

Abstract
Aims/hypothesis
IL-12 is an important cytokine in early inflammatory responses and is implicated in the immunemediated pathogenesis of pancreatic islets in diabetes. However, little is known about the direct effects of IL-12 on islets and beta cells.
Methods
In this study, beta cell function, gene expression and protein production were assessed in primary human donor islets and murine beta cell lines in response to stimulation with IL-12 or a pro-inflammatory cytokine cocktail (TNF-α, IL-1β and IFN-γ).
Results
The pro-inflammatory cytokine cocktail induced islet dysfunction and potently increased the expression and production of IL-12 ligand and IL-12 receptor in human islets. In human islets, the receptor for IL-12 co-localised to the cell surface of insulin-producing cells. Both IL-12 ligand and IL-12 receptor are expressed in the homogeneous beta cell line INS-1. IL-12 induced changes in gene expression, including a dose-dependent upregulation of IFNγ (also known as IFNG), in INS-1 cells. A neutralising antibody to IL-12 directly inhibited IFNγ gene expression in human donor islets induced by either IL-12 or pro-inflammatory cytokine stimulation. Functionally, IL-12 impaired glucose-stimulated insulin secretion (GSIS) in INS-1 cells and human donor islets. A neutralising antibody to IL-12 reversed the beta cell dysfunction (uncoupling of GSIS or induction of caspase-3 activity) induced by pro-inflammatory cytokines.
Conclusions/interpretation
These data identify beta cells as a local source of IL-12 ligand and suggest a direct role of IL-12 in mediating beta cell pathology.
Keywords: Diabetes, Human, Interleukin-12, Islets, Pro-inflammatory cytokines
Introduction
Type 1 diabetes, a T cell-mediated autoimmune disease, is characterised by active immune-mediated destruction of insulin-producing beta cells [1, 2]. Infiltration of the islets with inflammatory cells responding to islet-associated antigens [3], subsequent insulitis and promotion of a pro-inflammatory environment are hallmarks of the autoimmune attack in humans who spontaneously develop type 1 diabetes and in animal models of the disease. Manipulation in the balance of T cell subsets clearly shows that Th1-promoting factors enhance or accelerate the onset of type 1 diabetes [4, 5]. One of the key cytokine pathways that promotes the development of T helper 1 (Th1) cells is IL-12-driven expression of IFN-γ.
IL-12 is a heterodimeric ligand formed from a disulfide-linked protein 35 (p35) A-chain and a protein 40 (p40) B-chain. IL-12 binds the heterodimeric receptor of β1- and β2-plasma-membrane-traversing proteins to induce signalling pathways, including Janus kinase/signal transducer and activator of transcription (JAK/STAT) and mitogen-activated protein kinase/protein 38 (MAPK/p38 activation) [6, 7]. The primary intracellular mediator of IL-12-receptor ligation is phosphorylation of STAT4 [8, 9]. IL-12 has a critical role in the development and pathogenesis of autoimmune diseases by driving the recruitment of inflammatory cells. The accepted cell sources for IL-12 are monocytes, macrophages, dendritic cells, neutrophils and B lymphocytes [10]. Interestingly, IL-12 production is positively upregulated by IFN-γ, creating a potential reinforcing feedback loop, as IL-12 upregulates IFN-γ [7, 11]. IL-12 also contributes to the proliferation of differentiated Th1 cells by enhancement of IL-12 receptor expression [12]. Consequently, IL-12 has been identified as a key target for autoimmune modulation [13]. In NOD mice, which spontaneously develop diabetes, IL-12 plays a significant role in the transition from nondestructive to destructive insulitis [4, 5]. The importance of IL-12 in human diabetes has been raised in genetic studies. The type 1 diabetes susceptibility locus, IDDM18, is located near a regulatory allele of the IL-12p40 (also known as IL12B) gene [14], and single nucleotide polymorphisms in this region associate with an earlier age of onset of type 1 diabetes and accelerated deterioration in glycaemic control [15, 16].
Pro-inflammatory cytokines are associated with islet dysfunction and destruction. Chronic intracellular oxidative stress in islets can be activated by pro-inflammatory cytokines [17], leading to a loss of insulin secretion [18] and destructive protein or DNA modification [19]. Islets also show increased rates of apoptosis (reviewed by Mandrup-Poulsen et al [20]) and decreased proliferation indices [21] on exposure to pro-inflammatory cytokines. An increase in pro-inflammatory cytokines is contraindicative for graft survival in islet transplantation [22]. Pro-inflammatory cytokine levels are elevated in patients with type 2 diabetes (reviewed by Boden [23]). Significantly, a small molecule modulator of IL-12 signalling protects human islets from pro-inflammatory cytokine injury [24], suggesting a possible link between inflammatory cytokines and IL-12 in the pathogenesis of beta cell dysfunction.
Are IL-12 pathways also active directly in islet beta cells? Previous studies have found that the STAT4 signalling pathway is active in islet beta cells [25, 26]. In this study, we evaluated whether IL-12 has a direct role in islets and beta cells: an effect that is distinct from the established influence of IL-12 on immune cells. Our data reveal that pro-inflammatory cytokines upregulate IL-12 signalling components in islets and beta cells. We show that primary human islets and murine beta cell lines are responsive to IL-12. Islet and beta cell dysfunction is induced by IL-12 and a neutralising antibody that blocks IL-12 protects human islets from the destructive effects of pro-inflammatory cytokines.
Methods
Approvals and reagents
Institutional oversight committees approved studies involving animals or human donors. Institutional Animal Care and Use committee approved animal studies that were conducted in accordance with Principles of Laboratory Care, Institutional Review Board approved studies involving human tissue. Human donor islets (≥85% pure) were obtained from the Integrated Islet Distribution Program (IIDP), http://iidp.coh.org/. Tissue sections were obtained from the Network for Pancreatic Organ Donors with Diabetes (nPOD), www.jdrfnpod.org/.
Cell culture
INS-1 cells were cultured in RPM1 1640 (Invitrogen, Carlsbad, CA, USA) containing 10% v/v fetal calf serum, 100 units penicillin/100 μg/ml streptomycin, 10 mmol/l Hepes, 2 mmol/l l-glutamine, 1 mmol/l sodium pyruvate and 0.05 mmol/l 2-mercaptoethanol. β-TC3 cells were cultured in DMEM (Invitrogen) containing 2.4% v/v fetal calf serum, 15% v/v horse serum and 100 units penicillin/100 μg/ml streptomycin. Cells were cultured at 37°C and 5% CO2.
Real-time PCR
Primary human or mouse islets, INS-1 or βTC-3 beta cell lines were treated with IL-12 (1-10 ng/ml) or IL-1β (5 ng/ml), TNF-α (10 ng/ml), and IFN-γ (100 ng/ml) (R&D Systems, Minneapolis, MN, USA) with and without anti-IL-12 antibody (1 μg/ml, eBioscience, San Diego, CA, USA) for 4 or 24 h. cDNA was prepared using Moloney murine leukaemia virus reverse transcriptase (MMLV-RT; Invitrogen) and random hexamers (Invitrogen). All reactions were done in triplicate. Taqman primers for IL-12p40, IL-12p35 (also known as IL12A), IFNγ (also known as IFNG), STAT4, IL-12Rβ1 (also known as IL12RB1), IL-12Rβ2 (also known as IL12RB2), CCL5, CXCL10, β-actin and caspase-3 were purchased commercially from Applied Biosystems (Carlsbad, CA, USA). Results were normalised to the housekeeping gene β-actin and data were analysed using the 2−ΔΔCt method. If below the level of detection, basal expression was set at Ct=40.
For PCR array analysis, INS-1 or βTC-3 cells were incubated with 10 ng/ml IL-12 for 4 h.
