

Abstract
Ventilation at high tidal volume may cause lung inflammation and barrier dysfunction that culminates in ventilator-induced lung injury (VILI). However, the mechanisms by which mechanical stimulation triggers the inflammatory response have not been fully elucidated. This study tested the hypothesis that onset of VILI is triggered by activation of secretory group V phospholipase A2 (gVPLA2) in pulmonary vascular endothelium exposed to excessive mechanical stretch. High-magnitude cyclic stretch (18% CS) increased expression and surface exposure of gVPLA2 in human pulmonary endothelial cells (EC). CS-induced gVPLA2 activation was required for activation of ICAM-1 expression and polymorphonuclear neutrophil (PMN) adhesion to CS-preconditioned EC. By contrast, physiological CS (5% CS) had no effect on gVPLA2 activation or EC-PMN adhesion. CS-induced ICAM-1 expression and EC-PMN adhesion were attenuated by the gVPLA2-blocking antibody (MCL-3G1), general inhibitor of soluble PLA2, LY311727, or siRNA-induced EC gVPLA2 knockdown. In vivo, ventilator-induced lung leukocyte recruitment, cell and protein accumulation in the alveolar space, and total lung myeloperoxidase activity were strongly suppressed in gVPLA2 mouse knockout model or upon administration of MCL-3G1. These results demonstrate a novel role for gVPLA2 as the downstream effector of pathological mechanical stretch leading to an inflammatory response associated with VILI.
Keywords: endothelial cells, cyclic stretch, group V soluble phospholipase A2 inflammation, neutrophil adhesion, acute lung injury
acute respiratory distress syndrome (ARDS) is a severe pathological condition characterized by the formation of protein-rich pulmonary edema, hyaline membranes, and the influx of neutrophils into the airspace (31). Nearly all patients with ARDS require mechanical ventilation and are therefore at risk for ventilator-induced lung injury (VILI), which appears to be due in part to the uneven distribution of mechanical distension and lung injury in ARDS. Similarly to mechanically ventilated lungs, pathologically relevant levels of cyclic stretch (CS) applied to endothelial cell (EC) monolayers in vitro lead to exacerbation of agonist-induced endothelial barrier dysfunction, thus representing the two-hit model of VILI (5–7, 24).
Mechanical stress produced by mechanical ventilation also leads to the upregulation of an inflammatory response, the process known as biotrauma (13) and reflected by CS-induced cytokine production and leukocyte infiltration. CS-induced cytokine production by lung cells (macrophages, epithelium, endothelium) is well recognized and considered a prognostic factor of acute lung injury (ALI)/ARDS severity (2). The initial step of ventilator-induced leukocyte infiltration in the lung involves leukocyte adhesion to the lung vascular endothelium, which requires endothelial activation and surface expression of adhesion molecules intercellular adhesion molecule (ICAM)-1, vascular cell adhesion molecule (VCAM), and E-selectin (11). However, the effects of pathological CS on polymorphonuclear neutrophil (PMN)-EC interactions and CS-activated mechanisms driving these interactions in VILI settings remain poorly understood.
Secretory phospholipases A2 (PLA2) belong to a larger phospholipase A2 family of lipolytic enzymes that catalyze the cleavage of fatty acids from the sn-2 position of phospholipids, leading to the generation of free fatty acids and lysophospholipids (30). Elegant genetic studies demonstrated involvement of cytosolic phospholipase A2 in the ALI and pulmonary edema caused by lipopolysaccharide and zymosan administration or acid aspiration (23). Among the secreted PLA2s, gIIaPLA2, gVPLA2, and gXPLA2 have been implicated in a variety of inflammatory lung diseases, e.g., ALI, ARDS, sepsis, and asthma (1, 8, 22). However, the relative contributions of secretory PLA2s in these diseases remain unclear. Secretory gVPLA2 is a 14-kDa proinflammatory enzyme that has diverse biological effects, including airway inflammation, airway hyperresponsiveness, cell adhesion, transcellular communication, and generation of lipid mediators (22, 30). Among other cell types, secretory gVPLA2 is expressed in ECs (30). In activated state, gVPLA2 translocates from submembrane compartment to the outer cell membrane and cleaves membrane phosphatidyl choline, thus generating bioactive arachidonic metabolites (9, 22). However, the involvement of gVPLA2 in pathogenesis of VILI remains completely unknown.
This study investigated the mechanism initiating lung inflammatory response to pathological mechanical ventilation. We tested the hypothesis that, in addition to exacerbation of agonist-induced lung vascular leak leading to pulmonary edema, pathological CS may also trigger development of lung inflammation by stimulating endothelial cell-leukocyte interactions, leading to lung infiltration with inflammatory cells. We investigated a role of gVPLA2 as a mechanical stress-sensitive mediator of lung endothelial activation, stimulating EC-PMN interactions and lung injury.
MATERIALS AND METHODS
Reagents.
Recombinant human gVPLA2 protein and MCL-3G1, a mAb-directed against gVPLA2, were purchased from Cayman Chemical (Ann Arbor, MI). Immunofluorescent reagents were Texas red phalloidin (Invitrogen, Carlsbad, CA) and mouse anti-VE-cadherin antibody (Santa Cruz Biotechnology, Santa Cruz, CA). All other reagents (including LPS) were obtained from Sigma-Aldrich Chemical (St. Louis, MO), unless otherwise noted.
Cell culture.
Human pulmonary artery ECs (HPAECs) were obtained from Lonza (Walkersville, MD) and were cultured according to the manufacturer's instructions. ECs (passages 6–9) were grown in endothelial growth medium-2 at 37°C in a 5% CO2 incubator. The medium was changed 1 day before experimentation.
Cell culture under CS.
CS experiments were performed using FX-4000T Flexcell Tension Plus system (Flexcell International, McKeesport, PA) equipped with a 25-mm BioFlex Loading station (4, 5). Untreated EC with or without MCL-3G1 or 10 μM LY-311727 or cells after siRNA transfection were exposed to pathological or physiological CS (18% or 5% distension, respectively, sinusoidal wave, 25 cycles/min, 0.5, 1, 2, 4, or 24 h) to recapitulate a high- and low-tidal-volume mechanical ventilation regimen. Control BioFlex plates with static EC culture were placed in the same cell culture incubator and processed similarly to CS-preconditioned cells. At the end of experiment, cell lysates were collected for Western blot analysis or mRNA expression analysis.
siRNA transfection.
