verify the first result or to compare results over time.
Repeating tests for diagnosis/classification
Just as in research studies that involve classification, variation matters only at values near enough to the border between normal and abnormal, for which the risk of misclassification (i.e., misdiagnosis) increases (see Figure 2). For many but not all tests used for diagnosis, analytical imprecision and/or within-subject biological variation will contribute meaningfully to the risk of misclassification near a cut-off value that was originally chosen on the basis of studies comparing populations. If the analytical imprecision of the test and the within-subject biological variation are known, then the confidence interval (dispersion) of an individual test result can be calculated [2,104]:
where Z is the number of standard deviations associated with the level of confidence desired; for 95% confidence (equivalent to ± 2 CVs), Z = 1.96.
If the dispersion of the result falls fully outside the reference interval for the population, then the result is clearly abnormal, and more stringent levels of confidence than 95% could be applied.
More philosophically, the concern for misclassification arises primarily in a setting in which sensitivity and specificity would remain imperfect even in the absence of analytical imprecision or within-subject variation, and there may be little value in obtaining a more precise measurement of an uninterpretable finding. For example, I am seldom inclined to send another rheumatoid factor or antinuclear antibody test in follow-up of the test being positive at a very low titer. Thus, the decision to repeat a test with a borderline result may be driven by whether the result would continue to be viewed with skepticism or would instead be used to guide treatment, and that decision will in turn be driven by the clinical context.
Petersen and colleagues provide a quantitative justification for not routinely repeating tests in order to confirm or refute abnormal results [1,27,28]. For a mildly abnormal result for an analyte with a low index of individuality (relatively low within-subject variation), repeat testing is likely to give another result that is mildly abnormal or barely within the reference interval – that is, ‘borderline’. For an analyte with a high index of individuality, repeat testing is likely to yield a result that is within the reference interval regardless of whether the patient’s true result is abnormal or not, so that both true positives and false positives are decreased [1,27,28]. Conversely, a very abnormal result, such as described above, is unlikely to fall into the reference interval upon repetition. Thus, little is gained with repeat testing in any of these settings.
Repeating tests to detect change
A need to detect a change in a laboratory test over time is widespread in clinical medicine. Of course, in this case, the question is not whether to repeat the test, but how many times and how to interpret the result. The smaller the degree of change that one wishes to detect, the more likely that the combination of within-subject biological variation and analytical imprecision will make that task challenging. The reference change value (RCV), determined by these sources of variation, is widely used by clinical laboratories to identify and report changes that are statistically significant based on the dispersion of two results, usually at a 95% confidence level [1,36-38]. The RCV has been calculated and published for a large number of laboratory tests [34]. RCV can be calculated using either SD or CV, but usually the latter in order to express the RCV as a percentage change:
which reduces to:
in the usual case where no averaging of test results has occurred.
Although it has not been used this way, the RCV could be reduced by averaging multiple results (n1 and/or n2) obtained over time:
which reduces to:
If n1 and n2 are the same (= n), then the adjusted RCV, RCVadj, can be calculated quite simply from the unadjusted RCV, RCVunadj, which could be useful because the latter might be written [34]:
where RCVunadj is the RCV in the absence of repetition, n is the number of repeated tests to be averaged and RCVadj is the revised RCV obtained via this strategy.
If one were to want to use a cut-off value for CRP to determine whether a patient should receive a statin, as has been proposed [39], but remains controversial, then one might need to repeat the test several or many times and average the values in order to neutralize within-subject biological variation and be confident of classifying the patient as having a high CRP (i.e., reduce the false-positive rate). However, because CRP has a high index of individuality, that strategy would also reduce the detection of true positives [27,28].
If, in the future, one wanted to detect reduction in CRP in an individual patient as an indicator of effective drug treatment (statin or otherwise) and lower cardiovascular risk, the ability to do so with a small number of measurements before and after treatment would be severely limited by within-subject variation, which produces a one-sided RCV of approximately 140–190%.