Prepared cDNA was analysed with an RT2 Profiler PCR array of JAK/STAT Signalling Pathway genes (SAbiosciences, Frederick, MD, USA).
Immunofluorescence
Deparaffinised sections were prepared and blocked in DAKO peroxidase blocking reagent (DAKO, Carpinteria, CA, USA) for 30 min and permeabilised (0.3% v/v H2O2 80% v/v methanol) for 30 min. Sections were probed overnight at 4°C with 1:1,000 rabbit anti-IL-12 receptor β2 ([IL-12RB2] Sigma-Aldrich, St Louis, MO, USA). Following PBS washes, visualisation was achieved with incubation with 1:100 goat anti-rabbit-cyanine (CY)3 (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 3 h at room temperature. After PBS washes and exposure to DAKO blocking reagent, sections were incubated with 1:750 guinea pig anti-insulin antibody (Abcam, Cambridge, MA, USA) overnight at 4°C. The signal was visualised with 1:100 donkey anti-guinea pig-CY2 (Jackson ImmunoResearch Laboratories) for 3 h at room temperature. The antibody-incubation buffer was PBS/0.2% v/v Triton X-100/0.1% wt/v BSA. Signals were analysed on a Zeiss AxioObserver microscope (Zeiss, Thornwood, NY, USA).
β-TC3 cells were fixed (0.3% v/v H2O2 in 80% v/v methanol) for 20 min and permeabilised (0.1% Triton-X in PBS) for 10 min. Cells were stained overnight at 4°C with 1:40 goat anti-mouse IL-12p35 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or 1:40 rabbit anti-mouse IL-12p70 (Bioss, Woburn, MA, USA). The signal was visualised with either 1:100 donkey anti-goat horseradish peroxidase (HRP)-coupled antibody (Santa Cruz Biotechnology) or anti-rabbit Impress Reagent (Vector Laboratories, Burlingame, CA, USA) for 90 min and Vector SG (Vector Laboratories) substrate.
Glucose-stimulated insulin secretion
For each condition, 40 human islets were picked. Cytokine-treated islets were incubated with a pro-inflammatory cocktail of IL-1β (5 ng/ml), TNF-α (10 ng/ml) and IFN-γ (100 ng/ml) (R&D Systems) for 24 h in CMRL medium (Invitrogen) supplemented with 5% v/v FBS. After treatment, islets were placed in 1 ml serum-free Kreb–Ringer buffer (115 mmol/l NaCl, 24 mmol/l NaHCO3, 5 mmol/l KCl, 1 mmol/l MgCl2, 2.5 mmol/l CaCl2, 25 mmol/l HEPES, 0.001% wt/v BSA) and incubated for 1 h at 37°C. Glucose (Invitrogen) at low (3 mmol/l) or high (18 mmol/l) concentration was added for 1 h at 37°C. Media were sampled and insulin measured using species-specific insulin ELISA (Mercodia, Uppsala, Sweden).
Caspase-3 protease activity assay
INS-1 cells were treated with either murine IL-12 (10 ng/ml), or cytokine cocktail (5 ng/ml IL-1β, 10 ng/ml TNF-α, INF-γ 100 ng/ml [R&D Systems]) with or without 1 μg/ml anti-mouse IL-12 (eBioscience) for 4 h. Activity was assayed using the caspase-3 assay kit (BD Pharmigen, San Diego, CA, USA) as per the manufacturer’s instructions.
Western blot
Approximately 20 μg protein extracted from islet cells was loaded per lane. The polyvinylidene fluoride membranes were probed with primary antibodies to IL-12p35 or IL-12p40 (Santa Cruz Biotechnology) at 1:100 dilution, 1:250 phosphorylated-STAT4 (p-STAT4; B&D Scientific, Franklin Lakes, NJ, USA), or 1:3,000 β-actin (Santa Cruz Biotechnology). HRP-conjugated secondary antibody (GE Healthcare UK, Little Chalfont, UK) was added, and the signal was analysed with a ChemiDoc XRS System and associated densitometry software (Bio-Rad, Hercules, CA, USA).
Analysis
The data show fold induction over unstimulated control (defined as unity) and were analysed for significance (p<0.05) using an unpaired two-tailed t test with 95% CI (Graphpad Prism 4) and one-way ANOVA with Tukey post-hoc (Graphpad Prism 5). Data are provided as mean±SEM. All data shown are the result of a minimum of three separate experiments.
Results
Local expression of IL-12 in human islets exposed to pro-inflammatory cytokines
Incubation of donor human islets with a cocktail of pro-inflammatory cytokines (IL-1β, TNF-α and IFN-γ) induced islet dysfunction (electronic supplementary material [ESM] Fig. 1). Concomitant with human-islet dysfunction induced by pro-inflammatory cytokines were significant increases in target gene expression. Shown in Fig. 1a–d are fold changes from five human donors for the expression of the IL-12 ligand chains IL-12p40 and IL-12p35, IFNγ and MCP-1 (also known as CCL2, and encoding monocyte chemotactic protein-1 [MCP-1]), relative to donor islets that were time matched and incubated without pro-inflammatory cytokines. Inset for each graph is a representative response in a single human donor (insets, Fig. 1a–d). IL-12p40 expression (Fig. 1a) was at the limit of detection (Ct≥40) in control (untreated) islets and showed an exponential fold increase in expression following stimulation with the pro-inflammatory cocktail. In contrast, IL-12p35 (Fig. 1c) was detected (Ct=35±2) in untreated islets and expression increased upon stimulation with pro-inflammatory cytokines, reaching a plateau in fold expression at 4 h. Pro-inflammatory cytokines also induced the expression of IFNγ mRNA in an exponential manner (Fig. 1b). The expression of MCP-1 was rapidly upregulated by pro-inflammatory cytokines (Fig. 1d). A fold increase in MCP-1 expression was detectable at 2 h, with expression plateauing by 6 h.
Fig. 1.
