

Abstract
Neutrophilic inflammation is associated with chronic airway diseases. It has been observed that human neutrophil elastase (HNE), which is secreted by active neutrophils during inflammation, induces both mucin overproduction and goblet cell metaplasia. Several in vitro studies suggest that tumor necrosis factor-α converting enzyme (TACE) regulates the signaling axis that mediates HNE-induced mucin overproduction; however, it is unknown whether TACE performs a similar function in HNE-induced goblet cell metaplasia in vivo. We conducted this study to determine whether the inactivation of Tace gene expression attenuates HNE-induced goblet cell metaplasia in mice. Deletion of Tace is lethal shortly after birth in mice; therefore, we utilized Taceflox/floxR26CreER+/− mice and induced conditional deletion of Tace using a tamoxifen injection. Wild-type mice were given tamoxifen to control for its effect. Tace conditional deletion mice and wild-type mice were exposed to HNE via nasal instillation three times at 3-day intervals, and the lungs were harvested on day 11 after initial HNE exposure. Using periodic acid-Schiff staining and MUC5AC immunohistochemical staining to visualize goblet cells in the lungs, we found that HNE induced goblet cell metaplasia in the wild-type mice and that HNE-induced goblet cell metaplasia was significantly attenuated in the Tace conditional deletion mice. These findings suggest that TACE could be a potential target in the treatment of goblet cell metaplasia in patients with chronic airway diseases.
Keywords: airway mucin, airway epithelium, chronic airway disease, neutrophilic inflammation
neutrophilic inflammation is associated with chronic airway diseases such as chronic obstructive pulmonary disease, cystic fibrosis, and asthma (6, 24, 33). Mediators are secreted by active neutrophils during inflammation; one such mediator is neutrophil elastase (20). It has been observed that human neutrophil elastase (HNE) induces the expression of mucin genes such as MUC1, MUC4, and MUC5AC (8, 9, 15, 29) and the secretion of MUC5AC and MUC5B (13, 22) in various epithelial cells in vitro. Under normal conditions, mucin plays a protective role in the airway. When HNE induces the expression of mucin genes in patients with chronic airway diseases (7), mucin overproduction causes airway obstruction and enhanced bacterial colonization that promote chronic inflammation and impaired mucociliary clearance due to increased viscosity of mucus (26, 31).
Voynow and her colleagues (1, 10, 32) established a mouse model showing that HNE induces goblet cell metaplasia (GCM) in the airway, which results in mucin hypersecretion and overproduction. They found that the repeated exposure to HNE in mice induced GCM and mucin gene expression. They also determined that the proteolytic activity of HNE mediates HNE-induced GCM by observing that the administration of the HNE inhibitor Ala-Ala-Pro-Val chloromethylketone attenuates HNE-induced GCM. Additionally, Arai et al. (1) found that HNE-induced GCM is attenuated by a long-acting cholinergic antagonist, tiotropium.
Although these studies clearly show that HNE induces GCM, the underlying cellular signaling mechanism that enables HNE to induce GCM has not yet been fully elucidated in vivo. However, extensive studies performed in vitro have revealed that HNE-induced mucin gene expression is associated with the activation of the protein kinase C (PKC)-tumor necrosis factor-α (TNF-α) converting enzyme (TACE) pathway. TACE, also known as a disintegrin and metalloproteinase 17 (ADAM17), is a membrane-anchored enzyme that cleaves cell-surface proteins including epidermal growth factor receptor (EGFR) ligands, cytokines and their receptors, and adhesion molecules (2, 27). Among EGFR ligands, amphiregulin has been shown to contribute to GCM that occurs during naphthalene-induced lung injury in mice (18).
Furthermore, activation of the PKC-TACE-EGFR signaling cascade mediates HNE-induced MUC5AC expression and MUC5AC secretion in NCI-H292 cells (29) and activation of the PKC-TACE-tumor necrosis factor receptor 1 (TNFR1) signaling cascade mediates HNE-induced MUC1 expression in A549 cells (15).
