

Abstract
Chromosome 6q26–27 is linked to susceptibility to visceral leishmaniasis (VL) in Brazil and Sudan. DLL1 encoding the Delta-like 1 ligand for Notch 3 was implicated as the etiological gene. DLL1 belongs to the family of Notch ligands known to selectively drive antigen-specific CD4 T helper 1 cell responses, which are important in protective immune response in leishmaniasis. Here we provide further genetic and functional evidence that supports a role for DLL1 in a well-powered population-based study centred in the largest global focus of VL in India. Twenty-one single nucleotide polymorphisms (SNPs) at PHF10/C6orf70/DLL1/FAM120B/PSMB1/TBP were genotyped in 941 cases and 992 controls. Logistic regression analysis under an additive model showed association between VL and variants at DLL1 and FAM120B, with top associations (rs9460106, OR=1.17, 95%CI 1.01–1.35, P=0.033; rs2103816, OR=1.16, 95%CI 1.01–1.34, P=0.039) robust to analysis using caste as a covariate to take account of population substructure. Haplotype analysis taking population substructure into account identified a common 2-SNP risk haplotype (frequency 0.43; P=0.028) at FAM120B, while the most significant protective haplotype (frequency 0.18; P=0.007) was a 5-SNP haplotype across the interval 5’ of both DLL1 (negative strand) and FAM120B (positive strand) and extending to intron 4 of DLL1. Quantitative RT/PCR was used to compare expression of 6q27 genes in paired pre- and post-treatment splenic aspirates from VL patients (N=19). DLL1 was the only gene to show differential expression that was higher (P<0.0001) in pre- compared to post-treatment samples, suggesting that regulation of gene expression was important in disease pathogenesis. This well-powered genetic and functional study in an Indian population provides evidence supporting DLL1 as the etiological gene contributing to susceptibility to VL at Chromosome 6q27, confirming the potential for polymorphism at DLL1 to act as a genetic risk factor across the epidemiological divides of geography and parasite species.
Keywords: visceral leishmaniasis, DLL1, genetic association, Notch signalling
1. Introduction
Visceral leishmaniasis (VL) caused by the Leishmania donovani complex is endemic in the Indian subcontinent which forms the largest global focus of disease. Only about 10% of individuals infected with L. donovani develop clinical disease which is lethal without treatment (Badaro et al., 1986; Topno et al.; Zijlstra et al., 1995). Understanding parasite, host and environmental factors that determine asymptomatic infection versus lethal clinical disease is important in disease control. Host genetic factors are known to contribute to disease susceptibility (reviewed (Blackwell et al., 2009)), and in a recent report (Fakiola et al., 2011) we provided genetic and functional evidence to support DLL1, encoding the Delta-like 1 ligand for Notch 3, as the etiological susceptibility gene at the chromosomal region 6q27 previously linked to susceptibility to VL in both Sudan and Brazil (Jamieson et al., 2007; Miller et al., 2007).
Notch signalling is one of the most conserved pathways regulating cell differentiation/cell fate decisions (Artavanis-Tsakonas et al., 1999; Greenwald, 1998), playing an important role during development and in development and differentiation of immune cells. In bone marrow, Notch ligand Delta-1 completely inhibits differentiation of human hematopoietic progenitors into the B cell lineage while promoting emergence of cells with a T cell/natural killer precursor phenotype (Jaleco et al., 2001). Delta-1 also inhibits the differentiation of monocytes into macrophages and promotes their differentiation into dendritic cells (Ohishi et al., 2001). In spleen, Notch signalling is required to maintain CD8 positive dendritic cells (Sekine et al., 2009). Antigen presenting cells, including dendritic cells, also use Notch signaling to promote T helper cell differentiation in response to specific antigens (Amsen et al., 2004). The different Notch ligand families, Delta1,4 versus Jagged1,2, instruct antigen-driven CD4 T cell selection down Th1 versus Th2 fates, respectively (Amsen et al., 2004; Maekawa et al., 2003). The effect of polarized antigen-specific Th1 versus Th2 responses on the outcome of leishmaniasis is well established in murine models (Locksley and Scott, 1991; Scott et al., 1989) and human disease (Pirmez et al., 1993; Sundar et al., 1997). Clinical VL, in particular, has been associated with high Th2 cytokine responses (Sundar et al., 1997), while Th1-generated interferon-γ is higher in children infected with L. infantum chagasi that do not progress to clinical VL than those who do (Carvalho et al., 1992). The potential for Delta-1 driven Th1 differentiation to alter the course of infection has already been demonstrated for L. major infection in BALB/c mice (Maekawa et al., 2003), making genetic regulation of DLL1 expression a highly plausible explanation for the genetic associations and regulation of splenic expression we previously reported for this 6q27 gene (Fakiola et al., 2011).
The genetic studies undertaken in Sudan and Brazil were limited in terms of sample size and power (Fakiola et al., 2011), making it desirable to determine whether fine mapping of the Chromosome 6q27 region using a well-powered sample would replicate these earlier findings. To achieve this, and to determine the geographical extent of the influence of polymorphism at DLL1 on genetic susceptibility to VL, we turned to the largest global focus of VL in India. Fine mapping of disease association for genes across the 6q27 region using a well-powered population-based sample from India, together with a comparison of gene expression pre- and post-treatment in VL patients, confirms DLL1 as the likely etiological gene determining susceptibility to VL.
2. Materials and methods
2.1. Study population
The study was conducted in the highly endemic district of Muzaffarpur in Bihar State, India. Diagnosis of VL was made on the basis of clinical, parasitological and serological criteria as described (Fakiola et al., 2010; Mehrotra et al., 2011; Sundar and Benjamin, 2003). Further epidemiological and demographic details relating to the study samples and study site are described elsewhere (Mehrotra et al., 2011; Singh et al., 2006). The study was performed using a case-control design with a total of 2019 individuals comprising 990 cases and 1029 controls (Table 1) collected during 2009–2010. Information about caste was recorded as a proxy for population substructure.
