

Abstract
Hyperoxic lung injury is a major concern in critically ill patients who receive high concentrations of oxygen to treat lung diseases. Successful abrogation of hyperoxic lung injury would have a huge impact on respiratory and critical care medicine. Hydrogen can be administered as a therapeutic medical gas. We recently demonstrated that inhaled hydrogen reduced transplant-induced lung injury and induced heme oxygenase (HO)-1. To determine whether hydrogen could reduce hyperoxic lung injury and investigate the underlying mechanisms, we randomly assigned rats to four experimental groups and administered the following gas mixtures for 60 h: 98% oxygen (hyperoxia), 2% nitrogen; 98% oxygen (hyperoxia), 2% hydrogen; 98% balanced air (normoxia), 2% nitrogen; and 98% balanced air (normoxia), 2% hydrogen. We examined lung function by blood gas analysis, extent of lung injury, and expression of HO-1. We also investigated the role of NF-E2-related factor (Nrf) 2, which regulates HO-1 expression, by examining the expression of Nrf2-dependent genes and the ability of hydrogen to reduce hyperoxic lung injury in Nrf2-deficient mice. Hydrogen treatment during exposure to hyperoxia significantly improved blood oxygenation, reduced inflammatory events, and induced HO-1 expression. Hydrogen did not mitigate hyperoxic lung injury or induce HO-1 in Nrf2-deficient mice. These findings indicate that hydrogen gas can ameliorate hyperoxic lung injury through induction of Nrf2-dependent genes, such as HO-1. The findings suggest a potentially novel and applicable solution to hyperoxic lung injury and provide new insight into the molecular mechanisms and actions of hydrogen.
Keywords: NF-E2-related factor 2, heme oxygenase, inflammation, hydrogen
administration of high concentrations of oxygen is required to maintain sufficient blood oxygenation in some critically ill patients, such as patients with acute lung injury or acute respiratory distress syndrome (ARDS) (36). However, prolonged exposure to high concentrations of oxygen results in hyperoxic lung injury, which can lead to respiratory failure (3, 12). Excessive reactive oxygen species (ROS) generated during hyperoxic conditions are a main cause of hyperoxic lung injury (3). ROS have direct detrimental effects on cell structures, such as membranes, mitochondria, and nuclei, and modulatory effects on several cell signaling pathways (28). Successful abrogation of hyperoxic lung injury would have a huge impact on respiratory and critical care medicine. Developing therapeutic strategies based on the molecular pathogenesis of hyperoxic lung injury is critical.
Hydrogen can be used as a therapeutic medical gas and has been studied in clinical and experimental models of many different diseases in a variety of biomedical fields. Hydrogen exhibits potent antioxidant and anti-inflammatory properties and is a known scavenger of ROS (16). Hydrogen may also act as a gaseous signaling molecule, similar to nitric oxide (19). Our laboratory has explored the application of inhaled hydrogen for acute lung injuries and found that treatment with inhaled hydrogen mitigates mechanical ventilation-induced lung injury (15) and transplant-induced lung ischemia-reperfusion injury (23, 24). We discovered that inhaled hydrogen gas induces heme oxygenase (HO)-1 in the donor lungs before transplantation and leads to excellent preconditioning of lung allografts and improved transplant outcomes (23).
HO-1, a heme-degrading enzyme, is highly induced by oxidative stress and plays a critical role in defending the lung against inflammatory and oxidant-induced cellular and tissue injury (13, 32). HO-1 is induced by a number of signaling pathways and transcription factors (1, 41). In this study, we focused on the role of the nuclear factor E2-related factor (Nrf) 2-antioxidant response element (ARE) pathway. The Nrf2-ARE pathway transcriptionally regulates HO-1, as well as numerous other antioxidant and cytoprotective proteins. Thus, it is considered an essential pathway for protection against oxidative stress and resultant forms of lung injury (10, 25). We hypothesized that hydrogen gas could ameliorate hyperoxic lung injury by inducing the Nrf2-ARE pathway. We tested the role of hydrogen in migrating hyperoxic lung injury and its underlying mechanisms using rat and Nrf2-deficient (Nrf2−/−) mouse models.
MATERIALS AND METHODS
Animals.
Male Lewis (RT1l) rats (8–10 wk old) were purchased from Harlan Laboratories (Indianapolis, IN), and male C57BL/6J mice (7–9 wk old) were purchased from The Jackson Laboratory (Bar Harbor, ME). The Nrf2−/− mice (7–9 wk old) were maintained in a C57BL/6J background (18). All procedures were performed with approval from our Institutional Animal Care and Use Committee and in accordance with the National Research Council's Guide for the Humane Care and Use of Laboratory Animals.
Gas exposure.
Animals were exposed continuously to either normoxia (air) or hyperoxia (98% oxygen) with either 2% nitrogen or 2% hydrogen at a flow rate of 2 l/min in an exposure chamber (model no. 902-EC, 18 in. wide × 18 in. deep × 18 in. high; Plas Labs, Lansing, MI) for 60 h. When determining the effects of hydrogen on survival under prolonged hyperoxic conditions, rats were monitored every 8 h and exposed until death (>60 h). Gas samples were taken for analysis through a port in the top of the exposure chamber and analyzed by electrochemical detection. The oxygen concentrations in all experiments were confirmed to be >95% with a gas spectrometer (VTI Oxygen Monitor; Vascular Technology, Nashua, NH) once the chamber had equilibrated. Food and water were provided ad libitum. The chambers were opened one time a day for 5 min to replace food and water. Fewer than 10 mice and fewer than 4 rats were housed together in the chamber. Rats were weighed immediately before and immediately after exposure.
Assessment of lung function and injury and tissue collection.
After gas exposure, the animals were anesthetized, and lung function was assessed by analysis of blood gases on a fraction of inspired oxygen of 1.0 in blood drawn from the abdominal aorta. The left lung was used for bronchoalveolar lavage, as described previously (15). The pleural effusion volume was measured immediately after opening the chest cavity. The lungs were flushed through the main pulmonary artery with phosphate-buffered saline before tissue collection except in samples collected to assess the wet-to-dry (W/D) weight ratio. The W/D ratio was measured as described previously (23).