Because TACE regulates the signaling axis that mediates HNE-induced mucin overproduction in vitro, we hypothesized that TACE plays a important role in HNE-induced GCM in vivo. In this study, we used Taceflox/floxR26CreER+/− mice to demonstrate that deletion of TACE attenuates HNE-induced GCM in mice, which suggests that TACE plays a pivotal role in the regulation of GCM and could be a potential therapeutic target in chronic airway diseases.
MATERIALS AND METHODS
Animals.
R26CreER+/+ [B6;129-Gt(ROSA)26Sortm1(Cre/Esr1)Nat/J] mice were purchased from Jackson laboratory (Bar Harbor, ME). Taceflox/floxR26CreER+/− and Taceflox/flox R26CreER−/− mice were previously generated by crossing Taceflox/flox with R26CreER+/+ (30). In Taceflox/flox mice loxP sites flank exon 2 (11). Mice at ∼8–12 wk of age were used for all experiments.
Conditional deletion of Tace was generated by tamoxifen injection. Tamoxifen was prepared as a 10 mg/ml stock solution in sunflower oil containing 5% ethanol and was injected intraperitoneally at a dose of 0.04 mg/g body wt daily for 5 consecutive days, as described in Fig. 1. All procedures and protocols using mice were approved by the animal care committee of the Harvard School of Public Health.
Fig. 1.
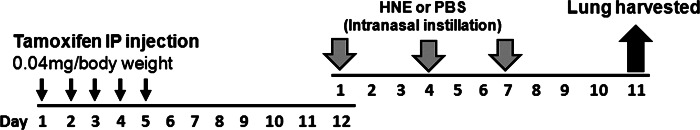
Experimental procedure for tamoxifen injection and human neutrophil elastase (HNE) exposure. Tamoxifen was injected intraperitoneally (IP) daily for 5 days. On day 7 after the last tamoxifen injection, mice were instilled intranasally with HNE (50 μg in 40 μl of PBS) or 40 μl of PBS 3 times at 3-day intervals. On day 4 after the last HNE exposure, lungs were harvested for histology and mRNA analysis of Muc5ac.
PCR genotyping.
Offspring of Taceflox/floxR26CreER+/− mice and Taceflox/floxR26CreER−/− mice were genotyped for Cre expression by using primers, IMR1084 and IMR1085, specified by Jackson Laboratory. Following a polymerase chain reaction (PCR) using genomic DNA prepared from a tail biopsy, amplified PCR product was loaded on 2% agarose gel and visualized under a UV transilluminator.
Exposure to HNE.
Mice were exposed to HNE three times at 3-day intervals starting on day 7 after the last tamoxifen injection and the lungs were harvested on day 11 after initial HNE exposure, as outlined in Fig. 1. A mouse model of HNE-induced GCM has been well established by Voynow et al. (32) and repeated by others (1). We used the process described by Voynow et al., including the suggested dosage of and duration between HNE exposures. Briefly, mice were anesthetized via inhalation of 4% isoflurane, and 50 μg of HNE dissolved in 40 μl of phosphate-buffered saline (PBS) was nasally instilled as described (1); 40 μl PBS was used as a vehicle control. HNE was purchased from Elastin Products (Owensville, MO).
Histology.
On the day of the lung harvest, the mice were euthanized via CO2 asphyxiation, the primary bronchial branch to the right lung was tied off, and the whole right lung was removed for RNA analysis. The left lung was inflated with 10% formalin at a pressure of 30 cmH2O, removed, and incubated in 10% formalin overnight on a gently rocking platform in the cold room. Fixed left lungs were embedded in paraffin blocks, and sectioned longitudinally at a thickness of 6 μm. Four serial sections were mounted per slide, and they were stained with periodic acid-Schiff (PAS) for goblet cell evaluation and with hematoxylin and eosin (H&E) for evaluation of tissue inflammation. Goblet cell and tissue inflammation indexes were scored on coded slides by a pathologist without knowledge of the treatment group.
GCM was scored by using five grades to estimate the proportion of airway epithelium showing changes (32) as follows: grade 0, no PAS staining; grade 1, 0–25%; grade 2, 26–50%; grade 3, 51–75%; grade 4, 76–100%. Tissue inflammation was also assessed semiquantitatively on a scale ranging from 0 to 4 to score presence and abundance of inflammatory cells within the lung.