Table 1.
Baseline characteristics of the Indian case-control cohort.
| Case-Control Sample | Number* |
|---|---|
| Cases | 958 |
| Male | 571 |
| Female | 387 |
| Mean age at study encounter ±SD (yr) | 31.2 ±16.7 |
| Range | 3–73 |
| Mean age at onset of VL ±SD (yr) | 26.8 ±15.3 |
| Religious Group Hindu Muslim† |
850 108 |
| Controls | 1015 |
| Male | 570 |
| Female | 445 |
| Mean age at study encounter ±SD (yr) | 31.8 ±15.9 |
| Religious Group Hindu Muslim |
885 130 |
Numbers are given for the individuals with DNA available for genotyping
Muslim forms a caste, such that religion is taken into account as a covariate in the caste analysis
2.2 Ethics statement
For collection and use of saliva samples and splenic aspirates in India, informed written consent in Hindi was obtained from all participating individuals and from parents of children under 18 years old. Approval for the study was provided by the Ethical Committee of the Institute of Medical Sciences, Banaras Hindu University, Varanasi, India. For the use of RNA samples from control spleens from necro organ transplantation healthy organ donors obtained in Sweden, ethical approval was provided by the Regionala Etiska Kommittén, Karolinska Institutet, Stockholm (Regional Ethical Committee, Karolinska Institute, Stockholm).
2.3 Genetic studies
Genomic DNA was extracted from saliva using the Oragene technology (DNA Genotek, Ontario, Canada) and SNPs genotyped using Sequenom iPLEX platform (Sequenom, San Diego, CA). A total of 21 SNPs at DLL1 and adjacent genes across the Chromosome 6q27 region were genotyped (Table 2). All SNPs met minimum quality control checks for call rate (>99.5%) across all individuals. Two SNPs were excluded from further analysis: PHF10_rs9371126 showed extreme deviation from HWE in controls and TBP_rs6937840 showed significant difference in missingness between cases and controls. A total of 59 cases and 37 controls for which there was >80% missing genotype data were removed from further analysis, providing a sample of 941 cases and 992 controls for genetic analysis. This cleaned dataset had 93.5 to 99.9% power for odds ratio 1.5, MAF=0.1 to 0.5, P=0.01, and 50 to 93.1% power for odds ratio 1.3. Allelic and haplotype association tests were performed in PLINK (Purcell et al., 2007) using logistic regression analysis under an additive model. Nominal P-values are presented throughout, i.e. without correction for multiple testing. Application of a strict Bonferroni correction for 19 independent (r2<0.8) SNPs provides a significance cut-off of P≤0.003 (i.e. P=0.05/19). A less stringent cut-off of P≤0.006 allows for non-independence of SNPs due to LD across the region (Supplementary Figure 1; P=0.05/8, 4 LD blocks and 4 independent SNPs). Haplotype analyses were undertaken using logistic regression analysis in PLINK (Purcell et al., 2007). Inclusion of caste as a covariate, which we have shown to provide a good surrogate for genetic substructure in genome-wide analyses of a separate sample of 989 cases and 1089 controls in this population (unpublished data), was used to take account of population substructure. Caste was converted into a set of binary dummy variables prior to being included as a covariate in the logistic regression. Permuted P-values were also determined in PLINK employing the label-swapping max(T) permutation method using the --mperm command. Empirical p-values (EMP1) were obtained for the most significant risk and protective haplotypes after performing 10,000 permutations. Conditional analyses and a version of stepwise regression based on estimating haplotypic effects (odds ratios) were carried out using the program UNPHASED (Dudbridge, 2008; Dudbridge et al., 2011). This allowed us to perform nested hypothesis testing (via likelihood ratio tests) of whether a particular SNP adds significantly to a model that already includes some other SNPs. LD patterns for D’ and r2 were determined in Haploview software v4.2 (Barrett et al., 2005) while LD blocks were generated using the default Gabriel et al. algorithm (Gabriel et al., 2002).
Table 2.
Details of polymorphisms genotyped and the minor allele frequency (MAF) of variants in the Indian study population. Bold indicates single SNPs for which allelic association at nominal P≤ 0.05 was observed (Table 3).