Histopathological analysis.
For histological evaluation, fixed lung sections were stained with hematoxylin and eosin. Acute lung injury was blindly scored according to previously described criteria (23). For immunofluorescent analysis, anti-HO-1 (Abcam, Cambridge, MA) and anti-aquaporin-5 (EMD Chemicals, Gibbstown, NJ) were used. For immunohistological staining, anti-HO-1 polyclonal antibody (Enzo Life Sciences, Farmingdale, NY), anti-cleaved caspase 3 polyclonal antibody (Cell Signaling Technology, Danvers, MA), and anti-8-hydroxy-2′-deoxyguanosine (8-OHdG) monoclonal antibody (NIKKEN SEIL, Shizuoka, Japan) were used, as described previously (29, 34, 45).
Protein analysis.
Western blot analysis was performed on 30 μg of whole cell protein from lung tissue as described previously (23). The following primary antibodies were used: anti-HO-1 (Enzo Life Sciences), anti-B cell lymphoma (Bcl)-2 (Cell Signaling Technology), anti-Bcl-2-associated X protein (Bax) (Cell Signaling Technology), and anti-β-actin (Sigma-Aldrich, St. Louis, MO). Tissue HO-1 activity was measured spectrophotometrically in microsomal fractions (29).
SYBR green real-time RT-PCR.
Rat and mouse mRNAs were quantified in duplicate using SYBR Green two-step, real-time RT-PCR, as previously described (20, 26, 34). The following mRNAs were quantitated: rat interleukin (IL)-1β, IL-6, tumor necrosis factor (TNF)-α, intercellular adhesion molecule (ICAM)-1, Bcl-2, Bax, HO-1, NAD(P)H dehydrogenase quinone (Nqo) 1, glutathione S-transferase (GST) A2, UDP-glucuronosyl transferase (UGT) 1A6 and peroxiredoxin (Prdx) 1, Kelch-like ECH-associated protein 1 (Keap 1), and Nrf2 and glyceraldehyde-3-phosphate dehydrogenase; and mouse HO-1, Nqo-1, GSTA2, and hypoxanthine phosphoribosyltransferase. Optimized and validated primer sets were obtained from realtimeprimers.com (Elkins Park, PA).
Measurement of malondialdehyde.
Lung tissues were harvested after 60 h of gas exposure, snap-frozen, and kept at −80°C until analysis. The tissue was homogenized, and tissue malondialdehyde (MDA) concentration was determined according to the manufacturer's instructions (Kit MDA-586; Oxidresearch, Portland, OR).
Statistical analysis.
Animal survival was plotted using the Kaplan-Meier method, and the differences between groups were analyzed using the log-rank test. Other data were expressed as means ± SE. Parametric data were analyzed with one-way analysis of variance followed by post hoc analysis with the Bonferroni correction. The lung injury score was analyzed with the Mann-Whitney U-test with the Bonferroni correction. The cell count was analyzed with Tukey-Kramer methods. JMP version 9 (SAS Institute, Cary, NC) was used for all statistical analyses.
RESULTS
Hydrogen gas ameliorated lung dysfunction after hyperoxic exposure and prolonged survival against lethal hyperoxia in rats.
Sixty hours of exposure to a high concentration (>95%) of oxygen impaired lung function, as was evident by the remarkable decrease in the partial pressure of oxygen (Po2) in rats exposed to 98% oxygen, 2% nitrogen (hyperoxic conditions) compared with rats exposed to normoxic conditions with 2% nitrogen (Fig. 1A). Administration of 2% hydrogen during hyperoxic exposure (hyperoxia/H2) significantly improved blood oxygenation (Fig. 1A). Body weight loss, a marker of general health status, was also evaluated. The rats exposed to hyperoxia with 2% nitrogen exhibited significant body weight loss compared with rats maintained in normoxia conditions. Hydrogen significantly reduced body weight loss during hyperoxic exposure (Fig. 1B). All rats exposed to hyperoxia with 2% nitrogen died within 64 h, whereas rats exposed to hyperoxia with 2% hydrogen survived a median of 72 h (range 72–120 h) (Fig. 1C).
Fig. 1.

Physiological effects of hydrogen on hyperoxic lung injury and assessment of lung permeability and edema in rats. A: blood oxygenation levels in blood drawn from the abdominal aorta of rats after 60 h of exposure to normoxic or hyperoxic conditions; n = 8 experiments, †P < 0.01 and *P < 0.05. B: body weight loss. Rats were weighed immediately before and immediately after 60 h of exposure to normoxic or hyperoxic conditions; n = 8, †P < 0.01 and *P < 0.05. C: animal survival during prolonged exposure to hyperoxic conditions. Survival was assessed every 8 h; n = 5 for each group, log-rank test, P = 0.0027.
Hydrogen reduced lung permeability, lung edema, and alveolar-capillary leakage induced by hyperoxia.
Exposure to hyperoxic conditions for 60 h substantially increased the pleural effusion volume (Fig. 2A) and the W/D ratio of the rat lung tissue (Fig. 2B). Hydrogen ameliorated hyperoxia-induced lung edema, as indicated by reduced pleural effusion volume (Fig. 2A) and by a significant decrease in the W/D ratio compared with lungs from hyperoxia/N2 rats (Fig. 2B). Taken together, the pleural effusion volume and W/D ratio findings provide consistent evidence that hydrogen reduced hyperoxia-induced lung edema.
Fig. 2.

Assessment of lung permeability and edema in rats. A: pleural effusion volume after 60 h of normoxia or hyperoxia; n = 8 for each group, *P < 0.05. B: wet-to-dry ratio of the lungs after 60 h of normoxia or hyperoxia; n = 5 for each group, †P < 0.01 and *P < 0.05. C: cell counts in the bronchoalveolar lavage fluid (BALF); n = 5 for each group, †P < 0.01 and *P < 0.05. D: protein concentration in BALF; n = 5 for each group, †P < 0.01 and *P < 0.05.