Immunohistochemical staining of MUC5AC.
To confirm that PAS-positive cells represent MUC5AC-positive cells, immunohistochemical staining was performed by using a MOM (mouse-on-mouse) Peroxidase Immunodetection Kit (Vector Laboratories, Burlingame, CA) following the manufacturer's instructions. Briefly, paraffin was removed from the slides and the slides were rehydrated with xylene and then decreasing grades of ethanol solution. Endogenously expressed peroxidase activity was quenched by incubation with 3% hydrogen peroxide solution for 30 min. A 50-μl aliquot of prediluted MUC5AC (45M1) antibody (NeoMarker, Fremont, CA) was gently dropped onto each tissue on the slides and incubated overnight in a humidified chamber at 4°C. Cells with positive stains were detected using streptavidin-peroxidase conjugate and diaminobenzidine (DAB). Slides were then mounted with a Vector Mount (Vector Laboratories) and imaged at ×200 magnification with a digital camera.
Western blot analysis.
To detect TACE protein, whole lungs were homogenized with liquid nitrogen and lysed with lysis buffer containing 1× protease inhibitor cocktails, 1 mM PMSF, and 5 mM phenanthroline. Western blot analysis was performed with 50 μg of protein lysates per lane. The protein concentration of cell lysates was quantified by a bicinchoninic acid protein assay (Pierce Biotechnology, Rockford, IL). Each sample was boiled in 2× sodium dodecyl sulfate-polyacrylamide gel electrophoresis sample buffer [125 mM Tris-Cl (pH 6.8), 25% glycerol, 4% sodium dodecyl sulfate, 10% β-mercaptoethanol, 0.04% bromophenol blue] for 10 min, loaded on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels, and transferred to a polyvinylidene difluoride membrane (PVDF) (Schleicher & Schuell BioScience, Keene, NH). After blocking with 5% skim milk, membranes were incubated with a primary antibody for TACE protein (12, 28) followed by a horseradish peroxidase-conjugated anti-mouse antibody.
After detecting TACE protein, PVDF membranes were stripped in a prewarmed solution as previously described (21). The deprobed membrane was blocked with 5% skim milk and reprobed with an antibody for α-tubulin as a loading control. Finally, the membranes were incubated with SuperSignal West Femto Maximum Sensitivity Substrate (Pierce Biotechnology) and protein bands were visualized with the ChemiGenius Bioimaging system (Syngene, Frederick, MD).
Real-time PCR analysis.
The right lung was cut into small pieces and incubated overnight in RNAlater Stabilizing Reagent (Qiagen, Valencia, CA) to prevent RNA degradation. RNA was purified from cell lysates by using the RNeasy Mini Kit (Qiagen) following the manufacturer's instructions. To synthesize cDNA, 2 μg of total RNA was used with MultiScribe Reverse Transcriptase (Applied Biosystems, Foster City, CA). In the real-time PCR analysis, 20 ng of cDNA was used in the mixture of primers and the 2× SYBR green PCR master mix. The forward and reverse primers for Muc5ac was used as described (32) and primers for Gapdh endogenous control were generated by Primer Express 3.0 software (Applied Biosystems) (Table 1). The level of Muc5ac expression was normalized to the expression of Gapdh and was plotted on a logarithmic scale.
Table 1.
Primers for real-time PCR
Enzyme-linked immunosorbent assay.
To quantify the concentration of TNF-α in bronchoalveolar lavage (BAL) fluid collected from mice, ELISA was performed by using a mouse TNF-α kit (eBioscience, San Diego, CA) following the manufacturer's instructions.
Statistical analysis.
Results are expressed as means ± SD. Two groups were compared using Student's t-test. A between-group comparison of means was examined by one-way ANOVA followed by Tukey's post hoc analysis. A P value of less than 0.05 was regarded as indicative of a significant difference (*P < 0.05).
RESULTS
Conditional deletion of Tace in floxed-Tace/Cre mice.
