

Abstract
Cyanobacterial aldehyde decarbonylase (cAD) is a non-heme di-iron oxygenase that catalyzes the conversion of fatty aldehydes to alkanes and formate. The mechanism of this chemically unusual reaction is poorly understood. We have investigated the mechanism of C1-C2 bond cleavage by cAD using a fatty aldehyde that incorporates a cyclopropyl group, which can act as a radical clock. When reacted with cAD, the cyclopropyl aldehyde produces 1-octadecene as the rearranged product, providing evidence for a radical mechanism for C-C bond scission. In an alternate pathway, the cyclopropyl aldehyde acts as a mechanism-based irreversible inhibitor of cAD, in which the alkyl chain becomes covalently bound to the enzyme.
The biosynthesis of long-chain aliphatic hydrocarbons is widely distributed in Nature, occurring in plants,1 animals2 and microbes,3 and is typified by the production of alkanes with an odd number of carbon atoms.4 These are derived from fatty acid biosynthesis in a two-step pathway in which fatty-acyl-CoA reductase reduces long-chain fatty-acyl-CoA esters to the corresponding fatty aldehydes.5 In the second step, aldehyde decarbonylase (AD) removes the formyl (HCO) group from fatty aldehydes to yield the long-chain aliphatic hydrocarbons.6 Recently, these biosynthetic processes have garnered increasing interest because of the potential for such pathways to be harnessed for the production of biofuels.7
The reaction catalyzed by aldehyde decarbonylase is unusual because it represents a rare biological case in which a completely unfunctionalized product is formed.8 Recently, it has become apparent that there are three different classes of aldehyde decarbonylases. In insects, the enzyme is a membrane-associated cytochrome P450 system and the aldehyde carbon is converted to CO2.2b,9 In plants and algae, the enzyme is also membrane bound and is most likely iron dependent; however, in this case the aldehyde carbon is converted to CO.5a,6,10 The most recently discovered cyanobacterial aldehyde decarbonylase3a (cAD) is a soluble protein, whose crystal structure11 reveals it to share the same non-heme di-iron metal site as enzymes such as methane monoxygenase, class I ribonucleotide reductase and ferritin.12
We recently demonstrated that in the cAD-catalyzed reaction the aldehyde carbon is converted to formate. Isotope-labeling studies established that the aldehyde C-H bond remains intact during the reaction and that the hydrogen in the newly formed methyl group of the alkane derives from the solvent (or a solvent-exchangeable group on the enzyme),13 findings that were independently arrived at by Warui et al. 14
Other di-iron enzymes that are structurally related to cAD utilize molecular oxygen, and although the substrate is not formally oxidized in the decarbonylation (deformylation) reaction, support for the involvement of molecular oxygen derives from labeling experiments in which 18O from 18O2 was found to be incorporated into formate.15 This observation implies that after reducing the diferric enzyme to the oxygen-reactive diferrous state two further electrons (supplied by the external reducing system) are required for the reaction, so that O2 is fully reduced to water (Scheme 1A). (Whereas our initial investigations of the reaction suggested that cAD may catalyze the reaction in an oxygen-independent manner,13,16 further experiments led us to conclude that we could not rule out the involvement of O2 due to trace oxygen contamination; therefore we currently consider an oxygen-dependent mechanism is more likely).
Scheme 1.