The importance of being right
The result of misclassification in patient care is incorrect treatment. “Five percent of patients given improper treatment” sounds a bit more serious than “sensitivity reduced from 85 to 80%”. This concern produces another reason to repeat tests, which is much more relevant to clinical practice than to research: detection of rare errors that are not related to analytical imprecision or within-subject variation. Errors in laboratory testing are classified based on whether they occur in the pre-analytic (ordering of test and collection of specimen), analytic (processing of specimen in the laboratory and performance of test) or post-analytic (reporting of result) phase [40,41]. Pre-analytic errors are the most common, but also seldom generate spurious data because most types (e.g., unlabeled tubes) are readily detected [41,42]. Post-analytic errors are the next most common, but again, the type that produce spurious data (e.g., data entry errors) are rare (five out of 40,490 analyses in one study [42]). Overt analytic errors related to instrument malfunction or poor assay performance are also rare (21 out of 40,490 analyses in the same study [42]).
Paradoxically, the better the test, as measured by sensitivity and specificity, the more likely it is to be relied upon for classification/diagnosis and to determine treatment decisions, and the more reasonable it is to consider repeating the test solely to rule out rare pre-analytic, analytic and post-analytic errors. The ‘repeat rules’ followed by clinical laboratories can detect many rare errors, but it has been argued that excessive resources are expended to do so (e.g., 20,844 repeated tests to detect 102 errors in one study [43]), and even then, such retesting always uses the same sample, meaning that many pre- and post-analytical errors could be missed. Put in the larger context of clinical decision-making, it would be reasonable and more cost effective to follow a strategy of repeating a test based on how surprising the finding is (whether normal or abnormal), but it is always more reassuring to see a critical result more than once. Perhaps it is my background in the research laboratory that causes me to deviate from quantitatively defensible arguments on this issue: a novel finding is not to be trusted until it is repeated. Fortunately, in many cases, it would be appropriate to repeat the test in order to detect change after treatment, but some tests, such as many autoantibody and genetic tests, are used only for the purpose of diagnosis. For example, when I was a resident, we admitted a patient on the basis of a serum creatinine of 2.4 mg/dl, identified on routine screening of a patient who felt well. As we considered further urgent work-up, we decided to repeat the test first. The value was 0.9, and the patient was discharged (in many cases, a third test would be indicated as a ‘tie-breaker’, but in this case the normal value matched the clinical setting). I expect that most practicing physicians have had at least one similar experience.
Should the test be repeated for individual patients?
The only reason to repeat tests in routine clinical practice is to prevent misclassification, or in other words, to avoid making an error. As a result, repeating a test is only a useful strategy if the test results will contribute to clinical decision-making. For a test that will be followed over time in order to detect change, averaging of repeated results may be necessary in order to reduce the RCV, which is more often driven by within-subject biological variation than by analytical imprecision. For a test that will be used only once in order to make a diagnosis, the impetus to repeat the test is different. Since the most influential diagnostic test results are those associated with very high sensitivity and/or specificity, I propose that repeating such a high-performing test is often more appropriate (to rule out rare errors independent of the ubiquitous sources of variation) than repeating a result that is ‘borderline’.
Conclusion & future perspective
If the principles outlined here become generally appreciated, then funding agencies and insurers may scrutinize whether routine repeated testing should be paid for on a case-by-case basis; determination and interpretation of normal within-subject biological variation will be included in guidelines for biomarker development; and practicing physicians will be educated about the interpretation of significant changes in test results as reported by clinical laboratories.
Executive summary.
Sources of variation
-
▪
Sources of variation in results of biological assays include pre-analytical variation, analytical imprecision, analytical bias, within-subject normal biological variation and between-subject variation.
Effects of averaging results
-
▪
Averaging the results of multiple measurements reduces subcomponents of the variance, in proportion to the number of replicates. Averaging replicates from the same sample reduces the effect of analytical imprecision; averaging values obtained repeatedly over time reduces the effects of both within-subject biological variation and analytical imprecision.
-
▪
When test results are used for classification, variation due to either analytical imprecision or within-subject variation can lead to misclassification near cut-off values, reducing sensitivity and specificity. Averaging multiple results can reverse this effect to some degree.
Effects of imprecision & repeated testing in research studies
-
▪
Precision in estimation and comparison of average values improves with increasing study size, so that in a research study with this goal, averaging results of repeated tests is only indicated for small studies.