| Variant | Physical Position1 |
Location | Code2 | Alleles3 | Strand | MAF |
|---|---|---|---|---|---|---|
| PHF10_rs4073926 | 170099398 | Intergenic | G/A | − | 0.476 | |
| PHF10_rs9371126 | 170108689 | Intron 9 | T/C | + | 0.401 | |
| PHF10_rs6459655 | 170123196 | Intergenic | A/G | + | 0.303 | |
| C6orf70_rs4145078 | 170173155 | Intron 12 | A/G | + | 0.317 | |
| C6orf70_rs4716396 | 170173914 | Intron 13 | T/C | + | 0.318 | |
| C6orf208_rs926714 | 170202517 | 3'UTR | T/G | + | 0.326 | |
| DLL1_rs1028488 | 170590475 | Intergenic | 7 | A/C | − | 0.361 |
| DLL1_rs1033583 | 170591923 | 3'UTR | 8 | A/C | − | 0.248 |
| DLL1_rs1884190 | 170596699 | Intron 4 | 9 | A/G | − | 0.076 |
| DLL1_rs2738820 | 170601259 | Intergenic | 10 | G/T | − | 0.379 |
| DLL1_rs9459988 | 170601548 | Intergenic | 11 | T/G | + | 0.063 |
| DLL1_rs17860704 | 170602429 | Intergenic | 12 | A/G | − | 0.181 |
| FAM120B_rs9366198 | 170608950 | intergenic | 13 | C/G | + | 0.270 |
| FAM120B_rs9460106 | 170615960 | Intron 1 | 14 | T/C | + | 0.436 |
| FAM120B_rs2103816 | 170676013 | Intron 6 | 15 | T/A | + | 0.491 |
| FAM120B_rs9460139 | 170712179 | Intron 9 | 16 | C/T | + | 0.060 |
| FAM120B_rs9295407 | 170713682 | 3’UTR | A/T | + | 0.054 | |
| PSMB1_rs6914744 | 170858520 | Intron 1 | A/C | + | 0.401 | |
| TBP_rs6456230 | 170867576 | Intron 2 | C/T | + | 0.406 | |
| TBP_rs13207114 | 170877444 | Intron 5 | A/G | + | 0.396 | |
| TBP_rs6937840 | 170882050 | Intergenic | G/A | + | 0.463 |
Physical positions of markers are given according to Build 37.2 of the human genome;
Number code for SNPs on Figure 1;
Major>minor alleles for Indian population
2.4 Functional studies
Splenic aspirates were taken as part of the routine diagnostic procedure for VL at the Kala Azar Medical Research Centre, Muzaffarpur, Bihar State, India. Paired pre- and post-treatment splenic samples were collected in 5×RNA Later (AMBION Inc., Austin, Texas, USA) from 19 VL patients treated during 2009–2010. Samples were transported from the Muzaffarpur back to the BHU laboratory in Varanasi at 4°C and stored at −80°C until RNA was isolated. Total RNA was isolated using the RNeasy tissue kit (Qiagen, GmbH, Hilden, Germany) according to the manufacturer’s instructions and eluted in 30µl of RNase free water. Sample quality and integrity was assessed by ND-2000 spectrophotometer (Thermo Fischer Scientific, Wilmington, DE, USA) and agarose (Sigma Aldrich Chemicals, St Louis, MO, USA) gel electrophoresis. RNA from control healthy spleen cells obtained from necro organ transplantation healthy organ donors (HODs; N=8) at Karolinska University Hospital, Huddinge, Sweden, were kindly provided by Dr. Suzane Nylen. 500ng of RNA was reverse transcribed using the High Capacity cDNA synthesis kit (Applied Biosystems, Foster City, CA, USA). Taqman predesigned gene expression assays (Applied Biosystems) for PHF10 (Hs01097834_m1), C6orf70 (Hs00858039_m1), DLL1 (Hs00194509_m1), FAM120B (Hs00921805_m1), PSMB1 (Hs00427357_m1) were used to perform expression studies (7500 HT Real Time PCR system, Applied Biosystems) with 18S rRNA (P/N 4319413E) used as an endogenous control to normalize expression data. Expression analysis was performed on 19 paired pre- and post-treatment splenic aspirates from VL patients with appropriate no RT and no template controls included in each plate. All samples were run in duplicate. Results were analysed by 7500 software v.2.0.1 (Applied Biosystems) and Graph pad prism 5. Paired two-tailed t-tests were used to compare pre- and post-treatment measures, unpaired two-tailed t-tests to compare control spleens against patient splenic aspirate data.
3. Results
3.1 Association at DLL1 and FAM120B in Indian VL
Previous analysis of genes at 6q27 and VL susceptibility in Sudan used large consanguineal pedigrees that provided evidence for associations across multiple genes from PHF10 to TBP (Fakiola et al., 2011). Analysis of Brazilian families narrowed the association down to DLL1. To determine whether this fine mapping of the VL susceptibility gene(s) at Chromosome 6q27 could be replicated in India, we analysed tag-SNPs in the full set of genes (Table 2) across the PHF10 to TBP region. Logistic regression analysis (Table 3) provides evidence for associations between Indian VL and SNPs at DLL1 and FAM120B, but not PHF10, C6orf70, PSMB1 or TBP. The top single point SNP signal was at rs9460106 (OR=1.22; 95%CI 1.07–1.39; nominal P=0.003) which lies in intron 1 of FAM120B. This SNP withstands correction for multiple SNPs tested (conservative cut-off P≤0.006; stringent cut-off P≤0.003), and remains significant (OR=1.17, 95%CI 1.01–1.35, P=0.033) when population substructure is taken into account using caste as a covariate. Conditioning on the top SNP (rs9460106) resulted in loss of significance at all other SNPs (data not shown), suggesting a single main genetic effect at 6q27 tagged by this SNP.
Table 3.
Population-based association between VL and Chromosome 6q27 SNPs. Results are shown for logistic regression analyses under an additive model comparing VL cases and controls. OR = odds ratio; L95 and U95 are lower and upper 95% confidence intervals. Allele counts are shown for minor/major allele for affected and unaffected individuals. Bold indicates single SNPs for which allelic association at nominal P≤ 0.05 was observed.