To suppress gVPLA2 transcription, cells were treated with gene-specific siRNA duplexes. Predesigned standard purity siRNA sets (Homo sapiens) were designed and synthesized by Invitrogen (Grand Island, NY). The corresponding target mRNA sequences for the siRNAs were as follows: 1, GGUGACAGGGAAGAACGCCCUGACA; 2, ACGAGCUUCCGGUCACAGGCACAGA; 3, UUUAGGUCCAGCAAGCCUCCUUGCA. Transfection of EC with siRNA was performed as previously described (29). After 48 h of transfection, cells were used for CS experiments. At the end of experiments, HPAEC were harvested for Western blot verification of specific protein depletion. Nonspecific, nontargeting siRNA (Dharmacon, Lafayette, CO) was used as a control treatment.
Immunoblotting analysis of gVPLA2 and ICAM-1.
Treated ECs were subsequently washed with cold Ca2+/Mg2+-free PBS and lysed with 0.3% SDS lysis buffer containing protease inhibitors (1 mM EDTA, 1 mM PMSF, 1 mM sodium orthovanadate, 1 mM sodium fluoride, 0.2 TIU/ml aprotinin, 10 mM leupeptin, and 5 mM pepstatin A). Sample proteins were separated with SDS-PAGE using 8% (ICAM-1) or 15% (gVPLA2) acrylamide gels and transferred into nitrocellulose membranes (Bio-Rad, Hercules, CA). The membrane was blocked with 1% BSA in TBS with Tween buffer and then immunoblotted with ICAM-1 mAb (Santa Cruz Biotechnology) or MCL-3G1 mAb (Cayman Chemicals), followed by secondary antibodies conjugated to horseradish peroxidase (1:5,000, room temperature, 30 min). Protein of interest was analyzed by an enhanced chemiluminescence system (Amersham, Arlington Heights, IL).
mRNA expression analysis.
Total RNA was isolated from ECs using TRIzol reagent, per manufacturer's instructions (Life Technology, Rockville, MD). RNA samples were quantified by measuring optical density at 260 nm using the NanoDrop ND 1,000 Spectrophotometer (Thermo Scientific, Wilmington, DE). cDNA was synthesized from 1 mg/ml of total RNA, per the manufacturer's instructions, using the Invitrogen SuperScript III First-Strand Synthesis SuperMix for qRT-PCR kit (no. 11752--50). The primers for gVPLA2 were as follows: sense, 59-TTGGTTCCTGGCTTGTAGT GTG-39; antisense, 59-TGGGTTGTAGCTCCGTAGGTTT-39. For 18S rRNA, the primers used were as follows: sense, 59-AAACGGCTACCACATCCAAG-39; antisense, 59-CCTCCAATGGATCCTCGTTA-39. Semiquantitative RT-PCR was performed using the Bio-Rad Thermal Cycler system (Bio-Rad) and AmpliTaq Gold(R) DNA Polymerase (cat. no. 4,311,806; Applied Biophysics, Troy, NY) using the following PCR conditions: 10 min at 93°C, followed by 40 cycles of 30 s at 95°C, 45 s at 60°C, 50 s at 72°C, and 10 min at 72°C. PCR products were separated by electrophoresis in a 1.5% agarose gel and then stained with ethidium bromide.
Analysis of membrane-bound gVPLA2 and ICAM-1 expression.
After mechanostimulation, ECs on each well were dispersed by trypsinization and resuspended in PBS. The cells were fixed with 1% paraformaldehyde in H2O and stored at 4°C. ECs were incubated with 10 mg/ml of mAb directed against gVPLA2 (MCL-3G1), mAb against ICAM-1, or isotype-matched control IgG1 Ab at 4°C. After 60 min, treated ECs were washed twice with PBS solution plus 0.2% BSA before addition of an excess of FITC-conjugated goat anti-mouse IgG for 45 min at 4°C. Final washing was performed to remove the excess Ab before flow cytometric analysis (BD Biosciences, Franklin Lakes, NJ). Fluorescence intensity was determined on at least 10,000 cells from each sample as acquired using FACscan flow cytometry (BD Bioscience). For quantitative evaluation, cell populations were gated manually, and mean fluorescence intensity was determined using FlowJo software (Tree Star, Ashland, OR).
Human PMN isolation.
Venous blood from normal human subjects was collected based on a protocol approved by the University of Chicago Institutional Review Board and was in compliance with the University of Chicago and U.S. governmental guidelines for studies in which donors are not participating subjects. Informed consent was obtained from all volunteers in this study before participation. PMNs were isolated by Ficoll-Paque sedimentation as we described previously (17). Cells were resuspended in HBSS buffer + Ca++/0.2% BSA before counting. Purity of PMN on hematoxylin and eosin(H and E)-stained cytoslides was ∼90–95%. Cells were kept on ice until use.
Neutrophil migration and adhesion assays.
Neutrophil chemotaxis was measured in a 96-well chemotaxis chamber (Neuroprobe, Gaithersburg, MD) as described previously (17). Briefly, freshly isolated neutrophils were placed in a 96-well chemotaxis chamber and incubated with 200 μl of preconditioned culture media, which was collected from static EC cultures or from pulmonary EC grown on BioFlex and exposed to 4-h CS at 18% amplitude. Preliminary experiments have established that the number of cells (4 × 104 cells) used allows the optimal cell migration without clogging the pores of the Transwell filter of the upper chamber. Data were expressed as the percentage of cell migration. PMN adhesion to the CS-preconditioned ECs was assessed at the end of CS sessions by adding the neutrophils freshly isolated from healthy donors to the EC monolayers grown in the six-well BioFlex plates right after the CS experiment. Neutrophil adhesion on HPAEC was assessed as described previously (18). Neutrophil adhesion data were expressed as a percentage of adhesion for all treated groups.
Animals.
Homozygous gVPLA2 knockout (KO) (pla2g5−/−) mice (28) and littermate control (pla2g5+/+) mice were derived from C57/BL6 mice. C57BL/6 mice, 10–12 wk old, were housed in a pathogen-free biohazard level 2 facility maintained by The University of Chicago Animal Resources Center. The studies reported here conform to the principles outlined by the Animal Welfare and the National Health Services guidelines for the care and use of animals in biomedical research and were approved by the University of Chicago Institutional Review Board. In protocols detailed below, animals were anesthetized and were subjected to increased mechanical stress-mediated lung injury via high tidal volume ventilation (HTV).
Murine model of lung injury.