Because hyperoxia increases the permeability of the barriers formed by the epithelial cells, the number of cells and the protein concentration in the bronchoalveolar lavage fluid (BALF) reflect the extent of lung injury (4). Total cell number and protein concentration were significantly higher in the BALF of hyperoxia/N2 rats compared with BALF from normoxia/N2 rats (Fig. 2, C and D). Hydrogen treatment during hyperoxic exposure (hyperoxia/H2) significantly reduced the cell number and the protein concentration in the BALF, indicating less lung injury.
Hydrogen reduced hyperoxic lung injury and the expression of proinflammatory cytokines.
The effects of hydrogen gas on hyperoxic injury were also evaluated by histological analysis. The lungs of rats exposed to hyperoxia showed marked cellular infiltration and edema in the interstitial area and associated thickening of the alveolar septum. In the presence of hydrogen, both edema and inflammatory cell infiltration were reduced despite exposure to hyperoxia (Fig. 3A). Additionally, the lung injury scores in hyperoxia/H2 rats were significantly lower than those of hyperoxia/N2 rats (Fig. 3B).
Fig. 3.

Assessment of pulmonary inflammation. A: representative images of hematoxylin-eosin (H&E) staining of the lung. Left: lungs exposed to normoxic conditions with 2% nitrogen or 2% hydrogen. Right: Lungs exposed to hyperoxic conditions with 2% nitrogen or 2% hydrogen. Magnification ×400. B: lung injury scores of H&E-stained lungs. Acute lung injury was scored according to 1) thickness of the alveolar wall, 2) infiltration or aggregation of neutrophils in airspace, the alveolar wall, or the vessel wall, and 3) alveolar congestion, and each item was graded on a four-point scale. Each component ranged from 0 to 3, with higher scores indicating more severe damage. A total lung injury score was calculated as the sum of the three components (from 0 to 9). At least 10 fields (median 17, range 13–20 fields) were chosen randomly from each section and were examined at ×400 magnification. Normoxia n = 4, hyperoxia n = 6. C–F: real-time RT-PCR for inflammatory mediators in lung tissue after 60 h normoxia or hyperoxia exposure. Relative levels of the mRNAs for interleukin (IL)-1β (C), IL-6 (D), tumor necrosis factor (TNF)-α (E), and intercellular adhesion molecule (ICAM)-1 (F) were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA; n = 4 for each group, †P < 0.01 and *P < 0.05. NS, not significant.
Histological hyperoxic lung injury is accompanied by increased expression of the mRNAs for several proinflammatory cytokines, including IL-1β, IL-6, TNF-α, and ICAM-1 (48). We evaluated the effects of hydrogen on proinflammatory cytokine expression in the lung using real-time RT-PCR on mRNA extracted from frozen lung tissues. After 60 h of exposure to hyperoxic conditions, the mRNAs for IL-1β, IL-6, TNF-α, and ICAM-1 were significantly upregulated compared with rats exposed to normoxic conditions. Treatment with 2% hydrogen during hyperoxic exposure significantly reduced the peak expression of the transcripts for these inflammatory mediators (Fig. 3, C–F). These results indicate that hydrogen reduced the inflammatory response by decreasing the expression of a variety of important proinflammatory cytokines.
Hydrogen mitigated hyperoxia-induced lung epithelial cell apoptosis.
Prolonged hyperoxic exposure induces apoptosis in the pulmonary epithelial cells and is one of the deleterious factors leading to lung dysfunction (38). Caspase 3 immunohistochemical staining was performed to evaluate apoptosis induced by hyperoxia in the rat lung tissue. While exposure to hyperoxia resulted in caspase 3-positive cells in hyperoxia/N2 rats, the presence of 2% hydrogen during hyperoxia decreased the number of caspase 3-positive cells (Fig. 4, A and B). Overexpression of the antiapoptotic protein Bcl-2 ameliorates lung injury by inhibiting apoptotic pathways (6). Hydrogen inhalation resulted in significant induction of Bcl-2 protein and upregulation of Bcl-2 mRNAs after 60 h of hyperoxic exposure, as demonstrated by Western blots and real-time RT-PCR, respectively (Fig. 4, C and E). The proapoptotic protein Bax can be induced in alveolar epithelial cells by oxidative stress (6). Hyperoxia resulted in an upregulation of Bax mRNA and protein in the lungs exposed with 2% nitrogen. Hydrogen inhalation reduced Bax protein levels and inhibited the upregulation of Bax mRNA (Fig. 4, D and F).
Fig. 4.

Assessment of pulmonary apoptosis. A: pulmonary epithelial cell apoptosis was determined by immunohistochemistry for cleaved caspase 3 after 60 h of exposure to normoxia or hyperoxia. Left: lungs exposed to normoxic conditions with 2% nitrogen or 2% hydrogen. Right: lungs exposed to hyperoxic conditions with 2% nitrogen or 2% hydrogen. Magnification ×400. B: cleaved caspase 3-positive cells were counted with the samples' identities masked and expressed as the number of positive cells per high-power field (HPF). More than 10 fields were chosen randomly from each section for quantitation. Normoxia n = 4, hyperoxia n = 6. C: Western blots for B cell lymphoma-2 (Bcl-2) and β-actin on protein extracts from lungs after a 60-h exposure to normoxia or hyperoxia with 2% nitrogen or hydrogen. The images are representative of 3 independent experiments; n = 4 for each group. D: Western blots for Bcl-2-associated X-protein (Bax) and β-actin on protein extracts from lungs after a 60-h exposure to normoxia or hyperoxia with 2% nitrogen or hydrogen. The images are representative of 3 independent experiments; n = 4 for each group. E: real-time RT-PCR for Bcl-2 mRNA in lung tissue after 60 h of normoxia or hyperoxia; n = 4 for each group, *P < 0.05. Expression was normalized to GAPDH mRNA. F: real-time RT-PCR for Bax mRNA in lung tissue after 60 h of normoxia or hyperoxia; n = 4 for each group, †P < 0.01. Expression was normalized to GAPDH mRNA.
Hydrogen induced HO-1.