| Variant | Minor Allele |
AFF | UNAFF | OR | L95 | U95 | P |
|---|---|---|---|---|---|---|---|
| PHF10_rs4073926 | A | 880/1002 | 958/1024 | 0.94 | 0.83 | 1.07 | 0.33 |
| PHF10_rs6459655 | G | 566/1316 | 604/1376 | 0.98 | 0.86 | 1.12 | 0.77 |
| C6orf70_rs4145078 | G | 581/1297 | 644/1338 | 0.93 | 0.82 | 1.07 | 0.31 |
| C6orf70_rs4716396 | C | 582/1298 | 645/1339 | 0.93 | 0.82 | 1.07 | 0.31 |
| C6orf208_rs926714 | G | 591/1289 | 668/1316 | 0.91 | 0.79 | 1.03 | 0.14 |
| DLL1_rs1028488 | C | 665/1217 | 728/1252 | 0.94 | 0.82 | 1.07 | 0.35 |
| DLL1_rs1033583 | C | 470/1400 | 478/1480 | 1.04 | 0.90 | 1.20 | 0.62 |
| DLL1_rs1884190 | G | 159/1719 | 134/1844 | 1.27 | 1.00 | 1.61 | 0.05 |
| DLL1_rs2738820 | T | 688/1188 | 773/1211 | 0.91 | 0. 80 | 1.03 | 0.14 |
| DLL1_rs9459988 | G | 134/1748 | 111/1873 | 1.29 | 1.00 | 1.68 | 0.05 |
| DLL1_rs17860704 | G | 359/1523 | 339/1637 | 1.13 | 0.97 | 1.33 | 0.13 |
| FAM120B_rs9366198 | G | 527/1317 | 485/1425 | 1.17 | 1.01 | 1.34 | 0.03 |
| FAM120B_rs9460106 | C | 853/997 | 806/1146 | 1.22 | 1.07 | 1.39 | 0.003 |
| FAM120B_rs2103816 | A | 960/912 | 929/1047 | 1.19 | 1.05 | 1.35 | 0.008 |
| FAM120B_rs9460139 | T | 106/1776 | 123/1843 | 0.89 | 0.68 | 1.17 | 0.41 |
| FAM120B_rs9295407 | T | 96/1786 | 112/1872 | 0.90 | 0.68 | 1.19 | 0.45 |
| PSMB1_rs6914744 | C | 756/1124 | 792/1192 | 1.01 | 0.89 | 1.15 | 0.85 |
| TBP_rs6456230 | T | 766/1110 | 799/1181 | 1.02 | 0.90 | 1.16 | 0.76 |
| TBP_rs13207114 | G | 740/1112 | 758/1176 | 1.03 | 0.91 | 1.17 | 0.64 |
3.2 LD and haplotype analysis across DLL1 and FAM120B
Demonstration of association at FAM120B in India was of interest given that this was the only gene at 6q27 analysed in the Sudanese study that did not show association with VL (Fakiola et al., 2011). SNPs at FAM120B were therefore not examined in the Brazilian family study. DLL1 and FAM120B lie adjacent to each other at 6q27 and are encoded on opposite strands. They share a 16.3kb region upstream of both genes which could contain regulatory elements influencing expression of either gene. Four of the SNPs analysed (Figure 1) lie in this intergenic region, three (rs2738820, rs9459988 and rs17860704) assigned to DLL1 on the basis of proximity, and one (rs9366198) assigned to FAM120B. To determine whether the associations at FAM120B could be tagging polymorphisms influencing expression of DLL1, we first looked at LD across the region in our control sample (Figure 1; Supplementary Figure 1), as well as in the larger dataset available for the HapMap Gujarati Indians living in Houston, Texas, USA (GIH) population (Supplementary Figure 2). The first thing to note is that pairwise LD between markers as defined by r2, which takes allele frequencies into account, mimics the association data. The two associated SNPs (rs1884190 in intron 4; rs9459988 intergenic) at DLL1 are in quite strong LD (r2=0.75) with each other but not with the intervening non-associated intergenic SNP rs2738820 or with the more distal non-associated intergenic SNP rs17860704. Similarly, the 2 most associated SNPs (rs9460106 and rs2103816) at FAM120B are in quite strong LD (r2=0.75), but show weaker LD (r2=0.49 and 0.38) with the less strongly associated FAM120B SNP rs9366198, and lower levels still (r2=0.17 and 0.15) with the more proximal intergenic SNP rs17860704 assigned to DLL1. The lack of LD (as defined by r2) between closely adjacent SNPs in the intergenic region (Figure 1), together with the observation that only 2 out of 4 of these SNPs showed single point association with disease, suggests a complex haplotype structure that is reflected in the stronger evidence for LD across the region as defined by D’ in both our control sample (Figure 1; Supplementary Figure 1) and the GIH population (Supplementary Figure 2). We therefore looked at haplotype associations across the region (Figure 1) bounded by the most proximal associated SNP at DLL1 (rs1884190) and the most distal associated SNP at FAM120B (rs2103816).
Figure 1. DLL1 and FAM120B genomic structure, SNP location, and pairwise LD.
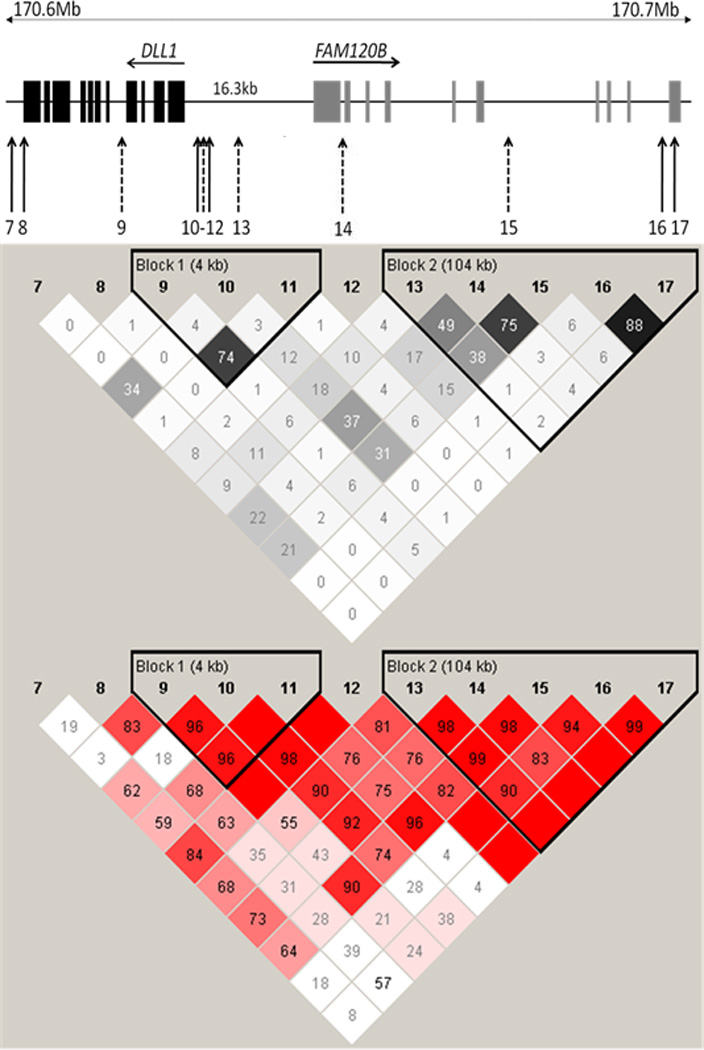
The upper diagram provides the intron/exon structure and location of DLL1 and FAM120B SNPs (numbered according to the code presented in Table 2) used in the study. SNPs associated with VL (nominal P<0.05) in this study are shown in dotted arrows. The lower diagram shows pairwise LD between SNPs for the control sample, as defined by r2 (above) and D’ (below) measures. r2 values are represented white for r2 = 0, with intermediate values for 0< r2< 1 indicated by shades of grey; D' values and confidence levels (LOD) are represented with shades of pink for high D', LOD>2; white for D'<1, LOD<2. The numbers within the squares represent the D' or r2 scores for pairwise LD.