Western blot analysis was used to detect TACE protein in Taceflox/flox R26CreER+/− mice (Tace conditional deletion mice) and Taceflox/floxR26CreER−/− mice (wild-type mice). Both genotypes received five consecutive tamoxifen injections to ensure that depletion of TACE was not caused by the toxicity of tamoxifen. In pilot experiments (not shown), depletion of TACE was detectable from day 3 after the last tamoxifen injection.
Tace mRNA expression was also determined by real-time PCR. The primers used for real-time PCR spanned Tace exon 2, which is removed upon activation of Cre because of the loxP sites flanking that exon. Tace mRNA expression in the Tace conditional deletion mice was 12.67% of that in the wild-type mice (Fig. 2A). Although the pro and mature forms of TACE protein were both detected in the lung lysates of both genotypes, the level of mature TACE was much higher in the wild-type mice than in the Tace conditional deletion mice after tamoxifen injection (Fig. 2B).
Fig. 2.

Detection of Tace mRNA and tumor necrosis factor-α converting enzyme (TACE) protein expression using real-time PCR and Western blot analysis, respectively. Intraperitoneal injection of tamoxifen (daily for 5 days, 0.04 mg/g of body wt) in Taceflox/flox/Cre+/− mice (Tace conditional deletion mice) reduced the levels of Tace mRNA expression (A; P < 0.01, mean ± SE, n = 3 independent experiments) and TACE protein (B) expression detected on day 7 after the last tamoxifen injection, compared with those detected in Taceflox/flox/Cre−/− mice (wild-type mice) injected with tamoxifen on the same dose schedule. Semiquantitative measurement of TACE protein normalized to α-tubulin from 3 independent experiments (C).
Goblet cell metaplasia in mice exposed to HNE.
We collected the lungs on study day 11 and used PAS staining to determine the goblet cell index in four sample groups: wild-type mice and Tace conditional deletion mice exposed to PBS (control groups), and wild-type mice and Tace conditional deletion mice exposed to HNE. The goblet cell index ranges from 0 to 4, with 4 indicating the highest concentration of goblet cells, as determined through blinded scoring performed by an investigator with anatomic pathology training and experience.
Under normal conditions, mice express a limited number of goblet cells in the lungs. The lungs of the wild-type mice and the Tace conditional deletion mice that were exposed to PBS had a goblet cell index level of 0, and there was no detectable difference in the number of goblet cells at the basal level between the two genotypes (Fig. 3, A and C).
Fig. 3.

Periodic acid-Schiff (PAS) staining of the airway. A–D: PAS staining was performed to visualize goblet cells in the lungs harvested on day 11 after the first exposure to HNE. Taceflox/flox/Cre−/− mice (wild-type mice) that were instilled with HNE developed goblet cell metaplasia, but HNE-mediated goblet cell metaplasia was attenuated in Taceflox/flox/Cre+/− mice (Tace conditional deletion mice). PBS instillation was used as a negative control and did not induce goblet cell metaplasia in either genotype. E: the goblet cell index determined by PAS staining shows that HNE significantly increased the number of goblet cells in the airways of wild-type mice (n = 10) compared with Tace conditional deletion mice (n = 14).
GCM was induced in the wild-type mice that were exposed to HNE (Fig. 3B), compared with the mice that were exposed to PBS; the goblet cell index level of these airways ranged from 0 to 4, with a median of 1.5 (Fig. 3E). However, in the Tace conditional deletion mice, HNE-induced GCM was significantly attenuated (Fig. 3D); the goblet cell index level ranged from 0 to 2, with a median of 0.4 (Fig. 3E) (P < 0.05, compared with wild-type mice).
To confirm that the PAS-positive cells were truly MUC5AC-positive cells, we performed immunohistochemical staining on the lung tissue samples using a monoclonal antibody against MUC5AC. Tissue from wild-type mice exposed to HNE, which was PAS positive, consistently showed MUC5AC-positive staining as well (representative image shown in Fig. 4A). In contrast, we did not detect MUC5AC-positive cells in the Tace conditional deletion mice that were exposed to HNE (representative image shown in Fig. 4B).