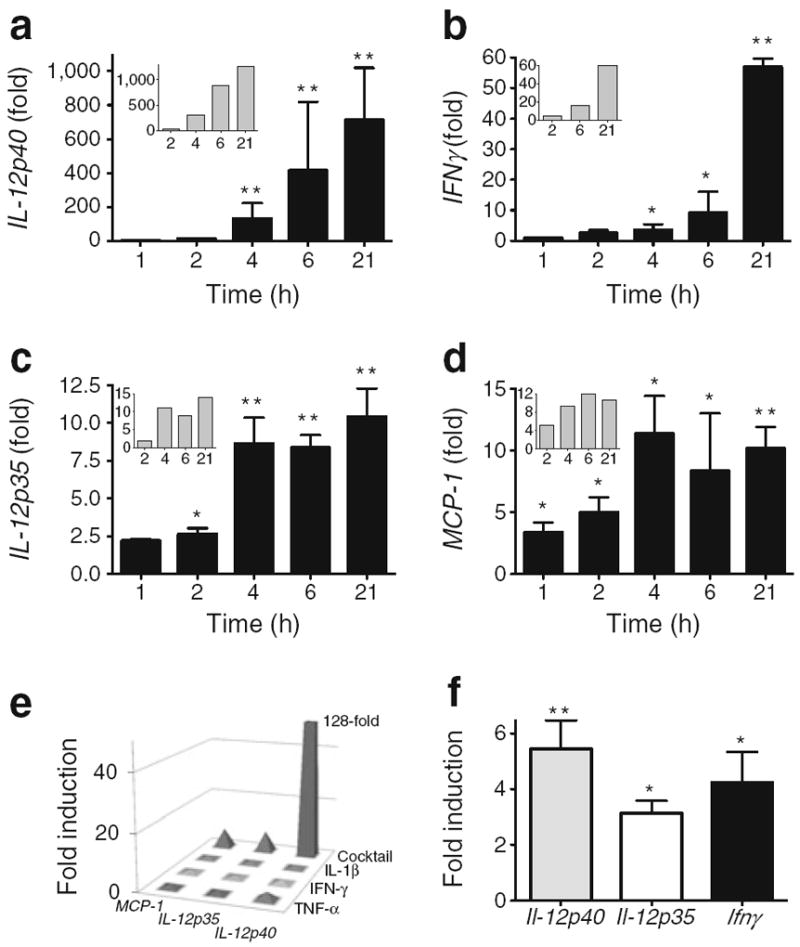
Changes in islet gene expression associated with pro-inflammatory cytokines. Pro-inflammatory cytokine (TNF-α, IL-1β, IFN-γ)-induced gene expression (fold induction over time-matched untreated) in human donor islets: IL-12p40 (a); IFNγ (b); IL-12p35 (c); and MCP-1 (d). The bar charts show the means of data from five different donors. Inset in (a–d) are representative responses from one donor. (e) Fold induction (over untreated) in islets from a single donor stimulated for 22 h with IL-1β, TNF-α and IFN-γ, either individually or combined (cocktail); the results are representative of findings in islets from three different donors (p<0.05). Gene changes were reproduced in primary mouse islets. (f) Induction of Il-12 ligand chains and Ifnγ following stimulation overnight with mouse cytokines (TNF-α, IL-1β and IFN-γ). The bars represent data from three or more experiments. *p<0.05; ** p<0.01 for increase over untreated time matched control
To explore if synergy between stimulatory cytokines was required for the observed IL-12 ligand gene induction, islets were stimulated with either the cocktail of pro-inflammatory cytokines or each pro-inflammatory cytokine alone. Relative fold changes in gene expression were determined (Fig. 1e). The cytokine cocktail resulted in sevenfold, 7.5-fold and ≥128-fold induction of gene expression (p>0.05) for MCP-1, IL-12p35 and IL-12p40, respectively. In contrast, negligible induction of gene expression was detected in islets stimulated with IL-1β, TNF-α or IFN-γ alone. Individual cytokines did not induce IFNγ (data not shown). This result was also observed in primary mouse islets (data not shown).
Analogous to the data from human donor islets, stimulation of primary mouse islets with a cocktail of murine pro-inflammatory cytokines (IL-1β, TNF-α and IFN-γ) increased gene expression of IL-12 ligand and Ifnγ. Pro-inflammatory cytokines stimulated the expression of mouse Il-12p40, Il-12p35 and Ifnγ, 5.4±2.5-fold (p<0.01), 3.1±0.8-fold (p<0.05) and 4.2±2.7-fold (p<0.05), respectively, relative to control (Fig. 1f).
Expression and regulation of IL-12 receptor subunits in human islets following treatment with pro-inflammatory cytokines
Immunohistochemistry detected the IL-12RB2 chain in human pancreas sections from non-diabetic donors. The IL-12RB2 was located predominantly in the islet, with limited detection in non-islet pancreatic regions (Fig. 2a). Co-staining with insulin was used to identify beta cells (Fig. 2b) and dual insulin- and IL-12RB2-positive cells were revealed in the merged image (Fig. 2c). These data suggest human islet beta cells produce the IL-12 receptor. The inset in Fig. 2c shows an expanded islet cell (location marked), the intracellular insulin (red) is contrasted with the IL-12RB2 staining (green) at the cell border (arrowhead in Fig. 2c). The ability of inflammatory cytokines to modulate expression of the IL-12 receptor gene was additionally explored (Fig. 2d). Stimulation of human donor islets with the pro-inflammatory cytokine cocktail induced IL-12 receptor chain expression (Fig. 2d). Shown in the graph are mean fold inductions in gene expression from three or more human islet donors relative to control. Expression of IL-12Rβ1 was increased 1.28±0.31-fold and 6.28±3.62-fold at 4 h and 24 h after stimulation, respectively (p<0.05). The expression of IL-12Rβ2 was also increased 1.70±1.18-fold and 7.63±2.35-fold at 4 h and 24 h after stimulation, respectively (p<0.05).
Fig. 2.
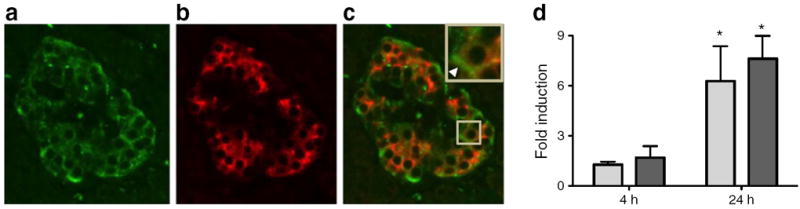
IL-12 receptor in human islets and induction following treatment with pro-inflammatory cytokines. Human pancreas sections were stained for IL-12RB2 (a) and insulin (b), with merged staining shown in (c). Inset in (c) is a magnification of the marked (square) islet cell. The arrow shows preferential cell surface staining. (d) Fold induction of IL-12 receptor chain expression in human donor islets following stimulation with pro-inflammatory cytokines; white bars, IL-12Rβ1; black bars, IL-12Rβ2. The results shown are representative of data from three or more human donors and over 20 islet fields from pancreatic sections from each of four donors. *p<0.05
Blocking IL-12 ligand inhibits pro-inflammatory-cytokine-stimulated gene expression in human islets
The direct role of IL-12 in mediating gene changes in human islets was explored using a neutralising antibody to the IL-12p40 ligand (Fig. 3). IL-12 stimulated a dose-dependent increase in IFNγ expression at 4 h (Fig. 3a, squares). This IL-12 stimulation of IFNγ gene expression was blocked with a neutralising antibody to IL-12 (Fig. 3a, triangles, p<0.05). The pro-inflammatory cytokine cocktail also induced IFNγ expression in human islets, as shown in Fig. 1. Inclusion of a neutralising antibody to IL-12 in the pro-inflammatory cytokine cocktail significantly (p<0.001) decreased the induced gene expression of IFNγ. Shown in Fig. 3b, the relative response with and without neutralising IL-12 antibody in seven donors is expressed as a percentage response. For comparison, gene induction of IFNγ by IL-12 with and without neutralising IL-12 antibody is also shown. Two irrelevant isotype-matched antibodies did not produce inhibition (% stimuli response ≥94%, p=NS). Fold induction of IFNγ expression in human islets was higher with 4 h of IL-12 stimulation than with 24 h (Fig. 3c). In contrast, the increase in IFNγ expression with pro-inflammatory cytokine stimulation was higher with 24 h stimulation than with 4 h stimulation (p<0.05).
Fig. 3.
