

Abstract
The copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) has become a commonly employed method for the synthesis of complex molecular architectures under challenging conditions. Despite the widespread use of copper-catalyzed cycloaddition reactions, the mechanism of these processes has remained difficult to establish due to the involvement of multiple equilibria between several reactive intermediates. Real-time monitoring of a representative cycloaddition process via heat flow reaction calorimetry revealed that monomeric copper acetylide complexes are not reactive toward organic azides unless an exogenous copper catalyst is added. Furthermore, crossover experiments with an isotopically enriched exogenous copper source illustrated the stepwise nature of the C–N bond-forming events and the equivalence of the two copper atoms within the cycloaddition steps.
Rigorous examination of catalytic reaction mechanisms remains a formidable but necessary pursuit for organic chemists. In-depth reaction analysis can aid in the optimization of reaction parameters, spur the development of new catalytic processes, and enhance understanding of fundamental reactivity. The status quo for analysis of catalytic systems usually relies upon one or a combination of the following methods: computational studies; isolation and reintroduction of an intermediate from the reaction mixture as proof of its involvement within the catalytic cycle; analysis of stereoelectronic effects of substituents present in the reactive intermediates; isotope crossover and kinetic isotope effects; and, most commonly, initial rate studies. Although these methods have shaped our current understanding of reaction mechanisms, they seldom provide a complete picture of complex catalytic reactions.
Among transition-metal catalyzed processes, reactions involving copper remain particularly difficult for rigorous mechanistic investigation due to the low reduction potential of copper (+0.159 V from Cu2+ to Cu+, and +0.520 V from Cu+ to Cu0 (1)), the propensity for Cu+ species to disproportionate in solution (2), the generally poor ability of copper to back-bond to ligands (3), and the well-documented tendency of copper complexes to aggregate (4). Despite these complications, copper catalysis has been employed successfully for decades in a variety of methods including carbon–heteroatom bond formation (5), carbon–carbon bond formation (6), oxidative processes (7, 8), and, most recently, dipolar cycloadditions (Fig. 1A, (9-12)). The latter of these includes the CuAAC, which has garnered widespread attention since its debut in 2001 as a facile and robust method for the creation of covalent linkages in a variety of environments. However, due to the aforementioned challenges in the direct study of copper catalysis, a mechanism of this reaction has yet to be fully elucidated; specifically, the identity and nuclearity of the copper species involved in catalysis has not been established (Fig. 1B). Herein, we propose from simple, deductive experiments a mechanism which necessitates two copper atoms within the active cycloaddition complex, and we describe their relationship to one another.
Fig. 1.

(A) Copper-catalyzed dipolar cycloadditions. (B) First proposed mechanism of the CuAAC (10). (C) Copper-catalyzed cycloaddition of terminal and 1-iodoalkynes.
Several studies towards elucidation of the mechanism of the CuAAC with terminal alkynes have been reported (13, 14), and experimental evidence of the possible involvement of polynuclear copper(I) intermediates (14-16) has been supported by theoretical studies (17, 18). Furthermore, recent advances in dipolar cycloadditions of 1-halo- and 1-metalloalkynes (formally internal alkynes) with organic azides have prompted additional investigation of the mechanism (19-22). In the case of 1-iodoalkynes, scission of the terminal alkyne-halogen bond to form a copper(I)-acetylide is not necessary for the cycloaddition to occur, suggesting that the copper catalyst effects the cycloaddition purely through weak π-interactions with the formally internal alkyne (Fig. 1C). Thus, we hypothesized that the classical CuAAC (with terminal alkynes) also proceeds through a similar reaction manifold, via: i) in-situ formation of the σ-bound copper acetylide; ii) recruitment of a second, π-bound copper atom, forming the catalytically active complex; iii) reversible coordination of the organic azide to the π-bound copper complex; and iv) the stepwise annulation events. Obtaining experimental support for this pathway would lead to a common mechanistic model involving activation of σ-acetylides (both terminal and formally internal) by weak and reversible π-interaction with a copper center (Fig. 1C).