Fig. 4.

Immunohistochemical staining of MUC5AC. Immunohistochemical staining of MUC5AC was performed to specifically immunolabel goblet cells (×200 magnification, A and B). Strong immunostaining was detected on the tissue from Taceflox/flox/Cre−/− mice (wild-type mice) that were exposed to HNE, whereas no staining was observed on the tissue from Taceflox/flox/Cre+/− mice (Tace conditional deletion mice) that were exposed to HNE. The dotted areas in A and B are magnified in C and D. Secondary antibody alone (E) was used to confirm the specificity of the MUC5AC antibody. Scale bars, 100 μm.
We also measured the expression of Muc5ac mRNA on day 4 after the last HNE exposure (day 11 after the initial HNE exposure). There was a slight induction of Muc5ac mRNA expression in the wild-type and Tace conditional deletion mice after HNE exposure, although this failed to reach statistical significance in either genotype. In addition, there was no significant difference in the transcript levels of Muc5ac between the two genotypes, in contrast to the clearly different levels of goblet cells present on day 11 after the initial HNE exposure (Fig. 4C).
Tissue inflammation in mice exposed to HNE.
As a secondary outcome, we performed H&E staining on the lung tissues to determine the level of inflammation in the mice exposed to HNE. The degree of inflammation was scored again in a blinded fashion; the scoring ranged from 0 to 5, with 5 indicating the highest level of inflammation. The range of inflammation was 0 to 2 in both the wild-type mice and the Tace conditional deletion mice (Fig. 5). There was no significant difference between the genotypes.
Fig. 5.

Muc5ac mRNA expression, tissue inflammation index, and concentration of TNF-α in bronchoalveolar lavage (BAL) fluid. A: Muc5ac mRNA expression in homogenized lung tissue was determined by real-time PCR analysis and normalized to the expression of Gapdh. B: tissue inflammation, which was determined by hematoxylin and eosin staining, was not significantly different between the genotypes. Number of mice used for each condition: Taceflox/flox/Cre−/− with PBS (wild-type-PBS), n = 6; Taceflox/flox/Cre−/− with HNE (wild-type-HNE), n = 9; Taceflox/flox/Cre+/− with PBS (Tace conditional deletion-PBS), n = 3; Taceflox/flox/Cre+/− with HNE (Tace conditional deletion-HNE), n = 14. C: concentration of TNF-α in BAL fluid. Number of mice used for each condition: Taceflox/flox/Cre−/− with PBS (wild-type-PBS), n = 2; Taceflox/flox/Cre−/− with HNE (wild-type-HNE), n = 3; Taceflox/flox/Cre+/− with PBS (Tace conditional deletion-PBS), n = 3; Taceflox/flox/Cre+/− with HNE (Tace conditional deletion-HNE), n = 5.
We also quantified the concentration of TNF-α in BAL collected from mice on the same day as we performed other observations. Surprisingly, TACE deletion did not attenuate the TNF-α levels in BAL fluid, as shown in Fig. 5C.
DISCUSSION
We conducted this study to determine whether the conditional deletion of Tace attenuates HNE-induced GCM in vivo. The strength of our study is the Cre-loxP conditional deletion system we utilized, which allowed us to study a gene deletion that otherwise results in perinatal lethality (23). We confirmed that HNE exposure in wild-type mice led to GCM, as measured by PAS and MUC5AC staining, and we now report that HNE-induced GCM is significantly attenuated in Tace conditional deletion mice, a finding that affirms our hypothesis that TACE is a regulator of HNE-induced GCM in vivo.