Figure 2 provides results of the haplotype association analysis undertaken using caste as a covariate to take account of population substructure. The most significant 2-SNP risk haplotype (P=0.028; permuted P= 0.029) was a common (frequency 0.432) CA haplotype for the risk alleles at the top intron 1 SNP rs9460106 and the more distal intron 6 SNP rs2103816 in FAM120B. This was mirrored by the protective 2-SNP TT haplotype (P=0.029) for these two SNPs. Interestingly, the most significant 2-, 3-, 4- and 5- SNP protective haplotypes extended proximally from these 2 SNPs across the intergenic region between FAM120B and DLL1. The most significant (P=0.007; permuted P=0.007) protective haplotype was the 4-SNP CTTC haplotype across the 4 intergenic SNPs rs2738820, rs9459988, rs17860704 and rs9366198, and extending to the DLL1 intron 4 SNP rs1884190 as a 5-SNP haplotype. Of particular interest is the role of SNP rs17860704, which was the single DLL1 SNP associated with VL in Brazil (Fakiola et al., 2011), in defining these haplotype associations. Although not significant on its own in India, it’s inclusion on 2- and 3-SNP haplotypes with rs9459988 and rs9366198 improved significance of the association with protection compared to either of these SNPs alone (Figure 2). We tested this formally using a stepwise regression model in UNPHASED. This demonstrated that a 2-SNP model that included rs17860704 provided significantly better fit (likelihood ratio test P=0.01) than a model in which rs9366198 was tested alone. Similarly, a 3-SNP model that included these two SNPs provided a significantly better fit (likelihood ratio test P=0.03) than a model in which rs9459988 was tested alone, whereas a 2-SNP model in which rs9366198 was added to rs9459988 did not provide a better fit. These data suggest that rs17860704 is contributing to resolution of protective haplotypes across the intergenic region, even though both risk and protective haplotypes are on the background of the same allele (T) at this SNP (Figure 2). The evolution of the etiological variant affecting VL susceptibility in the region clearly post-dates the evolution of variants at this SNP. In summary, the LD, haplotype and stepwise analyses suggest that the disease association is best defined by the protective CTTC haplotype for the 4 SNPs located in the intergenic region adjacent to the DLL1 gene, and extending as a 5-SNP haplotype into associated the intron 4 SNP within DLL1 (Figure 1).
Figure 2. Haplotype associations with Indian VL across DLL1 and FAM120B.
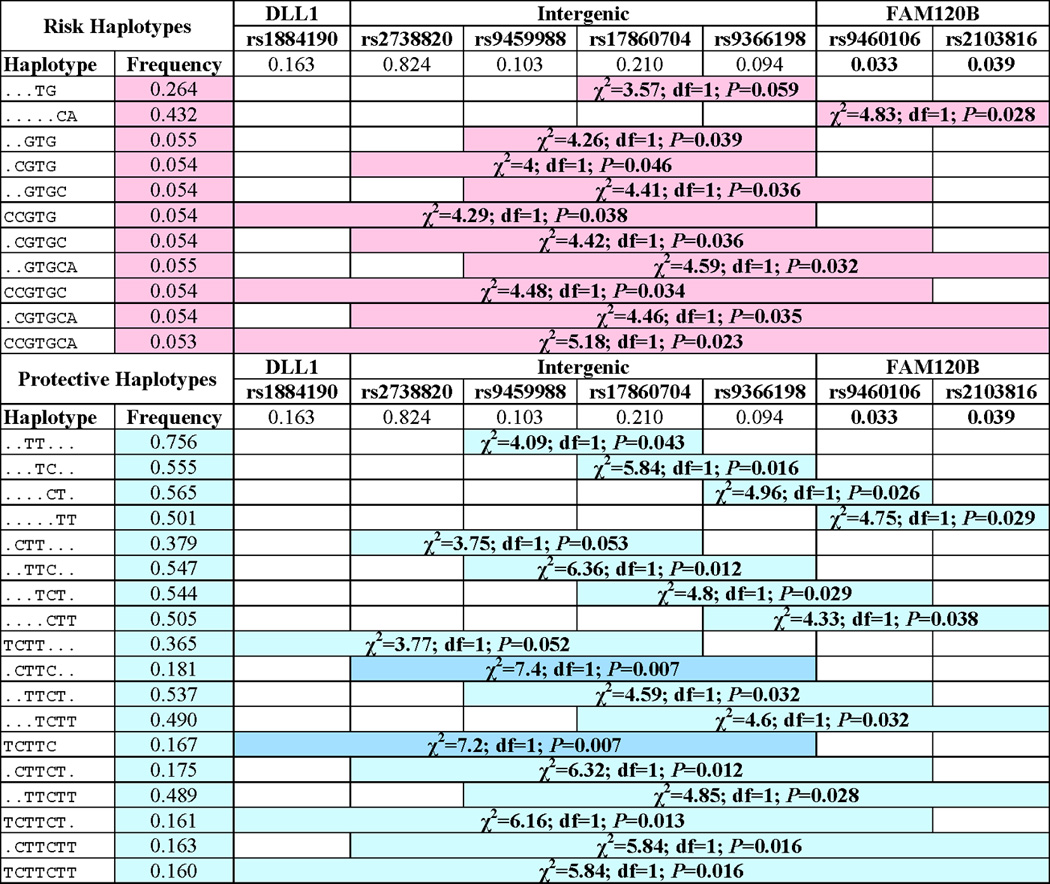
Chi-squared (χ2), degrees of freedom (df) and P-values for – single markers and 2-, 3-, 4- and 5-SNP haplotypes as determined in PLINK comparing cases and controls under an additive model and using caste as a covariate to take account of population substructure. Only haplotype associations significant at nominal P≤0.06 and with frequency of >0.05 are shown. Haplotypes are called on the positive strand across all SNPs.
