

Abstract
The GABAA receptors are ligand-gated chloride channels which are the targets for many clinically used sedatives, including the barbiturates. The barbiturate pentobarbital acts through multiple sites on the GABAA receptor. At low concentrations (μM), it acts as a positive allosteric modulator while at higher concentrations it can directly activate the receptor. This agonist action is influenced by the subunit composition of the receptor, and pentobarbital is a more effective agonist than GABA only at receptors containing an α6 subunit. The conformational change that translates GABA binding into channel opening is known to involve a lysine residue located in an extracellular domain between the 2nd and 3rd transmembrane domains. Mutations of this residue disrupt activation of the channel by GABA and have been linked to inherited epilepsy. Pentobarbital binds to the receptor at a different agonist site than GABA, but could use a common signal transduction mechanism to gate the channel. To address this question, we compared the effect of a mutating the homologous lysine residue in the α1 or α6 subunits (K278 or K277, respectively) to methionine on direct activation of recombinant GABAA receptors by GABA or pentobarbital. We found that this mutation reduced GABA sensitivity for both α1 and α6 subunits, but affected pentobarbital sensitivity only for the α1 subunit. This suggests that pentobarbital acts through a distinct signal transduction pathway at the α6 subunit, which may account for its greater efficacy compared to GABA at receptors containing this subunit.
Introduction
Ligand-gated channels such as the GABAA receptors (GABAARs) are complex, multimeric proteins. For the GABAARs, the pentameric complex can include subunits from seven different families and sixteen different subtypes (α1–6, β1–3, γ1–3, δ, ε, π and θ) [3, 14]. Neurotransmitter binding occurs in the large extracellular domain while the ion channel gate is controlled by transmembrane domains. The structural mechanisms that link the binding event with the channel gating process have been the subject of much investigation. A recent study suggests that electrostatic interactions between negatively charged residues within the extracellular N-terminal domain and a positively charged lysine in the TM2–TM3 extracellular region are important for GABA-mediated channel gating [9]. Mutations of this lysine in the α or β subunit reduce GABA sensitivity [8, 15] while an inherited mutation of this lysine to methionine in the γ2 subunit has been linked to epilepsy in humans [2].
In addition to GABA, a variety of compounds are known to act as agonists through different binding sites on the GABAAR. Agonist activity is particularly associated with the i.v. anesthetics, which include the barbiturates. At low concentrations (<10 μM, within the sedative therapeutic range of 0.5–3 μg/ml), barbiturates act at a positive allosteric site to increase the response to GABA but at higher concentrations (>100 μM) they can act as agonists, directly activating the receptor [6, 12, 13]. At most GABAARs, pentobarbital is a partial agonist. However, when the receptor contains an α6 subunit, pentobarbital produces larger maximum currents than GABA. [5, 16]. Pentobarbital acts as an agonist through a site distinct from the GABA binding site [16], and mutations within the extracellular N-terminal domain that reduce GABA binding do not typically influence activation by pentobarbital and vice versa [1, 5].
Although pentobarbital and GABA have separate binding sites, they may share a common structural mechanism to translate the binding signal into channel opening. To address this question, we compared the effect of mutating the M2–M3 lysine residue in the α1 and α6 subunits (Figure 1) on the ability of GABA or pentobarbital to activate the receptor. Wild-type or mutated α subunits were transiently transfected into HEK-293T cells along with β1 and γ2L subunits. The functional responses of the receptors to GABA or pentobarbital were measured using whole-cell patch clamp recordings.
Figure 1. Location of mutation site.