Our previous study demonstrated that hydrogen could increase HO-1 and mitigate lung ischemia-reperfusion injury in a rat lung transplantation model (23). Therefore, we examined whether hydrogen treatment increased expression of HO-1 in this model. In immunohistochemical analysis, more cells expressed HO-1 in the hyperoxia/H2 rats (Fig. 5, A and B) than in the hyperoxia/N2 rats. Immunofluorescent analysis for HO-1 and aquaporin-5, a lung epithelial cell marker, demonstrated that the HO-1-positive cells were lung epithelial cells (Fig. 5C).
Fig. 5.

Hydrogen increased heme oxygenase (HO)-1 in the lung. A: representative images of immunohistochemistry for HO-1 in the lungs after 60 h of normoxia or hyperoxia. Magnification = ×400. B: HO-1-positive cells were counted with the samples' identities masked and expressed as the number of positive cells per HPF. The cell count was analyzed with Tukey-Kramer methods. Normoxia n = 4, hyperoxia n = 6; >10 fields were chosen randomly from each section. C: representative images of immunofluorescent staining for aquaporin-5 (AQP-5), a marker for lung epithelial cells, and HO-1 in the lungs after 60 h of hyperoxia in the presence of 2% hydrogen (hyperoxia/H2). Top, AQP-5-positive cells. Middle, HO-1-positive cells. Bottom, merged image. D: Western blots for HO-1 and β-actin on protein extracts from the lungs taken after 30 and 60 h of normoxia or hyperoxia. The images are representative of 3 independent experiments; n = 4 for each group. E: real-time RT-PCR for HO-1 mRNA in lung tissues after 30 or 60 h normoxia or hyperoxia exposure. Gene expression was normalized to GAPDH mRNA; n = 4 for each group, †P < 0.01 and *P < 0.05. F: effect of hydrogen on HO-1 activity in the rat lung. HO-1 activity was expressed as pmol of bilirubin formed·mg of protein−1·60 min−1. *P < 0.05.
Consistent with the immunohistochemical analysis, hydrogen increased HO-1 protein (Fig. 5D) and HO-1 mRNA (Fig. 5E) expression after 60 h of exposure to hyperoxic conditions. HO-1 mRNA and protein were upregulated after 30 h of exposure to hydrogen and hyperoxia as well. To confirm that the increase in pulmonary HO-1 protein and mRNA expression was reflected in increased HO-1 activity, we examined HO-1 activity in the lungs after 60 h of hyperoxic exposure. Hydrogen increased HO-1 activity in the lungs of rats exposed to hyperoxia (Fig. 5F). Hydrogen treatment did not significantly induce HO-1 expression or HO-1 enzymatic activity under normoxic conditions.
Hydrogen modulated the Keap1/Nrf2 signaling pathway.
Nrf2 is one of the key transcription factors that regulates HO-1 gene expression as part of an adaptive response to oxidative stress (2). Nrf2 also plays key roles in protecting the lung from hyperoxic lung injury and allowing lung recovery after hyperoxic exposure (10, 43). To further explore the mechanisms underlying hydrogen's ability to mitigate hyperoxic lung injury, we performed real-time RT-PCR for several Nrf2-dependent, cytoprotective genes, including Nqo1 (Fig. 6A), GSTA2 (Fig. 6B), UGT1A6 (Fig. 6C), and Prdx 1 (Fig. 6D), after 60 h of exposure to hyperoxia. Hydrogen significantly upregulated these Nrf2-dependent transcripts in rats exposed to hyperoxic conditions.
Fig. 6.

Assessment of the NF-E2-related factor (Nrf) 2-dependent mRNA expression in the lung after 60 h of normoxia/hyperoxia exposure. Expression of Nrf2-dependent genes in lung tissues after 60 h of normoxia or hyperoxia. The levels of mRNAs for NAD(P)H dehydrogenase quinone (Nqo) 1 (A), GSTA2 (B), UDP-glucuronosyl transferase (UGT) 1A6 (C), and peroxiredoxin (Prdx) 1 (D) were quantitated by real-time RT-PCR. Gene expression was normalized to GAPDH mRNA; n = 4 for each group, †P < 0.01 and *P < 0.05.
Disruption of Nrf2 impaired the protective effect of hydrogen against hyperoxic lung injury.
To solidify a role for Nrf2 in the mitigation of hyperoxic lung injury by hydrogen, we examined if hydrogen protected against hyperoxic lung injury in Nrf2−/− mice. Sixty hours of exposure to a high concentration of oxygen in the absence of hydrogen (hyperoxia/N2) impaired lung function and resulted in a remarkable decrease of Po2 in both wild-type and Nrf2−/− mice (Fig. 7, A and B). Hydrogen treatment significantly improved blood oxygenation in the wild-type mice (Fig. 7A), as it did in the rat model (Fig. 1A). However, hydrogen did not significantly improve blood oxygenation in Nrf2−/− mice (Fig. 7B). Hydrogen reduced hyperoxic lung injury, apparent in gross pathological analysis, in wild-type mice after 60 h of hyperoxia (Fig. 7C) but did not protect against hyperoxic lung injury in Nrf2−/− mice (Fig. 7D). Histological analysis also revealed that the lungs of Nrf2−/− mice exposed to hyperoxia had more marked cellular infiltration and edema in the interstitial area and associated thickening of the alveolar septum, even in the presence of hydrogen, compared with wild-type mice (Fig. 7, E and F). These results suggested that disruption of the Nrf2 gene abrogates hydrogen's ability to protect against hyperoxic lung injury.
Fig. 7.

Assessment of the effects of hydrogen on hyperoxic lung injury in Nrf2−/− mice. A and B: blood oxygenation levels were measured in the blood drawn from the abdominal aorta of the Nrf2+/+ (A) and Nrf2−/− (B) mice after 60 h of exposure to normoxia or hyperoxia; n = 4 for each group, *P < 0.05. C and D: representative images of the gross anatomy of lungs from the Nrf2+/+ (C) and Nrf2−/− (D) mice 60 h after hyperoxia with 2% hydrogen. E and F: representative images of H&E staining of lungs from Nrf2+/+ and Nrf2−/− mice after 60 h of normoxia (E) or hyperoxia (F) with and without hydrogen.