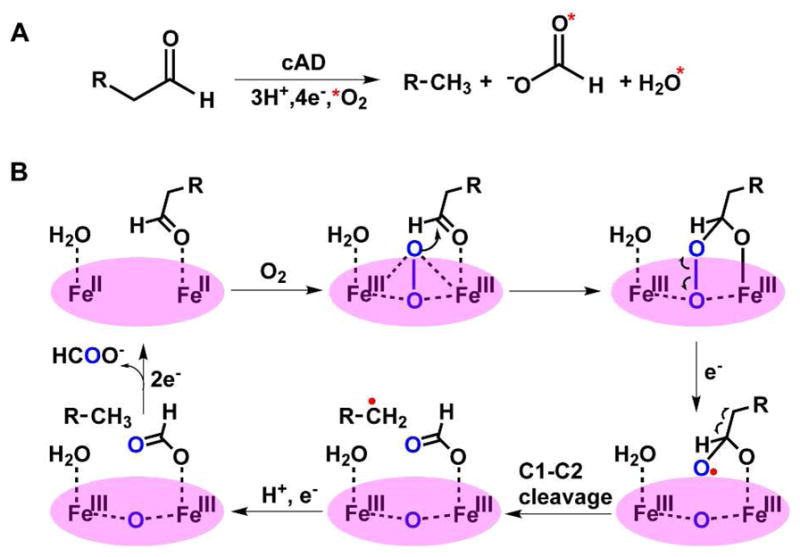
A) Reaction Catalyzed by cAD B) Proposed Mechanism for cAD
A central mechanistic question, which remains to be answered for all classes of aldehyde decarbonylase, is how the bond between the aldehyde carbon (C1) and the α-carbon (C2) is cleaved. One plausible mechanism15a is shown in Scheme 1B. This involves initial formation of a reactive iron-peroxo species that attacks the aldehyde carbon. A 1-electron reduction leads to the formation of a hemiacetal radical followed by scission of the C1-C2 bond. A subsequent proton-coupled electron transfer step reduces the alkyl radical to the alkane. Tentative evidence that the reaction may involve radicals comes from spin-trapping experiments performed on cAD incubated with substrates in the absence of a reducing system.13 Therefore, to gain insights into the C1-C2 bond scission step we have investigated the reaction of cAD with a substrate that incorporates a strategically placed cyclopropyl group that can act as a “radical clock”.
Cyclopropylcarbinyl radicals, formed when radicals are generated adjacent to the cyclopropyl ring, undergo rapid and very well characterized ring-opening reactions.17 Cyclopropyl compounds have been extensively employed to investigate the mechanisms of cytochrome P450 enzymes17–18 and non-heme iron enzymes including methane monooxygenase,19 isopenicillin N synthase20 and, most recently, HppE that catalyzes epoxide formation in the biosynthesis of fosfomycin.21 For cAD, if the lifetime of the postulated alkyl radical intermediate is relatively long one expects to observe ring opening, leading to the formation of octadecene. On the other hand, if either the radical is very short lived, or the reaction is concerted, then retention of the cyclopropyl ring in the product alkane is expected.
As a potential radical clock substrate, we synthesized the cyclopropyl analog of octadecanal, 2-(2-tetradecylcyclopropyl)-acetaldehyde 6, in which the cyclopropyl group is positioned β-to the carbonyl group, as outlined in Scheme 2. Briefly, a Wittig reaction between pentadecanal, 1, and 3-hydroxypropyl-triphenylphosphonium bromide was employed to form octadec-3-ene-1-ol, 2, as the (E) stereoisomer.22 After protection of the alcohol as its TBDMS silyl ether, the double bond was converted to a cyclopropyl group using diethylzinc and diiodomethane, to give the trans stereoisomer, 4, as the major product.23 Finally, deprotection of the TBDMS group by TBAF, followed by oxidation of the alcohol with TEMPO, yielded the cyclopropyl aldehyde, 6.24
Scheme 2.
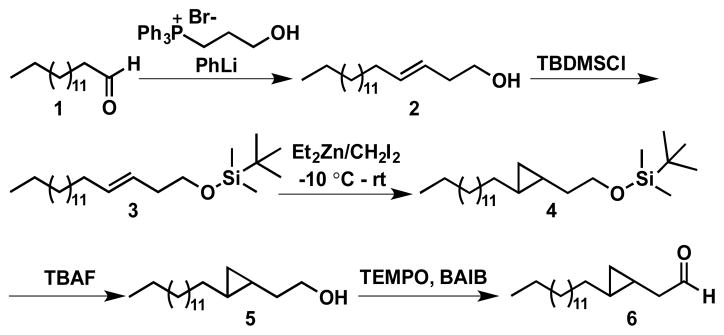
Synthesis of Cyclopropyl aldehyde 6
We investigated the reaction of 6 with cAD from Nostoc punctiformes, which was recombinantly over-expressed in E. coli and purified as previously described.16 Assays were performed at 37°C and typically contained 400 μM 6, 10 μM cAD, 20 μM Fe(NH3)6SO4, 1 mM NADH and 100 μM phenazine methosulfate (PMS) in 100 mM HEPES buffer, 100 mM KCl, 10 % glycerol, pH 7.2 and 4 % DMSO to improve substrate solubility. The reaction products were extracted with ethyl acetate and analyzed by GC-MS.13,16 The chromatograph revealed a new peak at 9.84 min, which co-chromatographed with an authentic standard of 1-octadecene and was characterized by a molecular ion of m/z = 252.3, confirming its identity (Figure S7–S8). The formation of 1-octadecene was strictly dependent on the presence of PMS, NADH and molecular oxygen. Prolonged incubation of the assay mixture under rigorously anaerobic conditions (pO2 < 0.5 ppm) gave no reaction products (Figure S10). This observation supports a radical mechanism for C-C bond scission, in which the cyclopropylcarbinyl radical rearranges to the octadecenyl radical, leading to the formation of 1-octadecene as the product, as shown in Scheme 3. The rate constant for ring opening of cyclopropylcarbinyl radicals is known; k = 8.6 × 107 s−1 at 298 K.25 This implies that the radical generated at the α-carbon of 6 has a lifetime of ≥ 10 ns. The aldehyde carbon was, as expected, converted to formate in the reaction (Figure S11).