3.3 Expression of 6q27 genes in splenic aspirates from VL patients
The top associated SNP for Indian VL lies in intron 1 of FAM120B, but analysis of LD patterns and haplotype associations are consistent with the hypothesis that the etiological variant lies within the intergenic region and could regulate expression of either gene. To gain some insight into the role of 6q27 genes in VL, QRT/PCR of mRNA from paired splenic aspirates taken from VL patients before and after drug treatment was undertaken. Only DLL1 showed a significant (paired t-test, 2-sided, P<0.0001) reduction in expression following treatment (Figure 3), with levels of expression returning toward those observed in control spleen (unpaired t-test, 2-sided, P=0.02). This suggests that expression of DLL1 is being regulated in response to changes in pathogenesis of the disease. For all other 6q27 genes, including FAM120B, no change in expression was observed pre- or post-treatment or in comparison to control spleens (Figure 3, Supplementary Figure 3).
Figure 3. DLL1 and FAM120B expression.

Relative expression of (A) DLL1 and (B) FAM120B in mRNA from paired splenic aspirates harvested from VL patients (N= 19) pre- (Day 0) and post-treatment (Day 30) with antileishmanial treatment. HOD= Healthy organ donor.
4. Discussion
Here we present data from a well-powered case-control sample from India that shows single point genetic associations between VL and SNPs at DLL1 and FAM120B on Chromosome 6q27. Whilst haplotype analyses point to the most significant protective haplotypes occurring across the intergenic region between these two genes, we could not formally discount FAM120B as the etiological gene on the basis of the genetic data alone. FAM120B encodes a constitutive coactivator of peroxisome proliferator-activated receptor (PPAR-gamma) that promotes adipogenesis (Li et al., 2007). Although the major disease roles associated with polymorphisms at PPAR-gamma affect lipodystrophy, body mass index, and diabetes, a molecular pathway by which PPAR-gamma represses the transcriptional activation of inflammatory response genes in mouse macrophages by promoter-specific repression of nuclear factor kappa-B has been described (Jennewein et al., 2008). Hence, a role for FAM120B as a co-activator of PPAR-gamma influencing L. donovani infection in macrophages cannot be completely discounted. We therefore turned to analysis of gene expression in patient biopsies to guide us in determining which of these two genes relates to susceptibility to VL on Chromosome 6q27. In particular to find evidence as to which of DLL1 or FAM120B was more likely to be differentially expressed as a result of putative regulatory polymorphisms located in the intergenic region between them. In the event, expression of DLL1 but not FAM120B (or any of the other 6q27 genes) was significantly altered pre- and post-treatment, suggesting a role in disease pathogenesis and providing further support for DLL1 as the etiological gene influencing VL susceptibility in this region. Current research is designed to refine these observations in relation to specific cell types expressing DLL1 in the splenic aspirates ex vivo, and to determine the impact of L. donovani infection on DLL1 expression in experimental systems in vitro. On present evidence we conclude that DLL1 is the more likely candidate to be under genetic regulation that associates with VL.
As outlined above, Notch signalling via DLL1 has many pleiotropic effects on immune cell function, and a case could be made for the involvement of any one of these functions in the pathogenesis of VL disease. Of these it seemed likely that the ability of DLL1 to drive Th1 development might be crucial in protection from VL. This seems at odds with our observation that DLL1 expression went down in the spleen post-treatment. However, in active VL disease, high levels of TNF-α contribute to fever and cachexia, and are detrimental (Barral-Netto et al., 1991). This is indicative of a pro-inflammatory response that could be mediated by CD4+ Th1 cells. Until recently it was thought that active VL patients failed to make interferon-γ responses to recall antigen in cells from peripheral blood (Sacks et al., 1987), and that this anergy may be the main reason for susceptibility. More recently, whole blood quantiferon assays carried out in our study area (Gidwani et al., 2011) have demonstrated very high levels of antigen-driven CD4+ T cell-derived interferon-γ in active VL patients. The high level of DLL1 could be driving this response. Further research is required to determine the precise functional link between DLL1 expression and susceptibility to VL.
In summary, we have demonstrated association between SNPs and haplotypes across the DLL1 to FAM120B region on Chromosome 6q27 in Indian VL, which support earlier findings in Sudan and Brazil. Functional studies suggest DLL1 is the etiological candidate gene, confirming the potential for polymorphism at DLL1 to act as a genetic risk factor across the epidemiological divides of geography and parasite species and providing the potential for targeted therapeutic intervention to alter disease progression in humans.
Supplementary Material
Highlights.
Chromosome 6q26–27 is linked to susceptibility to visceral leishmaniasis.
Delta-like 1 ligand (DLL1) for Notch 3 was implicated as the etiological gene.
Here we find genetic association with DLL1 for L. donovani in India.