Wild-type control (pla2g5+/+) or gVPLA2 KO (pla2g5−/−) mice were weighed, and anesthesia was introduced by intraperitoneal injection of ketamine-xylazine (100 mg/kg:20 mg/kg). Tracheostomy was performed, and then the animal was connected to a small-computerized animal ventilator (Flexivent; Scireq, Vancouver, Canada) in room air, with a tidal volume of 30 ml/kg, 70 breaths per min for 4 h. Nonmechanically ventilated mice were used as controls. In separate experiments, pla2g5+/+ mice were pretreated with 20 μg MCL-3G1 intraperitoneally before mechanical ventilation.
BAL protein and cellular analysis.
Mice underwent BAL (bronchial airway lavage) of both lungs with 0.5 ml PBS while gently aspirating the fluid. The procedure was performed three times, and the recovered BAL fluid was used to measure total protein according to the manufacturer's manual (BCA Protein Assay Kit; Bio-Rad). Cell pellets were examined for total cell count using a hemocytometer and differential cell count using cytocentrifugation and Diff-Quik staining (Dade Diagnostics, Deerfield, IL).
Total lung myeloperoxidase content.
This assay was determined from homogenized lungs and normalized based on its protein contents. Homogenates were then added with 100 μl of HBSS + 10% FBS buffer and 100 μl developing solution [8 ml 100 nM NaH2PO4 (pH = 5.5), 1,000 μl 10% hexadecyl trimethyl ammonium bromide, 3 μl 30% hydrogen peroxide, 1,000 μl 10% o-dianisidine dihydrochloride]. The reaction mixture was terminated with introduced 50 μl sulfuric acid, and absorbance was measured at 405 nm in a microplate reader (Thermomax; Molecular Devices, Menlo Park, CA). A standard curve was generated on each sample plate, and a linear regression curve was generated.
Lung histology.
For histopathological lung analysis, left lung was harvested from each treated mouse and immediately fixed with 10% buffered formalin overnight, followed by embedding in paraffin. Airway microsections were used for histological evaluation by H and E staining. Cellular infiltration in treated lungs was examined by light microscopy.
gVPLA2 mRNA expression.
Total RNA was isolated from whole lungs of treated animals for gVPLA2 expression using TRIzol reagent (Life Technologies, Rockville, MD) as previously described (21). cDNA was synthesized using 0.5 μg of total RNA in a 20-μl reverse transcriptase reaction using iScript cDNA synthesis kit (Bio-Rad Laboratories). The amount of gVPLA2 mRNA expression was normalized to the housekeeping gene 18S rRNA and calculated according to the MyiQ Single Color Real-Time PCR Detection System Software (Bio-Rad).
Lung capillary leakage.
Treated animals were injected with Evans blue dye (30 mg/kg) into the external jugular vein 2 h before the termination of the experiment. Due to high binding affinity of Evans blue dye to serum albumin, albumin-bound Evans blue moves into the lung parenchyma when the vascular barrier in the lung is compromised. Homogenized lungs were incubated with 2 ml formamide and centrifuged at 12,000 g × 20 min. The optical density of the supernatant was then determined spectrophotometrically at 620 nm. The extravasated Evans blue concentration in lung homogenate was calculated against a standard curve and was expressed as micrograms of Evans blue dye per gram of lung.
Measurement of cytokines and chemokines.
The concentrations of keratinocyte chemokine (KC), macrophage inflammatory protein (MIP)-1, TNF-α, and IL-13 in mouse BAL fluid samples were measured using a Mouse Cytokine Multiplex Panel according to the manufacturer's protocol (Millipore, Billerica, MA). For IL-8 measurements in preconditioned medium of human pulmonary EC cultures, supernatants from treated EC were collected and centrifuged to remove debris. IL-8 levels were determined by ELISA (ELISA MAX Deluxe set; Biolegend, San Diego, CA) following manufacturer's protocol. Absorbance was read at 450 nm within 30 min in microplate reader (Thermomax; Molecular Devices, Menlo Park, CA). Standard curves were generated with expected minimum detectable concentration of 8 pg/ml.
Statistical analysis.
The data are expressed as the means ± SE for each group. Individual statistical comparisons of paired data were assessed by Student's t-test, and P < 0.05 was considered to be statistically significant. Where multiple comparisons were made, differences among the populations were evaluated by ANOVA followed by Bonferroni correction.
RESULTS
Pathological CS increases gVPLA2 expression in human pulmonary EC.
Exposure of HPAEC to 18% CS (4 h) resulted in 3.5-fold increase in gVPLA2 mRNA levels. Accordingly, 18% CS induced time-dependent increase in gVPLA2 protein expression (Fig. 1, A and B). Because gVPLA2 functional activity is associated with its translocation to the cell surface, the next experiments used flow cytometric analysis to examine effects of 18% CS on gVPLA2 surface expression. 18% CS increased gVPLA2 surface expression in HPAEC in a time-dependent manner (Fig. 1C). Increased gVPLA2 surface expression was observed after 30 min of 18% CS stimulation and reached maximal levels by 24 h of 18% CS stimulation. To evaluate specific involvement of gVPLA2 in signaling induced by pathological mechanical stimulation, we compared gVPLA2 surface expression in pulmonary EC exposed to physiological (5% CS) and pathological (18% CS) stretch levels. In contrast to activation of gVPLA2 surface expression by 18% CS, EC stimulation with 5% CS had no effect on surface-expressed gVPLA2 (Fig. 1D).
Fig. 1.

Effect of pathological cyclic stretch (CS) on secretory group V phospholipase A2 (gVPLA2) mRNA and protein content and surface expression. Human pulmonary endothelial cells (EC) were exposed to 18% CS (4 or 24 h). A: analysis of gVPLA2 mRNA levels in static and 18% CS-exposed EC was performed by RT-PCR, as described in materials and methods. B: analysis of CS-induced gVPLA2 protein expression by Western blot. C: time-dependent effect of 18% CS stimulation on gVPLA2 surface expression was evaluated by fluorescence-activated cell sorting (FACS) scan analysis as described in materials and methods. Top: distribution curves of gVPLA2 intensity in gVPLA2-positive EC. Bottom: bar graph presents pooled data of 6 independent FACS measurement experiments; *P < 0.05 vs. static. D: gVPLA2 surface expression was evaluated as described in materials and methods. *P < 0.05 vs. static. MFI, mean fluorescence intensity.
gVPLA2 mediates PMN adhesion to mechanically stimulated human pulmonary EC.