Hydrogen reduced oxidative injury but did not induce Nrf2-dependent cytoprotective genes in Nrf2−/− mice.
In lungs exposed to hyperoxic conditions without hydrogen for 60 h, tissue MDA levels markedly increased in both wild-type and Nrf2−/− mice. Hydrogen supplementation significantly reduced lung MDA levels in both wild-type and Nrf2−/− mice (Fig. 8A). 8-OHdG immunohistochemical staining was performed to evaluate DNA damage induced by hyperoxia in the lung tissue. Although DNA damage was scarce in the lungs of normoxia animals (data not shown), 60 h exposure to hyperoxia resulted in 8-OHdG-positive cells. Consistent with the MDA assay, the presence of hydrogen during hyperoxia significantly decreased the number of 8-OHdG-positive cells in both wild-type and Nrf2−/− mice (Fig. 8, B and C). Expression of Nrf2-dependent genes (HO-1, Nqo1, GSTA2) was assessed by real-time RT-PCR after 60 h of exposure to hyperoxia in the presence and absence of hydrogen in Nrf2−/− and wild-type mice. HO-1 (Fig. 8D), Nqo1 (Fig. 8E), and GSTA2 (Fig. 8F) were not induced in response to hydrogen treatment in Nrf2−/− mice.
Fig. 8.
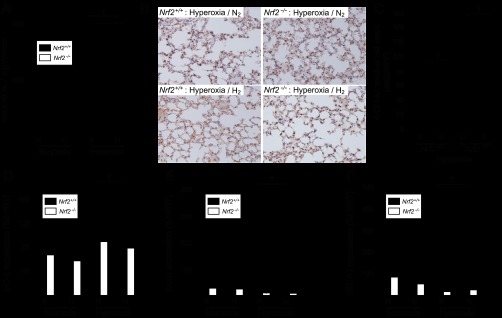
Assessment of the oxidative stress markers and the Nrf2-dependent mRNA expression in Nrf2−/− mice. A: assessment of malondialdehyde (MDA) in the lung tissue of Nrf2+/+ and Nrf2−/− mice after 60 h normoxia or hyperoxia exposure; n = 4 for each group, *P < 0.05. B: representative images of immunohistochemistry for 8-hydroxy-2′-deoxyguanosine (8-OHdG) in the lungs after 60 h of hyperoxia in Nrf2+/+ and Nrf2−/− mice. Magnification ×400. C: 8-OHdG-positive cells were counted with the samples' identities masked and expressed as the number of positive cells per HPF. More than 10 fields were chosen randomly from each section for quantitation; n = 4 for each group, †P < 0.01. D–F: assessment of the Nrf2-dependent mRNA expression in Nrf2+/+ and Nrf2−/− mice after 60 h normoxia/hyperoxia exposure. Nrf2-dependent genes, HO-1 (D), Nqo1 (E), and GSTA2 (F), were quantitated by real-time RT-PCR after 60 h of exposure to hyperoxia; n = 4 for each group, †P < 0.01. Gene expression was normalized to hypoxanthine phosphoribosyltransferase (HPRT) mRNA.
Taken together with the results in Fig. 8, the MDA assay and 8-OHdG immunohistochemical staining results suggest that hydrogen may protect against hyperoxic lung injury both by decreasing the extent of oxidative injury caused by ROS, through hydrogen's free radical scavenging activities, and by inducing Nrf2-dependent protective signaling pathways.
DISCUSSION
This study demonstrated that treatment with 2% hydrogen via inhalation ameliorated hyperoxic lung injury by modulating the Nrf2 pathway. Treatment with hydrogen during hyperoxic exposure induced several Nrf2-dependent genes, including HO-1. The study suggests that hydrogen protects against hyperoxic lung injury both by decreasing the extent of oxidative injury caused by ROS, perhaps through hydrogen's free radical scavenging activities, and by inducing Nrf2-dependent protective signaling pathways. Although the molecular mechanisms underlying hydrogen's actions are largely undefined, we demonstrated that hydrogen is a novel activator of the Nrf2 pathway with therapeutic potential.
Since discovery of the antioxidant effects of hydrogen (37), a number of experimental and clinical studies have indicated that hydrogen gas may be a useful new therapeutic modality in a variety of biomedical fields (16, 19, 21, 22). Our laboratory has extensively explored the application of hydrogen for the treatment of various diseases (7, 9, 33), including acute lung injuries (15, 23, 24). Hydrogen is a selective radical scavenger for hydroxyl radicals and peroxynitrite (14, 37), and this aspect of hydrogen's chemistry may explain its therapeutic effects. However, our studies suggest that hydrogen exerts indirect antioxidative effects by triggering the activation or upregulation of additional antioxidant enzymes or cytoprotective proteins. Hydrogen may act as a modulator of signal transduction, similar to nitric oxide, carbon monoxide, and hydrogen sulfide, which are well-characterized gaseous signaling molecules (19).
Hydrogen treatment during oxidative stress induces HO-1. Our recent study in a rat lung transplantation model demonstrated that induction of HO-1 by inhaled hydrogen gas in the donor lung before transplantation was associated with reduced ischemia-reperfusion injury in the lungs after transplantation (23). We also demonstrated that hydrogen enrichment during preservation induced HO-1 in intestinal grafts using a rat intestinal-transplant model (7). In the present study, hydrogen induced HO-1 expression and ameliorated hyperoxic lung injury in rat and mouse models, consistent with our other recent observations (7, 23). HO-1 is a rate-limiting anti-inflammatory/antiapoptotic enzyme that catalyzes the conversion of heme into equimolar quantities of biliverdin (further reduced to bilirubin through biliverdin reductase), iron, and carbon monoxide (46). Animal studies have demonstrated that HO-1 plays a critical protective role in several different lung diseases, including ARDS, pulmonary hypertension, asthma, chronic obstructive pulmonary disease (COPD), and hyperoxic lung injury (13). In vivo, exogeneous administration of HO-1 to rats via a recombinant adenovirus significantly attenuated hyperoxic lung injury. Rats overexpressing HO-1 in their lungs had reduced pulmonary edema, parenchymal inflammation, and apoptosis after hyperoxia (39). Although possible therapeutic approaches to modulate HO-1 expression in patients include the use of pharmacological agents or gene therapy (13), hydrogen treatment to induce HO-1 might be more easily translated into clinical practice than other therapies.