Scheme 3.
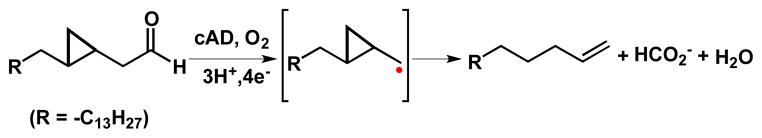
Reaction of Cyclopropyl Substrate 6 with Np cAD
Interestingly, a smaller peak at 10.0 min with m/z = 252.3 was also evident that co-chromatographed with an authentic standard of 1-methyl-2-tetradecylcyclopropane, the non-rearranged product from the decarbonylation of 6 (Figure S7). The amount of cyclopropyl compound formed increased with time, but was independent of the amount enzyme in the assay and, furthermore, was present in similar amounts in control experiments in which the enzyme was omitted (Figure S9). It must therefore arise from non-enzymatic decarbonylation of the cyclopropyl aldehyde (simple aldehydes do not undergo non-enzymatic decarbonylation under these conditions). The non-enzymatic reaction required the presence of NADH, PMS and O2: the non-rearranged product was not observed if either the reducing system or O2 was omitted (Figure S10). Nor was the non-rearranged product formed if a “biological” reducing system comprising NADPH, ferredoxin, and ferredoxin reductase was substituted for NADH and PMS. The fate of the C1 carbon in this reaction remains to be determined. Further discussion of this unusual side reaction is included in the Supporting Information.
Despite varying the concentrations of reagents and the length of assay, no more than about one equivalent of 1-octadecene was observed in the reaction of 6 with cAD (Figure 1A). This observation suggested that 6 may partition between turn-over and acting as an irreversible inhibitor of the enzyme. Inactivation of cAD was confirmed by incubating cAD with 6 (400 μM) in assay buffer containing PMS, NADH and O2 for 1 hour. The activity of the enzyme was then assayed using octadecanal (400 μM) as the substrate. Essentially no activity remained, as shown in Figure 1B (I). In contrast, if the alternate substrate, pentadecanal (400 μM), was substituted for 6 a significant amount of enzyme activity remained after 1 hour when the enzyme was assayed with octadecanal (Figure 1B (II)). The formation of 1-octadecene appeared to be described by first order kinetics. The partitioning of substrate between turnover and enzyme inactivation is described by Equation 1:
| (1) |
Figure 1.
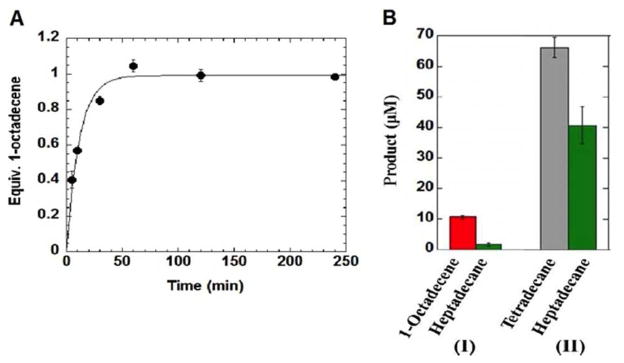
Inactivation of cAD by 6. A: Time course for formation 1-octadecene by cAD. B: (I) Reaction of cAD with 6 for 1 h followed by addition of octadecanal results in negligible heptadecane being formed; (II) as a control, reaction of cAD with pentadecanal for 1 h followed by addition of octadecanal demonstrates that significant enzyme activity remains.