Despite definitive histological evidence of GCM and positive MUC5AC immunostaining, we did not observe significant changes in Muc5ac mRNA expression at day 11, 4 days after the last HNE exposure. This is likely the result of transient changes of message level returning to baseline at 4 days after the last HNE exposure. Alternatively, the difference between MUC5AC immunostaining and Muc5ac mRNA expression could be accounted for by a decrease in mucin secretion or an increase in mucin synthesis in Tace conditional deletion mice. In addition to GCM, infiltration of inflammatory cells is often present in patients with chronic airway diseases and has been previously noted in response to HNE exposure (32). However, at day 4 after the last HNE exposure (day 11 after the first HNE exposure) an assessment of tissue inflammation, using H&E staining to visualize infiltrating inflammatory cells, showed indistinguishable levels of tissue inflammation in Tace conditional deletion mice and control mice. Similarly, TNF-α levels were indistinguishable in the BAL fluid obtained from wild-type and Tace conditional deletion mice. These results suggest the conditional deletion of Tace acted selectively on GCM without altering the inflammatory response to HNE, although caution is warranted in interpreting our findings because a previous study by Voynow et al. (32) using the same HNE exposure noted a maximal inflammatory response at day 8 that was greatly diminished by day 11 after the first HNE exposure.
TNF-α is an important proinflammatory cytokine that is synthesized as a membrane-anchored precursor form and shed by TACE. TNF-α has previously been linked to mucin gene expression in vitro and in vivo (4, 15), making it a candidate to mediate the functional effects of the TACE deletion observed in our study. However, our analysis of BAL fluid revealed that TNF-α levels were increased after HNE exposure in both wild-type and Tace-deficient mice, in contrast to the divergent effects of HNE on goblet cell metaplasia observed in these two genotypes. Thus, although TNF-α is one of the major substrates of TACE, our results strongly suggest that HNE-mediated GCM does not depend on TNF-α levels in vivo. Although somewhat surprising, the increased TNF-α levels in Tace-deficient mice could be explained by the activation of other enzymes capable of processing TNF-α processing, including MMP7, ADAM10, and ADAM19 (11). In particular, ADAM10 is known to function as a TNF-α sheddase when TACE is deficient in vivo and in vitro (17, 19).
Taken together, these results demonstrate that TACE, but not TNF-α, plays a necessary role in HNE-induced GCM. Previous evidence has linked GCM to EGFR activation (5, 16, 25) and TACE to shedding of bioactive EGFR ligands (2). Our results are consistent with conditional deletion of Tace attenuating GCM through inhibition of EGFR activation, which builds on evidence that EGFR is a prominent mediator in airway remodeling events, including GCM (3). TACE proteolytically sheds a variety of EGFR ligands, such as amphiregulin (18), TGF-α (14, 29), and heparin-binding epidermal growth factor-like growth factor (30), which together contribute to the activation of EGFR and the development of GCM in different model systems. We previously showed that TACE deletion attenuated EGFR-mediated responses in cultured mouse tracheal epithelial cells in vitro (30), demonstrating that TACE acts as a common upstream mediator of EGFR ligand shedding in these cells. Thus our results are consistent with a critical role for TACE in HNE-induced airway remodeling and suggest that further efforts should focus on the specific ligands that link TACE to GCM in vivo.
In conclusion, the conditional deletion of TACE by use of a Cre-loxP system significantly attenuated the number of goblet cells induced by HNE exposure, which demonstrates that TACE is a regulator of HNE-induced GCM in mice. This finding suggests that TACE could be a potential target in the treatment of GCM in patients with chronic airway diseases.
GRANTS
This research was funded by National Heart, Lung, and Blood Institute Grants HL88028 (J. M. Drazen) and T32-HL007118 (J. Park).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
J.-A.P. and J.M.D. conception and design of research; J.-A.P., A.S.S., T.S., and D.I.K. performed experiments; J.-A.P. and L.K. analyzed data; J.-A.P., L.K., D.J.T., and J.A.V. interpreted results of experiments; J.-A.P. prepared figures; J.-A.P. drafted manuscript; J.-A.P., L.K., D.J.T., and J.M.D. edited and revised manuscript; J.-A.P., A.S.S., T.S., L.K., D.I.K., D.J.T., J.A.V., and J.M.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Carl Blobel at Weill Cornell Medical College for providing Taceflox/flox mice, Dr. Shore at Harvard School of Public Health for helpful discussions, and Dr. Joo-Hyeon Lee at Boston Children's Hospital for helping to acquire histological images.
