

Abstract
TLE1 is a Groucho related transcriptional repressor protein that exerts survival and anti-apoptotic function in several cellular systems and has been implicated in the pathogenesis of cancer. In the present study, we found that TLE1 is a regulator of anoikis in normal mammary epithelial and breast carcinoma cells. The induction of apoptosis following loss of cell attachment to the extracellular matrix (anoikis) in untransformed mammary epithelial MCF10A cells was associated with significant downregulation of TLE1 expression. Forced expression of exogenous TLE1 in these cells promoted resistance to anoikis. In breast cancer cells, TLE1 expression was significantly upregulated following detachment from the ECM. Genetic manipulation of TLE1 expression via overexpression and downregulation approaches indicated that TLE1 promotes the anoikis resistance and anchorage-independent growth of breast carcinoma cells. Mechanistically, we show that TLE1 inhibits the Bit1 anoikis pathway by reducing the formation of the pro-apoptotic Bit1-AES complex in part through sequestration of AES in the nucleus. The mitochondrial release of Bit1 during anoikis as well as exogenous expression of the cytoplasmic localized Bit1 or its cell death domain (CDD) induced cytoplasmic translocation and degradation of nuclear TLE1 protein. These findings indicate a novel role for TLE1 in the maintenance of anoikis resistance in breast cancer cells. This conclusion is supported by an immunohistochemical analysis of a breast cancer tissue array illustrating that TLE1 is selectively upregulated in invasive breast tumors relative to noninvasive ductal carcinoma in situ (DCIS) and normal mammary epithelial tissues.
INTRODUCTION
The Groucho (Gro)/TLE family of transcriptional co-respressors represents a group of gene regulators that influence the transcriptional activity of a wide range of genes (1). As co-repressors, Gro proteins do not bind to DNA directly, but instead bind to other DNA-binding transcription factors to form large multi-protein complexes (2). The binding of Gro to such a regulatory complex most often results in the decreased translation of the target gene. For instance, in Wnt gene regulation, binding of Gro to the transcription factor LEF results in the displacement of the Wnt-activator beta catenin and in decreased translation of the Wnt gene (3). This is accompanied by the recruitment of histone deacetylase to the gene and the subsequent removal of acetyl groups from the DNA bound histones, which results in further gene silencing.
Two groups of the Gro/TLE protein family have been identified. The first group, TLE1-TLE4, shares homologous structural motifs including an N-terminal oligomerization domain (Q-domain), a CcN linker, and a C-terminal Histone deacetylase (HDAC) binding WD domain (1). Normally, TLE1 is required to be in a homotetramer in order to be active. The second group, which includes the Amino Enhancer of Split (AES), retains the same N-terminal oligomerization domain as the other TLE family members, but importantly lacks the WD domain. Because of this, AES is considered a dominant-negative for TLE1-4 family members since AES is able to oligomerize and sequester TLE proteins, preventing them from forming homotetramers and repressing gene transcription.
As transcriptional repressors, the Gro/TLE proteins play important roles in several processes including regulation of neurogenesis and a number of developmental processes (4,5). Recently, data have begun to emerge indicating a prosurvival and/or anti-apoptotic role of groucho proteins, TLE1 in particular. Overexpression of TLE1 in chicken embryo fibroblast led to significant growth stimulation and conferred anchorage-independent growth (6). In mature neurons, exogenous expression of TLE1 prevented cell death and apoptosis (7). Our previous studies have shown that TLE1 is antiapoptotic by blocking the apoptosis induced by the release of mitochondrial Bit1 (Bcl2-inhibitor of transcription 1) protein to the cytoplasm (8). Recently, Seo et al. 2011 demonstrated that TLE1 functions to inhibit apoptosis induced by doxorubicin in synovial sarcoma cells (9). Taken together, these observations provide a case for the role of TLE1 in promoting cell survival via its antiapototic function. It is conceivable that TLE1 may suppress an apoptosis gene transcription program or alternatively it may upregulate a survival-promoting gene transcription program. Consistent with this notion, TLE1 positively regulates Bcl2 expression (8) and ErbB1 and ErbB2 signaling (10), two survival pathways that influence tumorigenesis.
TLE1 appears to be a part of the novel integrin-dependent Bit1 apoptotic pathway (8). Following loss of cell attachment, mitochondrial bound Bit1 protein is released to the cytoplasm and initiates a caspase-independent apoptosis which is unresponsive to various anti-apoptotic treatment including Bcl-2, Bcl-xl, akt. Importantly, Bit1 is unique among cell death inducers in that it is negatively regulated by integrin engagement and therefore represents a critical pathway in understanding cell death caused by ECM detachment (anoikis). Considering that anoikis resistance is a determinant of cancer metastasis, this also places Bit1 as a regulator of metastasis (11). However, the molecular mechanisms underlying Bit1 anoikis function and its regulation by integrin have not been fully elucidated. Bit1, once released from the mitochondria or ectopically expressed in the cytoplasm, forms a complex with AES, and the Bit1-AES complex represents the pro-apoptotic component that mediates the effect of Bit1 on apoptosis (8). One of the downstream targets of the Bit1 signaling pathway appears to be the Extracellular signal-regulated kinase (Erk) (12). Normal and transformed cells in which Bit1 expression is downregulated exhibit increased Erk activation and such elevated Erk activity contributes in part to the enhanced anoikis resistance of the Bit1 knockdown cells. Recently, we have also demonstrated that the serine/threonine kinase PKD acts as an upstream activating kinase that promotes the apoptotic and anoikis function of Bit1 (13).
In light of previous reports documenting the antiapoptotic and growth promoting functions of TLE1, we examined in the present study the effect of TLE1 expression in the survival and anoikis resistance of breast carcinoma cells. We discovered a critical role for TLE1 in suppressing anoikis in breast cancer cells, and that its inhibitory effect on anoikis is in part through circumventing the Bit1-mediated anoikis pathway. Interestingly, the nuclear TLE1 protein is targeted and channelled to degradation in the cytoplasm by Bit1 during anoikis, indicating that turning off the TLE1 transcriptional machinery may represent an important mechanism in the induction of anoikis by Bit1.
Materials and Methods
Cell culture and transfection assays
MCF7, MDA-MB-231, MCF10A, and HEK 293T from American Type Culture Collection (ATCC) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with glutamine containing 10% fetal bovine serum, penicillin, and streptomycin. Stable MDA-MD-231 control and DDK-TLE1 pool of cells were generated by transfection with DDK-TLE1 or empty vector construct (Origene) as described previously (11,13). Briefly, transfections were carried out with lipofectamine 2000 (Invitrogen) in OPTI-MEM (Invitrogen) according to the manufacturer’s protocol with cells plated 18 hr before transfection. 48 hr post transfection, transfected cells were cultured in the 1000 ug/ml G418 to select for positive DDK-TLE expressing clones. Several G418 resistant control and DDK-TLE1 clones were harvested by ring-cloning, and the level of DDK-TLE1 expression was confirmed by immunoblotting against a specific DDK antibody. Two control clones and three positive DDK-TLE1 clones were pooled together to generate the control and DDK-TLE1 pools, respectively. In generating the MCF10A control and GFP-TLE1 pool of cells, the MCF01A cell line was transfected with GFP-TLE1 or empty GFP-tagged vector (Origene) using the Lipofectamine 2000 Plus reagent (Invitrogen), and transfected cells were then selected in the presence of 400 ug/ml G418 to generate the control and GFP-TLE1 pools as described above. In transient transfection assays, the total amount of plasmid used per transfection was normalized with the corresponding empty vector constructs.