Consistent with stretch-induced activation of gVPLA2, 18% CS preconditioning of pulmonary EC stimulated neutrophil adhesion, which increased with duration of EC exposure to pathological stretch. In contrast, EC exposure to 5% CS did not stimulate EC-PMN interactions (Fig. 2A). Previous studies demonstrated stimulation of IL-8 production by lung epithelial cells and ECs (15, 26). Increased IL-8 levels generate chemotaxic signal and further promote neutrophil recruitment to inflammatory sites. We found that IL-8 production was stimulated by 18% CS, whereas 5% CS treatment was without effect (Fig. 2B).
Fig. 2.

Differential effects of 5% CS and 18% CS on gVPLA2 surface expression, EC-polymorphonuclear neutrophil (PMN) adhesion and IL-8 production. Human pulmonary EC were exposed to 5% CS or 18% CS (4 h). A: PMN adhesion to CS-preconditioned EC. B: IL-8 production in EC preconditioned medium was evaluated as described in materials and methods. *P < 0.05 vs. static.
gVPLA2 mediates activation of EC-neutrophil adhesion and neutrophil chemotaxis induced by stimulation of pulmonary EC with 18% CS.
Involvement of gVPLA2 mechanism in 18% CS-induced PMN adhesion and PMN chemotaxis response to preconditioned medium from CS-stimulated EC was evaluated in the next experiments. EC were exposed to 4 h of 18% CS in the presence or absence of gVPLA2 blocking antibody MCL-3G1 or secretory phospholipase A2 inhibitor LY311727 (12). Both these interventions abolished the PMN adhesion to 18% CS-stimulated EC (Fig. 3A), IL-8 production by 18% CS-stimulated EC (Fig. 3B), and PMN migration stimulated by preconditioned medium from CS-stimulated EC (Fig. 3C).
Fig. 3.

gVPLA2 mediates EC-PMN adhesion, EC IL-8 production, and PMN migration stimulated by preconditioned medium from EC exposed to pathological CS. Human pulmonary EC were exposed to 18% CS (4 h) with our without addition of gVPLA2 blocking antibody MCL-3G1 (25 μg/ml) and PLA2 inhibitor LY311727 (10 μg/ml). A: PMN adhesion to CS-preconditioned EC. B: IL-8 production by CS-stimulated EC. C: PMN adhesion to CS-preconditioned EC was evaluated as described in materials and methods. *P < 0.05; ND, data not statistically different; ST, static.
siRNA-induced knockdown of gVPLA2 in pulmonary EC was used as an alternative approach to test a role of gVPLA2 in stretch-induced activation of PMN-EC interactions. Preliminary testing showed potent inhibition of gVPLA2 protein expression by all three gene-specific siRNA duplexes (Fig. 4A). Further studies used a combination of all three siRNAs (total concentration 50 nM) to efficiently downregulate gVPLA2 expression. Depletion of endogenous gVPLA2 significantly reduced basal levels of gVPLA2 surface expression detected by fluorescence-activated cell sorting (FACS) scanning approach and completely abolished upregulation of gVPLA2 surface expression in EC exposed to 4 h of 18% CS (Fig. 4B). Consistent with these results, gVPLA2 knockdown abolished PMN adhesion to 18% CS-preconditioned HPAEC (Fig. 4C).
Fig. 4.
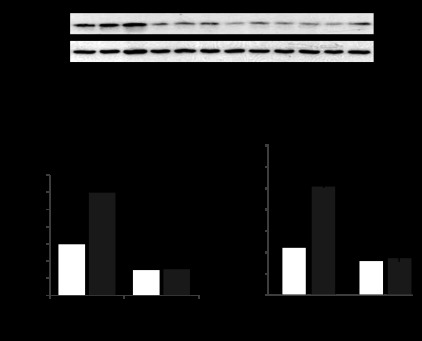
Effect of gVPLA2 knockdown in EC on stretch-induced PMN adhesion. Human pulmonary EC were treated with gVPLA2-specific siRNA as described in materials and methods before exposure to 18% CS (4 h). A: validation of siRNA-induced gVPLA2 protein depletion by Western blot. B: siRNA-induced gVPLA2 knockdown abrogates CS-induced gVPLA2 surface expression. C: EC-specific gVPLA2 knockdown abrogates PMN adhesion to 18% CS-stimulated EC. *P < 0.05.
gVPLA2 mediates activation of endothelial ICAM-1 expression by 18% CS.
Because ICAM-1 plays an important role in initiation of PMN-EC adhesion, we examined cell surface ICAM-1 expression and total protein levels in CS-stimulated pulmonary EC. 18% CS induced a time-dependent increase in the ICAM-1 surface expression evaluated by FACS scan (Fig. 5A). ICAM-1 protein levels were elevated after 4 h of 18% CS and continued to grow during 24 h of 18% CS (Fig. 5B). 18% CS stimulation in the presence of gVPLA2 blocking antibody abolished CS-induced activation of ICAM-1 surface expression in pulmonary EC (Fig. 5C).
Fig. 5.

Role of gVPLA2 in stretch-induced intercellular adhesion molecule (ICAM)-1 expression by pulmonary EC. A: human pulmonary EC were exposed to 18% CS (4 and 24 h), and ICAM-1 surface expression was evaluated by FACS scan analysis as described in materials and methods. B: Western blot analysis of CS-induced ICAM-1 protein expression by EC. C: ICAM-1 surface expression in EC exposed to 18% CS (4 h) in the absence or presence of gVPLA2 blocking antibody MCL-3G1. *P < 0.05.
Inhibition of ICAM-1 attenuates PMN adhesion but not stretch-induced gVPLA2 activation in pulmonary EC.
To examine whether CS-induced activation of ICAM-1 is upstream or downstream of gVPLA2, ICAM-1 blocking antibody or IgG Ab, an isotype matched control, were added to HPAEC before the 4-h stimulation with 18% CS. ICAM-1 blocking antibody attenuated increase in PMN adhesion to CS-preconditioned HPAEC, whereas control IgG Ab was without effect (Fig. 6A). In contrast, CS stimulation of HPAEC in the presence of ICAM-1 blocking antibody failed to inhibit stretch-induced gVPLA2 cell surface expression (Fig. 6B). These results demonstrate that CS-induced gVPLA2 activation is upstream of ICAM-1 in the pathway of activation of PMN-EC adhesion caused by VILI-relevant mechanical forces. The pathophysiological significance of CS-induced upregulation in VILI was further tested in vivo.
Fig. 6.