One possible explanation for the protective role of HO-1 induced by hydrogen seen in our study may be a removal of free heme. HO-1 degrades heme, which, when released from damaged cells, is highly lipophilic and detrimental. Free heme not only directly induces tissue injury of the lung cells but is also a major source of iron, which generates highly detrimental hydroxyl radicals through the Fenton reaction (8). The breakdown of heme to three byproducts has its own significance in essential cellular metabolism and contributes to the suppression of oxidative lung injury, as shown by Otterbein et al. who demonstrated that carbon monoxide inhalation prevents hyperoxic lung injury (40).
One key finding of this study was that, although hydrogen decreases the extent of oxidative injury caused by ROS even in the absence of Nrf2, Nrf2 is critical for the amelioration of hyperoxic lung injury by hydrogen. Disruption of Nrf2 completely abrogated the protective effects of hydrogen against hyperoxic lung injury. HO-1 induction in response to hydrogen treatment was greatly attenuated in Nrf2−/− mice, suggesting that HO-1 induction by hydrogen is at least partially dependent on Nrf2. Moreover, the Nrf2-ARE pathway is activated by many different electrophilic and oxidative stresses and regulates a large battery of genes encoding proteins with antioxidant activities (25, 47). Nrf2 signaling plays a critical protective role in pulmonary diseases such as carrageenin-induced acute lung injury (31), COPD (30), elastase-induced lung emphysema (17), and asthma (42). In several models, Nrf2-deficient mice exhibited more severe outcomes than their wild-type counterparts (17, 31, 42). Nrf2 is also an important determinant of susceptibility to hyperoxic lung injury (11). Nrf2−/− mice exhibit increased protein permeability, macrophage infiltration, and epithelial injury compared with Nrf2+/+ control mice following exposure to hyperoxia (10). The activation of Nrf2 by pharmacological or endogeneous molecules, such as CDDO-imidazole {1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl] imidazole} and 15-deoxy-Δ12,14-prostaglandin J2, protects wild-type, but not Nrf2 knockout, mice from a variety of pulmonary diseases (31, 44). Recently, clinical trials have been initiated to determine the therapeutic value of inducers of Nrf2 in the treatment of pulmonary diseases (http://clinicaltrials.gov).
To elucidate the detailed mechanisms of the Nrf2-ARE pathway activation by hydrogen, we performed real-time RT-PCR analysis for Nrf2 and Keap1. We found that transcriptional activation of Nrf2 is decreased under hyperoxic conditions and recovers to baseline (normoxic) levels with hydrogen treatment (data not shown), and Keap1 mRNA expression did not change in normoxia and hyperoxia with H2 or N2 inhalation (data not shown). Thus, hydrogen may at least partially modulate the Nrf2-ARE pathway by modulating transcriptional activation of Nrf2. Although there is some evidence that transcriptional regulation of Nrf2 is one way of modulating the Nrf2-ARE signaling pathway (27), most evidence supports the model that Keap1 modulation of Nrf2 subcellular localization and protein stability controls Nrf2 activity. Nrf2 phosphorylation and ubiquitination are believed to play key regulatory roles (5). We performed exploratory studies but were unable to gain further insight into the mechanisms by which hydrogen modulates the Nrf2 pathway and how this modulation protects against hyperoxic lung injury in the current study. Our study suggests that hydrogen may also be worth investigation in this context. Additionally, studies to determine the detailed mechanisms of the Nrf2-ARE pathway activation by hydrogen and how pathway activation ameliorates hyperoxic lung injury are warranted.
We used 2% hydrogen for this study and did not see any adverse effects during the study period. Hydrogen gas is physiological and safe for humans. Although the use of hydrogen with a ventilator raises technical and safety issues that will need to be resolved before we can use this mixture of gases in ventilated patients, hydrogen has no risk of explosion at concentrations <4% in the presence of oxygen (35). With appropriate precautions, there should be little risk of explosion in the intensive care unit or if administered in conjunction with oxygen therapy at home. Thus, hydrogen inhalation therapy is a straightforward approach to lung disease, which can be administered by simply providing gas for the patient to inhale using a ventilator circuit, facemask, or nasal cannula. In addition, hydrogen may be relatively easily incorporated into our current interventional or surgical procedures without increasing their complexity.
In conclusion, this study demonstrated, for the first time, that hydrogen ameliorates hyperoxic lung injury by modulating the Nrf2 pathway. The findings suggest a potentially novel and easily applicable solution to hyperoxic lung injury and provide new insight into our scientific knowledge of hydrogen. Although further investigation is required, hydrogen is expected to be an innovative therapeutic tool for unmet medical needs that currently cause considerable health burdens, in particular for critically ill patients with lung diseases. Our findings in this study may pave the way for successful translation of hydrogen inhalation therapy into clinical practice.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant R21-HL-02528-01 (A. Nakao) and by research funds of the Department of Cardiothoracic Surgery, Thomas E. Starzl Postdoctoral fellowship (T. Kawamura).
DISCLOSURES
None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
AUTHOR CONTRIBUTIONS
Author contributions: T.K., N.W., N.S., T.R.B., M.O., Y. Toyoda, and A.N. conception and design of research; T.K., C.-S.H., K.M., Y. Tanaka, K.N., X.P., T.T., and A.N. performed experiments; T.K., N.S., C.-S.H., K.M., Y. Tanaka, K.N., X.P., T.T., T.R.B., M.O., Y. Toyoda, and A.N. analyzed data; T.K., N.W., K.M., and A.N. interpreted results of experiments; T.K. and A.N. prepared figures; T.K., N.S., and A.N. drafted manuscript; T.K., N.S., T.W.K., and A.N. edited and revised manuscript; T.K., N.S., T.W.K., and A.N. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Shannon L. Wyszomierski for editing the manuscript and Michiko Aoyama for technical support.