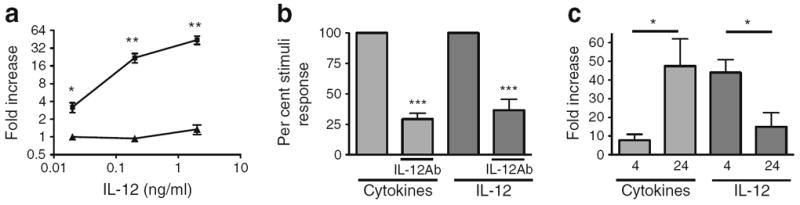
IL-12 neutralising antibody inhibits human islet gene expression induced by pro-inflammatory cytokines. (a) Induction of IFNγ expression in human islets stimulated by IL-12 for 4 h in the presence (triangles) or absence (squares) of a neutralising antibody to IL-12. (b) Pro-inflammatory cytokine or IL-12 induced an increase in IFNγ expression in the absence or presence of a neutralising antibody to IL-12. (c) Pro-inflammatory cytokines or IL-12 induced an increase in IFNγ expression after stimulation for 4 and 24 h. The graphs are representative of three (a), six (b) or five (c) experiments in separate donors. *p<0.05, **p<0.01, ***p<0.001. IL-12Ab, antibody to IL-12
Il-12 pathways are present and functional in murine beta cell lines
To remove immune-cell-derived IL-12 and explore if isolated beta cells are functionally responsive to IL-12, studies were performed on homogeneous beta cell lines (Fig. 4). INS-1 and βTC-3 (rat and mouse, respectively) are beta cell lines that retain insulin responsiveness to glucose. The data for INS-1 cells are shown in Fig. 4. Stimulation with the pro-inflammatory cytokine cocktail for 4 h induced the expression of Il-12 ligand chains Il-12p35 and Il-12p40, 2.6±1.5 (p=0.1) and 4.3±2 (p<0.05), respectively (Fig. 4a). Induction of IL-12 ligand chains was not detected with IL-12 stimulation for 4 h (Fig. 4a). Stimulation for 4 h with pro-inflammatory cytokines, but not IL-12, increased the expression of both chains of the IL-12 receptor, 15.3±4.1 (p<0.05) and 6.1±0.6 (p<0.01) for Il-12rβ1 and Il-12rβ2, respectively (Fig. 4b). Cellular production of IL-12p35 was upregulated in βTC-3 cells treated with cytokines (ESM Fig. 2a–d) and IL-12p70 was detected in lysates from INS-1 cells treated with cytokines (ESM Fig. 2e). To determine if the beta cell lines were responsive to IL-12 signalling, expression analysis was performed using a targeted JAK/STAT PCR gene array. The expression of several genes was specifically elevated in IL-12-treated INS-1 cells (ESM Fig. 3 and ESM Table 1) and βTC-3 cells (inset, ESM Fig. 3). Ifnγ was identified as being upregulated in both arrays (broken circle in ESM Fig. 3). Quantitative real-time PCR of cytokine- or IL-12-stimulated INS-1 cells showed a 3.9±1.5- and 2.6±0.9-fold induction in Ifnγ expression, respectively (p<0.05 for both) (Fig. 4c). Protein secretion of IFN-γ was detected in cytokine-stimulated INS-1 cell culture media (ESM Fig. 2f). The induced gene expression of Ifnγ by IL-12 in INS-1 cells showed an IL-12 dose response that is consistent with the reported half-maximal effective concentration (EC50) for IL-12 (Fig. 4d).
Fig. 4.
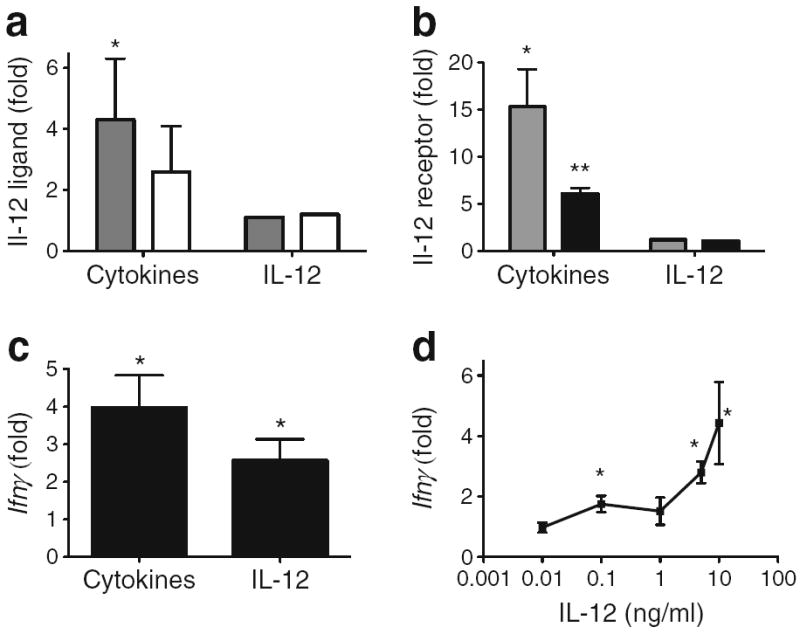
IL-12 pathways are present and functional in murine beta cell lines. Fold increase in gene expression for IL-12 ligand chains (a) or receptor chains (b) in INS-1 cells stimulated with pro-inflammatory cytokines or IL-12 (2.5 ng/ml). In (a): grey bars, Il-12p40; white bars, Il-12p35. In (b): grey bars, Il-12rb1; black bars, Il-12rb2. (c) Fold increase in Ifnγ (detected by real-time PCR) stimulated with proinflammatory cytokines or IL-12 (2.5 ng/ml). (d) Dose-dependent IL- 12 stimulation of Ifnγ gene expression in INS-1 cells. Graphs in (a), (b), (c) and (d) are representative of data from three or more experiments. *p<0.05, **p<0.01
Induction of INS-1 beta cell dysfunction by IL-12
To assess the direct functional effects of IL-12 in beta cells, the homogeneous beta cell line INS-1 was studied (Fig. 5). Static glucose-stimulated insulin secretion (GSIS) in INS-1 cells was measured in control cells (Fig. 5a, light grey bars) or cells treated with 2.5 ng/ml IL-12 for 6 h (Fig. 5a, dark grey bars). A significant (p<0.05) increase in insulin secretion was seen in control cells incubated with 16.7 mmol/l glucose (high) related to 1 mmol/l glucose (low). In cells pre-treated with IL-12, the high-glucose-stimulated insulin response was attenuated (p<0.01). IL-12 also inhibited GSIS in the homogeneous mouse beta cell line MIN6 (S. Green-Mitchell, unpublished data). To discover if loss of GSIS by IL-12 in INS-1 cells was associated with induced beta cell apoptosis, induction of caspase-3 protease activity was assessed (Fig. 5b) together with cell death (ESM Fig. 4). Activity of caspase-3, determined by fluorescent substrate conversion, was increased in INS-1 cells treated with IL-12 or pro-inflammatory cytokine cocktail (p<0.05). Addition of a neutralising antibody to IL-12 inhibited both IL-12-and cytokine-induced caspase-3 activity (p<0.05). No significant inhibition was seen with an irrelevant antibody (143.1±0.9, 144.8±0.5; IL-12, Cyto, p=NS). Caspase-3 activity was not significantly different from control in all conditions containing neutralising antibody.
Fig. 5.
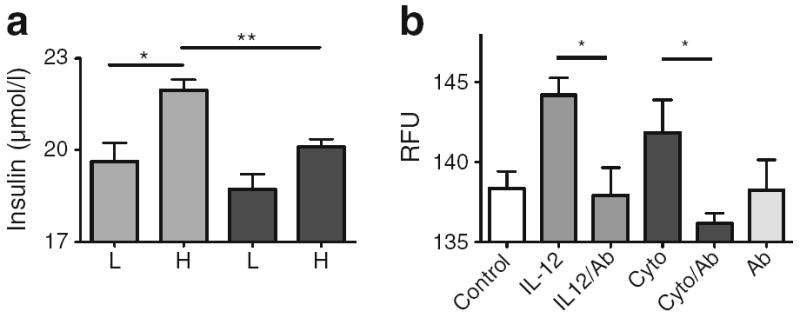
Impaired beta cell function in INS-1 cells treated with IL-12. (a) Static GSIS in INS-1 cells untreated (light grey bars) or treated for 6 h with 2.5 ng/ml IL-12 (dark grey bars). Insulin secreted in response to 1 mmol glucose (low) or 16.7 mmol glucose (high) was determined by ELISA. (b) Induction of caspase-3 protease activity in INS-1 cells treated with IL-12 or pro-inflammatory cytokines for 4 h in the presence or absence of a neutralising antibody to IL-12. Data are from three separate experiments. *p<0.05, **p<0.01. Cyto, cytokine; H, high glucose; IL-12Ab, antibody to IL-12; L, low glucose; RFU, relative fluorescence units
Modulation of human beta cell function by IL-12
GSIS in human donor islets was contrasted in islets untreated for 24 h, islets treated with the pro-inflammatory cytokine cocktail, or IL-12 alone (Fig. 6). Cytokine or IL-12 treatment of human islets resulted in a loss of insulin secretion in response to high (16.7 mmol/l) glucose stimulation (p<0.05). To determine if induced IL-12 is a component of cytokine-induced islet dysfunction, human donor islets were additionally treated with the pro-inflammatory cytokine cocktail in the presence of a neutralising antibody to IL-12 (Fig. 7). The neutralising antibody to IL-12 partially restored the GSIS response that was inhibited by cytokines (p<0.05).