Involvement of ICAM-1 in CS-induced EC-PMN adhesion and gVPLA2 upregulation. A: human pulmonary EC were exposed to 18% CS (4 h) with or without addition of ICAM-1 blocking antibody (25 μg/ml), and PMN adhesion assay at the end of CS experiment was performed as described in materials and methods. B: gVPLA2 surface expression in EC exposed to 18% CS with or without ICAM-1 blocking antibody was evaluated by FACS scan analysis as described in materials and methods. *P < 0.05 vs. static.
gVPLA2 expression in vivo.
We first examined the mRNA expression of gVPLA2 in lung tissues obtained from control and HTV-exposed mice. In pla2g5+/+ mice, the gVPLA2 mRNA level was significantly increased after 4 h of HTV (0.33 ± 0.05 AU) compared with baseline control expression of 0.19 ± 0.05 AU for nonventilated pla2g5+/+ mice (P < 0.05) (Fig. 7A). As expected, gVPLA2 mRNA level in pla2g5−/− mice was negligible (0.01 ± 0.002 AU). All data were normalized per 18S housekeeping gene and expressed as the ratio of gVPLA2 mRNA/18S.
Fig. 7.
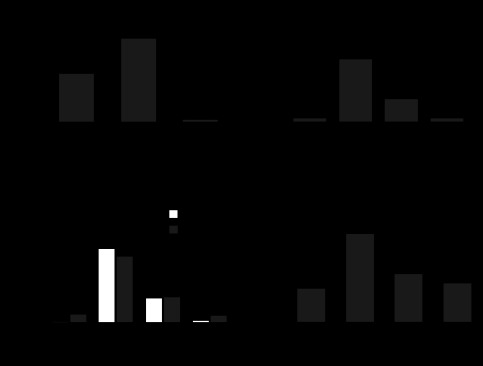
Role of gVPLA2 in leukocyte accumulation in bronchoalveolar lavage (BAL) fluid of mice exposed to high-tidal-volume mechanical ventilation (HTV). A: gVPLA2 mRNA levels in control and HTV-exposed wild-type littermate control (pla2g5+/+) with or without injection with gVPLA2 blocking antibody or gVPLA2 knockout (pla2g5−/−) mice were measured in lung homogenates by RT-PCR as described in materials and methods. The upregulation of mRNA was normalized as the ratio of mRNA/18S compared with vehicle-stimulated control airways. RT-PCRs were performed in triplicates. The data represent the means ± SD of 6 samples; *P < 0.05. Effect of gene knockout and gVPLA2 blocking antibody on total cell count in BAL fluid (B) from mice exposed to HTV. C: number of neutrophils and macrophages in BAL fluid. *P < 0.05 vs. HTV alone; **P < 0.01 vs. HTV alone. D: effects of gene knockout and gVPLA2 blocking antibody on myeloperoxidase (MPO) activity in mice exposed to HTV. *P < 0.01 vs. HTV alone in wild-type mice.
Pulmonary inflammation.
Evaluation of the BAL in pla2g5+/+ mice after 4 h of HTV revealed a significant increase in total cell count compared with the nonventilated control mice (Fig. 7B). By contrast, no significant difference was found between HTV-treated pla2g5−/− mice and control mice. Treatment of pla2g5+/+ mice with MCL-3G1 before HTV caused significant reduction of cellular infiltration (P < 0.05 vs. HTV-treated pla2g5+/+ mice). In contrast to the models of LPS-induced lung injury and inflammation, exposure to pathological mechanical ventilation induced much a lower extent of leukocyte recruitment to the BAL of ventilated mice with almost equal proportion of neutrophils and monocytes/macrophages in the mix (Fig. 7C). Although pla2g5−/− mice were seemingly protected from HTV-induced injury and increase in BAL cell count, treatment of pla2g5+/+ mice with MCL-3G1 decreased the infiltration of both neutrophils and macrophages after 4 h of HTV by 2.5-fold (P < 0.05 vs. HTV-treated pla2g5+/+ mice). HTV caused a significant increase in lung myeloperoxidase (MPO) activity compared with the control group (Fig. 7D). Lung MPO concentration, expressed as optical density per 150 μg/ml protein, increased to 0.22 ± 0.03 AU after 4 h of HTV compared with 0.08 ± 0.02 AU for nonventilated pla2g5+/+ control mice (P < 0.01). Intraperitoneal injection of MCL-3G1 before HTV significantly reduced MPO activity to 0.12 ± 0.01 AU compared with the HTV-treated pla2g5+/+ mice (P < 0.01), whereas HTV-treated pla2g5−/− mice had no significant effect (P = NS vs. nonventilated pla2g5+/+ control mice).
Lung histology, analysis of lung vascular leakage, and cytokine production.
Compared with nonventilated pla2g5+/+ control mice, HTV induced inflammatory cell infiltration (predominantly neutrophils and mononuclear cells) in both lung interstitium and alveolar compartments (Fig. 8A). Pretreatment with MCL-3G1 blocked HTV-induced inflammatory cell infiltration in lung tissue. Remarkably, lungs of pla2g5−/− mice were preserved against HTV-induced injury, and morphological patterns of HTV-treated pla2g5−/− mice were comparable with the nonventilated pla2g5+/+ mice.
Fig. 8.

Histological assessment of the effect of gVPLA2 inhibition on ventilator-induced lung injury and analysis of lung vascular leak. Wild-type littermate control (pla2g5+/+) with or without injection with gVPLA2 blocking antibody or gVPLA2 knockout (pla2g5−/−) were exposed to HTV. Spontaneously breathing animals were used as controls. A: histological analysis of lung tissue (×40 magnification). Whole lungs (4 to 6 animals from each experimental group) were agarose inflated in situ, fixed with 10% formalin, and used for histological evaluation by hematoxylin and eosin staining as described in materials and methods. B: protein concentration in BAL fluid from HTV-exposed wild-type littermate control (pla2g5+/+) mice with or without injection with gVPLA2 blocking antibody or gVPLA2 knockout (pla2g5−/−) mice. C: lung vascular leak was assessed by measurements of Evans blue leakage into lung tissues, as described in materials and methods. Quantitative analysis was performed by spectrophotometric measurement of Evans blue extracted from the lung tissue samples. The data represent the means ± SE of 6 samples; *P < 0.05 vs. HTV alone in wild-type mice. D: images of lung preparations depicting Evans blue accumulation in the lung tissues.
The BAL total protein concentration, a gross measurement of capillary permeability, was also found to be significantly increased in HTV-treated pla2g5+/+ mice (1,223.83 ± 36.2 μg/ml) than nonventilated pla2g5+/+ control mice (Fig. 8B; P < 0.01). BAL protein concentration was significantly decreased in pla2g5+/+ mice treated with gVPLA2 blocking antibody MCL-3G1 (795.01 ± 95.2 μg/ml) and in HTV-challenged pla2g5−/− mice (504.26 ± 36.2 μg/ml; P < 0.01 vs. HTV-treated pla2g5+/+ mice).