In which Pt is the concentration of product at time t; E is enzyme concentration and kin is the rate constant for inactivation. Fitting the data to Equation 1, kcat ≈ kin = 0.088 ± 0.011 min−1; for comparison kcat = ~ 0.4 min−1 with octadecanal as the substrate.13
To gain further insight into the mechanism of inactivation, we analyzed the inactivated cAD by LC-ES-MS. The mass of cAD prior to reaction with 6 was determined as 28911 ± 0.5 Da (Figure S12), in excellent agreement with the calculated molecular weight. Reaction of cAD with octadecanal, resulted in no change in Mr (Figure 2A). However, reaction of cAD with 6 resulted in between 60 – 80 % of the recovered enzyme eluting from the column as a species characterized by a slightly longer retention time and a molecular weight of 29162 ± 0.5 Da (Figure 2B). The increase in molecular weight of 251 ± 0.5 Da is consistent with the formation of a covalent adduct between decarbonylated 6 and cAD. Covalent modification of cAD by 6 provides a plausible mechanism for inactivation.
Figure 2.

LC-MS of cAD. A: No modification occurs after reaction of cAD with octadecanal (blue); Mr of cAD = 28911 Da; B: reaction of cAD with 6 results in covalently modified protein (red), Mr = 29162 Da.
To determine the location of the covalent modification, samples of the inactivated and the unmodified enzyme were subjected to proteolytic digestion with either trypsin or Glu-C (see Supporting Information). The proteolytic fragments were analyzed by MALDI-TOF mass spectrometry and the spectra of the modified and unmodified enzyme digests compared (Figures S15, S16). Analysis of the spectra identified two peptides, one Glu-C-derived, the other trypsin-derived, which were absent from the spectra of the covalently modified enzyme. Significantly, the peptides overlapped in sequence and encompass a 20-residue segment, CFAIAAYNIYIPVADDFARK, that forms part of the hydrophobic substrate-binding channel of cAD.
Unfortunately, we were unable to detect the alkylated peptides directly by MALDI-TOF mass spectrometry. However, it is commonly observed that hydrophobic peptides are under-represented or absent in “bottom up” proteomic analyses of proteins.26 This may be attributed to loss of hydrophobic peptides during sample preparation steps prior to MS analysis; there is also the potential for the modification to interfere with the proteolytic digestion of the peptide, and/or adversely affect its ability to ionize in the mass spectrometer.
The proteolytic digests of covalently modified cAD were subjected to more extensive analysis using ES-MS-MS. Samples were analyzed using an ion-trap mass spectrometer (LTQ-XL, ThermoFisher) equipped with a nano-spray ion source; the resulting mass spectra were analyzed using Trans-Proteomic Pipeline (TPP) software including PeptideProphet and ProteinProphet.27 This analysis succeeded in identifying one covalently modified peptide, present in low-abundance, in the tryptic digest. The secondary ion mass spectrum of this peptide displayed a fragmentation pattern that was consistent with F107 being modified by an additional mass of 251 ± 0.5 Da (Figure S17). F107 forms part of the hydrophobic substrate channel of cAD (Figure S19) and would be within ~ 5 Å of the putative alkyl radical formed by the opening of the cyclopropyl ring of 6. These results suggest that the reaction of the product alkyl radical with the phenylalanine ring results in the covalent attachment of the alkyl fragment to the protein, thereby inactivating the enzyme (Scheme 4, pathway II).
Scheme 4.

Alternate pathways for reaction of 6 with cAD
Although 6 completely inactivated cAD, a significant fraction of the protein escaped covalent modification by 6 (Figure 2B), suggesting that another inactivation mechanism might be operating. Further insights into the mechanism of inactivation came from deuterium labeling experiments. We previously determined that for the decarbonylation of octadecanal, the hydrogen in heptadecane is derived from the solvent.13 However, the rearrangement of the cyclopropylcarbinyl radical derived from 6 would place the presumed radical intermediate at C-4, rather than at C-1, of the product (Scheme 4). We were therefore interested to know whether the new hydrogen in 1-octadecene was derived from the solvent or some other source. To avoid complications arising from exchange of aldehyde proton during the deuterium labeling experiment, we prepared 6 in which the α-carbon was di-deuterated (see supporting information). When di-deuterated 6 was reacted with cAD in deuterated buffer the predominant molecular ion for 1-octadecene had m/z = 255.3 (Figure S21), corresponding to tri-deuterated 1-octadecene. This is consistent with the proton coming from the solvent or a solvent-exchangeable group on the enzyme.