Chemical reagents, antibodies, and plasmids
Poly(2-hydroxyethyl methacrylate (Polyhema) and the mouse monoclonal anti-β-actin antibody were obtained from Sigma Chemical Co. (St. Louis, Mo.). The mouse monoclonal anti-FLAG, anti-AES, anti-GFP and anti-B-actin antibodies were purchased from Sigma (St. Louis, MO). The polyclonal anti-TLE1 antibody was obtained from Abcam (Cambridge, MA). The proteasomal inhibitor MG132 and the caspase inhibitor zVad-fmk were purchased from Calbiochem (La Jolla, CA) while the chemical reagent Bortezomib and was obtained from LC Laboratories (Woburn, MA). The anti-caspase-3, anti-cleaved caspase-3, anti-caspase-8, anti-cleaved caspase-8, and anti-PARP antibodies were obtained from Cell Signaling Technology (Beverly, MA). The anti-c-myc antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The mammalian expression vector encoding for mitochondrial Bit1 were generated as described previously (8,13). The FLAG-TLE1 plasmids were obtained from Origene (Rockville, MD). The myc-Bit1CDD plasmids were provided by Dr. Renwei Chen.
SiRNA and shRNA transfection
The TLE1 specific siRNAs (#186345, #186346, and #110786) and the control, non-targeting siRNAs were purchased from Invitrogen (Carlsbad, CA). For transient transfection experiments, MDA-MB-231 cells (2 x 105) were transfected with 25 μM of each siRNA by using the Lipofectamine RNAiMAX transfection reagent (Invitrogen). 48 hrs post-transfection, cells were harvested and subjected to immunoblotting, anoikis, or soft agar assays as described below.
To generate stable MDA-MB-231 TLE1 knockdown and control pools, MDA-MB-231 parental cells were transfected with pRS vector containing the short hairpin RNA against TLE1 (Origene) or the non-targeting scrambled shRNA (Origene). 48 h post-transfection, 1 μg/ml puromycin (Invitrogen) was added to the medium to select for puromycin-resistant clones. Individual puromycin-resistant clones were screened for TLE1 downregulation by immunoblotting using a specific antibody to TLE1. Three TLE1 knockdown-positive clones were pooled as well two control clones were also pooled for further characterization.
Analysis of apoptosis, anoikis, and cell viability
Apoptosis was assessed by determining the level of cytosolic nucleosomal fragments with the use of Cell Death Detection ELISA kit (Roche Molecular Biochemicals), according to the manufacturer’s instructions. To assess for anoikis cell death, cells were plated onto Polyhema coated 96 well plates in complete growth medium containing 0.5% methylcellulose at a density of 1.0 x104/well at various time points as previously described (8,11). Detached cells were then collected and subjected to the Cell Death ELISA apoptosis assay (13). In some experiments, cell viability was quantified by alarmar blue staining (Invitrogen) and subsequent fluorescence reading (485 nm excitation wavelength and 520 nm emission wavelength) using the microplate reader.
Cell proliferation and soft agar assays
To determine anchorage-dependent growth, cells were plated in a volume of 150 μl at a density of 2,000 cells per well in 96-well plates. At each indicated time, the number of metabolically active cells was measured with the use of MTT assay as described previously (11). Briefly, MTT reagent (Sigma) was dissolved in sterile PBS at 5 mg/ml. Ten microliters of this solution was added to each well in a 96-well plate (1:10 dilution). The plate was then incubated at 37°C, 5% CO2 for 3h. Afterwards, the medium was gently aspirated away and the MTT precipitate was dissolved in 100 ul of a 50% MeOH-50% DMSO solution. The precipitate was allowed to dissolve at room temperature for ten minutes with gentle shaking. The resulting 550 nm absorbance was read on a microplate reader (BioTek Instruments). The anchorage-independent growth of cells was measured using the 96-well plate format (25). Briefly, 5,000 cells in 0.3% agar solution was plated onto wells precoated with 0.6% agar in culture medium. The anchorage-independent growth of cells was then quantified by alarmar blue staining (Invitrogen) and fluorescence reading at 485 nm excitation wavelength and 520 nm emission wavelength with a microplate plate reader.
Immunofluorescence microscopy
Immunofluorescent staining was performed as previously described (8). Briefly, cultured cells were transfected with FLAG-tagged TLE1 and myc-tagged Bit1 or CDD. 24h post-transfection, cells were fixed in 4% paraformaldehyde and permeabilized in 0.5% Triton X-100, followed by blocking with 20% fetal bovine serum. The cells were then stained with anti-Flag (mouse, Sigma), and secondary antibodies (Alexa 488, Molecular Probes). The specimens were mounted with DAPI-containing media (Vectashield) and analyzed on a confocal the microscope (BioRad 1024 equipped with Lasersharp software OS2).
Protein preparation and western blotting assays
Protein preparation and western blotting were performed as described previously (11,13). Briefly, cells were harvested 24-48 hr after transfection with various constructs or siRNAs by adding ice-cold NP-40 lysis buffer (1% NP-40; 20mM Tris-HCL [pH7.4]; 150 mM NaCl; 10% glycerol, 2 mM sodium vanadate; 1 mM henylmethylsulfonyl fluoride; 10 μg/ml leupeptine; and 5 μg/ml aprotinin) and incubating at 4°C for 20 min. For immunoblot analysis, equal amounts of proteins were resolved on 4–20% gradient Tris-glycine gels (Invitrogen) and electrophoretically transferred to nitrocellulose membrane. The membranes were incubated with primary antibodies overnight at 4°C followed by secondary antibodies conjugated with horseradish peroxidase. Membranes were developed using the ECL detection system. Densitometric analysis was performed using Image J software (13).
Coimmunoprecitation Assay
Coimmunoprecipitation assay was performed as described previously (13). Briefly, transfected cells cultured in attached or detached conditions were harvested by washing once with PBS and resuspended in ice-cold Nonidet P-40 lysis buffer (1% Nonidet P-40, 20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 10% glycerol, 2 mm sodium vanadate, 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, and 5 μg/ml aprotinin) followed by a 20-min incubation at 4 °C. Cell debris was removed by centrifugation. Myc-tagged Bit1 was immunoprecipitated with anti-Myc-agarose conjugate (Abcam) and thoroughly washed with lysis buffer. Bound proteins were resolved by SDS-PAGE, and Western blotting was performed using anti-AES antibody.
Subcellular Fractionation
Subcellular fractionation was performed as described previously (13). Briefly, transfected cells cultured in attached or detached conditions for 12 hr were harvested washed once with PBS, resuspended in 1 ml of isotonic mitochondrial buffer (250 mM mannitol, 70 mM sucrose, 1 mM EDTA, 10 mM HEPES [pH 7.5]), and homogenized with 40 strokes in a Dounce homogenizer. The lysates were centrifuged at 500 × g for 5 min to eliminate nuclei and unbroken cells. The supernatant was further centrifuged at 10,000 × g for 30 min at 4°C to isolate the mitochondrial enriched pellet, which was resuspended in isotonic mitochondrial buffer. Both the cytosolic supernatant and mitochondrial fraction were subjected to SDS-PAGE electrophoresis and immunoblotting. For separation of cytosol and the nuclear fraction, the NE-PER nuclear isolation kit (Pierce, No. 78833) was used and performed as prescribed by the manufacturer. The protein concentration in the different fractions was quantified using the Bio-Rad protein assay kit with BSA as the standard.