HTV-induced lung injury caused significant lung vascular leak detected by Evans blue dye accumulation in the lung parenchyma, which was evident in pla2g5+/+ mice after 4 h of HTV. Quantitative analysis of Evans blue-labeled albumin extravasation further confirmed these results (Fig. 8C). HTV treatment of pla2g5+/+ mice caused substantial increase in Evans blue accumulation in the lungs (2.15 ± .06 μg/ml vs. 1.18 ± .02 μg/ml in control animals; P < 0.05). In turn, pretreatment with MCL-3G1 caused substantial decrease in Evans blue accumulation (1.26 ± 0.7 μg/ml; P < 0.05 vs. HTV-treated pla2g5+/+ mice). Similarly, decreased Evans blue content after 4 h of HTV was also observed in pla2g5−/− mice (P < 0.05 vs. HTV-treated pla2g5+/+ mice). Images of original lung preparations showing Evans blue extravasation are shown in Fig. 8D.
Because gVPLA2 was involved in 18% CS-induced upregulation of IL-8 production by human pulmonary EC cultures, involvement of gVPLA2 mechanism in HTV-induced cytokine production was further tested in a murine model of VILI using MCL-3G1 blocking antibody. The levels of mouse cytokines were measured in BAL samples using ELISA assay, as described in materials and methods. Exposure of wild-type mice to HTV (30 ml/kg, 4 h) caused significant increase in KC, MIP-1, TNF-α, and IL-13 levels detected in BAL samples (Fig. 9). Importantly, HTV-induced cytokine production was significantly attenuated by pretreatment with MCL-3G1. Altogether, these results suggest a major role of gVPLA2 in pathogenesis of VILI.
Fig. 9.

Effect of gVPLA2 blocking antibody on HTV-induced cytokine production in the lung. Wild-type mice we exposed to HTV with our without injection with gVPLA2 blocking antibody. The levels of mouse cytokines keratinocyte chemokine (KC), macrophage inflammatory protein (MIP)-1, TNF-α, and IL-13 were measured in BAL samples using ELISA assay, as described in materials and methods. The data represent the means ± SE of 6 samples; *P < 0.05, **P < 0.01.
DISCUSSION
This study demonstrates for the first time a major role of gVPLA2 as a CS magnitude-sensitive activator of EC surface molecule expression, chemokine production, EC-PMN adhesion, and VILI-associated lung inflammation. Pathological CS of pulmonary EC caused a rapid increase in cell population bearing gVPLA2 on the cell surface, which was detected as early as 30 min and remained increased after 24 h of CS, causing increased gVPLA2 protein expression. CS-induced gVPLA2 activation triggered ICAM-1 expression and cytokine production by CS-preconditioned EC, resulting in stimulation of EC-PMN adhesion and PMN migration activity. The gVPLA2-dependent mechanism of CS-induced neutrophil-EC adhesion was supported by experiments with blocking of gVPLA2 activity using MCL-3G1 antibody, which abolished CS-induced neutrophil-EC interactions. By contrast to 18% CS, EC exposure to physiologically relevant 5% CS neither induced gVPLA2 activation nor stimulated EC-PMN adhesion.
CS-induced leukocyte-EC interactions mediated by gVPLA2 were critical for PMN and monocyte sequestration in the mechanically ventilated lungs and defined severity of lung injury caused by mechanical ventilation. Similarly to cell culture conditions, blocking gVPLA2 function in vivo dramatically improved parameters of inflammation and lung injury in mice exposed to HTV.
This study demonstrates a direct role for gVPLA2 activation in lung dysfunction caused by pathological mechanical factors. Interestingly, observations made in newborn transgenic mice overexpressing gVPLA2 are consistent with our findings. Mice overexpressing gVPLA2 died within several hours of birth as a result of respiratory failure that pathophysiologically resembles ALI (25). The lungs of these mice have extensive atelectasis, hyaline membrane formation in the airspaces, and thickened alveolar walls similar to the lungs of patients with ALI. The fact that these mice develop respiratory failure after birth, when lungs become ventilated and undergo mechanical stretch, may suggest that gVPLA2 overexpression renders the lungs of newborn mice vulnerable to mechanical injury even during normal respiration. Of note, overexpression of another member of this enzyme family, gXPLA2, does not cause lung injury of transgenic mice (25).
Precise mechanisms of CS-induced activation and synthesis of gVPLA2 in pulmonary endothelium remain to be elucidated. Unlike other signaling enzymes, gVPLA2 activation may be achieved by increased gVPLA2 exposure to the outer cell membrane surface as well as by transcriptional activation of protein expression. It is possible that membrane translocation and surface exposure of gVPLA2 may be mediated by the same cellular mechanisms, which control exocytosis or release of microparticles by activated endothelium (10). Among them, Rho kinase signaling, activation of calpain system, and nonapoptotic caspase activity have been shown to be involved in plasma membrane shedding by chemically or mechanically stimulated cells (27, 32). These potential mechanisms warrant further investigation.
Our results show CS-induced activation of ICAM-1 as an essential step promoting PMN-EC adhesion because gVPLA2 blocking antibody suppressed CS-induced ICAM-1 surface expression and PMN adhesion. By contrast, inhibition of ICAM-1 using ICAM-1 blocking antibody inhibited CS-induced HPAEC-PMN adhesion but was without effect on CS-induced gVPLA2 activation. These results indicate that gVPLA2 acts upstream of induction of ICAM-1 expression and increased EC-PMN adhesion. An important result of this study is inhibition of CS-induced PMN adhesion, which was achieved by gVPLA2 blocking antibody and general soluble PLA2 inhibitor, LY-311727 (12). These modalities can be tested in further studies as potential therapeutic strategies for VILI treatment.
How is stretch-induced gVPLA2 activation linked to ICAM-1 and cytokine production? Previous studies suggest a role for lysophosphatidyl choline and its derivates generated by activated gVPLA2 from membrane phosphatidyl choline as intermediate steps of downstream inflammatory signaling. Indeed, gVPLA2 is involved in the inflammatory processes by its capacity to support the generation and release of lysophospholipids (30), ecosanoids (14, 20), and another inflammatory mediator, platelet-activating factor (3, 16). These mediators stimulate NF-κB inflammatory signaling cascade, leading to expression of cytokines and adhesion molecules (i.e., ICAM-1, CD11b). Although lysophospholipids at low doses were not effective for rapid EC barrier disruption (19), they might activate intracellular inflammatory signaling, leading to induction of ICAM-1 expression and cytokine production by pulmonary EC.