However, a less abundant but still significant peak at m/z = 254.3 was also observed, corresponding to di-deuterated 1-octadecene (Figure S21). This suggests that some hydrogen may derive from a non-exchangeable position on the protein. (Some protium may also come from the 1 – 2 % of protium remaining in the buffer, but it seems unlikely that this would account for all the di-deuterated product). Abstraction of hydrogen from a non acidic side-chain by the alkyl radical derived from 6 would certainly be energetically feasible. Such oxidative damage might plausibly result in inactivation of the enzyme. This may provide an alternative pathway for the inactivation of cAD by 6 that does not involve in the formation of a covalent adduct between the protein and substrate (Scheme 4, pathway III).
In conclusion, these experiments provide support for a radical mechanism for C-C bond scission in the unusual decarbonylation reaction catalyzed by cAD. Based on the well-documented lifetimes for the ring-opening of cyclopropylcarbinyl radicals, the reaction of cAD with 6 supports the formation of a relatively long-lived radical, i.e. one with a lifetime greater than 10 ns, on the α-carbon during the reaction. The opening of the cyclopropyl ring results in the migration of the radical initially generated at C-1 of the product to C-4. The shift in the position of the radical appears to cause the reaction to partition between completing the catalytic cycle, i.e. producing 1-octadecene as product, and reacting with the protein to inactivate cAD.
Supplementary Material
Acknowledgments
We thank Dr. V. Basrur, University of Michigan Proteomic Facility, for performing MS analyses and advice on interpretation of MS data and Dr. Benjamin C. Buer for assistance with MALDI experiments. This work was supported in part by grants from European Union FP-7 256808, National Science Foundation CHE 1152055 and National Institute of Health GM 093088 to E.N.G.M.
Footnotes
Supporting Information. Experimental details for: the synthesis and characterization of 6; the reaction of Np cAD with 6 and characterization of reaction products; proteolytic digestion and ES-MS-MS and MALDI-TOF analysis of covalently modified Np cAD. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Aarts MGM, Keijzer CJ, Stiekema WJ, Pereira A. Plant Cell. 1995;7:2115. doi: 10.1105/tpc.7.12.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Cheesbrough TM, Kolattukudy PE. J Biol Chem. 1988;263:2738. [PubMed] [Google Scholar]; (b) Reed JR, Vanderwel D, Choi SW, Pomonis JG, Reitz RC, Blomquist GJ. Proc Natl Acad Sci U S A. 1994;91:10000. doi: 10.1073/pnas.91.21.10000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Schirmer A, Rude MA, Li XZ, Popova E, del Cardayre SB. Science. 2010;329:559. doi: 10.1126/science.1187936. [DOI] [PubMed] [Google Scholar]; (b) Ladygina N, Dedyukhina EG, Vainshtein MB. Process Biochem. 2006;41:1001. [Google Scholar]
- 4.Kunst L, Samuels AL. Prog Lipid Res. 2003;42:51. doi: 10.1016/s0163-7827(02)00045-0. [DOI] [PubMed] [Google Scholar]
- 5.(a) Bourdenx B, Bernard A, Domergue F, Pascal S, Leger A, Roby D, Pervent M, Vile D, Haslam RP, Napier JA, Lessire R, Joubes J. Plant Physiol. 2011;156:29. doi: 10.1104/pp.111.172320. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rowland O, Zheng HQ, Hepworth SR, Lam P, Jetter R, Kunst L. Plant Physiol. 2006;142:866. doi: 10.1104/pp.106.086785. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang X, Kolattukudy PE. FEBS Lett. 1995;370:15. doi: 10.1016/0014-5793(95)00781-4. [DOI] [PubMed] [Google Scholar]
- 6.Cheesbrough TM, Kolattukudy PE. Proc Natl Acad Sci U S A. 1984;81:6613. doi: 10.1073/pnas.81.21.6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Ghim CM, Kim T, Mitchell RJ, Lee SK. Biotechnol Bioprocess Eng. 2010;15:11. [Google Scholar]; (b) Somerville C, Youngs H, Taylor C, Davis SC, Long SP. Science. 2010;329:790. doi: 10.1126/science.1189268. [DOI] [PubMed] [Google Scholar]; (c) Rude MA, Schirmer A. Curr Opin Microbiol. 2009;12:274. doi: 10.1016/j.mib.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 8.Buist PH. Nat Prod Rep. 2007;24:1110. doi: 10.1039/b508584p. [DOI] [PubMed] [Google Scholar]
- 9.Qui Y, Tittiger C, Wicker-Thomas C, Le Goff G, Young S, Wajnberg E, Fricaux T, Taquet N, Blomquist GJ, Feyereisen R. Proc Natl Acad Sci U S A. 2012;109:14858. doi: 10.1073/pnas.1208650109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dennis M, Kolattukudy PE. Proc Natl Acad Sci U S A. 1992;89:5306. doi: 10.1073/pnas.89.12.5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Unpublished structure solved by Joint Center of Structural Genomics (protein database entry PDB|2OC5|A).