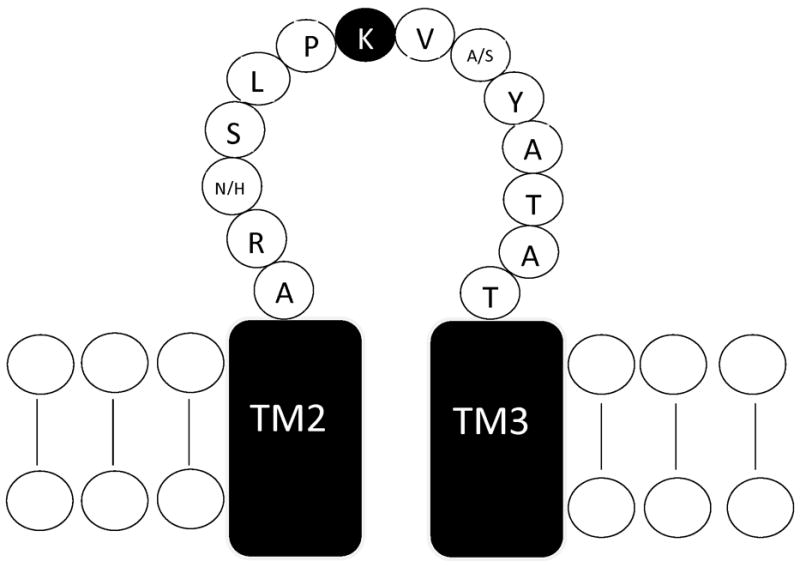
Schematic representation of the TM2–TM3 extracellular domain of the GABAAR subunit. The conserved lysine residue mutated in this study is indicated by the filled circle. The α1 and α6 subunits differ at only two residues within in this domain, shown as α1/α6. Sequence from [17].
Materials and Methods
Transfection of mammalian cells
Full-length cDNAs encoding rat GABAAR subunits in mammalian expression vectors were transiently transfected into the human embryonic kidney cell line HEK-293T. For selection of transfected cells, the plasmid pHook™-1 (Invitrogen) encoding the surface antibody sFv was also transfected into the cells. Cells were maintained in DMEM supplemented with 10% fetal bovine serum, 100 IU/ml penicillin and 100 μg/ml streptomycin. The cells were transfected using calcium phosphate precipitation with 2 μg of each of the GABAAR subunit cDNAs along with 1μg of the pHook plasmid. After a 2–5 hour incubation at 3% CO2, the cells were treated with a 15% glycerol solution for 30 seconds. Cells were selected for pHook expression 44–52 hours after transfection [4]. Cells were resuspended into supplemented DMEM following a 2 min. incubation with 0.025% trypsin/0.01% EDTA solution in phosphate-buffered saline (10 mM Na2HPO4, 150 mM NaCl, pH = 7.3), incubated with antigen-coated magnetic beads (~6 × 105 beads), for 30–60 min. and then isolated with a magnetic stand. The isolated cells were plated onto glass coverslips coated with poly-lysine and collagen, and used for recording 20–28 hours later.
Electrophysiological recording of transfected cells
External solution for all recordings consisted of (in mM): 142 NaCl, 8.1 KCl, 6 MgCl2, 1 CaCl2 and 10 HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), (295–305 mOsm, pH=7.4). Recording electrodes were pulled from borosilicate glass on a two-stage puller (Narishige, Japan) to a resistance of 5–10 MΩ and filled with an internal solution consisting of (in mM); 153 KCl, 1 MgCl2, 5 K-EGTA (ethylene glycol-bis (β-aminoethyl ether N,N,N′N′-tetraacetate) and 10 HEPES (295–305 mOsm, pH = 7.4). Drugs were applied to cells using a stepper solution exchanger (SF-77B, Warner Instruments) with a complete exchange time of <50 msec and currents were recorded with an Axon 200B patch clamp amplifier. GABA and pentobarbital were diluted into external solution from stock solutions in water.
Construction of mutations
Single point amino acid mutations were generated using the QuikChange procedure (Agilent Tech.). Oligonucleotide primers were synthesized and sequencing was performed by the University of South Carolina DNA core facility (Columbia, SC).
Data Analysis
Whole-cell currents were analyzed using Clampfit (pClamp 8.2 suite, Axon Instruments, Foster City CA) and Prism v.3.03 (Graphpad, San Diego, CA). Concentration response data were fit with a four-parameter logistic equation (Current = [Maximum current + (Maximum current − Minimum current]/1 + (10^(log EC50−log[GABA]n) where n represents the Hill number. Fits were made to data normalized to the maximum response for each cell. Because of the onset of inhibition, the response to 1 mM pentobarbital was excluded from the fit for the α6β3γ2L isoform. To determine statistical significance, unpaired t-tests were performed using the Instat program (Graphpad) with a significance level of p<0.05. The logs of the GABA EC50 measurements were used for statistical comparison.