DLL1 is the only 6q27 gene differentially expressed in spleen before treatment.
DLL1 is a determinant of visceral leishmaniasis across continents/parasite species.
Acknowledgments
We would like to thank the families from the state of Bihar, northeast India for their participation in this study. This research was funded by grants from The Wellcome Trust in the UK (grant numbers: 074196/Z/04/Z and 085475/Z/08/Z) and The National Institutes of Health in the USA (grant numbers: R01 AI076233-01 and 1P50AI074321-01). Authors SM, MS and PT are thankful to Council of Scientific and Industrial Research (CSIR), New Delhi for providing financial assistance. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authors' contributions
AM and SM carried out the field collection and/or preparation of the samples. SM performed the genotyping, and participated in the statistical analysis and interpretation of the data. MF cross-checked statistical analyses and carried out additional statistical tests, including the haplotype, conditional and step-wise logistic regression analyses. MR oversaw laboratory-based work in Varanasi. DSR and KT oversaw the Sequenom genotyping undertaken by SM in Hyderabad. MS and PT assisted with RNA preparation. SM designed and carried out the QRT/PCR. SS helped conceive the study, was responsible for clinical care of cases at the Kala Azar Medical Research Centre, Muzaffarpur, Bihar State, India, and provided the logistical support to make the study possible. SM prepared the first draft of the manuscript. JMB designed the study, conceived the specific hypothesis to be tested, made the final interpretation of the data, and prepared the final manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that no competing interests exist.
The authors declare no conflict of interest.
Contributor Information
Sanjana Mehrotra, Email: anniebhu19@gmail.com.
Michaela Fakiola, Email: mf300@cam.ac.uk.
Anshuman Mishra, Email: anshuindia@gmail.com.
Medhavi Sudharshan, Email: medhavisudarshan@gmail.com.
Puja Tiwary, Email: tiwarypuja@gmail.com.
Deepa Selvi Rani, Email: deepa@ccmb.res.in.
Kumarasamy Thangaraj, Email: thangs@ccmb.res.in.
Madhukar Rai, Email: upicon2007@gmail.com.
Shyam Sundar, Email: drshyamsundar@hotmail.com.
Jenefer M. Blackwell, Email: jmb37@cam.ac.uk, jblackwell@ichr.uwa.edu.au.
References
- Amsen D, Blander JM, Lee GR, Tanigaki K, Honjo T, Flavell RA. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell. 2004;117:515–526. doi: 10.1016/s0092-8674(04)00451-9. [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- Badaro R, Jones TC, Carvalho EM, Sampaio D, Reed SG, Barral A, Teixeira R, Johnson WD., Jr New perspectives on a subclinical form of visceral leishmaniasis. J Infect Dis. 1986;154:1003–1011. doi: 10.1093/infdis/154.6.1003. [DOI] [PubMed] [Google Scholar]
- Barral-Netto M, Badaro R, Barral A, Almeida RP, Santos SB, Badaro F, Pedral-Sampaio D, Carvalho EM, Falcoff E, Falcoff R. Tumor necrosis factor (cachectin) in human visceral leishmaniasis. J. Infect. Dis. 1991;163:853–857. doi: 10.1093/infdis/163.4.853. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Blackwell JM, Fakiola M, Ibrahim ME, Jamieson SE, Jeronimo SB, Miller EN, Mishra A, Mohamed HS, Peacock CS, Raju M, Sundar S, Wilson ME. Genetics and visceral leishmaniasis: of mice and man. Parasite Immunol. 2009;31:254–266. doi: 10.1111/j.1365-3024.2009.01102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho EM, Barral A, Pedral-Sampaio D, Barral-Netto M, Badaro R, Rocha H, Johnson WD., Jr Immunologic markers of clinical evolution in children recently infected with Leishmania donovani chagasi. J. Infect. Dis. 1992;165:535–540. doi: 10.1093/infdis/165.3.535. [DOI] [PubMed] [Google Scholar]
- Dudbridge F. Likelihood-based association analysis for nuclear families and unrelated subjects with missing genotype data. Hum Hered. 2008;66:87–98. doi: 10.1159/000119108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudbridge F, Holmans PA, Wilson SG. A flexible model for association analysis in sibships with missing genotype data. Ann.Hum.Genet. 2011;75:428–438. doi: 10.1111/j.1469-1809.2010.00636.x. [DOI] [PubMed] [Google Scholar]
- Fakiola M, Miller EN, Fadl M, Mohamed HS, Jamieson SE, Francis RW, Cordell HJ, Peacock CS, Raju M, Khalil EA, Elhassan A, Musa AM, Silveira F, Shaw JJ, Sundar S, Jeronimo SM, Ibrahim ME, Blackwell JM. Genetic and functional evidence implicating DLL1 as the gene that influences susceptibility to visceral leishmaniasis at chromosome 6q27. J. Infect. Dis. 2011;204:467–477. doi: 10.1093/infdis/jir284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakiola M, Mishra A, Rai M, Singh SP, O'Leary RA, Ball S, Francis RW, Firth MJ, Radford BT, Miller EN, Sundar S, Blackwell JM. Classification and regression tree and spatial analyses reveal geographic heterogeneity in genome wide linkage study of Indian visceral leishmaniasis. PLoS ONE. 2010;5:e15807. doi: 10.1371/journal.pone.0015807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- Gidwani K, Jones S, Kumar R, Boelaert M, Sundar S. Interferon-gamma release assay (modified QuantiFERON) as a potential marker of infection for Leishmania donovani, a proof of concept study. PLoS Negl Trop Dis. 2011;5:e1042. doi: 10.1371/journal.pntd.0001042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald I. LIN-12/Notch signaling: lessons from worms and flies. Genes & development. 1998;12:1751–1762. doi: 10.1101/gad.12.12.1751. [DOI] [PubMed] [Google Scholar]