- 12.Feig AL, Lippard SJ. Chem Rev. 1994;94:759. [Google Scholar]
- 13.Das D, Eser BE, Han J, Sciore A, Marsh ENG. Angew Chem Intl Ed. 2011;50:7148. doi: 10.1002/anie.201101552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warui DM, Li N, Norgaard H, Krebs C, Bollinger JM, Booker SJ. J Am Chem Soc. 2011;133:3316. doi: 10.1021/ja111607x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Li N, Norgaard H, Warui DM, Booker SJ, Krebs C, Bollinger JM. J Am Chem Soc. 2011;133:6158. doi: 10.1021/ja2013517. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li N, Chang WC, Warui DM, Booker SJ, Krebs C, Bollinger JM. Biochemistry. 2012;51:7908. doi: 10.1021/bi300912n. [DOI] [PubMed] [Google Scholar]
- 16.Eser BE, Das D, Han J, Jones PR, Marsh EN. Biochemistry. 2011;50:10743. doi: 10.1021/bi2012417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newcomb M, Toy PH. Acc Chem Res. 2000;33:449. doi: 10.1021/ar960058b. [DOI] [PubMed] [Google Scholar]
- 18.Bowry VW, Ingold KU. J Am Chem Soc. 1991;113:5699. [Google Scholar]
- 19.Valentine AM, LeTadic-Biadatti MH, Toy PH, Newcomb M, Lippard SJ. J Biol Chem. 1999;274:10771. doi: 10.1074/jbc.274.16.10771. [DOI] [PubMed] [Google Scholar]
- 20.Baldwin JE, Adlington RM, Domaynehayman BP, Knight G, Ting HH. J Chem Soc, Chem Commun. 1987:1661. [Google Scholar]
- 21.Huang H, Chang WC, Pai PJ, Romo A, Mansoorabadi SO, Russell DH, Liu HW. J Am Chem Soc. 2012;134:16171. doi: 10.1021/ja3078126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlosser M, Christmann KF. Angew Chem Intl Ed. 1966;5:126. [Google Scholar]
- 23.(a) Furukawa J, Kawabata N, Nishimura J. Tetrahedron. 1968;24:53. [Google Scholar]; (b) Lebel Hl, Marcoux J-F, Molinaro C, Charette AB. Chem Rev. 2003;103:977. doi: 10.1021/cr010007e. [DOI] [PubMed] [Google Scholar]
- 24.De Mico A, Margarita R, Parlanti L, Vescovi A, Piancatelli G. J Org Chem. 1997;62:6974. [Google Scholar]
- 25.Bowry VW, Lusztyk J, Ingold KU. J Am Chem Soc. 1991;113:5687. [Google Scholar]
- 26.Souda P, Ryan CM, Cramer WA, Whitelegge J. Methods. 2011;55:330. doi: 10.1016/j.ymeth.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao F, Chen Y, Cierpicki T, Liu Y, Basrur V, Lei M, Dou Y. PLoS One. 2010;5:e14102. doi: 10.1371/journal.pone.0014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.