The results of this study are summarized in Fig. 10. Excessive CS stimulates gVPLA2 activity by increasing gVPLA2 cell surface presentation and protein expression. Activated gVPLA2 stimulates production of proinflammatory lipid mediators, leading to activation of inflammatory signaling and increased expression of ICAM-1, which promotes HPAEC-PMN adhesion. In vivo, these mechanisms lead to PMN infiltration in the lung and propagation of VILI-associated lung inflammation and barrier dysfunction. It is also possible that interaction of naïve PMNs with CS-activated HPAEC may cause an additional activation of PMNs, leading to further amplification of inflammatory process and facilitating transmigration of activated neutrophils through the endothelial monolayer.
Fig. 10.

Proposed role of gVPLA2 as a trigger of ventilator-induced lung inflammation and barrier dysfunction. Pathological mechanical stimulation of lung tissue associated with mechanical ventilation at high tidal volume increases expression of gVPLA2 at the surface of CS-stimulated vascular endothelium leading to activation of bioactive lipid production resulting in EC inflammatory activation characterized by increased ICAM-1 surface expression and production of chemotaxis cytokines. These events stimulate adhesion of leukocytes (neutrophils and monocytes) to endothelial beds exposed to pathological mechanical forces leading to leukocyte transmigration and activation of inflammatory process in the lung parenchyma and local barrier dysfunction. Inhibition of gVPLA2 function at the early stage of ventilator-induced lung injury using genetic approach, pharmacological inhibitor, or blocking antibody abrogates pathological signaling cascade leading to lung injury.
Based on these findings, we propose a novel paradigm of leukocyte recruitment to mechanically ventilated lungs directly via stretch-induced activation of endothelial gVPLA2. In this mechanism, gVPLA2 serves as a mechanically induced signal transmitter that stimulates endothelial-neutrophil interactions induced by pathological mechanical forces in the absence of classical septic pathogens (Gram-positive or Gram-negative cell wall lipopolysaccharides, viral antigens, bacterial particles, etc.). We speculate that termination of mechanical ventilation downregulates gVPLA2 activity, leading to normalization of lung function. Thus, giving the added beneficial effects of gVPLA2 inhibition in protection from endotoxin-induced ALI (21), pharmacological approaches to inhibit gVPLA2 activity may be useful for developing future therapies for the treatment of VILI and ARDS.
GRANTS
This work was supported by NIH grants HL076259, HL089257, HL087823, and a grant from the Glaxo Smith Kline Center of Excellence to A. Leff.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: A.Y.M. and L.N.M. performed experiments; A.Y.M., N.M.M., A.A.B., and K.G.B. analyzed data; A.Y.M., N.M.M., A.A.B., A.R.L., and K.G.B. interpreted results of experiments; A.Y.M. and A.A.B. prepared figures; A.Y.M., N.M.M., L.N.M., A.A.B., A.R.L., and K.G.B. approved final version of manuscript; N.M.M., A.A.B., A.R.L., and K.G.B. edited and revised manuscript; A.A.B., A.R.L., and K.G.B. conception and design of research; A.A.B. and K.G.B. drafted manuscript.
ACKNOWLEDGMENTS
The authors acknowledge technical assistance of Nicolene Sarich and Oleksii Dubrovskyi with cell culture and cyclic stretch experiments. We thank Dr. Jonathan P. Arm, Harvard Medical School and Brigham and Women's Hospital, Boston, MA, for providing the gVPLA2 knockout (pla2g5−/−) mice.
REFERENCES
- 1. Attalah HL, Wu Y, Alaoui-El-Azher M, Thouron F, Koumanov K, Wolf C, Brochard L, Harf A, Delclaux C, Touqui L. Induction of type-IIA secretory phospholipase A2 in animal models of acute lung injury. Eur Respir J 21: 1040–1045, 2003 [DOI] [PubMed] [Google Scholar]
- 2. Belperio JA, Keane MP, Lynch JP, 3rd, Strieter RM. The role of cytokines during the pathogenesis of ventilator-associated and ventilator-induced lung injury. Semin Respir Crit Care Med 27: 350–364, 2006 [DOI] [PubMed] [Google Scholar]
- 3. Bernatchez PN, Winstead MV, Dennis EA, Sirois MG. VEGF stimulation of endothelial cell PAF synthesis is mediated by group V 14 kDa secretory phospholipase A2. Br J Pharmacol 134: 197–205, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Birukov KG, Jacobson JR, Flores AA, Ye SQ, Birukova AA, Verin AD, Garcia JG. Magnitude-dependent regulation of pulmonary endothelial cell barrier function by cyclic stretch. Am J Physiol Lung Cell Mol Physiol 285: L785–L797, 2003 [DOI] [PubMed] [Google Scholar]
- 5. Birukova AA, Chatchavalvanich S, Rios A, Kawkitinarong K, Garcia JG, Birukov KG. Differential regulation of pulmonary endothelial monolayer integrity by varying degrees of cyclic stretch. Am J Pathol 168: 1749–1761, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Birukova AA, Fu P, Xing J, Birukov KG. Rap1 mediates protective effects of iloprost against ventilator induced lung injury. J Appl Physiol 107: 1900–1910, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Birukova AA, Rios A, Birukov KG. Long-term cyclic stretch controls pulmonary endothelial permeability at translational and post-translational levels. Exp Cell Res 314: 3466–3477, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Luca D, Baroni S, Vento G, Piastra M, Pietrini D, Romitelli F, Capoluongo E, Romagnoli C, Conti G, Zecca E. Secretory phospholipase A2 and neonatal respiratory distress: pilot study on broncho-alveolar lavage. Intens Care Med 34: 1858–1864, 2008 [DOI] [PubMed] [Google Scholar]
- 9. Dennis EA. Diversity of group types, regulation, and function of phospholipase A2. J Biol Chem 269: 13057–13060, 1994 [PubMed] [Google Scholar]