-
▪
In a study in which results are used to classify individual subjects, averaging multiple results may improve sensitivity and specificity, but increasing the number of subjects cannot.
-
▪
It may be important to incorporate replicate and repeated measurements in a study in order to estimate the analytical imprecision of the assay and normal within-person biological variation if these are not already known, particularly if one anticipates wanting to interpret small differences between subjects or small changes in values longitudinally.
Test repetition in clinical medicine
-
▪
In clinical care, repeating a test result is only indicated if the result will contribute meaningfully to decision-making. Repeating a ‘borderline’ result has little value, since both the true- and false-positive rates decrease with such a strategy. Repeating a test that is critical to decision-making may be indicated in order to eliminate the possibility of a rare error that has nothing to do with the ubiquitous sources of variation.
Acknowledgments
The author is grateful to the members of the Clinical Epidemiology Unit at the Boston University School of Medicine, MA, USA, particularly D Felson, P Grayson and P Merkel, for critical review of the manuscript.
RC1 AR058303 (NIH/NIAMS), Arthritis Investigator Award (Arthritis Foundation).
Footnotes
Financial & competing interests disclosure
The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
-
▪
of interest
-
▪▪
of considerable interest
- 1▪▪.Fraser CG. Biological Variation: From Principles to Practice. AACC Press; Washington, DC, USA: 2001. Comprehensive summary of within-subject biological variation that relates to other sources of variation in laboratory testing. [Google Scholar]
- 2.Fraser CG. Test result variation and the quality of evidence-based clinical guidelines. Clin Chim Acta. 2004;346(1):19–24. doi: 10.1016/j.cccn.2003.12.032. [DOI] [PubMed] [Google Scholar]
- 3.Bland JM, Altman DG. Measurement error. BMJ. 1996;313(7059):744. doi: 10.1136/bmj.313.7059.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris EK. Statistical principles underlying analytic goal-setting in clinical chemistry. Am J Clin Pathol. 1979;72(Suppl. 2):S374–S382. [PubMed] [Google Scholar]
- 5.Harris EF, Smith RN. Accounting for measurement error: a critical but often overlooked process. Arch Oral Biol. 2009;54(Suppl. 1):S107–S117. doi: 10.1016/j.archoralbio.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 6▪.Petersen PH, Fraser CG, Kallner A, Kenny D. Editors: strategies to set global quality specifications in laboratory medicine. Scand J Clin Lab Invest. 1999;59(7):475–585. Proceedings of an international consensus conference in which guidelines incorporating within-subject variation were developed. [PubMed] [Google Scholar]
- 7.Fraser CG. Improved monitoring of differences in serial laboratory results. Clin Chem. 2011;57(12):1635–1637. doi: 10.1373/clinchem.2011.175026. [DOI] [PubMed] [Google Scholar]
- 8.Ricos C, Iglesias N, Garcia-Lario JV, et al. Within-subject biological variation in disease: collated data and clinical consequences. Ann Clin Biochem. 2007;44(Pt 4):343–352. doi: 10.1258/000456307780945633. [DOI] [PubMed] [Google Scholar]
- 9.Carlsen S, Petersen PH, Skeie S, Skadberg O, Sandberg S. Within-subject biological variation of glucose and HbA(1c) in healthy persons and in Type 1 diabetes patients. Clin Chem Lab Med. 2011;49(9):1501–1507. doi: 10.1515/CCLM.2011.233. [DOI] [PubMed] [Google Scholar]
- 10.Karanicolas PJ, Bhandari M, Kreder H, et al. Evaluating agreement: conducting a reliability study. J Bone Joint Surg Am. 2009;91(Suppl. 3):S99–S106. doi: 10.2106/JBJS.H.01624. [DOI] [PubMed] [Google Scholar]