Despite the multiple equilibria of unstable, non-isolable, and highly reactive intermediates within the catalytic cycle of the CuAAC, the copper(I)-acetylide and the copper(I)-triazolide intermediates can be reliably isolated and characterized (fig. S1, 23). Therefore, to investigate the mechanistic model proposed above, we envisioned a simple experiment in which a preformed, stable, mononuclear σ-bound copper(I)-acetylide is treated with a stoichiometric amount of an organic azide in the presence or absence of an added copper(I) catalyst, allowing us to directly verify the requirement of the putative π-bound copper intermediate. To this end, the σ-bound copper(I)-acetylide complex 2 was synthesized following the procedure reported by Straub and co-workers (23) on a large scale (>20g) in five overall steps (fig. S2). Subsequently, azide 1 was treated with acetylide 2 (0.090 M and 0.082 M in tetrahydrofuran (THF), respectively) under two sets of conditions: with and without soluble exogenous copper catalyst 3 (Cu(PPh3)2NO3, 0.0041 M), and reaction progress was tracked by real-time heat flow reaction calorimetry (Fig. 2, (24)). The contrast in rate was drastic: the catalyzed cycloaddition rapidly reached completion, producing copper triazolide 4 within 20 minutes (table S2, table S3), whereas the non-catalyzed reaction showed no appreciable conversion to the triazolide (table S4). Lowering the catalyst concentration resulted in a corresponding decrease in the maximum rate, showing a positive-order dependence on the exogenous copper species (fig. S3). Moreover, multiple soluble exogenous copper sources exhibited similar catalytic efficacy (fig. S4) and organic azides bearing sterically encumbered or aliphatic functional groups showed comparable rate acceleration in the presence of an added copper catalyst (25, fig. S5, table S5). Similarly striking difference in the rates of the catalyzed and non-catalyzed reactions initially shown in THF was observed in other solvents, chloroform and dimethylformamide (25, fig. S6).
Fig. 2.

Global heat (mW) output graph for the catalyzed and uncatalyzed formation of triazolide 4. Bn = Benzyl. Ar = p-tBuPh. NHC = 1,3-bis(2,6-diisopropyl)phenyl-4,5-dihydroimidazol-2-ylidene.
With the necessity for a second copper atom within the active cycloaddition complex established, we sought to determine the respective roles of each copper atom. Given the well-documented stability of the copper–carbene complexes (26) and the robust nature of acetylide 2, we hypothesized that the two copper atoms act in discrete and specialized roles, with copper in acetylide 2 acting purely as a strongly σ-bound ligand while the second copper atom operates exclusively through weak π-complexation (Fig. 1C, where X=Cu). To probe this hypothesis, we prepared an isotopically pure 63Cu exogenous catalyst to differentiate it from the naturally abundant distribution of copper isotopes (63Cu:65Cu, 69:31) contained in the preformed copper(I)-acetylide. The copper(I) coordination complex [Cu(MeCN)4]PF6 5 (27), an air-stable, crystalline solid, was selected for the stoichiometric crossover studies and synthesized in short order from the isotopically pure, commercially available copper(II) oxide (99.9% 63Cu).
As both the acetylide reactant 2 and the copper triazolide product 4 contain a copper atom, we designed a stoichiometric crossover experiment in which the absence of isotopic enrichment of the metal center of the resulting triazolide would support the hypothesized independent roles of the two copper species. Thus azide 1, acetylide 2 (natural isotopic ratio of 63/65Cu), and isotopically enriched copper complex 5 (isotopically pure 63Cu) were combined, and the resultant triazolide was analyzed using direct injection time-of-flight mass spectrometry (TOF-MS) (Fig. 3A). To our surprise, statistical enrichment of the resulting triazolide was observed, yielding complex 6 (with the 63Cu:65Cu ratio of 85:15) and disproving the discrete bonding hypothesis of the two copper atoms postulated above (fig. S9). This enrichment event would require the migration of the N-heterocyclic carbene (NHC) from one copper atom to another; given the established strength of the copper–NHC bond (energetically on the order of a C–C alkane bond (26)), this is indeed an unexpected result.
Fig. 3.