- 10. Dignat-George F, Boulanger CM. The many faces of endothelial microparticles. Arterioscler Thromb Vasc Biol 31: 27–33, 2011 [DOI] [PubMed] [Google Scholar]
- 11. Dosquet C, Weill D, Wautier JL. Molecular mechanism of blood monocyte adhesion to vascular endothelial cells. Nouv Rev Fr Hematol 34, Suppl 1 S55–S59, 1992 [PubMed] [Google Scholar]
- 12. Fleisch JH, Armstrong CT, Roman CR, Mihelich ED, Spaethe SM, Jackson WT, Bobbitt JL, Draheim S, Bach NJ, Dillard RD, Martinelli M, Fouts R, Snyder DW. Recombinant human secretory phospholipase A2 released thromboxane from guinea pig bronchoalveolar lavage cells: in vitro and ex vivo evaluation of a novel secretory phospholipase A2 inhibitor. J Pharmacol Exp Ther 278: 252–257, 1996 [PubMed] [Google Scholar]
- 13. Halbertsma FJ, Vaneker M, Scheffer GJ, van der Hoeven JG. Cytokines and biotrauma in ventilator-induced lung injury: a critical review of the literature. Neth J Med 63: 382–392, 2005 [PubMed] [Google Scholar]
- 14. Han SK, Kim KP, Koduri R, Bittova L, Munoz NM, Leff AR, Wilton DC, Gelb MH, Cho W. Roles of Trp31 in high membrane binding and proinflammatory activity of human group V phospholipase A2. J Biol Chem 274: 11881–11888, 1999 [DOI] [PubMed] [Google Scholar]
- 15. Iwaki M, Ito S, Morioka M, Iwata S, Numaguchi Y, Ishii M, Kondo M, Kume H, Naruse K, Sokabe M, Hasegawa Y. Mechanical stretch enhances IL-8 production in pulmonary microvascular endothelial cells. Biochem Biophys Res Commun 389: 531–536, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marchand C, Favier J, Sirois MG. Role of MSK1 in the signaling pathway leading to VEGF-mediated PAF synthesis in endothelial cells. J Cell Biochem 98: 1095–1105, 2006 [DOI] [PubMed] [Google Scholar]
- 17. Meliton AY, Munoz NM, Meliton LN, Binder DC, Osan CM, Zhu X, Dudek SM, Leff AR. Cytosolic group IVa phospholipase A2 mediates IL-8/CXCL8-induced transmigration of human polymorphonuclear leukocytes in vitro. J Inflamm 7: 14, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meliton AY, Munoz NM, Zhu X, Leff AR. Attenuated translocation of group IVa phospholipase A2 and up-regulated annexin-1 synthesis by glucocorticoid blocks beta 2-integrin adhesion in neutrophils. J Leukoc Biol 83: 344–351, 2008 [DOI] [PubMed] [Google Scholar]
- 19. Munoz NM, Desai A, Meliton LN, Meliton AY, Zhou T, Leff AR, Dudek SM. Group V phospholipase A(2) increases pulmonary endothelial permeability through direct hydrolysis of the cell membrane. Pulm Circ 2: 182–192, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Munoz NM, Kim YJ, Meliton AY, Kim KP, Han SK, Boetticher E, O'Leary E, Myou S, Zhu X, Bonventre JV, Leff AR, Cho W. Human group V phospholipase A2 induces group IVA phospholipase A2-independent cysteinyl leukotriene synthesis in human eosinophils. J Biol Chem 278: 38813–38820, 2003 [DOI] [PubMed] [Google Scholar]
- 21. Munoz NM, Meliton AY, Meliton LN, Dudek SM, Leff AR. Secretory group V phospholipase A2 regulates acute lung injury and neutrophilic inflammation caused by LPS in mice. Am J Physiol Lung Cell Mol Physiol 296: L879–L887, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murakami M, Kudo I. Diversity and regulatory functions of mammalian secretory phospholipase A2s. Adv Immunol 77: 163–194, 2001 [DOI] [PubMed] [Google Scholar]
- 23. Nagase T, Uozumi N, Ishii S, Kume K, Izumi T, Ouchi Y, Shimizu T. Acute lung injury by sepsis and acid aspiration: a key role for cytosolic phospholipase A2. Nat Immunol 1: 42–46, 2000 [DOI] [PubMed] [Google Scholar]
- 24. Nonas S, Birukova AA, Fu P, Xing J, Chatchavalvanich S, Bochkov VN, Leitinger N, Garcia JG, Birukov KG. Oxidized phospholipids reduce ventilator-induced vascular leak and inflammation in vivo. Crit Care 12: R27, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ohtsuki M, Taketomi Y, Arata S, Masuda S, Ishikawa Y, Ishii T, Takanezawa Y, Aoki J, Arai H, Yamamoto K, Kudo I, Murakami M. Transgenic expression of group V, but not group X, secreted phospholipase A2 in mice leads to neonatal lethality because of lung dysfunction. J Biol Chem 281: 36420–36433, 2006 [DOI] [PubMed] [Google Scholar]
- 26. Oudin S, Pugin J. Role of MAP kinase activation in interleukin-8 production by human BEAS-2B bronchial epithelial cells submitted to cyclic stretch. Am J Respir Cell Mol Biol 27: 107–114, 2002 [DOI] [PubMed] [Google Scholar]
- 27. Sapet C, Simoncini S, Loriod B, Puthier D, Sampol J, Nguyen C, Dignat-George F, Anfosso F. Thrombin-induced endothelial microparticle generation: identification of a novel pathway involving ROCK-II activation by caspase-2. Blood 108: 1868–1876, 2006 [DOI] [PubMed] [Google Scholar]
- 28. Satake Y, Diaz BL, Balestrieri B, Lam BK, Kanaoka Y, Grusby MJ, Arm JP. Role of group V phospholipase A2 in zymosan-induced eicosanoid generation and vascular permeability revealed by targeted gene disruption. J Biol Chem 279: 16488–16494, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Singleton PA, Chatchavalvanich S, Fu P, Xing J, Birukova AA, Fortune JA, Klibanov AM, Garcia JG, Birukov KG. Akt-mediated transactivation of the S1P1 receptor in caveolin-enriched microdomains regulates endothelial barrier enhancement by oxidized phospholipids. Circ Res 104: 978–986, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Valentin E, Lambeau G. Increasing molecular diversity of secreted phospholipases A(2) and their receptors and binding proteins. Biochim Biophys Acta 1488: 59–70, 2000 [DOI] [PubMed] [Google Scholar]
- 31. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 342: 1334–1349, 2000 [DOI] [PubMed] [Google Scholar]
- 32. Yano Y, Shiba E, Kambayashi J, Sakon M, Kawasaki T, Fujitani K, Kang J, Mori T. The effects of calpeptin (a calpain specific inhibitor) on agonist induced microparticle formation from the platelet plasma membrane. Thromb Res 71: 385–396, 1993 [DOI] [PubMed] [Google Scholar]