Fig. 6.
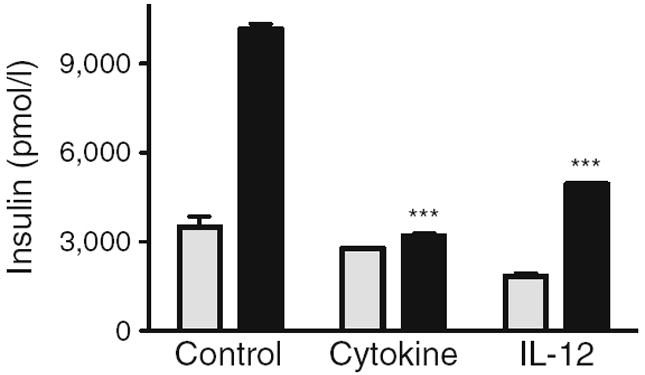
Impaired beta cell function in human islets treated with IL-12. Static GSIS in human donor islets untreated (control) or treated overnight with pro-inflammatory cytokines or 2.5 ng/ml IL-12. Insulin secreted in response to 3.3 mmol/l glucose (low, grey bars) or 16.7 mmol/l glucose (high, black bars) was determined by ELISA. Data are from four human donor islet preparations. ***p<0.001
Fig. 7.
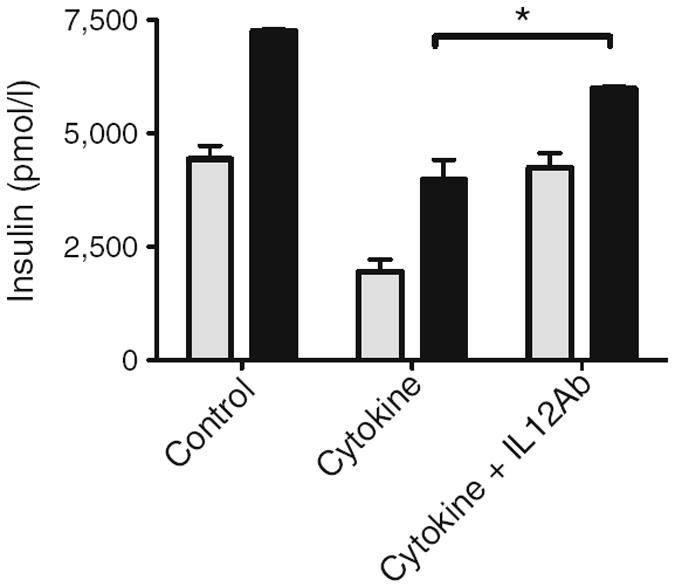
Human beta cell dysfunction induced by pro-inflammatory cytokines is partially reversed by a neutralising IL-12 antibody. Static GSIS in human donor islets untreated (control) or treated overnight with pro-inflammatory cytokines (cytokine) in the presence or absence of a neutralising antibody to IL-12. Insulin secreted in response to 3.3 mmol/l glucose (low, grey bars) or 16.7 mmol/l glucose (high, black bars) was determined by ELISA. Data are from three human donor islet preparations. *p<0.05. IL-12Ab, antibody to IL-12
Comparison of protein production between control and type 2 diabetic human islets
The relative protein production of IL-12 ligand and p-STAT4 were compared between human islets from non-diabetic donors and islets from type 2 diabetic donors (Fig. 8). Protein production for IL-12p35 (Fig. 8a) and IL-12p40 (Fig. 8b) was increased in islets from type 2 diabetic donors (p<0.05). Consistent with an upregulation of IL-12 ligand production, an increase in p-STAT4 was seen in the islets from the type 2 diabetic donors (Fig. 8c).
Fig. 8.
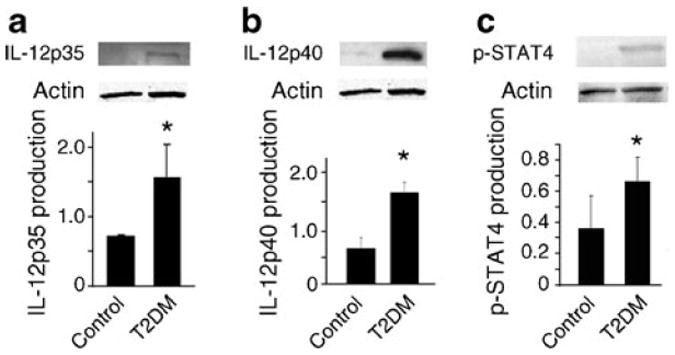
Comparison of protein production for IL-12p35 (a), IL-12p40 (b) and STAT4 phosphorylation (c) between islets from non-diabetic and type 2 diabetic donors. Representative western blots and densitometry analyses of islets from three donors are shown. T2DM, islets from donors with type 2 diabetes. *p<0.05
Collectively, these data show that IL-12 ligand upregulation in islets follows acute stimulation with pro-inflammatory cytokines. Beta cells directly produce IL-12 ligand, express IL-12 receptor and are responsive to IL-12 signalling. IL-12 directly contributes to beta cell dysfunction. Modulation of IL-12 with a neutralising antibody can protect human islets from the destructive effects of pro-inflammatory cytokines.
Discussion
This study describes the local production and function of IL-12 in human islets and rodent beta cells. The results provide evidence that IL-12 participates in islet dysfunction induced by pro-inflammatory cytokines. Recent studies have identified inflammation as an underlying component of diabetes. Elevation of pro-inflammatory cytokines is a feature of both type 1 [27] and type 2 diabetes [28-30]. Exposure of islets to elevated pro-inflammatory cytokines induces cellular dysfunction and increases intracellular reactive oxygen species [31, 32], to which islet beta cells are especially susceptible [19, 33, 34].
An acute exposure of human donor islets to a cocktail of three pro-inflammatory cytokines (TNF-α, IFN-γ and IL-1β) at pathological concentrations induced islet dysfunction [17, 35]. Coincident with the loss of islet function and viability was a significant elevation in the expression of genes encoding the IL-12 ligand and the downstream effector of IL-12, IFN-γ. Pro-inflammatory cytokines also induced the expression of MCP-1 by beta cells, an established marker of islet dysfunction that is used as a clinical measure of islet viability in islet transplantation [36, 37]. Rapid changes in islet gene expression followed pro-inflammatory cytokine stimulation. MCP-1 was detected within 2 h and kinetic profiling showed that pro-inflammatory cytokine induction of IL-12 function, as measured by the induction of IFNγ, was rate limited by the expression of IL-12p40.
Cytokine synergy in human islets has previously been reported [38]. Synergy between TNF-α, IL-1β and IFN-γ was required to upregulate IL-12 in human donor islets. IL-1β has been reported as a prime mediator of islet damage. Consequently, inhibition of IL-1β is the focus of several therapeutic strategies to preserve islets [39, 40]. In the current study, IL-1β alone was not sufficient to induce the expression of MCP-1 or IL-12 ligand chains.