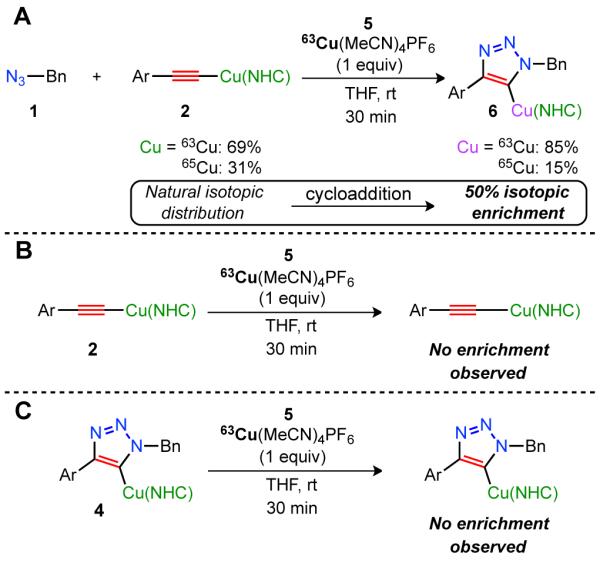
(A) Observed isotopic enrichment of triazolide 6. (B) No isotopic enrichment of acetylide 2 observed. (C) No isotopic enrichment of triazolide 4 observed. Bn = Benzyl. Ar = p-tBuPh. (NHC) = 1,3-bis(2,6-diisopropyl)phenyl-4,5-dihydroimidazol-2-ylidene. rt = room temperature.
The enrichment of triazolide 4 must occur in one of the following three steps: i) via the acetylide intermediate 2; ii) via the triazolide intermediate 4; or iii) within the cycloaddition steps. Due to the polynuclear nature of copper(I)-acetylides in solution, and multiple equilibria between the various aggregates (28), acetylide 2 could be a candidate for the facile exchange of the embedded copper atom. Hence, acetylide 2 was treated with a stoichiometric amount of the enriched copper complex 5 (tracked by TOF-MS), from which no isotopic enrichment of the acetylide was observed (Fig. 3B, fig. S10). Although this control experiment showed no crossover, azide 1 could theoretically act as a Lewis base and mediate the enrichment observed in the initial experiment. To address this possibility, acetylide 2, isotopically enriched copper complex 5, and azide 1 were combined (in analogy to the initial crossover experiment), and the reaction mixture was analyzed by TOF-MS. Again, no enrichment of acetylide 2 in the presence of azide 1 was noted (fig. S11, fig. S12). Thus, while organic azides could act as ligands for the copper(I)-complex, evidently this interaction is not sufficient to mediate the observed enrichment event.
With acetylide 2 excluded as a possible intermediate for exchange, we next examined whether the isotopic enrichment occurred after the formation of triazolide 4. Accordingly, azide 1, acetylide 2, and a catalytic amount of [Cu(MeCN)4]PF6 (natural isotopic distribution, 5 mol%) were combined in THF to fully form the non-enriched triazolide 4. To this solution, a stoichiometric equivalent of the enriched copper complex 5 was added (tracked by TOF-MS), from which no enrichment of triazolide 4 was observed (Fig. 3C, fig. S13), excluding it as a venue for the observed enrichment.
By process of elimination, the observed 50% isotopic enrichment must occur within the cycloaddition steps (boxed intermediates in Fig. 4A). We propose the following mechanistic interpretation to account for these results (Fig. 4A): first, the σ-bound copper acetylide bearing the π-bound enriched copper atom 7 reversibly coordinates an organic azide, forming complex 8. Following this step, nucleophilic attack at N–3 of the azide by the β-carbon of the acetylide forms the first covalent C–N bond, producing intermediate 9. The ligand exchange in this intermediate (isomers shown in Fig. 4B) is faster than the formation of the second covalent C–N bond, which results in ring closure, accounting for the statistical (50%) incorporation of 63Cu into triazolide 6. The exclusive formation of triazolide 6 indicates a thermodynamic preference for the NHC-bound copper triazolide; however, rapid ligand exchange renders equal the probability for either copper atom (natural or enriched) to be incorporated in the resulting intermediate. We attribute the unprecedented lability and rapid exchange of the NHC ligand between the two copper atoms in intermediate 9 to the weakened copper–carbene backbonding (29-31) as the formal oxidation state of copper increases upon the formation of the first C–N bond. The degenerate, proximal copper centers in 9 facilitate this oxidation event, with the second copper atom acting as a stabilizing donor ligand to the otherwise highly energetic and unstable mononuclear metallacycle intermediate ((13), Fig. 1B). Taken together with prior studies of the full catalytic cycle, these experiments support the mechanistic model for the CuAAC featuring two chemically equivalent copper atoms working in concert for the regioselective formation of 1,4-substituted-1,2,3-triazoles (Fig. 4C, fig. S14).