RESULTS
Mutation of a TM2–TM3 lysine residue to methionine in either the α1 or α6 subunit reduces GABA sensitivity
All GABAAR subunits contain a conserved lysine residue within the TM2–TM3 extracellular domain (Figure 1). Electrostatic interactions between its positively charged sidechain and negatively charged residues within the extracellular N-terminal domain have been suggested to mediate part of the signal transduction pathway from agonist binding to channel opening [9]. This lysine residue was changed to methionine to produce the α1(K278M) and α6(K277M) subunits. Each α subunit was co-expressed with wild-type β1 and γ2L subunits to form heteromeric receptors. The β1 subunit was used because it reduces the agonist activity of pentobarbital compared to the β3 subunit and therefore produces a greater differentiation between α1- and α6-containing receptors [5, 16].
The mutations in either the α1 or the α6 subunit produced comparable effects on GABA sensitivity (Figure 2). Receptors containing α1(K278M) had an average GABA EC50 of 70.0 ± 8.4 μM (N=4), significantly different (p≤0.05) from the wild-type α1β1γ2L receptors (17.6 ± 3.7 μM, N=5). Receptors containing the α6 subunit had an increased sensitivity to GABA compared to α1-containing receptors, but the mutation produced a similar shift in GABA sensitivity, from 2.3 ± 0.3 μM (N=5) for the wild-type receptors to 10.7 ± 1.5 μM (N=5) for α6(K277M)β1γ2L (p≤0.001 compared to wild-type). Except for the change in GABA sensitivity, the mutations had no obvious effect on other properties of the whole-cell current. Consistent with the change in EC50, currents from receptors with mutated subunits typically showed modestly faster deactivation and slower onset of desensitization. Quantification of these effects would require rapid application recordings from excised patches.
Figure 2. GABA sensitivity is reduced by the mutation of lysine to methionine.

A. Representative current traces from receptors containing the subunits indicated in response to 5 sec applications of GABA(solid line). Whole-cell recordings were obtained at a membrane potential of −50 mV.
B. Concentration-response relationships were constructed by normalizing the peak response to each concentration of GABA to the maximum current response for each cell. Points shown are the mean ± SEM. Averaged data were fit with a four-parameter logistic equation represented by the solid (wild type) or dashed (mutated) line. EC50s for the fits shown are 16.2 μM (α1β1γ2L, N=5), 69.1 μM (α1(K278M)β1γ2L, N=4), 2.2 μM (α6β1γ2L, N=5) and 9.3 μM (α6(K277M)β1γ2L, N=5).
These data suggest that this lysine residue performs a similar role in signal transduction in response to GABA for both the α1 and α6 subunits. Since pentobarbital is a partial agonist at α1-containing receptors, but is more efficacious than GABA at α6-containing receptors, we compared the effect of the mutation in each subunit on the agonist activity of pentobarbital.
Mutation of lysine to methionine in the α1, but not the α6 subunit, reduced pentobarbital sensitivity
The mutation in the α1 subunit reduced its sensitivity to direct activation by pentobarbital (Figure 3). Pentobarbital is a relatively poor agonist at α1β1γ2L receptors, producing a response to 1 mM pentobarbital averaging 66.3 ± 4.2% of the response to 1 mM GABA (N=7). The mutation significantly reduced the relative response to pentobarbital from the lowest effective concentration (100 μM) to 1 mM. Inclusion of responses from higher concentrations of pentobarbital is limited by the onset of inhibition, making it difficult to accurately fit the concentration-response relationship for individual cells. Therefore, we were unable to statistically compare EC50 values. However, the EC50 to the fit of the averaged data (Figure 3B) was increased by the mutation from 630.9 μM to 1.69 mM. These results are consistent with those reported by Sigel et al., [15], who also found reduced responsiveness to pentobarbital when the homologous lysine in the α1 or β2 subunit was mutated to alanine.