This study questioned whether IL-12 production and response are functionally relevant for islet beta cells. Immunofluorochemistry identified that IL-12RB2 chain was localised at the cell surface of human islet beta cells, suggesting beta cells may be directly responsive to IL-12. Genes for the IL-12 receptor were upregulated by pro-inflammatory cytokines. Both pro-inflammatory cytokines and IL-12 directly induced IFNγ in human islets. Inclusion of a neutralising antibody to IL-12p40 inhibited the expression of IFNγ induced by pro-inflammatory cytokines. IL-12p40 is a shared ligand chain, forming IL-23 when heterodimerised with IL-23p19 [41]. Thus, neutralising IL-12p40 does not eliminate a contribution by IL-23 [42]. The importance of IL-12 is supported by expression of IL-12p35 and the direct effects mediated by exogenous IL-12. These data indicate that IL-12 is a mediator of the effects of pro-inflammatory cytokines in human islets. Peak induction of IFNγ occurred earlier with IL-12 stimulation than with pro-inflammatory cytokines. This temporal shift is consistent with the concept that pro-inflammatory cytokines induce IL-12 in islets that subsequently contributes to increased IFNγ expression.
While IL-12 has been linked to diabetes onset in previous reports, a direct action of IL-12 on beta cells has not been resolved. During the development of diabetes in NOD mice and biobreeding (BB) rats the expression of endogenous Il-12p40 and Ifnγ increased prior to diabetes onset [43, 44]. Systemic daily administration of IL-12 to NOD mice increased diabetes incidence [5], whereas sustained, but not intermittent [45], addition of an IL-12 antagonist decreased diabetes incidence [46, 47]. To address if local expression of IL-12 in beta cells could initiate diabetes, Holz et al generated transgenic mice producing IL-12 or monomeric IL-12 chains in the beta cell [48]. Rat insulin promoter IL-12 (RIP-IL-12) transgenic mice developed pancreatic islet inflammation that was associated with an elevation in IFN-γ. Conversion to diabetes did not progress in these mice. Ectopic production of IL-12 may not induce a concomitant upregulation of the IL-12 receptor as we describe for cytokine stimulation, thus limiting paracrine stimulation of the beta cell.
Islets are heterogeneous cell structures containing endocrine, neuronal, vascular and surveying immune cells. One potential source of IL-12 in islets is the surveying immune cell pool. IL-12 expression and production has not been described in endocrine islet cells. To resolve if endocrine cells are a source of IL-12, homogeneous beta cell lines were studied. Genes for IL-12 ligand and receptor were detected in INS-1 cells and these were upregulated following stimulation with the pro-inflammatory cytokine cocktail. Functional IL-12 receptor–ligand interactions were detected by PCR array and quantitative RT-PCR analysis. Study of beta cell lines showed cytokine-induced production of IL-12 ligand chains and cytokine or IL-12 induction of IFN-γ. These data suggest beta cells are a contributory source of IL-12 ligand and IFN-γ. As beta cells are responsive to IL-12, local paracrine production of IL-12 may facilitate a microenvironment of sufficient ligand concentration to promote beta cell dysfunction.
Exogenous IL-12 directly promoted beta cell dysfunction. In INS-1 beta cells, using GSIS as the indicator of beta cell function, IL-12 significantly reduced the GSIS response (p<0.01), a result also observed in mouse MIN6 cells (S. Green-Mitchell, unpublished results). Further, IL-12 induced caspase-3 protease activity in INS-1 cells. Caspase-3 activity is a marker of induced cell apoptosis. A neutralising antibody to IL-12 blocked caspase-3 activity induced by IL-12 and also caspase-3 activity induced by the pro-inflammatory cytokine cocktail. In human donor islets, IL-12 attenuated the GSIS response and a neutralising antibody to IL-12 partially restored the loss of GSIS response by the pro-inflammatory cytokine cocktail (p<0.05). In RIP-IL-12 mice, the expression of the chemokine genes Ccl5 and Cxcl10 was upregulated [48]. IL-12 increased the expression of these chemokine genes in βTC-3 cells, and this was inhibited by an IL-12-neutralising antibody (ESM Fig. 5). These data show that IL-12 is a component of beta cell dysfunction induced by pro-inflammatory cytokines in human donor islets and beta cell lines. IL-12 directly and negatively modulates islet beta cell function.
Protein production of IL-12p40, IL-12p35 and p-STAT4 was upregulated in islets from type 2 diabetic donors compared with non-diabetic islets in a small study (p<0.05). This would be consistent with elevated local and systemic concentrations of pro-inflammatory cytokines associated with type 2 diabetes [28-30]. Serum concentrations of IL-12 have a positive correlation with insulin resistance and pro-inflammatory cytokine expression in individuals with newly diagnosed type 2 diabetes [49]. STAT4 is a principal second messenger associated with IL-12 receptor ligation [8]. STAT4 activation is an important factor in the development of rodent models of type 1 diabetes [8, 26, 32, 50]. Genetic deletion of Stat4 completely prevented the spontaneous development of type 1 diabetes in the NOD mouse model [25, 50]. In human type 1 diabetes genetic studies, polymorphisms in IL-12p40 have been identified from linkage analysis [14-16]. Consolidating the data from this study, modulation of IL-12/STAT4 activity may be an attractive therapeutic target for human diabetes.
In summary, this study describes a direct role of IL-12 on pancreatic islets and beta cells. Both produce IL-12 and are functionally responsive to IL-12. IL-12 directly promotes islet dysfunction and is a mediator of beta cell dysfunction induced by pro-inflammatory cytokines. Modulation of IL-12, either through molecule sequestration, pharmaceutical inactivation of IL-12 signalling or small molecule/biological blockade of IL-12 receptor–ligand interaction, may provide a new therapeutic approach to preserve and protect pancreatic beta cell mass in diabetes.
Supplementary Material
Acknowledgments
Islets were obtained from the IIDP (http://iidp.coh.org/) and tissue sections from the nPOD (www.jdrfnpod.org/).
Funding Work was supported in part by grants from Department of Defense (PR093521, D. A. Taylor-Fishwick), Juvenile Diabetes Research Foundation (D. A. Taylor-Fishwick, J. L. Nadler) and NIH (DK090490, Y. Imai; HL112605, J. L. Nadler).
Abbreviations
- GSIS
Glucose-stimulated insulin secretion
- HRP
Horseradish peroxidase
- IIDP
Integrated Islet Distribution Program
- IL-12RB2
IL-12 receptor β2
- JAK/STAT
Janus kinase/signal transducer and activator of transcription
- MAPK/p38
Mitogen-activated protein kinase/protein 38
- MCP-1
Monocyte chemotactic protein-1
- MMLV-RT
Moloney murine leukaemia virus reverse transcriptase
- nPOD
Network for Pancreatic Organ Donors
- p35
Protein 35
- p40
Protein 40
- p-STAT4
Phosphorylated STAT4
- RIP-IL-12
Rat insulin promoter IL-12
- STAT4
Signal transducer and activator of transcription 4
- Th1
T helper 1
Footnotes
Contribution statement DAT-F wrote the article, provided research data and interpretation, and directed the work. JRW, SC, WG, SG-M, NK, YI provided research data and helped to draft and/or review the article. JLN helped review the article and contributed to data interpretation. All authors approved the final manuscript version.
Electronic supplementary material The online version of this article (doi:10.1007/s00125-012-2732-9) contains peer-reviewed but unedited supplementary material, which is available to authorised users.
Duality of interest The authors confirm there is no duality of interest associated with this manuscript.
Contributor Information
D. A. Taylor-Fishwick, Email: Taylord@evms.edu, Strelitz Diabetes Center, Department of Internal Medicine, Eastern Virginia Medical School, 700 W. Olney Road, Norfolk, VA 23501, USA; Internal Medicine, Microbiology and Molecular Cell Biology, Eastern Virginia Medical School, Norfolk, VA, USA.