REFERENCES
- 1. Alam J, Cook JL. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am J Respir Cell Mol Biol 36: 166–174, 2007 [DOI] [PubMed] [Google Scholar]
- 2. Alam J, Cook JL. Transcriptional regulation of the heme oxygenase-1 gene via the stress response element pathway. Curr Pharm Des 9: 2499–2511, 2003 [DOI] [PubMed] [Google Scholar]
- 3. Altemeier WA, Sinclair SE. Hyperoxia in the intensive care unit: why more is not always better. Curr Opin Crit Care 13: 73–78, 2007 [DOI] [PubMed] [Google Scholar]
- 4. Awasthi S, Gyurasics A, Knight SA, Welty SE, Smith CV. Protein oxidation biomarkers in hyperoxic lung injury in rats: effects of U-74389. Toxicol Lett 95: 47–61, 1998 [DOI] [PubMed] [Google Scholar]
- 5. Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol 85: 241–272, 2011 [DOI] [PubMed] [Google Scholar]
- 6. Buccellato LJ, Tso M, Akinci OI, Chandel NS, Budinger GR. Reactive oxygen species are required for hyperoxia-induced Bax activation and cell death in alveolar epithelial cells. J Biol Chem 279: 6753–6760, 2004 [DOI] [PubMed] [Google Scholar]
- 7. Buchholz BM, Masutani K, Kawamura T, Peng X, Toyoda Y, Billiar TR, Bauer AJ, Nakao A. Hydrogen-enriched preservation protects the isogeneic intestinal graft and amends recipient gastric function during transplantation. Transplantation 92: 985–992, 2011 [DOI] [PubMed] [Google Scholar]
- 8. Bysani GK, Kennedy TP, Ky N, Rao NV, Blaze CA, Hoidal JR. Role of cytochrome P-450 in reperfusion injury of the rabbit lung. J Clin Invest 86: 1434–1441, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cardinal JS, Zhan J, Wang Y, Sugimoto R, Tsung A, McCurry KR, Billiar TR, Nakao A. Oral hydrogen water prevents chronic allograft nephropathy in rats. Kidney Int 77: 101–109, 2010 [DOI] [PubMed] [Google Scholar]
- 10. Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, Kleeberger SR. Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol 26: 175–182, 2002 [DOI] [PubMed] [Google Scholar]
- 11. Cho HY, Jedlicka AE, Reddy SP, Zhang LY, Kensler TW, Kleeberger SR. Linkage analysis of susceptibility to hyperoxia. Nrf2 is a candidate gene. Am J Respir Cell Mol Biol 26: 42–51, 2002 [DOI] [PubMed] [Google Scholar]
- 12. Claireaux AE. The effect of oxygen on the lung. J Clin Pathol Suppl 9: 75–80, 1975 [PMC free article] [PubMed] [Google Scholar]
- 13. Fredenburgh LE, Perrella MA, Mitsialis SA. The role of heme oxygenase-1 in pulmonary disease. Am J Respir Cell Mol Biol 36: 158–165, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hanaoka T, Kamimura N, Yokota T, Takai S, Ohta S. Molecular hydrogen protects chondrocytes from oxidative stress and indirectly alters gene expressions through reducing peroxynitrite derived from nitric oxide (Abstract). Med Gas Res 1: 18, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang CS, Kawamura T, Lee S, Tochigi N, Shigemura N, Buchholz BM, Kloke JD, Billiar TR, Toyoda Y, Nakao A. Hydrogen inhalation ameliorates ventilator-induced lung injury (Abstract). Crit Care 14: R234, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang CS, Kawamura T, Toyoda Y, Nakao A. Recent advances in hydrogen research as a therapeutic medical gas. Free Radic Res 44: 971–982, 2010 [DOI] [PubMed] [Google Scholar]
- 17. Ishii Y, Itoh K, Morishima Y, Kimura T, Kiwamoto T, Iizuka T, Hegab AE, Hosoya T, Nomura A, Sakamoto T, Yamamoto M, Sekizawa K. Transcription factor Nrf2 plays a pivotal role in protection against elastase-induced pulmonary inflammation and emphysema. J Immunol 175: 6968–6975, 2005 [DOI] [PubMed] [Google Scholar]
- 18. Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236: 313–322, 1997 [DOI] [PubMed] [Google Scholar]
- 19. Itoh T, Fujita Y, Ito M, Masuda A, Ohno K, Ichihara M, Kojima T, Nozawa Y. Molecular hydrogen suppresses FcepsilonRI-mediated signal transduction and prevents degranulation of mast cells. Biochem Biophys Res Commun 389: 651–656, 2009 [DOI] [PubMed] [Google Scholar]
- 20. Itoh T, Itoh A, Pleasure D. Bcl-2-related protein family gene expression during oligodendroglial differentiation. J Neurochem 85: 1500–1512, 2003 [DOI] [PubMed] [Google Scholar]
- 21. Kajiya M, Silva MJ, Sato K, Ouhara K, Kawai T. Hydrogen mediates suppression of colon inflammation induced by dextran sodium sulfate. Biochem Biophys Res Commun 386: 11–15, 2009 [DOI] [PubMed] [Google Scholar]
- 22. Kamimura N, Nishimaki K, Ohsawa I, Ohta S. Molecular hydrogen improves obesity and diabetes by inducing hepatic FGF21 and stimulating energy metabolism in db/db mice. Obesity (Silver Spring) 19: 1396–13403, 2011 [DOI] [PubMed] [Google Scholar]
- 23. Kawamura T, Huang CS, Peng X, Masutani K, Shigemura N, Billiar TR, Okumura M, Toyoda Y, Nakao A. The effect of donor treatment with hydrogen on lung allograft function in rats. Surgery 150: 240–249, 2011 [DOI] [PubMed] [Google Scholar]
- 24. Kawamura T, Huang CS, Tochigi N, Lee S, Shigemura N, Billiar TR, Okumura M, Nakao A, Toyoda Y. Inhaled hydrogen gas therapy for prevention of lung transplant-induced ischemia/reperfusion injury in rats. Transplantation 90: 1344–1351, 2010 [DOI] [PubMed] [Google Scholar]
- 25. Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol 47: 89–116, 2007 [DOI] [PubMed] [Google Scholar]