Present address of T. Shiomi: Department of Pulmonary Medicine, Kawasaki Municipal Ida Hospital, 2-27-1 Ida, Nakahara-ku, Kawasaki City, Kanagawa 211-0035, Japan.
REFERENCES
- 1. Arai N, Kondo M, Izumo T, Tamaoki J, Nagai A. Inhibition of neutrophil elastase-induced goblet cell metaplasia by tiotropium in mice. Eur Respir J 35: 1164–1171, 2010 [DOI] [PubMed] [Google Scholar]
- 2. Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol 6: 32–43, 2005 [DOI] [PubMed] [Google Scholar]
- 3. Burgel PR, Nadel JA. Roles of epidermal growth factor receptor activation in epithelial cell repair and mucin production in airway epithelium. Thorax 59: 992–996, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Busse PJ, Zhang TF, Schofield B, Kilaru S, Patil S, Li XM. Decrease in airway mucous gene expression caused by treatment with anti-tumor necrosis factor alpha in a murine model of allergic asthma. Ann Allergy Asthma Immunol 103: 295–303, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cohn L. Mucus in chronic airway diseases: sorting out the sticky details. J Clin Invest 116: 306–308, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fahy JV, Kim KW, Liu J, Boushey HA. Prominent neutrophilic inflammation in sputum from subjects with asthma exacerbation. J Allergy Clin Immunol 95: 843–852, 1995 [DOI] [PubMed] [Google Scholar]
- 7. Fahy JV, Schuster A, Ueki I, Boushey HA, Nadel JA. Mucus hypersecretion in bronchiectasis. The role of neutrophil proteases. Am Rev Respir Dis 146: 1430–1433, 1992 [DOI] [PubMed] [Google Scholar]
- 8. Fischer BM, Cuellar JG, Diehl ML, deFreytas AM, Zhang J, Carraway KL, Voynow JA. Neutrophil elastase increases MUC4 expression in normal human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 284: L671–L679, 2003 [DOI] [PubMed] [Google Scholar]
- 9. Fischer BM, Voynow JA. Neutrophil elastase induces MUC5AC gene expression in airway epithelium via a pathway involving reactive oxygen species. Am J Respir Cell Mol Biol 26: 447–452, 2002 [DOI] [PubMed] [Google Scholar]
- 10. Foster WM, Adler KB, Crews AL, Potts EN, Fischer BM, Voynow JA. MARCKS-related peptide modulates in vivo the secretion of airway Muc5ac. Am J Physiol Lung Cell Mol Physiol 299: L345–L352, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11. Horiuchi K, Kimura T, Miyamoto T, Takaishi H, Okada Y, Toyama Y, Blobel CP. Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J Immunol 179: 2686–2689, 2007 [DOI] [PubMed] [Google Scholar]
- 12. Horiuchi K, Morioka H, Takaishi H, Akiyama H, Blobel CP, Toyama Y. Ectodomain shedding of FLT3 ligand is mediated by TNF- converting enzyme. J Immunol 182: 7408–7414, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim KC, Wasano K, Niles RM, Schuster JE, Stone PJ, Brody JS. Human neutrophil elastase releases cell surface mucins from primary cultures of hamster tracheal epithelial cells. Proc Natl Acad Sci USA 84: 9304–9308, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kohri K, Ueki IF, Nadel JA. Neutrophil elastase induces mucin production by ligand-dependent epidermal growth factor receptor activation. Am J Physiol Lung Cell Mol Physiol 283: L531–L540, 2002 [DOI] [PubMed] [Google Scholar]