J. R. Weaver, Strelitz Diabetes Center, Department of Internal Medicine, Eastern Virginia Medical School, 700 W. Olney Road, Norfolk, VA 23501, USA
W. Grzesik, Strelitz Diabetes Center, Department of Internal Medicine, Eastern Virginia Medical School, 700 W. Olney Road, Norfolk, VA 23501, USA
S. Chakrabarti, Strelitz Diabetes Center, Department of Internal Medicine, Eastern Virginia Medical School, 700 W. Olney Road, Norfolk, VA 23501, USA
S. Green-Mitchell, Strelitz Diabetes Center, Department of Internal Medicine, Eastern Virginia Medical School, 700 W. Olney Road, Norfolk, VA 23501, USA
Y. Imai, Strelitz Diabetes Center, Department of Internal Medicine, Eastern Virginia Medical School, 700 W. Olney Road, Norfolk, VA 23501, USA
N. Kuhn, Strelitz Diabetes Center, Department of Internal Medicine, Eastern Virginia Medical School, 700 W. Olney Road, Norfolk, VA 23501, USA
J. L. Nadler, Strelitz Diabetes Center, Department of Internal Medicine, Eastern Virginia Medical School, 700 W. Olney Road, Norfolk, VA 23501, USA
References
- 1.Katz JD, Benoist C, Mathis D. T helper cell subsets in insulin-dependent diabetes. Science. 1995;268:1185–1188. doi: 10.1126/science.7761837. [DOI] [PubMed] [Google Scholar]
- 2.Tisch R, McDevitt H. Insulin-dependent diabetes mellitus. Cell. 1996;85:291–297. doi: 10.1016/s0092-8674(00)81106-x. [DOI] [PubMed] [Google Scholar]
- 3.Serreze DV, Chapman HD, Varnum DS, Gerling I, Leiter EH, Shultz LD. Initiation of autoimmune diabetes in NOD/Lt mice is MHC class I-dependent. J Immunol. 1997;158:3978–3986. [PubMed] [Google Scholar]
- 4.Trembleau S, Germann T, Gately MK, Adorini L. The role of IL-12 in the induction of organ specific autoimmune diseases. Immunol Today. 1995;16:383–386. doi: 10.1016/0167-5699(95)80006-9. [DOI] [PubMed] [Google Scholar]
- 5.Trembleau S, Penna G, Bosi E, Mortara A, Gately MK, Adorini L. Interleukin 12 administration induces T helper type I cells and accelerates autoimmune diabetes in NOD mice. J Exp Med. 1995;181:817–821. doi: 10.1084/jem.181.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Athie M, Flotow H, Hilyard KL, Cantrell DA. IL-12 selectively regulates STAT4 via phosphatidylinositol 3-kinase and Ras-independent signal transduction pathways. Eur J Immunol. 2000;30:1425–1434. doi: 10.1002/(SICI)1521-4141(200005)30:5<1425::AID-IMMU1425>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 7.Visconti R, Gadina M, Chiariello M, et al. Importance of the MKK6/p38 pathway for interleukin-12-induced STAT4 serine phosphorylation and transcriptional activity. Blood. 2000;96:1844–1852. [PubMed] [Google Scholar]
- 8.Kaplan MH, Sun YI, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 9.Watford WT, Hissong BD, Bream JH, Kanno Y, Muul L, O’Shea JJ. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol Rev. 2004;202:139–156. doi: 10.1111/j.0105-2896.2004.00211.x. [DOI] [PubMed] [Google Scholar]
- 10.Watford WT, Moriguchi M, Morinobu A, O’Shea JJ. The biology of IL-12: coordinating innate and adaptive immune responses. Cytokine Growth Factor Rev. 2003;14:361–368. doi: 10.1016/s1359-6101(03)00043-1. [DOI] [PubMed] [Google Scholar]
- 11.Morinobu A, Gadina M, Strober W, et al. STAT4 serine phosphorylation is critical for IL-12-induced IFN-gamma production but not for cell proliferation. Proc Natl Acad Sci USA. 2002;99:12281–12286. doi: 10.1073/pnas.182618999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen T, Wang R, Russell JH. IL-12 enhances IL-2 function by inducing CD25 expression through a p38 mitogen-activated protein kinase pathway. Eur J Immunol. 2000;30:1445–1452. doi: 10.1002/(SICI)1521-4141(200005)30:5<1445::AID-IMMU1445>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 13.Vandenbroeck K, Alloza I, Gadina M, Matthys P. Inhibiting cytokines of the interleukin-12 family: recent advances and novel challenges. J Pharm Pharmacol. 2004;56:145–160. doi: 10.1211/0022357022962. [DOI] [PubMed] [Google Scholar]
- 14.Morahan G, Huang D, Ymer SI, et al. Linkage disequilibrium of a type 1 diabetes susceptibility locus with a regulatory IL12B allele. Nat Genet. 2001;27:218–221. doi: 10.1038/84872. [DOI] [PubMed] [Google Scholar]
- 15.Altinova AE, Engin D, Akbay E, et al. Association of polymorphisms in the IL-18 and IL-12 genes with susceptibility to Type 1 diabetes in Turkish patients. J Endocrinol Invest. 2010;33:451–454. doi: 10.1007/BF03346623. [DOI] [PubMed] [Google Scholar]
- 16.Morahan G, McKinnon E, Berry J, et al. Evaluation of IL12B as a candidate type I diabetes susceptibility gene using data from the Type I Diabetes Genetics Consortium. Genes Immun. 2009;10(Suppl 1):S64–S68. doi: 10.1038/gene.2009.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgan D, Oliveira-Emilio HR, Keane D, et al. Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia. 2007;50:359–369. doi: 10.1007/s00125-006-0462-6. [DOI] [PubMed] [Google Scholar]
- 18.Pi J, Bai Y, Zhang Q, et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes. 2007;56:1783–1791. doi: 10.2337/db06-1601. [DOI] [PubMed] [Google Scholar]
- 19.Modak MA, Parab PB, Ghaskadbi SS. Pancreatic islets are very poor in rectifying oxidative DNA damage. Pancreas. 2009;38:23–29. doi: 10.1097/MPA.0b013e318181da4e. [DOI] [PubMed] [Google Scholar]
- 20.Mandrup-Poulsen T. Apoptotic signal transduction pathways in diabetes. Biochem Pharmacol. 2003;66:1433–1440. doi: 10.1016/s0006-2952(03)00494-5. [DOI] [PubMed] [Google Scholar]
- 21.Blandino-Rosano M, Perez-Arana G, Mellado-Gil JM, Segundo C, Aguilar-Diosdado M. Anti-proliferative effect of pro-inflammatory cytokines in cultured beta cells is associated with extracellular signal-regulated kinase 1/2 pathway inhibition: protective role of glucagon-like peptide-1. J Mol Endocrinol. 2008;41:35–44. doi: 10.1677/JME-07-0154. [DOI] [PubMed] [Google Scholar]
- 22.Lund T, Fosby B, Korsgren O, Scholz H, Foss A. Gluco-corticoids reduce pro-inflammatory cytokines and tissue factor in vitro and improve function of transplanted human islets in vivo. Transpl Int. 2008;21:669–678. doi: 10.1111/j.1432-2277.2008.00664.x. [DOI] [PubMed] [Google Scholar]