Total RNA Extraction and Quantitative Real Time PCR
Total RNA was extracted from 1.0x106 cultured cells using the RNeasy kit (Qiagen), quantified by spectrophotometry (NanoDrop 8000, Thermo Scientific). Total RNA was reversed transcribed using the Superscript First-Strand Synthesis Kit for RT-PCR (Invitrogen) as prescribed by the supplier. cDNA was quantified by real-time PCR on the ABI Prism 7900 Sequence Detection System (Applied Biosystems). Human TLE1 forward primer: CCCATATCCTGCTCCTTTTG and reverse primer: GGTTGAGGGTGTTGATCTGG were used. PCR was performed using the Sybr Green PCR Core reagents (Applied Biosystems, Foster City, CA) based on manufacturer’s instructions. Amplification of the same cDNAs with human GAPDH primers was used for internal normalization.
Human breast tumor array analysis
Breast tumor tissue array slides containing intraductal carcinomas, invasive ductal carcinoma, and matched normal breast epithelial tissues were obtained from US Biomax, Inc. (Rockville, MD). The immunohistochemistry procedure was performed by Biomax Inc. on two tissue microarray slides. As described previously (11), tissue array slides were deparaffinised, hydrated and subjected to antigen retrieval. The slides were then incubated in 2.5% normal horse serum for 30 min at room temperature followed by incubation with the primary antibody (1:100 dilution) for 1 h at room temperature. The affinity purified rabbit anti-TLE1 antibody (M-101) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, 11) while the affinity purified rabbit anti-Bit1 antibody (HPA012897) was purchased from Sigma (11). Rabbit normal serum was used as negative control antibody to replace the primary antibody on control slide with 1 hr incubation. Tissue array slides were then washed and incubated with ImmPRESS reagent (Vector Laboratories) followed by treatment with peroxidise substrate DAB solution (DAKO Cytomation). Each of the experimental and control slide was scored for average staining intensity by two investigators with no knowledge of the pathologic status of the samples. These investigators scored staining intensity as 0, no staining; 1 low staining; 2 medium staining; or 3 high staining. The intensity of TLE1 or Bit1 staining was further measured as the average staining intensity of the majority of epithelial cells (75% to 100%) in each sample. The average staining was graded as 0, no staining; 1, slight staining; 2, moderate staining; and 3, strong staining.
Statistical Analysis
Data are presented as means (±S.E.). For western blots and anoikis assays, experiments were performed at least three times with duplicates. Statistical differences between groups were established at a P value < 0.05 using the two-tailed Student’s t test. For breast tumor tissue array analysis, a one-way ANOVA with subsequent post hoc testing using the Tukey-Kramer multiple comparison test was used to compare the average staining intensity of each case type (11). All calculations were done using the NCSS statistical software (NCSS, Kasville, UT).
RESULTS
Induction of anoikis in normal mammary epithelial cells is associated with downregulation of TLE1
To address the hypothesis that TLE1 plays a role in the anoikis resistance of breast carcinoma cells, we first examined the effects of TLE1 on anoikis of normal mammary epithelial cell line MCF10A. As previously described (14,15), forced suspension of MCF10A cells resulted in a significant induction of apoptosis (Fig. 1A) which was associated with the downregulation of TLE1 expression both at the mRNA (Fig. 1B) and protein levels (Fig. 1C and 1D). To examine the significance of TLE1 suppression in MCF10A following loss of attachment, we stably expressed exogenous GFP-tagged TLE1 or the GFP alone in MCF10A cells. Exogenous TLE1 expression was confirmed by immunoblotting using anti-GFP antibody (Fig. 1E) and anti-TLE1 antibody (Supplementary Fig. 1). Immunofluorescent imaging also confirmed the nuclear localization of GFP-TLE1 (Fig. 1F) as previously described (1, 16). While the control and TLE1 expressing pools displayed similar levels of basal apoptosis in adherent conditions, the TLE1 expressing pool of cells showed significantly decreased apoptosis in suspension culture as compared to control cells (Fig. 1G). These findings indicate that TLE1 promotes survival of normal mammary epithelial cells in response to detachment from the ECM, and its downregulation following loss of cell attachment may contribute to induction of anoikis cell death program in breast epithelial cells.
Fig. 1.


TLE1 expression is downregulated during anoikis and expression of exogenous TLE1 inhibits anoikis in nontransformed breast epithelial cell line MCF10A. A, B, C, and D. MCF10A cells were plated onto a polyhema coated or uncoated tissue culture plates for 24 h in culture. In A, the cells were harvested and analyzed for apoptosis using the Cell Death Elisa ELISA. In B, harvested cells were subjected to total RNA isolation and TLE1 mRNA expression levels were quantified by reverse transcription and real time PCR analysis. Data represent relative values, normalized to the levels of GAPDH mRNA (see materials and methods) In C, harvested cells were subjected to total cell lysate isolation, SDS-PAGE, and immunoblotting against specific antibodies to TLE1 and B-actin. In D, the TLE1/B-actin ratios were determined by densitometry analysis of immunoblots in C from three independent experiments. E, F and G. Stable MCF10A derived GFP-TLE1 and control overexpressing pools of cells were generated as described in materials and methods. In E. and F., expression of exogenous GFP-TLE1 was confirmed by immunoblotting (E) using the total cell lysates derived from control and TLE1 overexpressing pools against specific antibodies to GFP and B-actin and immunofluorescent imaging (F). In G. control and TLE1 overexpressing cells were plated onto a polyhema coated or uncoated tissue culture plates. Following 24 h in culture, cells were harvested and analyzed for apoptosis by measuring the amount of DNA histone fragments (Cell Death Elisa). In A, B, D, and G, three independent experiments were performed in triplicates, * indicates p<0.05 by Student’s t test.
Anoikis resistance of breast carcinoma cells is associated with increased expression of TLE1
We next examined the role of TLE1 in the anoikis resistance of human breast cancer cells. Consistent with the previous finding (17), the highly aggressive MDA-MB-231 cells showed significant resistance to anoikis as evidenced by the lack of apoptosis following 24 h culture in suspension (Fig. 2A). In marked contrast to that found in the highly anoikis sensitive MCF10A cells, TLE1 expression at both the mRNA (Fig. 2B) and protein (Fig. 2C and 2D) levels was significantly increased in the MDA-MB-231 cells following 24 h in suspension culture as compared to the level in attached cells. Similar results were also obtained in another anoikis resistant mammary carcinoma MCF7 cells (Fig. 2A) which exhibited significantly high levels of TLE1 protein following culture in suspension for 24 h (Fig. 2C and 2D). These findings show that loss of cell-substrate attachment triggers induction of TLE1 expression in breast cancer cells.
Fig. 2.

Detachment induced upregulation of TLE1 expression in breast cancer cells. A. MCF10A and breast cancer (MDA-MB-231 and MCF-7) cell lines were plated onto a polyhema coated or uncoated tissue culture plates. Following 24 h in culture, cells were harvested and analyzed for apoptosis by measuring the amount of DNA histone fragments (Cell Death Elisa). In B, MDA-MB-231 cells cultured in attached and detached conditions for 24 h were subjected to total RNA isolation, and TLE1 mRNA expression levels were quantified by reverse transcription and real time PCR analysis. C. MDA-MB-231 and MCF-7 cells cultured in attached and detached conditions for 24 h were subjected to total cell lysate isolation, SDS-PAGE, and immunoblotting against specific antibodies to TLE1 and B-actin. Total cell lysates of MCF10A cells in attached and detached conditions were also run on the same gel for comparison. D. The TLE1/B-actin ratios were determined by densitometry analysis of immunoblots in C from three independent experiments. In B and D, three independent experiments were performed in triplicates, * indicates p<0.05 as compared to attached conditions (Student’s t test).