- 15. Kuwahara I, Lillehoj EP, Koga T, Isohama Y, Miyata T, Kim KC. The signaling pathway involved in neutrophil elastase stimulated MUC1 transcription. Am J Respir Cell Mol Biol 37: 691–698, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Le Cras TD, Acciani TH, Mushaben EM, Kramer EL, Pastura PA, Hardie WD, Korfhagen TR, Sivaprasad U, Ericksen M, Gibson AM, Holtzman MJ, Whitsett JA, Hershey GK. Epithelial EGF receptor signaling mediates airway hyperreactivity and remodeling in a mouse model of chronic asthma. Am J Physiol Lung Cell Mol Physiol 300: L414–L421, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Le Gall SM, Bobe P, Reiss K, Horiuchi K, Niu XD, Lundell D, Gibb DR, Conrad D, Saftig P, Blobel CP. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor alpha, L-selectin, and tumor necrosis factor alpha. Mol Biol Cell 20: 1785–1794, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Manzo ND, Foster WM, Stripp BR. Amphiregulin-dependent mucous cell metaplasia in a model of non-allergic lung injury. Am J Respir Cell Mol Biol 47: 349–357, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mezyk-Kopec R, Bzowska M, Stalinska K, Chelmicki T, Podkalicki M, Jucha J, Kowalczyk K, Mak P, Bereta J. Identification of ADAM10 as a major TNF sheddase in ADAM17-deficient fibroblasts. Cytokine 46: 309–315, 2009 [DOI] [PubMed] [Google Scholar]
- 20. Nadel JA. Protease actions on airway secretions. Relevance to cystic fibrosis. Ann NY Acad Sci 624: 286–296, 1991 [DOI] [PubMed] [Google Scholar]
- 21. Park JA, Crews AL, Lampe WR, Fang S, Park J, Adler KB. Protein kinase C delta regulates airway mucin secretion via phosphorylation of MARCKS protein. Am J Pathol 171: 1822–1830, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Park JA, He F, Martin LD, Li Y, Chorley BN, Adler KB. Human neutrophil elastase induces hypersecretion of mucin from well-differentiated human bronchial epithelial cells in vitro via a protein kinase Cδ-mediated mechanism. Am J Pathol 167: 651–661, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Peschon JJ. An essential role for ectodomain shedding in mammalian development. Science 282: 1281–1284, 1998 [DOI] [PubMed] [Google Scholar]
- 24. Pettersen CA, Adler KB. Airways inflammation and COPD: epithelial-neutrophil interactions. Chest 121: 142S–150S, 2002 [DOI] [PubMed] [Google Scholar]
- 25. Puddicombe SM, Polosa R, Richter A, Krishna MT, Howarth PH, Holgate ST, Davies DE. Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J 14: 1362–1374, 2000 [DOI] [PubMed] [Google Scholar]
- 26. Rogers DF. Physiology of airway mucus secretion and pathophysiology of hypersecretion. Respir Care 52: 1134–1146; discussion 1146–1139, 2007 [PubMed] [Google Scholar]
- 27. Scheller J, Chalaris A, Garbers C, Rose-John S. ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol 32: 380–387, 2011 [DOI] [PubMed] [Google Scholar]
- 28. Schlondorff J, Becherer JD, Blobel CP. Intracellular maturation and localization of the tumour necrosis factor alpha convertase (TACE). Biochem J 347: 131–138, 2000 [PMC free article] [PubMed] [Google Scholar]
- 29. Shao MX, Nadel JA. Dual oxidase 1-dependent MUC5AC mucin expression in cultured human airway epithelial cells. Proc Natl Acad Sci USA 102: 767–772, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shiomi T, Tschumperlin DJ, Park JA, Sunnarborg SW, Horiuchi K, Blobel CP, Drazen JM. TNF-alpha-converting enzyme/a disintegrin and metalloprotease-17 mediates mechanotransduction in murine tracheal epithelial cells. Am J Respir Cell Mol Biol 45: 376–385, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thai P, Loukoianov A, Wachi S, Wu R. Regulation of airway mucin gene expression. Annu Rev Physiol 70: 405–429, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Voynow JA, Fischer BM, Malarkey DE, Burch LH, Wong T, Longphre M, Ho SB, Foster WM. Neutrophil elastase induces mucus cell metaplasia in mouse lung. Am J Physiol Lung Cell Mol Physiol 287: L1293–L1302, 2004 [DOI] [PubMed] [Google Scholar]
- 33. Voynow JA, Rubin BK. Mucins, mucus, sputum. Chest 135: 505–512, 2009 [DOI] [PubMed] [Google Scholar]