- 26. Kohmoto J, Nakao A, Kaizu T, Tsung A, Ikeda A, Tomiyama K, Billiar TR, Choi AM, Murase N, McCurry KR. Low-dose carbon monoxide inhalation prevents ischemia/reperfusion injury of transplanted rat lung grafts. Surgery 140: 179–185, 2006 [DOI] [PubMed] [Google Scholar]
- 27. Kwak MK, Itoh K, Yamamoto M, Sutter TR, Kensler TW. Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1, 2-dimethiole-3-thione. Mol Med 7: 135–145, 2001 [PMC free article] [PubMed] [Google Scholar]
- 28. Lee PJ, Choi AM. Pathways of cell signaling in hyperoxia. Free Radic Biol Med 35: 341–350, 2003 [DOI] [PubMed] [Google Scholar]
- 29. Maeshima K, Takahashi T, Uehara K, Shimizu H, Omori E, Yokoyama M, Tani T, Akagi R, Morita K. Prevention of hemorrhagic shock-induced lung injury by heme arginate treatment in rats. Biochem Pharmacol 69: 1667–1680, 2005 [DOI] [PubMed] [Google Scholar]
- 30. Malhotra D, Thimmulappa R, Navas-Acien A, Sandford A, Elliott M, Singh A, Chen L, Zhuang X, Hogg J, Pare P, Tuder RM, Biswal S. Decline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1. Am J Respir Crit Care Med 178: 592–604, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31. Mochizuki M, Ishii Y, Itoh K, Iizuka T, Morishima Y, Kimura T, Kiwamoto T, Matsuno Y, Hegab AE, Nomura A, Sakamoto T, Uchida K, Yamamoto M, Sekizawa K. Role of 15-deoxy delta(12,14) prostaglandin J2 and Nrf2 pathways in protection against acute lung injury. Am J Respir Crit Care Med 171: 1260–1266, 2005 [DOI] [PubMed] [Google Scholar]
- 32. Morse D, Lin L, Choi AM, Ryter SW. Heme oxygenase-1, a critical arbitrator of cell death pathways in lung injury and disease. Free Radic Biol Med 47: 1–12, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nakao A, Kaczorowski DJ, Wang Y, Cardinal JS, Buchholz BM, Sugimoto R, Tobita K, Lee S, Toyoda Y, Billiar TR, McCurry KR. Amelioration of rat cardiac cold ischemia/reperfusion injury with inhaled hydrogen or carbon monoxide, or both. J Heart Lung Transplant 29: 544–553, 2010 [DOI] [PubMed] [Google Scholar]
- 34. Nakao A, Kimizuka K, Stolz DB, Neto JS, Kaizu T, Choi AM, Uchiyama T, Zuckerbraun BS, Nalesnik MA, Otterbein LE, Murase N. Carbon monoxide inhalation protects rat intestinal grafts from ischemia/reperfusion injury. Am J Pathol 163: 1587–1598, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. NASA Safety Standard for Hydrogen and Hydrogen Systems. Washington, DC: National Aeronautics and Space Administration, 2005 [Google Scholar]
- 36. O'Driscoll BR, Howard LS, Davison AG. BTS guideline for emergency oxygen use in adult patients. Thorax 63, Suppl 6: vi1–vi68, 2008 [DOI] [PubMed] [Google Scholar]
- 37. Ohsawa I, Ishikawa M, Takahashi K, Watanabe M, Nishimaki K, Yamagata K, Katsura K, Katayama Y, Asoh S, Ohta S. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med 13: 688–694, 2007 [DOI] [PubMed] [Google Scholar]
- 38. Otterbein LE, Chin BY, Mantell LL, Stansberry L, Horowitz S, Choi AM. Pulmonary apoptosis in aged and oxygen-tolerant rats exposed to hyperoxia. Am J Physiol Lung Cell Mol Physiol 275: L14–L20, 1998 [DOI] [PubMed] [Google Scholar]
- 39. Otterbein LE, Kolls JK, Mantell LL, Cook JL, Alam J, Choi AM. Exogenous administration of heme oxygenase-1 by gene transfer provides protection against hyperoxia-induced lung injury. J Clin Invest 103: 1047–1054, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Otterbein LE, Mantell LL, Choi AM. Carbon monoxide provides protection against hyperoxic lung injury. Am J Physiol Lung Cell Mol Physiol 276: L688–L694, 1999 [DOI] [PubMed] [Google Scholar]
- 41. Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Can J Gastroenterol 80: 1895–1903, 2010 [DOI] [PubMed] [Google Scholar]
- 42. Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, Biswal S. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med 202: 47–59, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reddy NM, Kleeberger SR, Kensler TW, Yamamoto M, Hassoun PM, Reddy SP. Disruption of Nrf2 impairs the resolution of hyperoxia-induced acute lung injury and inflammation in mice. J Immunol 182: 7264–7271, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reddy NM, Suryanaraya V, Yates MS, Kleeberger SR, Hassoun PM, Yamamoto M, Liby KT, Sporn MB, Kensler TW, Reddy SP. The triterpenoid CDDO-imidazolide confers potent protection against hyperoxic acute lung injury in mice. Am J Respir Crit Care Med 180: 867–874, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shintani M, Sangawa A, Yamao N, Miyake T, Kamoshida S. Immunohistochemical analysis of cell death pathways in gastrointestinal adenocarcinoma. Biomed Res 32: 379–386, 2011 [DOI] [PubMed] [Google Scholar]
- 46. Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci USA 61: 748–755, 1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wakabayashi N, Slocum SL, Skoko JJ, Shin S, Kensler TW. When NRF2 Talks, Who's Listening? Antioxid Redox Signal 13: 1649–1663, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zaher TE, Miller EJ, Morrow DM, Javdan M, Mantell LL. Hyperoxia-induced signal transduction pathways in pulmonary epithelial cells. Free Radic Biol Med 42: 897–908, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]