TLE1 is a regulator of anoikis resistance in breast cancer cells
To examine the role of TLE1 upregulation in the anoikis resistance of breast carcinoma cells, we down-regulated TLE1 expression in MDA-MB-231 cells via short-interfering RNAs (siRNAs). Two TLE1 specific siRNAs #1(186345) and #3(117086) showed a significant 50–70% downregulation of TLE1 expression (Fig. 3A). Suppression of TLE1 expression by these two specific TLE1 siRNAs did not induce a change in the morphology (Supplementary Fig. 2A) or anchorage-dependent growth of MDA-MB-231 cells (Supplementary Fig. 2B). In contrast, culturing cells transfected with the TLE1siRNAs in suspension showed reduced growth (Fig. 3B) and increased apoptosis (Fig. 3C) as compared with the parental and control-siRNA treated cells. It is noteworthy that no significant difference in basal apoptosis was observed between the TLE1-siRNA and control siRNA treated cells in adherent conditions (Fig. 3C). The observed enhanced anoikis sensitivity is specific to TLE1 downregulation since forced overexpression of exogenous TLE1 attenuated the increased anoikis sensitivity of TLE1-siRNA treated cells (Supplementary Fig. 3A and 3B). To complement the results from the transient transfection studies, we also generated stable TLE1 knockdown clones by infecting MDA-MB-231 cells with lentiviral expression vectors for TLE1 specific shRNAs or control shRNAs. As shown in Fig. 3D, TLE1 expression was reduced by 70% in a pool of TLE1 shRNA clones as compared with control shRNA pool of clones. The control shRNA and TLE1 shRNA pools exhibited similar growth kinetics (Supplementary Fig. 2C) and basal apoptosis (Fig. 3E) in monolayer culture. In contrast, when cultured without anchorage, the TLE1 shRNA pool displayed a significantly more apoptosis than the control shRNA pool (Fig. 3E). Consistent with the enhanced anoikis sensitivity induced by TLE1 downregulation, both transient and stable suppression of TLE1 resulted in decreased growth in soft agar (Supplementary Fig. 4A). TLE1 knockdown cells exhibited fewer and smaller colonies compared to the control cells (Supplementary Fig. 4B and 4C). Taken together, these results indicate that suppression of TLE1 induces detachment-induced apoptosis and attenuates the anchorage-independent growth of breast carcinoma cells.
Fig. 3.


TLE1 regulates anoikis sensitivity of breast cancer cells. MDA-MB-231 cells were transfected with control- or TLE1 specific siRNAs, and 48 h post-transfection, cells were harvested and subjected to immunoblotting (A) with antibodies against TLE1 and B-actin. In B and C, control- and TLE1 siRNA treated cells were plated onto a polyhema coated or uncoated tissue culture plates and cultured for 24 h. In B, the growth of detached cells was measured by alamar blue staining and fluorescent reading. In C, the level of apoptosis in attached and detached cells was quantified by measuring the amount of DNA histone fragments (Cell Death Elisa). In B and C, results are representative of three independent experiments, *p<0.05 (Student’s t test) as compared with control siRNA transfected cells. D. Stable MDA-MB-231 controlshRNA and TLE1shRNA knockdown pools were generated as described in materials and methods, and the total cell lysates derived from controlshRNA and Bit1shRNA knockdown pools were subjected to immunobloting using specific antibodies to TLE1 and B-actin. E. Stable MDA-MB-231 controlshRNA and TLE1shRNA knockdown pools were plated onto a polyhema coated or uncoated tissue culture plates. Following 24 h in culture, cells were harvested and analyzed for apoptosis by measuring the amount of DNA histone fragments (Cell Death Elisa). F. Stable MDA-MB-231 derived DDK-TLE1 and control overexpressing pool of cells were generated as described in materials and methods. The expression of exogenous DDK-TLE1 was confirmed by immunoblotting using the total cell lysates derived from control and TLE1 overexpressing pools against a specific antibody to DDK and B-actin. G. The control and TLE1 overexpressing cells were plated onto a polyhema coated or uncoated tissue culture plates. Following 72 h in culture, cells were harvested and analyzed for apoptosis by measuring the amount of DNA histone fragments (Cell Death Elisa). In E and G, three independent experiments were performed in triplicates, * indicates p<0.05 by Student’s t test.
To confirm the results obtained with the downregulation approaches, we stably overexpressed TLE1 in MDA-MB-231 cells. Several TLE1 overexpressing and vector transfected clones were selected and combined to generate the stable TLE1 and control pool, respectively (Fig. 3F). In monolayer culture, no significant differences in the growth rate (Supplementary Fig. 5A) and basal apoptosis (Fig. 3G) were observed between the control and TLE1 overexpressing pools. After a prolonged culture in suspension for 72 h, the control pool underwent significant apoptosis while the stable TLE1 pool exhibited significantly low degree of apoptosis (Fig. 3G). Consistent with this enhanced anoikis resistance, the stable TLE1 pool showed greater ability to grow in soft agar than the control pool (Supplementary Fig. 5B). These findings indicate that TLE1 provides protection from apoptosis under anchorage-independent conditions.
TLE1 inhibits Bit1-induced anoikis
Based on our previous finding that TLE1 counteracts Bit1 apoptosis function (8), we explored the notion that TLE1 is a target of mitochondrial Bit1 when this protein causes anoikis. To address this possibility, the highly aggressive and anoikis resistant MDA-MB-231 cells were transfected with the C-terminally myc tagged Bit1, which associates with mitochondria, or vector construct. The cells were then grown in suspension or left attached in culture dish. Consistent with previous findings (8,11,13), exogenous expression of mitochondrial Bit1 dose-dependently enhanced apoptosis following culture in suspension for 24 hr (Fig. 4A). In contrast, the Bit1 and control transfected cells had the same level of spontaneous apoptosis when grown attached to a culture dish. As described previously (8), the Bit1-induced anoikis was independent of caspase activation as evidenced by the inability of the pan-caspase inhibitor z-Vad-fmk to block the Bit1 anoikis function (Fig. 4B). In addition, we also found no significant increase in the 85-kD PARP apoptotic fragment and in the active cleaved caspase-3 and cleaved caspase-8 fragments in the lysates of Bit1 transfected cells (Fig. 4C). Instead, we observed that the Bit1-induced anoikis was associated with significant downregulation of the detachment-induced TLE1 expression (Fig. 4D and 4E). The effect of Bit1 on TLE1 expression occurred primarily at the level of protein accumulation since Bit1 had no effect on the detachment-induced TLE1 mRNA (Fig. 4F).
Fig. 4.

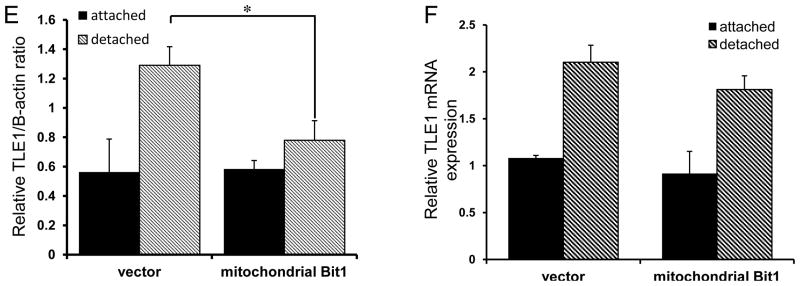
Induction of anoikis by Bit1 is associated with downregulation of TLE1 expression. A. MDA-MB-231 cells were transfected with the indicated amounts of C-terminally myc-tagged mitochondrial localized Bit1 construct. The total amount of DNA was normalized with the empty vector in each transfection. 24 h post-transfection, cells were plated onto polyhema coated or uncoated tissue culture plates. Following 24 h in culture, cells were harvested and analyzed for apoptosis by measuring the amount of DNA histone fragments (Cell Death Elisa). Three independent experiments were performed in triplicates; *, p<0.05 as compared to the corresponding attached condition (Student’s t test). B. Mitochondrial Bit1 transfected cells were cultured in attached or detached conditions in the presence of z-Vad-fmk (50 uM) or DMSO for 24 h. Cells were then harvested and subjected to Cell Death ELISA. C and D. MDA-MB-231 cells transfected with C-terminally myc-tagged Bit1or vector construct were plated onto polyhema coated or uncoated tissue culture plates. Following 24 h in culture, cells were harvested and subjected to total cell lysate isolation, SDS-PAGE, and immunoblotting against specific antibodies to caspase-3, cleaved caspase-3, caspase-8, cleaved caspase-8, PARP, TLE1, myc-tag, and B-actin. E. The TLE1/B-actin ratios were determined by densitometry analysis of immunoblots in D from three independent experiments; *, p<0.05 by Student’s t test. F. MDA-MB-231 cells transfected with mitochondrial Bit1 or vector construct were plated onto polyhema coated or uncoated tissue culture plates and cultured for 24 h. Cells were then harvested and subjected to total RNA isolation, and TLE1 mRNA expression levels were quantified by reverse transcription and real time PCR analysis.