- 11.Bland JM, Altman DG. Measurement error and correlation coefficients. BMJ. 1996;313(7048):41–42. doi: 10.1136/bmj.313.7048.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cotlove E, Harris EK, Williams GZ. Biological and analytic components of variation in long-term studies of serum constituents in normal subjects. 3. Physiological and medical implications. Clin Chem. 1970;16(12):1028–1032. [PubMed] [Google Scholar]
- 13.Cummings J, Ward TH, Greystoke A, Ranson M, Dive C. Biomarker method validation in anticancer drug development. Br J Pharmacol. 2008;153(4):646–656. doi: 10.1038/sj.bjp.0707441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maksymowych WP, Landewe R, Tak PP, et al. Reappraisal of OMERACT 8 draft validation criteria for a soluble biomarker reflecting structural damage endpoints in rheumatoid arthritis, psoriatic arthritis, and spondyloarthritis: the OMERACT 9 v2 criteria. J Rheumatol. 2009;36(8):1785–1791. doi: 10.3899/jrheum.090346. [DOI] [PubMed] [Google Scholar]
- 15.Clark GH, Fraser CG. Biological variation of acute phase proteins. Ann Clin Biochem. 1993;30(Pt 4):373–376. doi: 10.1177/000456329303000404. [DOI] [PubMed] [Google Scholar]
- 16.Macy EM, Hayes TE, Tracy RP. Variability in the measurement of C-reactive protein in healthy subjects: implications for reference intervals and epidemiological applications. Clin Chem. 1997;43(1):52–58. [PubMed] [Google Scholar]
- 17.Cho LW, Jayagopal V, Kilpatrick ES, Atkin SL. The biological variation of C-reactive protein in polycystic ovarian syndrome. Clin Chem. 2005;51(10):1905–1907. doi: 10.1373/clinchem.2005.052753. [DOI] [PubMed] [Google Scholar]
- 18.Ockene IS, Matthews CE, Rifai N, Ridker PM, Reed G, Stanek E. Variability and classification accuracy of serial high-sensitivity C-reactive protein measurements in healthy adults. Clin Chem. 2001;47(3):444–450. [PubMed] [Google Scholar]
- 19.Lacher DA, Hughes JP, Carroll MD. Estimate of biological variation of laboratory analytes based on the third national health and nutrition examination survey. Clin Chem. 2005;51(2):450–452. doi: 10.1373/clinchem.2004.039354. [DOI] [PubMed] [Google Scholar]
- 20.Bland JM, Altman DG. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet. 1986;1(8476):307–310. [PubMed] [Google Scholar]
- 21.Klee GG. Establishment of outcome-related analytic performance goals. Clin Chem. 2010;56(5):714–722. doi: 10.1373/clinchem.2009.133660. [DOI] [PubMed] [Google Scholar]
- 22.Westgard JO, Groth T. Design and evaluation of statistical control procedures: applications of a computer “quality control simulator” program. Clin Chem. 1981;27(9):1536–1545. [PubMed] [Google Scholar]
- 23.Petersen PH, Fraser CG, Westgard JO, Larsen ML. Analytical goal-setting for monitoring patients when two analytical methods are used. Clin Chem. 1992;38(11):2256–2260. [PubMed] [Google Scholar]
- 24.Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997;336(14):973–979. doi: 10.1056/NEJM199704033361401. [DOI] [PubMed] [Google Scholar]
- 25.Ridker PM, Buring JE, Shih J, Matias M, Hennekens CH. Prospective study of C-reactive protein and the risk of future cardiovascular events among apparently healthy women. Circulation. 1998;98(8):731–733. doi: 10.1161/01.cir.98.8.731. [DOI] [PubMed] [Google Scholar]
- 26.Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Plasma concentration of C-reactive protein and risk of developing peripheral vascular disease. Circulation. 1998;97(5):425–428. doi: 10.1161/01.cir.97.5.425. [DOI] [PubMed] [Google Scholar]
- 27.Petersen PH, Fraser CG, Sandberg S, Goldschmidt H. The index of individuality is often a misinterpreted quantity characteristic. Clin Chem Lab Med. 1999;37(6):655–661. doi: 10.1515/CCLM.1999.102. [DOI] [PubMed] [Google Scholar]