Figure 3. Direct activation by pentobarbital is reduced by the mutation of the α1 subunit but not of the α6 subunit.

A. Representative whole-cell traces in response to a 5 sec applications (solid line) of 300 μM pentobarbital or 1 mM GABA at a membrane potential of −50 mV. Traces were obtained from the same cell for each subunit combination.
B. Concentration-response relationships were constructed by dividing the peak response to each concentration of pentobarbital by the response to 1 mM GABA for each cell. The maximum response to GABA (100%) is indicated by the dotted line. Points shown are the mean ± SEM. Averaged data were fit with a four-parameter logistic equation represented by the solid (wild type) or dashed (mutated) line. Because of the onset of inhibition, the response to 1 mM pentobarbital was not included in the fit for the wild-type α6β3γ2L isoform. EC50s for the fits shown are 630.9 μM (α1β1γ2L, N=7), 1.69 mM (α1(K278M)β1γ2L, N=6), 143.9 μM (α6β1γ2L, N=6) and 140.2 μM (α6(K277M)β1γ2L, N=6). *(p≤0.05) or ***(p≤0.001) indicate a significant difference between the wild-type and mutated counterparts (unpaired, 2-tailed t-test).
C. Whole-cell traces in response to a 5 sec applications (solid line) of 3 mM pentobarbital at a membrane potential of −50 mV. Rebound current following the removal of applied drug is characteristic of rapid open channel block.
D. The peak current response during pentobarbital application was measured. Symbols and bars represent the average ± SEM. Data from 5–10 cells for each concentration **(p≤0.01) or ***(p≤0.001) indicate a significant difference from the mutated counterpart (unpaired, 2-tailed t-test).
In contrast, the K277M mutation in the α6 subunit had no apparent effect on the activation of the receptor by pentobarbital (Figure 3A, 3B). Pentobarbital is more efficacious than GABA at these receptors, and the mutation did not alter the maximum response. 300 μM pentobarbital produced a current 204.0 ± 7.4% (wild-type, N=6) or 243.7 ± 32.5% (α6(K277M), N=6) of the response to 1 mM GABA (p>0.05). The mutation also had no effect on the sensitivity of the receptor to direct activation by pentobarbital, with EC50’s averaging 125.5 ± 20.6 μM (wild-type, N=6) and 139.9 ± 28.9 μM (α6(K277M), N=6) (p>0.5).
Interestingly, the mutation did appear to reduce sensitivity to the inhibitory effects of pentobarbital, normally prominent at mM levels in these receptors (Figure 3B). To further characterize this observation, we tested the response to concentrations up to 10 mM. The mutations did not eliminate the inhibitory effect, as the reduced amplitude and rebound current were apparent for receptors containing all subunits in response to pentobarbital levels of 1 mM and higher (Figure 3C). However, for both α subunits, the mutation caused a slight shift in the concentration dependence of inhibition. For the α6(K277M) subunit, the amplitude of the current was significantly higher than the wild-type receptor at concentrations that produce inhibition (1–10 mM) although the maximum response still decreased with higher pentobarbital concentrations. For the α1(K278M) subunit, which shows reduced sensitivity to the agonist action of pentobarbital, the amplitude of the response was similar at 1 and 3 mM, suggesting that it also exhibits a lower sensitivity to inhibition. Therefore, unlike the effect on activation, the reduction in inhibition is observed in both α1 and α6 subunits.
These data suggest that this highly conserved lysine residue in the extracellular M2–M3 domain is important for agonist activity of both GABA and pentobarbital for the α1 subunit, but that it does not play a significant role in activation of α6-containing receptors by pentobarbital. The ability of pentobarbital to utilize a distinct pathway for channel activation may be responsible for its greater efficacy compared to GABA at these receptors.