- Jaleco AC, Neves H, Hooijberg E, Gameiro P, Clode N, Haury M, Henrique D, Parreira L. Differential effects of Notch ligands Delta-1 and Jagged-1 in human lymphoid differentiation. J Exp Med. 2001;194:991–1002. doi: 10.1084/jem.194.7.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson SE, Miller EN, Peacock CS, Fakiola M, Wilson ME, Bales-Holst A, Shaw MA, Silveira F, Shaw JJ, Jeronimo SM, Blackwell JM. Genome-wide scan for visceral leishmaniasis susceptibility genes in Brazil. Genes Immun. 2007;8:84–90. doi: 10.1038/sj.gene.6364357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennewein C, Kuhn AM, Schmidt MV, Meilladec-Jullig V, von Knethen A, Gonzalez FJ, Brune B. Sumoylation of peroxisome proliferator-activated receptor gamma by apoptotic cells prevents lipopolysaccharide-induced NCoR removal from kappaB binding sites mediating transrepression of proinflammatory cytokines. J.Immunol. 2008;181:5646–5652. doi: 10.4049/jimmunol.181.8.5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Kang Q, Wang DM. Constitutive coactivator of peroxisome proliferator-activated receptor (PPARgamma), a novel coactivator of PPARgamma that promotes adipogenesis. Mol Endocrinol. 2007;21:2320–2333. doi: 10.1210/me.2006-0520. [DOI] [PubMed] [Google Scholar]
- Locksley RM, Scott P. Helper T-cell subsets in mouse leishmaniasis induction, expansion and effector function. Immunol. Today. 1991;12:A58–A61. doi: 10.1016/S0167-5699(05)80017-9. [DOI] [PubMed] [Google Scholar]
- Maekawa Y, Tsukumo S, Chiba S, Hirai H, Hayashi Y, Okada H, Kishihara K, Yasutomo K. Delta1-Notch3 interactions bias the functional differentiation of activated CD4+ T cells. Immunity. 2003;19:549–559. doi: 10.1016/s1074-7613(03)00270-x. [DOI] [PubMed] [Google Scholar]
- Mehrotra S, Oommen J, Mishra A, Sudharshan M, Tiwary P, Jamieson SE, Fakiola M, Rani DS, Thangaraj K, Rai M, Sundar S, Blackwell JM. No evidence for association between SLC11A1 and visceral leishmaniasis in India. BMC Med Genet. 2011;12:71. doi: 10.1186/1471-2350-12-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EN, Fadl M, Mohamed HS, El Zein A, Jamieson SE, Cordell HJ, Peacock CS, Fakiola M, Raju M, Khalil EA, El Hassan AM, Ibrahim ME, Blackwell JM. Y chromosome lineage- and village-specific genes on chromosomes 1p22 and 6q27 that control visceral leishmaniasis in The Sudan. PLoS Genet. 2007;3:679–688. doi: 10.1371/journal.pgen.0030071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohishi K, Varnum-Finney B, Serda RE, Anasetti C, Bernstein ID. The Notch ligand, Delta-1, inhibits the differentiation of monocytes into macrophages but permits their differentiation into dendritic cells. Blood. 2001;98:1402–1407. doi: 10.1182/blood.v98.5.1402. [DOI] [PubMed] [Google Scholar]
- Pirmez C, Yamamura M, Uyemura K, Paes-Oliveira M, Modlin RL. Cytokine patterns in the pathogenesis of human leishmaniasis. J Clin Invest. 1993;91:1390–1395. doi: 10.1172/JCI116341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacks DL, Lal SL, Shrivastava SN, Blackwell JM, Neva FA. An analysis of T cell responsiveness in Indian Kala-azar. J. Immunol. 1987;138:908–913. [PubMed] [Google Scholar]
- Scott P, Pearce E, Cheever AW, Coffman RL, Sher A. Role of cytokines and CD4+ T-cell subsets in the regulation of parasite immunity and disease. Immunol. Rev. 1989;112:162–182. doi: 10.1111/j.1600-065x.1989.tb00557.x. [DOI] [PubMed] [Google Scholar]
- Sekine C, Moriyama Y, Koyanagi A, Koyama N, Ogata H, Okumura K, Yagita H. Differential regulation of splenic CD8- dendritic cells and marginal zone B cells by Notch ligands. Int Immunol. 2009;21:295–301. doi: 10.1093/intimm/dxn148. [DOI] [PubMed] [Google Scholar]
- Singh SP, Reddy DC, Mishra RN, Sundar S. Knowledge, attitude, and practices related to Kala-azar in a rural area of Bihar state, India. Am J Trop Med Hyg. 2006;75:505–508. [PubMed] [Google Scholar]
- Sundar S, Benjamin B. Diagnosis and treatment of Indian visceral leishmaniasis. J Assoc Physicians India. 2003;51:195–201. [PubMed] [Google Scholar]
- Sundar S, Reed SG, Sharma S, Mehrotra A, Murray HW. Circulating T helper 1 (Th1) cell- and Th2 cell-associated cytokines in Indian patients with visceral leishmaniasis. Am. J. Trop. Med. Hyg. 1997;56:522–525. doi: 10.4269/ajtmh.1997.56.522. [DOI] [PubMed] [Google Scholar]
- Topno RK, Das VN, Ranjan A, Pandey K, Singh D, Kumar N, Siddiqui NA, Singh VP, Kesari S, Bimal S, Kumar AJ, Meena C, Kumar R, Das P. Asymptomatic infection with visceral leishmaniasis in a disease-endemic area in bihar, India. Am J Trop Med Hyg. 83:502–506. doi: 10.4269/ajtmh.2010.09-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zijlstra EE, el-Hassan AM, Ismael A. Endemic kala-azar in eastern Sudan: post-kala-azar dermal leishmaniasis. Am J Trop Med Hyg. 1995;52:299–305. doi: 10.4269/ajtmh.1995.52.299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.