- 28▪.Petersen PH, Sandberg S, Fraser CG, Goldschmidt H. Influence of index of individuality on false positives in repeated sampling from healthy individuals. Clin Chem Lab Med. 2001;39(2):160–165. doi: 10.1515/CCLM.2001.027. Exploration of how the index of individuality determines the value (or lack thereof) of repeated testing. [DOI] [PubMed] [Google Scholar]
- 29.Harris EK. Effects of intra- and interindividual variation on the appropriate use of normal ranges. Clin Chem. 1974;20(12):1535–1542. [PubMed] [Google Scholar]
- 30.Ridker PM, Danielson E, Fonseca FA, et al. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. 2009;373(9670):1175–1182. doi: 10.1016/S0140-6736(09)60447-5. [DOI] [PubMed] [Google Scholar]
- 31.Peters SA, Palmer MK, Grobbee DE, et al. C-reactive protein lowering with rosuvastatin in the METEOR study. J Intern Med. 2010;268(2):155–161. doi: 10.1111/j.1365-2796.2010.02230.x. [DOI] [PubMed] [Google Scholar]
- 32.Rudez G, Meijer P, Spronk HM, et al. Biological variation in inflammatory and hemostatic markers. J Thromb Haemost. 2009;7(8):1247–1255. doi: 10.1111/j.1538-7836.2009.03488.x. [DOI] [PubMed] [Google Scholar]
- 33.Ricos C, Alvarez V, Cava F, et al. Current databases on biological variation: pros, cons and progress. Scand J Clin Lab Invest. 1999;59(7):491–500. doi: 10.1080/00365519950185229. [DOI] [PubMed] [Google Scholar]
- 34▪.Ricos C, Cava F, Garcia-Lario JV, et al. The reference change value: a proposal to interpret laboratory reports in serial testing based on biological variation. Scand J Clin Lab Invest. 2004;64(3):175–184. doi: 10.1080/00365510410004885. Calculation of reference change values based on biological variation for 261 analytes, intended for use in interpreting serial changes. [DOI] [PubMed] [Google Scholar]
- 35.Fraser CG. Making better use of differences in serial laboratory results. Ann Clin Biochem. 2012;49(Pt 1):1–3. doi: 10.1258/acb.2011.011203. [DOI] [PubMed] [Google Scholar]
- 36.Fraser CG. Optimal analytical performance for point of care testing. Clin Chim Acta. 2001;307(1–2):37–43. doi: 10.1016/s0009-8981(01)00429-6. [DOI] [PubMed] [Google Scholar]
- 37.Fraser CG. Reference change values. Clin Chem Lab Med. 2011;50(5):807–812. doi: 10.1515/CCLM.2011.733. [DOI] [PubMed] [Google Scholar]
- 38.Plebani M, Lippi G. Biological variation and reference change values: an essential piece of the puzzle of laboratory testing. Clin Chem Lab Med. 2012;50(2):189–190. doi: 10.1515/cclm.2011.751. [DOI] [PubMed] [Google Scholar]
- 39.Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 40.Hollensead SC, Lockwood WB, Elin RJ. Errors in pathology and laboratory medicine: consequences and prevention. J Surg Oncol. 2004;88(3):161–181. doi: 10.1002/jso.20125. [DOI] [PubMed] [Google Scholar]
- 41.Bonini P, Plebani M, Ceriotti F, Rubboli F. Errors in laboratory medicine. Clin Chem. 2002;48(5):691–698. [PubMed] [Google Scholar]
- 42.Plebani M, Carraro P. Mistakes in a stat laboratory: types and frequency. Clin Chem. 1997;43(8 Pt 1):1348–1351. [PubMed] [Google Scholar]
- 43.Deetz CO, Nolan DK, Scott MG. An examination of the usefulness of repeat testing practices in a large hospital clinical chemistry laboratory. Am J Clin Pathol. 2012;137(1):20–25. doi: 10.1309/AJCPWPBF62YGEFOR. [DOI] [PubMed] [Google Scholar]
Websites
- 101.Westgard QC. Biologic Variation Database, the 2012 update. www.westgard.com/biodatabase-2012-update.htm.
- 102.Westgard QC. Biologic Variation Database, the 2010 update. www.westgard.com/biodatabase-2010-update.htm.
- 103.Westgard QC. www.westgard.com.
- 104.Westgard QC. Are ‘Scientific Statements’ the scientific truth? www.westgard.com/are-scientific-statements-the-scientific-truth.htm.