Considering that TLE1 confers significant protection from anoikis in breast cancer cells and has been previously shown to inhibit Bit1 apoptosis function (8), we examined the notion that loss of TLE1 may contribute to Bit1-induced anoikis in breast carcinoma cells. To address this possibility, MDA-MB-231 cells were cotransfected with C-terminally myc-tagged Bit1 and/or full length DDK-tagged TLE1. The cells were then cultured on tissue culture plates, or on poly-HEMA coated plates to induce anoikis. The ectopic expression of mitochondrial Bit1 and TLE1 was confirmed via immunoblotting (Fig. 5A). Consistent with our previous findings (8,11,13), detachment induced a higher level of apoptosis in Bit1 transfected cells than in control cells (Fig. 5B). Importantly, ectopic TLE1 levels attenuated the high level of detachment-induced apoptosis in mitochondrial Bit1 transfected cells. Consistent with the inhibitory effect of TLE1 on Bit1 anoikis, the apoptotic activity of cytoplasmic localized Bit1 (cyto) was inhibited by TLE1 (Supplementary Fig. 6) as shown previously (8). It is noteworthy that similar attenuation of Bit1 anoikis was also observed in stable TLE1 expressing cells (Fig. 5C). To complement our results with ectopic TLE1 expression studies, we also examined the effect of decreased TLE1 expression on Bit1-mediated anoikis. Knockdown of TLE1 further increased the sensitivity of MDA-MB-231 cells to Bit1 anoikis (Fig. 5D).
Fig. 5.


TLE1 antagonizes Bit1 anoikis function. A. MDA-MB-231 cells were transfected with C-terminally myc-tagged Bit1 together with DDK-tagged TLE1. 24 h post transfection, the expression of exogenous Bit1 and TLE1 in transfected cells was confirmed by immunoblotting using specific antibodies to myc and DDK tags. The amount of plasmid transfected into cells was normalized with the vector construct. B. MDA-MB-231 cells transfected with the indicated construct were grown for 24h in suspension, and cells were then harvested and subjected to Cell Death ELISA assay to measure the amount of DNA histone fragments. C. Stable MDA-MB-231 derived DDK-TLE1 and control overexpressing pool of cells were transfected with mitochondrial Bit1 or vector construct, and 24 h post-transfection cells were plated onto polyhema coated tissue culture plates. Following 48 hr in suspension culture, cells were harvested and analyzed for apoptosis by measuring the amount of DNA histone fragments (Cell Death Elisa). D. Stable MDA-MB-231 controlshRNA and TLE1shRNA knockdown cells transfected with mitochondrial Bit1 or vector construct were subjected to anoikis and Cell Death Elisa assays as described in C. E. MDA-MB-231 cells were transfected with the indicated plasmids. 24h post-transfection, cells were grown for another 24h in attached or detached conditions. Cell extracts were prepared, immunoprecipitated with agarose-immobilized anti-myc, and immunoblotted with anti-AES. F. MDA-MB-231 cells transfected with the indicated plasmids were grown in suspension for 24h, and cells were then harvested and subjected to nuclear isolation as indicated in the materials and methods. The resulting nuclear fraction was run on SDS-PAGE and immunoblotted with an anti-AES antibody. The Histone H2B was used as a nuclear marker. In B, C, and D, three independent experiments were performed in triplicates, * indicates p<0.05 by Student’s t test.
To address the mechanism underlying the inhibitory effect of TLE1 on Bit1 anoikis function, we examined whether TLE1 can inhibit the formation of the Bit1-AES complex, which is the pro-apoptotic component that mediates the effect of Bit1 on apoptosis (8). As shown in Fig. 5E, AES co-precipitated with mitochondrial Bit1 in detached conditions while no Bit1-AES complexes were detected in attached cells. Importantly, the ability of mitochondrial Bit1 to interact with AES during anoikis is inhibited by exogenous TLE1 (Fig. 5E). Since Bit1 complexes with AES in the cytoplasm (8), we speculate that TLE1 inhibits Bit1-AES complex formation via sequestration of AES in the nucleus through hetero-oligomerization (1). Indeed, concomitant with the observed reduction of Bit1-AES complexes by exogenous TLE1 is the significant enrichment of AES in the nucleus (Fig. 5F). Collectively, these findings indicate that the induction of anoikis by Bit1 may involve suppression of the antiapoptotic TLE1 and that Bit1 anoikis activity can be inhibited by overexpression of TLE1 in breast cancer cells.
Bit1 induces cytoplasmic translocation and degradation of nuclear TLE1 protein during anoikis
Mitochondrial Bit1 markedly downregulates the level of endogenous TLE1 protein during anoikis (Fig. 4D and 4E). Consistent with this observation, we also found that the level of exogenous TLE1 protein in a stable TLE1 expressing pool of cells was significantly reduced by Bit1 in detached conditions, while the ectopic TLE1 expression was unaffected by Bit1 in attached conditions (Fig. 6A and 6B). These findings indicate that translocation of mitochondrial Bit1 to the cytoplasm during anoikis (8,13, Fig. 6C) is required to decrease TLE1 protein levels. Consistent with the effect of mitochondrial release of Bit1 on TLE1 during anoikis, the cytoplasmic expression of pro-apoptotic N-terminally myc tagged Bit1 (8, 13, Supplementary Fig. 6) and of the highly cytotoxic cell death domain (CDD) of Bit1 (localized to amino acids 1-62 of the molecule, Chen et al., 2012) resulted in a significant reduction in the TLE1 protein levels (Fig. 6D and 6E). We previously created negatively charged phosphomimetic mutation in the Ser5 amino acid within the mitochondrial localization sequence of the C-terminally GFP tagged mitochondrial Bit1 (Bit1S5D-GFP and Bit1S5E-GFP), and such Bit1 phosphomimetic mutants exhibited enhanced cytoplasmic localization as a result reduced mitochondrial import efficiency and displayed a more potent anoikis-sensitization activity (13). Indeed, these Bit1S5D-GFP and Bit1S5E-GFP mutants induced a higher reduction of TLE1 protein as compared to wild type Bit1 (Bit1-GFP) and Bit1S5A-GFP mutant during anoikis (Fig. 6F). The observed reduction of TLE1 during anoikis is specific to Bit1 since the basal anoikis activity in HEK 293T cells which we have previously shown to have a negligible amount of endogenous Bit1 expression (8) was not associated with alteration of TLE1 protein levels (Supplementary Fig. 7A).
Fig. 6.