- 23.Boden G. Obesity, insulin resistance and free fatty acids. Curr Opin Endocrinol Diabetes Obes. 2011;18:139–143. doi: 10.1097/MED.0b013e3283444b09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma K, Nunemaker CS, Wu R, Chakrabarti SK, Taylor-Fishwick DA, Nadler JL. 12-Lipoxygenase products reduce insulin secretion and β cell viability in human islets. J Clin Endocrinol Metab. 2010;95:887–893. doi: 10.1210/jc.2009-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Z, Chen M, Ellett JD, et al. Autoimmune diabetes is blocked in Stat4-deficient mice. J Autoimmun. 2004;22:191–200. doi: 10.1016/j.jaut.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Yang Z, Chen M, Fialkow LB, Ellett JD, Wu R, Nadler JL. The novel anti-inflammtory compound, lisofylline, prevents diabetes in multiple low-dose streptozotocin-treated mice. Pancreas. 2003;26:e99–e104. doi: 10.1097/00006676-200305000-00021. [DOI] [PubMed] [Google Scholar]
- 27.Jorns A, Gunther A, Hedrich HJ, Wedekind D, Tiedge M, Lenzen S. Immune cell infiltration, cytokine expression, and beta-cell apoptosis during the development of type 1 diabetes in the spontaneously diabetic LEW.1AR1/Ztm-iddm rat. Diabetes. 2005;54:2041–2052. doi: 10.2337/diabetes.54.7.2041. [DOI] [PubMed] [Google Scholar]
- 28.Catalan V, Gomez-Ambrosi J, Ramirez B, et al. Proinflammatory cytokines in obesity: impact of type 2 diabetes mellitus and gastric bypass. Obes Surg. 2007;17:1464–1474. doi: 10.1007/s11695-008-9424-z. [DOI] [PubMed] [Google Scholar]
- 29.Igoillo-Esteve M, Marselli L, Cunha DA, et al. Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia. 2010;53:1395–1405. doi: 10.1007/s00125-010-1707-y. [DOI] [PubMed] [Google Scholar]
- 30.Steinberg GR. Inflammation in obesity is the common link between defects in fatty acid metabolism and insulin resistance. Cell Cycle. 2007;6:888–894. doi: 10.4161/cc.6.8.4135. [DOI] [PubMed] [Google Scholar]
- 31.Michalska M, Wolf G, Walther R, Newsholme P. Effects of pharmacological inhibition of NADPH oxidase or iNOS on pro-inflammatory cytokine, palmitic acid or H2O2-induced mouse islet or clonal pancreatic beta cell dysfunction. Biosci Rep. 2010;30:445–453. doi: 10.1042/BSR20090138. [DOI] [PubMed] [Google Scholar]
- 32.Yang ZD, Chen M, Wu R, McDuffie M, Nadler JL. The anti-inflammatory compound lisofylline prevents Type 1 diabetes in non-obese diabetic mice. Diabetologia. 2002;45:1307–1314. doi: 10.1007/s00125-002-0901-y. [DOI] [PubMed] [Google Scholar]
- 33.Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med. 1996;20:463–466. doi: 10.1016/0891-5849(96)02051-5. [DOI] [PubMed] [Google Scholar]
- 34.Lenzen S. Oxidative stress: the vulnerable beta-cell. Biochem Soc Trans. 2008;36:343–347. doi: 10.1042/BST0360343. [DOI] [PubMed] [Google Scholar]
- 35.Azevedo-Martins AK, Lortz S, Lenzen S, Curi R, Eizirik DL, Tiedge M. Improvement of the mitochondrial antioxidant defense status prevents cytokine-induced nuclear factor-kappaB activation in insulin-producing cells. Diabetes. 2003;52:93–101. doi: 10.2337/diabetes.52.1.93. [DOI] [PubMed] [Google Scholar]
- 36.Chen MC, Proost P, Gysemans C, Mathieu C, Eizirik DL. Monocyte chemoattractant protein-1 is expressed in pancreatic islets from prediabetic NOD mice and in interleukin-1 beta-exposed human and rat islet cells. Diabetologia. 2001;44:325–332. doi: 10.1007/s001250051622. [DOI] [PubMed] [Google Scholar]
- 37.Marzorati S, Antonioli B, Nano R, et al. Culture medium modulates proinflammatory conditions of human pancreatic islets before transplantation. Am J Transplant. 2006;6:2791–2795. doi: 10.1111/j.1600-6143.2006.01512.x. [DOI] [PubMed] [Google Scholar]
- 38.Rabinovitch A, Sumoski W, Rajotte RV, Warnock GL. Cytotoxic effects of cytokines on human pancreatic islet cells in monolayer culture. J Clin Endocrinol Metab. 1990;71:152–156. doi: 10.1210/jcem-71-1-152. [DOI] [PubMed] [Google Scholar]
- 39.Larsen CM, Faulenbach M, Vaag A, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356:1517–1526. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- 40.Mandrup-Poulsen T, Pickersgill L, Donath MY. Blockade of interleukin 1 in type 1 diabetes mellitus. Nat Rev Endocrinol. 2010;6:158–166. doi: 10.1038/nrendo.2009.271. [DOI] [PubMed] [Google Scholar]
- 41.Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 42.Leonardi CL, Kimball AB, Papp KA, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1) Lancet. 2008;371:1665–1674. doi: 10.1016/S0140-6736(08)60725-4. [DOI] [PubMed] [Google Scholar]
- 43.Rabinovitch A, Suarez-Pinzon WL, Sorensen O. Interleukin 12 mRNA expression in islets correlates with beta-cell destruction in NOD mice. J Autoimmun. 1996;9:645–651. doi: 10.1006/jaut.1996.0084. [DOI] [PubMed] [Google Scholar]
- 44.Zipris D, Greiner DL, Malkani S, Whalen B, Mordes JP, Rossini AA. Cytokine gene expression in islets and thyroids of BB rats IFN-gamma and IL-12p40 mRNA increase with age in both diabetic and insulin-treated nondiabetic BB rats. J Immunol. 1996;156:1315–1321. [PubMed] [Google Scholar]
- 45.O’Hara RM, Jr, Henderson SL, Nagelin A. Prevention of a Th1 disease by a Th1 cytokine: IL-12 and diabetes in NOD mice. Ann NY Acad Sci. 1996;795:241–249. doi: 10.1111/j.1749-6632.1996.tb52673.x. [DOI] [PubMed] [Google Scholar]
- 46.Rothe H, O’Hara RM, Jr, Martin S, Kolb H. Suppression of cyclophosphamide induced diabetes development and pancreatic Th1 reactivity in NOD mice treated with the interleukin (IL)-12 antagonist IL-12(p40)2. Diabetologia. 1997;40:641–646. doi: 10.1007/s001250050728. [DOI] [PubMed] [Google Scholar]
- 47.Trembleau S, Penna G, Gregori S, Gately MK, Adorini L. Deviation of pancreas-infiltrating cells to Th2 by interleukin-12 antagonist administration inhibits autoimmune diabetes. Eur J Immunol. 1997;27:2330–2339. doi: 10.1002/eji.1830270930. [DOI] [PubMed] [Google Scholar]
- 48.Holz A, Brett K, Oldstone MB. Constitutive beta cell expression of IL-12 does not perturb self-tolerance but intensifies established autoimmune diabetes. J Clin Invest. 2001;108:1749–1758. doi: 10.1172/JCI13915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mishra M, Kumar H, Bajpai S, Singh RK, Tripathi K. Level of serum IL-12 and its correlation with endothelial dysfunction, insulin resistance, proinflammatory cytokines and lipid profile in newly diagnosed T2DM. Diabetes Res Clin Pract. 2011;94:255–261. doi: 10.1016/j.diabres.2011.07.037. [DOI] [PubMed] [Google Scholar]
- 50.Boyton RJ, Davies S, Marden C, et al. Stat4-null non-obese diabetic mice: protection from diabetes and experimental allergic encephalomyelitis, but with concomitant epitope spread. Int Immunol. 2005;17:1157–1165. doi: 10.1093/intimm/dxh293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.