Discussion
The goal of this work was to determine whether pentobarbital and GABA rely upon the same structural mechanisms to induce the conformational changes that lead to channel gating. We compared the effect of mutating a conserved lysine residue in the extracellular TM2–TM3 domain of the α subunit on the response to GABA and pentobarbital. This residue is known to interact with negatively charged residues in the extracellular N-terminal domain of the subunit [9]. We found that mutating this residue to methionine in the α1 subunit had similar effects on the sensitivity to activation by either GABA or pentobarbital. However, when created in the α6 subunit, the mutation reduced only the response to GABA, and not that to pentobarbital. At most GABAARs, including the α1-containing receptors, pentobarbital is a weak partial agonist. However, at GABAARs containing the α6 subunit, pentobarbital is a better agonist, producing a maximum current nearly twice that seen in response to GABA. Our results suggest that this greater efficacy may be achieved through a unique signal transduction pathway, accessible by pentobarbital in the α6, but not the α1 subunit. Our finding that mutation of K278 in the α1 subunit reduced sensitivity to the agonist activity of both GABA and pentobarbital are in line with the findings of Hales et al. [8], who found that agonist activity of the anaesthetic propofol was also reduced by this mutation. Together these results suggest that all three of these agonists rely upon a common signal transduction pathway for the α1 subunit.
Our findings also suggest that the inhibitory effect of pentobarbital, apparent as a rapid channel block at mM levels, is also affected by this lysine residue. Unlike the agonist action, however, the mutation in either the α1 or α6 subunit had equivalent effects on the onset of inhibition. In this study we did not examine allosteric modulation by pentobarbital, but it may be of interest to determine if mutations at sites known to alter agonist properties also influence the activity of modulators. An earlier study found that modulation by phenobarbital was enhanced in receptors containing a γ2 subunit mutated at the equivalent lysine residue [10]. However, this may have been related to the reduced sensitivity to inhibition that we observed in our study.
A substantial body of evidence suggests that the agonist binding sites for barbiturates and GABA are distinct [1, 5, 16]. Results from structural studies of α1 and β2 subunits indicate that the conformational changes induced by these different agonists differ as well [11, 12]. The structural differences may reflect stabilization of distinct open or desensitized states of the channel. Interestingly, single channel studies have shown that for the α1β3δ isoform, pentobarbital-gated openings have a longer mean duration than GABA-gated openings [6]. In particular, pentobarbital was able to induce openings to an additional, longer duration, open state that was not observed in response to GABA. Similar studies have not yet been performed with receptors containing the α6 subunit.
While our results indicate that the M2–M3 lysine residue is not critical for pentobarbital activation of α6-containing receptors, it is not known what other structural interactions might be utilized by barbiturates to gate this channel. Amino acid residues within the N-terminal extracellular domain, the pre-MI domain, the M2–M3 extracellular domain and the transmembrane domains have all been implicated in the signal transduction process for various members of the cys-loop family of ligand-gated channels (see [7] for review). Comparing the effects of mutations in these regions on activation of α6-containing receptors by various agonists may provide insight into the multiple mechanisms by which signal transduction can occur at the GABAARs.
Acknowledgments
This work was supported by funds from NIH-NINDS (RO1-NS045950) and by the University of South Carolina Mini-grant program. Thanks to Laura Heidelberg and Shana Dykema for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Matthew T. Fisher, College of Arts and Sciences, University of South Carolina-Columbia