Bit1 induces cytoplasmic translocation and degradation of nuclear TLE1 protein during anoikis. A. Stable DDK-TLE1 overexpressing MDA-MB-231 cells were transfected with C-terminally myc-tagged Bit1 or vector construct. 24 h post transfection, cells were then plated onto polyhema coated or uncoated tissue culture plates. Following 24 h in culture, cells were harvested and subjected to total cell lysate isolation, SDS-PAGE, and immunoblotting against specific antibodies to DDK, myc-tag, and B-actin. B. The DDK-TLE1/B-actin ratios were determined by densitometry analysis of immunoblots in A from three independent experiments. C. MDA-MB-231 cells transfected with C-terminally myc tagged Bit1 were plated onto polyhema coated or uncoated tissue culture plates. After 12 h, cells were harvested and subjected to subcellular fractionation (see materials and methods). The resulting cytosolic and mitochondrial fractions were run on SDS-PAGE and immunoblotted with an anti-myc antibody. The mitochondrial protein HSP 70 (mtHsp70) and B-actin were used as mitochondrial and cytosolic marker, respectively. D. MDA-MB-231 cells were transfected with N-terminally myc tagged Bit1(cyto) or vector construct and 48h later, cells were harvested and subjected to SDS-PAGE and immunoblotting against TLE1 and B-actin antibodies. E HEK293T cells were co-transfected with Bit1 CDD and FLAG-TLE1 constructs and 24 h later, cells were collected and subjected to immunoblotting against antibodies to FLAG-tag and B-actin. F. MDA-MB-231 cells were transfected with various forms of C-terminally GFP-tagged Bit1 (Bit1-GFP, 15) and plated onto polyhema coated or uncoated tissue culture plates. After 24h, total cell lysates were prepared and subjected to SDS-PAGE and immunoblotting with anti-TLE1, anti-GFP, and B-actin antibodies. G. MDA-MB-231 cells were transfected with C-terminally myc tagged Bit1 or vector construct and 24h later, the cells were plated onto polyhema coated or uncoated tissue culture plates. After 24h, cells were harvested and subjected to nuclear and cytosolic fractionation (see materials and methods). The resulting cytosolic and nuclear fractions were run on SDS-PAGE and immunoblotted with an anti-TLE1 antibody. The Histone H2B and B-actin were used as nuclear and cytosolic marker, respectively. H. HEK293T cells were transfected with FLAG-TLE1 together with N-terminally myc tagged Bit1 (cyto) or Bit1 CDD. The amount of DNA was normalized with the empty vector in each transfection. 24h post-transfection, cells were subjected to immunofluorescent staining and confocal imaging. Representative images are shown to visualize FLAG-TLE1 (green). I. MDA-MB-231 cells transfected with C-terminally myc tagged Bit1(mito) or vector construct were plated onto polyhema coated or uncoated tissue culture plates. Cell in suspension were treated in the presence of MG132 or DMSO (solvent) at the indicated concentration. Following culture for 24 h, cells were harvested and subjected to SDS-PAGE and immunoblotting against specific antibodies to TLE1, myc-tag, and B-actin. J. The TLE1/B-actin ratios were determined by densitometry analysis of immunoblots in I from three independent experiments. In B and J, three independent experiments were performed in triplicates, * indicates p<0.05 by Student’s t test.
Despite the reduction of TLE1 protein levels in Bit1 transfected cells during anoikis, mitochondrial Bit1 expression has no significant effect on the detachment-induced upregulation of TLE1 mRNA (Fig. 4F), suggesting that Bit1 may regulate TLE1 at the level of protein accumulation. We first examined whether Bit1 may regulate the intracellular localization of TLE1. Translocation of mitochondrial Bit1 to the cytoplasm during anoikis (Fig. 6C) coincided with the cytoplasmic re-localization of the nuclear TLE1 protein (Fig. 6G and Supplementary Fig. 7C). Consistent with this finding, exogenous expression of the cytoplasmic localized Bit1 or its CDD domain also led to TLE1 accumulation in the cytoplasm (Fig. 6H). The observed cytoplasmic re-localization of TLE1 is likely specific to Bit1 since the basal anoikis in low Bit1 expressing HEK 293T cells did not show alteration of TLE1 nuclear localization (Supplementary Fig. 7B). The cytoplasmic enrichment of TLE1, however, became evident upon induction of Bit1-mediated anoikis (Supplementary Fig. 7C and 7D). Given that a number of critical anoikis regulators are subject to proteasomal degradation in response to detachment (18,19), we then explored whether TLE1 is subsequently channelled to proteasomal degradation in the presence of Bit1 during anoikis. Cells transfected with mitochondrial Bit1 were plated in suspension culture in the presence or absence of proteasomal inhibitor, MG132. Fig. 6I and 6J show that treatment of Bit1-transfected cells grown in suspension with MG132 exhibited increased levels of TLE1 protein compared to untreated Bit1 transfected cells, indicating that Bit1 may channel TLE1 to proteasomal degradation.
TLE1 is overexpressed in invasive breast tumors
Based on our in vitro evidence indicating that TLE1 promotes breast cancer cell anoikis resistance, which is a primary determinant of transformation and tumor aggressiveness, we explored the possibility that TLE1 may contribute to breast tumorigenesis in humans by screening a breast tumor tissue microarray for overexpression of TLE1. Both the normal breast epithelium tissue (Fig. 7Ai) and the non-invasive ductal carcinoma in situ (DCIS) lesions (Fig. 7Aiii) similarly showed low nuclear TLE1 staining In contrast, the majority of tumor cells in a significant fraction of invasive breast carcinoma lesions consistently displayed moderate to strong TLE1 nuclear immunoreactivity (Fig. 7Av). Quantification of the average TLE1 nuclear staining (Fig. 7B) confirmed the similar TLE1 expression between the normal and DCIS subgroups and the upregulation of TLE1 immunoreactivity in invasive breast carcinoma tissues as compared to normal/DCIS subgroups (ANOVA and subsequent Tukey post-hoc analysis). We have previously shown that Bit1 is expression is downregulated in a significant fraction of advanced invasive breast carcinoma tissues as compared to the normal breast epithelial tissue and DCIS lesions (11). To assess the relationship between TLE1 and Bit1 expression in breast carcinogenesis, immunohistochemical staining of Bit1 was performed in the above studied breast tumor tissue arrays (Fig. 7Aii, iv, vi). Both the normal breast tissue and DCIS lesions, which exhibited low nuclear TLE1 staining, displayed moderate to strong cytoplasmic Bit1 immunoreactivity (Fig. 7Aii and iv), as previously observed (16). Importantly, a significant number of breast carcinoma tissues (43.2%) examined showed strong nuclear TLE1 and low cytoplasmic Bit1 staining (Fig. 7Avi, Table 1). Only a minor fraction (24.2%) of breast tumor tissue samples showed both strong nuclear TLE1 and cytoplasmic Bit1 staining (Table 1). The majority of the low TLE1 expressing breast carcinoma tissues displayed high cytoplasmic Bit1 immunoreactivity (21.2%, Table 1), and a small number of breast carcinoma tissues (10.6%, Table 1) display low levels of both TLE1 and Bit1 immunostains. Taken together, these data indicate that TLE1 is overexpressed in a fraction of human invasive breast carcinomas and that upregulation of TLE1 expression coupled with Bit1 downregulation may accompany the transition from DCIS to invasive carcinoma during breast cancer progression.
Fig. 7.


TLE1 is overexpressed in invasive breast carcinoma tissues. A. Breast tumor tissue array slides were stained with a rabbit polyclonal antibody to TLE1 (i, iii, v) or the affinity purified rabbit anti-Bit1 antibody (ii, iv, vi). Images are representative of each respective case type: normal breast (i, ii 10x), Ductal carcinoma in situ (DCIS) (iii, iv 10x), and invasive breast carcinoma (v, vi 10X). B. The average staining intensity of each subgroup was determined as described in materials and methods. While no significant difference was found between normal and DCIS subgroups, the normal/DCIS was statistically significant from the invasive breast carcinoma tissues using the ANOVA and subsequent Tukey post-hoc analysis (see Materials and Methods) (*, P<0.05).