Fig. 4.

(A) Mechanistic rationale for the isotopic enrichment of triazolide 6. (B) Rapidly interconverting isomers of intermediate 9. (C) Proposed catalytic model for the CuAAC with two copper atoms. Bn = Benzyl. Ar = p-tBuPh. NHC = 1,3-bis(2,6-diisopropyl)phenyl-4,5-dihydroimidazol-2-ylidene.
The in situ reaction calorimetry and metal isotope crossover methods used in this study have permitted us to deduce the involvement of unstable, non-isolable intermediates that have to date eluded more conventional mechanistic studies. Similarly innovative studies that delineate reactivity patterns of more capricious but abundant metals (Cu, Fe, Ni) are needed to further understand and develop new catalytic processes, reducing synthetic reliance on well-behaved but rare and expensive transition metals (Pd, Rh, Ru). Moreover, the mechanistic insights presented in this study, specifically the formation of the active cycloaddition complex with two differentiable copper atoms, imply a unified reactivity of electronically rich σ-acetylides (1-halo- or 1-metallo-) with 1,3-dipoles (e.g., azides, nitrile oxides, and nitrones). As such, we propose a common reactivity trend whereby any σ-acetylide that can effectively recruit a π-bound copper atom will undergo annulation with a compatible dipolar partner.
Supplementary Material
One Sentence Summary.
Mechanistic interrogation of the CuAAC establishes two copper atoms within the active catalytic species and isotopic crossover experiments illustrate their equivalency.
Acknowledgments
This work was supported by a grant from the National Science Foundation (CHE-0848982) and National Institute of General Medical Sciences, the National Institutes of Health (GM-087620). We also gratefully acknowledge Mr. Bill Webb for his assistance with mass spectrometry analyses, Dr. Jason Kwok (MIT) for his support with reaction calorimetry, and Prof. Jason Hein (University of California, Merced) for helpful scientific discussions.
Footnotes
Supplementary Materials: Materials and Methods Figures S1-S14 Tables S1-S5 NMR Spectra
References and Notes
- 1.CRC Handbook of Chemistry and Physics. 93rd Edition W. M. Haynes; 2012. [Google Scholar]
- 2.Moen A, Nicholson DG. J. Chem. Soc., Faraday Trans. 1995;91:3529. [Google Scholar]
- 3.Dias HVR, Flores JA, Wu J, Kroll P. J. Am. Chem. Soc. 2009;131:11249. doi: 10.1021/ja904232v. [DOI] [PubMed] [Google Scholar]
- 4.Hathaway BJ. In: Comprehensive Coordination Chemistry. Wilkinson G, editor. vol. 5. Pergamon; Oxford: 1987. pp. 533–757. [Google Scholar]
- 5.Ley SV, Thomas AW. Angew. Chem. Int. Ed. 2003;42:5400. doi: 10.1002/anie.200300594. [DOI] [PubMed] [Google Scholar]
- 6.Monnier F, Taillefer M. Angew. Chem. Int. Ed. 2009;48:6954. doi: 10.1002/anie.200804497. [DOI] [PubMed] [Google Scholar]
- 7.Schindler S. Eur. J. Inorg. Chem. 2000;2000:2311. [Google Scholar]
- 8.Nigh WG. In: Oxidation in Organic Chemistry. Part B. Trahanovsky WS, editor. Academic Press; New York: 1973. pp. 1–95. [Google Scholar]