Janet L. Fisher, Department of Pharmacology, Physiology, and Neuroscience, University of South Carolina, School of Medicine
References
- 1.Amin J, Weiss DS. GABAA receptor needs two homologous domains of the β-subunit for activation by GABA but not by pentobarbital. Nature. 1993;366:565–569. doi: 10.1038/366565a0. [DOI] [PubMed] [Google Scholar]
- 2.Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud’homme J, Baulac M, Brice A, Bruzzone R, LeGuern E. First genetic evidence of GABAA receptor dysfunction in epilepsy: a mutation in the γ2-subunit gene. Nature Genetics. 2001;28:46–48. doi: 10.1038/ng0501-46. [DOI] [PubMed] [Google Scholar]
- 3.Baumann SW, Baur R, Sigel E. Forced subunit assembly in α1β2γ2 GABAA receptors. J Biol Chem. 2002;277:46020–46025. doi: 10.1074/jbc.M207663200. [DOI] [PubMed] [Google Scholar]
- 4.Chesnut JD, Baytan AR, Russell M, Chang MP, Bernard A, Maxwell IH, Hoeffler JP. Selective isolation of transiently transfected cells from a mammalian cell population with vectors expressing a membrane anchored single-chain antibody. J Immunol Methods. 1996;193:17–27. doi: 10.1016/0022-1759(96)00032-4. [DOI] [PubMed] [Google Scholar]
- 5.Drafts BC, Fisher JL. Identification of structures within GABAA receptor α subunits that regulate the agonist action of pentobarbital. J Pharm Exp Ther. 2006;318:1094–1096. doi: 10.1124/jpet.106.104844. [DOI] [PubMed] [Google Scholar]
- 6.Feng H, Bianchi MT, Macdonald RL. Pentobarbital differentially modulates α1β3δ and α1β3γ2L GABAA receptor currents. Mol Pharm. 2004;66:988–1003. doi: 10.1124/mol.104.002543. [DOI] [PubMed] [Google Scholar]
- 7.Gay EA, Yakel JL. Gating of nicotinic ACh receptors; new insights into structural transitions triggered by agonist binding that induce channel opening. J Physiol. 2007;584:727–733. doi: 10.1113/jphysiol.2007.142554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hales TG, Deeb TZ, Tang H, Bollan KA, King DP, Johnson SJ, Connolly CN. An asymmetric contribution to γ-aminobutyric type A receptor function of a conserved lysine within TM2–3 of α1, β2, and γ2 subunits. J Biol Chem. 2006;281:17034–17043. doi: 10.1074/jbc.M603599200. [DOI] [PubMed] [Google Scholar]
- 9.Kash TL, Jenkins A, Kelley JC, Trudell JR, Harrison NL. Coupling of agonist binding to channel gating in the GABAA receptor. Nature. 2003;421:272–275. doi: 10.1038/nature01280. [DOI] [PubMed] [Google Scholar]
- 10.Krivoshein AV, Hess GP. On the mechanism of alleviation by phenobarbital of the malfunction of an epilepsy-linked GABAA receptor. Biochem. 2006;45:11632–11641. doi: 10.1021/bi061207t. [DOI] [PubMed] [Google Scholar]
- 11.Mercado J, Czajkowski C. γ-aminobutyric acid (GABA) and pentobarbital induce different conformational rearrangements in the GABAA receptor α1 and β2 pre-M1 regions. J Biol Chem. 2008;283:15250–15257. doi: 10.1074/jbc.M708638200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muroi Y, Theusch CM, Czajkowski C, Jackson MB. Distinct structural changes in the GABAA receptor elicited by pentobarbital and GABA. Biophys J. 2009;96:499–509. doi: 10.1016/j.bpj.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicoll R, Wojtowicz J. The effects of pentobarbital and related compounds on frog motoneurons. Brain Research. 1980;191:225–237. doi: 10.1016/0006-8993(80)90325-x. [DOI] [PubMed] [Google Scholar]
- 14.Sieghart W, Fuchs K, Tretter V, Ebert V, Jechlinger M, Hoger H, Adamiker D. Structure and subunit composition of GABAA receptors. Neurochem Int. 1999;34:379–385. doi: 10.1016/s0197-0186(99)00045-5. [DOI] [PubMed] [Google Scholar]
- 15.Sigel E, Buhr A, Baur R. Role of the conserved lysine residue in the middle of the predicted extracellular loop between M2 and M3 in the GABAA receptor. J Neurochem. 1999;73:1758–1763. doi: 10.1046/j.1471-4159.1999.731758.x. [DOI] [PubMed] [Google Scholar]
- 16.Thompson SA, Whiting PJ, Wafford KA. Barbiturate interactions at the human GABAA receptor: dependence on receptor subunit combination. Br J Pharmacol. 1996;117:521–527. doi: 10.1111/j.1476-5381.1996.tb15221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tyndale RF, Olsen RW, Tobin AJ. GABAA receptors. In: North RA, editor. Ligand- and Voltage-Gated Ion Channels. CRC Press; Boca Raton: 1995. pp. 265–290. [Google Scholar]