Table 1.
Immunohistochemical analysis for nuclear TLE1 and cytoplasmic Bit1 expression in the breast tumor tissue population
| Nuclear TLE1 | cytoplasmic Bit1 | Number of tissues(%) |
|---|---|---|
| Moderate to strong | low | 29 (43.2) |
| Moderate to strong | high | 16 (24.2) |
| No or low | low | 7 (10.6) |
| No or low | high | 14 (21.2) |
Discussion
In this report, we have provided evidence that the Groucho family member TLE1 is a regulator of mammary epithelial anoikis cell death program. Our findings also reveal an important link between cell-ECM interaction and the transcriptional regulation of TLE1 expression. In normal breast epithelial cells, loss of integrin engagement results in downregulation of TLE1 expression, and such TLE1 suppression may contribute to the anoikis sensitivity of untransformed epithelial cells. Conversely, in breast carcinoma cells, loss of matrix interaction results in the upregulation of TLE1 expression. The detachment-induced TLE1 expression functions to protect breast cancer cells from anoikis and promotes anchorage-indepedent growth, at least in part by inhibiting the Bit1 anoikis pathway. Importantly, we also show that the nuclear TLE1 is a target of the anoikis effector Bit1 and is channelled to the cytoplasm for proteasomal degradation by Bit1 during anoikis. The induction of TLE1 loss may represent a critical step in the anoikis function of Bit1. Taken together, these findings indicate a novel mechanism by which breast cancer cells evade anoikis via TLE1 expression which is regulated downstream of matrix attachment.
Gro proteins act as repressors for a wide range of gene targets (1,2). Changes in Gro protein levels or repressor activity have a dramatic influence on the developmental fate of a cell. In neurons, TLE1 exerts an anti-differentiation or anti-neurogenic function wherein ectopic expression of TLE in primary cultures of neural progenitor cells decreased neuronal differentiation and induced accumulation of proliferating progenitor cells (4,5). TLE1 is also expressed in the proliferative stage of epithelial cells and is downregulated during differentiation (16). In addition to its role in cellular differentiation, our results indicate that TLE1 plays a critical role in protecting mammary epithelial and breast carcinoma cells from apoptosis induced by loss of matrix adhesion. Such findings are reminiscent of previous studies demonstrating a potent pro-survival or antiapoptotic function of TLE1 in other cellular models. TLE1 has a strong growth promoting function in chicken embryo fibroblasts (6) while exogenous expression of TLE1 is sufficient to prevent apoptosis in neurons (7). Interestingly, neurons that are primed to die exhibit reduced expression of TLE1, which mimics our observation of loss of TLE1 expression in normal breast epithelial cells undergoing anoikis.
We have previously reported the Bit1 anoikis effector whose apoptotic function is uniquely regulated by integrin-mediated cell adhesion and is independent from caspases (8). The apoptotic activity of mitochondrial Bit1 is dependent on its ability to form a complex with AES in the cytoplasm. Here, we show that the release of mitochondrial Bit1 channels the nuclear TLE1 protein to the cytoplasm for proteasomal degradation during anoikis. This finding is supported by our studies with the use of the apoptotic cytoplasmic localized Bit1 and its apoptotic fragment CDD. Importantly, the Bit1-induced TLE1 loss upon cellular detachment from ECM is critical for the mitochondrial Bit1 anoikis function since enforced expression of TLE1 is sufficient to attenuate Bit1-mediated anoikis. Thus, our previous and current data raises a possibility that the specific role of Bit1 is to turn off a survival-promoting gene transcription program regulated by TLE1. In this regard, ECM-mediated cell survival may involve complex transcriptional regulation of target apoptotic and anti-apoptotic gene(s) through the suppressive effect of Bit1 on TLE1.
The downstream target gene(s) of the TLE1-mediated survival promoting transcriptional program remains to be determined. In light of our findings, the TLE1 corepressor may function to suppress the expression of pro-apoptotic factors that are intricately involved in anoikis program. In mammary epithelial cells, BH3-only factors Bim and Bmf are upregulated following loss of cell matrix attachment and required for induction of anoikis (15). These proapoptotic factors are potential candidate downstream target genes that are suppressed by TLE1 during anoikis. Intriguingly, there are data to suggest that transcriptional suppression of Bmf requires histone deacytelase (HDAC) activity (20). Considering that HDAC function requires recruitment of TLE1, it will be interesting to determine whether TLE1 plays a critical role in the basal repression of Bmf. In this scenario, the loss of TLE1 upon loss of matrix adhesion in breast epithelial cells as evidenced in this paper (Fig. 1B, 1C, and 1D) may be sufficient to relieve the transcriptional repression of Bmf, leading to its upregulation during anoikis as previously observed (15). We are currently investigating the downstream target gene(s) regulated by TLE1 through microarray analysis and determining which of these gene(s) specifically regulate the apoptotic program during anoikis.
The ability of TLE1 to impair anoikis in breast epithelial cells may have a profound influence on breast tumorigenesis. Anoikis resistance is a determinant of transformation and tumor aggressiveness (21). Indeed, we found here that while exogenous TLE1 expression confers increased anchorage-independent growth, specific TLE1 downregulation attenuates breast cancer cells’ anchorage-independent growth capacity. Sonderegger et al. 2003 have previously shown that TLE1 is required for Qin-induced transformation in chicken embryo fibroblasts (6), but the mechanism for this TLE1 effect remains to be determined. Our results reveal a critical role of TLE1 in anoikis suppression which may be the basis for its stimulatory effect on anchorage-independent growth and transformation. It is therefore not surprising that TLE1 has been found to be overexpressed in various types of human solid malignancies including cervical (16), lung (10), soft tissue (22), brain (23) and in breast cancinoma in the present study (Fig. 7A-7B). In lymphoma, elevated TLE1 has been identified as a marker for poor prognosis (24). Importantly, Grg1 (the mouse homologue of TLE1) transgenic mice developed lung tumors (10). Taken together, these findings implicate TLE1 in the development of human malignancy and raise the possibility of TLE1 as a putative oncogene.
As a summary, we found that found that TLE1 expression is significantly downregulated in a normal mammary epithelial cell MCF10A line following loss of cell attachment, and the loss of TLE1 expression contributes in part to the anoikis sensitivity of these non-malignant cells. In contrast, TLE1 expression is induced in breast carcinoma cells during anoikis. The upregulation of TLE1 protects malignant breast cells from anoikis at least in part by inhibiting the caspase-independent anoikis function of Bit1. We further provide evidence that Bit1 targets TLE1 to cytoplasm for proteasomal degradation during anoikis, and that the TLE1 degradation in part contributes to Bit1-induced anoikis. Based on our observation that TLE1 expression is an important determinant of anoikis resistance in breast carcinoma cells, Bit1 may be an important therapeutic target to induce anoikis sensitivity in part by suppressing the TLE1 pathway.
Supplementary Material
Acknowledgments
We thank Dr. Eva Engvall for comments on the manuscript. This work was supported by Louisiana Cancer Research Consortium (LCRC) Start up Grant (HB), NIH RCMI G12RR026250-03 Grant (to Xavier University of Losuiana), NIH SC2CA153382 grant (HB), Deparment of Defense Breast Cancer Program Grant W81XWH-08-10727 (ER), and Cancer Center Support Grant CA30199 (to the Sanford-Burnham Medical Research Institute).