- 9.Tornoe CW, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 10.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 11.Meldal M, Tornøe CW. Chem. Rev. 2008;108:2952. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 12.Hein JE, Fokin VV. Chem. Soc. Rev. 2010;39:1302. doi: 10.1039/b904091a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Himo F, et al. J. Am. Chem. Soc. 2005;127:210. doi: 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]
- 14.Rodionov VO, Fokin VV, Finn MG. Angew. Chem. Int. Ed. 2005;44:2210. doi: 10.1002/anie.200461496. [DOI] [PubMed] [Google Scholar]
- 15.Rodionov VO, Presolski SI, Diaz DD, Fokin VV, Finn MG. J. Am. Chem. Soc. 2007;129:12705. doi: 10.1021/ja072679d. [DOI] [PubMed] [Google Scholar]
- 16.Kuang G-C, et al. J. Am. Chem. Soc. 2011;133:13984. doi: 10.1021/ja203733q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Straub BF. Chem. Comm. 2007:3868. doi: 10.1039/b706926j. [DOI] [PubMed] [Google Scholar]
- 18.Ahlquist M, Fokin VV. Organometallics. 2007;26:4389. [Google Scholar]
- 19.Kuijpers BHM, et al. Synlett. 2005:3059. [Google Scholar]
- 20.Hein JE, Tripp JC, Krasnova LB, Sharpless KB, Fokin VV. Angew. Chem., Int. Ed. 2009;48:8018. doi: 10.1002/anie.200903558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Partyka DV, et al. Organometallics. 2009;28:6171. [Google Scholar]
- 22.Zhou Y, Lecourt T, Micouin L. Angew. Chem. Int. Ed. 2010;49:2607. doi: 10.1002/anie.200907016. [DOI] [PubMed] [Google Scholar]
- 23.Nolte C, Mayer P, Straub BF. Angew. Chem. Int. Ed. 2007;46:2101. doi: 10.1002/anie.200604444. [DOI] [PubMed] [Google Scholar]
- 24.Blackmond DG. Angew. Chem. Int. Ed. 2005;44:4302. doi: 10.1002/anie.200462544. [DOI] [PubMed] [Google Scholar]
- 25.Although the starting acetylide complex 2 is stable to atmospheric moisture, acidic or strongly protic solvents or azides can sever the Cu-acetylide bond. The liberated Cu(I) will in turn catalyze the formation of triazolide 4, albeit at a significantly lower rate than observed by copper catalyst 3. Therefore, it is best to use non-protic solvents/azides to observe the highest difference in rate between the catalyzed and non-catalyzed experiments.
- 26.Boehme C, Frenking G. Organometallics. 1998;17:5801. [Google Scholar]
- 27.Kubas GJ. Inorg. Synth. 1979;19:90. [Google Scholar]
- 28.Mykhalichko BM, Temkin ON, Mys’kiv MG. Russ. Chem. Rev. 2001;69:957. [Google Scholar]
- 29.Although complexation of NHCs to transition metals is strongly governed by σ-donation, recent investigations have shown that π-backbonding can account for as much as 30% of the overall bonding character.
- 30.Sanderson MD, Kamplain JW, Bielawski CW. J. Am. Chem. Soc. 2006;128:16514. doi: 10.1021/ja067475w. [DOI] [PubMed] [Google Scholar]
- 31.Comas-Vives A, Harvey JN. Eur. J. Inorg. Chem. 2011;2011:5025. [Google Scholar]
- 32.Bates CG, Saejueng P, Murphy JM, Venkataraman D. Org. Lett. 2002;4:4727. doi: 10.1021/ol0272040. [DOI] [PubMed] [Google Scholar]
- 33.Wang X-L, Wan K, Zhou C-H. Eur. J. Med. Chem. 2010;10:4631. doi: 10.1016/j.ejmech.2010.07.031. [DOI] [PubMed] [Google Scholar]
- 34.Kuang G-C, Michaels HA, Simmons JT, Clark RJ, Zhu L. J. Org. Chem. 2010;75:6540. doi: 10.1021/jo101305m. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.