Abbreviations
- TLE1
References
- 1.Chen G, Courey AJ. Groucho/TLE family proteins and transcription repression. Gene. 2000;249:1–16. doi: 10.1016/s0378-1119(00)00161-x. [DOI] [PubMed] [Google Scholar]
- 2.Gasperowicz M, Otto F. Mammalian Groucho homologs: redundancy or specificity? J Cell Biochem. 2005;95:670–687. doi: 10.1002/jcb.20476. [DOI] [PubMed] [Google Scholar]
- 3.Arce L, Pate KT, Waterman ML. Groucho binds two conserved regions of LEF-1 for HDAC-dependent repression. BMC Cancer. 2009;9:159. doi: 10.1186/1471-2407-9-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buscarlet M, Hermann R, Lo R, Tang Y, Joachim K, Stifani S. Cofactor-activated phosphorylation is required for inhibition of cortical neuron differentiation by Groucho/TLE1. PLoS One. 4:e8107. doi: 10.1371/journal.pone.0008107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flowers EB, Poole RJ, Tursun B, Bashllari E, Pe’er I, Hobert O. The Groucho ortholog UNC-37 interacts with the short Groucho-like protein LSY-22 to control developmental decisions in C. elegens. Development. 2010;137:1799–17805. doi: 10.1242/dev.046219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sonderegger CK, Vogt PK. Binding of the corepressor TLE1 to Qin enhances Qin-mediated transformation of chicken embryo fibroblasts. Oncogene. 2003;22:1749–1757. doi: 10.1038/sj.onc.1206308. [DOI] [PubMed] [Google Scholar]
- 7.Zhang X, Chen HM, Jaramillo E, Wang L, D’Mello SR. Histone deacetylase-related protein inhibits AES-mediated neuronal cell death by direct interaction. J Neuroscience Res. 2008;86:2423–2431. doi: 10.1002/jnr.21680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jan Y, Matter M, Pai JT, Chen YL, Pilch J, Komatsu M, Ong E, Fukuda M, Ruoslahti E. A mitochondrial protein, Bit1, mediates apoptosis regulated by integrins and Groucho/TLE corepressors. Cell. 2004;116:751–762. doi: 10.1016/s0092-8674(04)00204-1. [DOI] [PubMed] [Google Scholar]
- 9.Seo SW, Lee H, Lee HI, Kim HS. The role of TLE1 in synovial sarcoma. J Orthop Res. 2011;29:1131–1136. doi: 10.1002/jor.21318. [DOI] [PubMed] [Google Scholar]
- 10.Allen T, van Tuyl M, Iyengar P, Jothy S, Post M, Tsao MS, Lobe CG. Grg1 acts as a lung-specific oncogene in a transgenic mouse model. Cancer Res. 2006;66:1294–1301. doi: 10.1158/0008-5472.CAN-05-1634. [DOI] [PubMed] [Google Scholar]
- 11.Karmali PP, Brunquell C, Tram H, Ireland SK, Ruoslahti E, Biliran H. Metastasis of tumor cells is enhanced by downregulation of Bit1. PLoS One. 2011;6:e23840. doi: 10.1371/journal.pone.0023840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kairouz-Wahbe R, Biliran H, Luo X, Khor I, Wankell M, et al. Anoikis effector Bit1 negatively regulates Erk activity. Proc Natl Acad Sci USA. 2008;105:1528–1532. doi: 10.1073/pnas.0711357105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biliran H, Jan Y, Chen R, Pasquale EB, Ruoslahti E. Protein kinase D is a positive regulator of Bit1 apoptotic function. J Biol Chem. 2008;283:28029–28037. doi: 10.1074/jbc.M803139200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parsons JM, Patel P, Brat DJ, Colbert L, Vertino PM. Silencing of TMS1/ASC promotes resistance to anoikis in breast epithelial cells. Cancer Res. 2009;69:1706–1711. doi: 10.1158/0008-5472.CAN-08-2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmelzle T, Mailleux AA, Overholtzer M, Carroll JS, Solimini NL, Lightcap ES, Veiby OP, Brugge JS. Functional role and oncogene-regulated expression of the BH3-only factor Bmf in mammary epithelial anoikis and morphogenesis. PNAS. 2007;104:3787–3792. doi: 10.1073/pnas.0700115104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Dehni G, Purcell KJ, Sokolow J, Carcangiu ML, Artavanis-Tsakonas S, Stifani S. Epithelial expression and choromosomal location of human TLE genes: implications for notch signaling and neoplasia. Genomics. 1996;31:58–64. doi: 10.1006/geno.1996.0009. [DOI] [PubMed] [Google Scholar]
- 17.Bharadwaj S, Thanawala R, Bon G, Falcioni R, Prasad GL. Resensitization of breast cancer cells to anoikis by Tropomyosin-1: role of Rho kinase-dependent cytoskeleton and adhesion. Oncogene. 2005;24:8291–8303. doi: 10.1038/sj.onc.1208993. [DOI] [PubMed] [Google Scholar]
- 18.Yang JM, O’Neill P, Jin W, Foty R, Medina DJ, Xu Z, Lomas M, Arndt GM, Tang Y, Nakada M, Yan L, Hait WN. Extracellular matrix metalloproteinase inducer (CD147) confers resistance of breast cancer cells to anoikis through inhibition of Bim. J Biol Chem. 2006;281:9719–9727. doi: 10.1074/jbc.M508421200. [DOI] [PubMed] [Google Scholar]
- 19.Woods NT, Yamaguchi H, Lee FY, Bhalla KN, Wang HG. Anoikis, initiated by Mcl-1 degradation and Bim induction, is deregulated during oncogenesis. Cancer Res. 2007;67:10744–10752. doi: 10.1158/0008-5472.CAN-07-3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Adachi M, Kawamura R, Imai K. Bmf is a possible mediator in histone deacetylase inhibitors FK228 and CBHA-induced apoptosis. Cell Death Differ. 2006;13:129–140. doi: 10.1038/sj.cdd.4401686. [DOI] [PubMed] [Google Scholar]
- 21.Frish SM, Frances H. Disruption of epithelial cell-matrix interactions induce apoptosis. J Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Terry J, Saito T, Subramanian S, Ruttan C, Antonescu CR, Goldblum JR, Downs-Kelly E, Corless CL, Rubin BP, van de Rijn M, Ladanyi M, Nielson TO. TLE1 as a diagnostic immunohistochemical marker for synovial sarcoma emerging from gene expression profiling studies. Am J Surg Pathol. 2007;31:240–246. doi: 10.1097/01.pas.0000213330.71745.39. [DOI] [PubMed] [Google Scholar]
- 23.Cuevas IC, Slocum AL, Jun P, Costello JF, Bollen AW, Riggins GJ, McDermott MW, Lal A. Meningioma transcript profiles reveal deregulated Notch signaling pathway. Cancer Res. 2005;65:5070–5075. doi: 10.1158/0008-5472.CAN-05-0240. [DOI] [PubMed] [Google Scholar]
- 24.Shipp MA, Ross KN, Tamayo P, Weng AP, Kutok JL, Aguiar RC, Gaasenbeek M, Angelo M, Reich M, Pinkus GS, Ray TS, Koval MA, Last KW, Norton A, Lister TA, Mesirov J, Neuberg DS, Lander ES, Aster JC, Golub TR. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat Med. 2002;8:68–74. doi: 10.1038/nm0102-68. [DOI] [PubMed] [Google Scholar]
- 25.Ke N, Albers A, Claassen G, Yu DH, Chatterton JE, Hu X, Meyhack B, Wong-Stall F, Li QX. One-week 96-well soft agar growth assay for cancer target validation. Biotechniques. 2004;36:826–8. 830, 832–3. doi: 10.2144/04365ST07. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.