

Abstract
Pulmonary insufficiency (PI) is a common long-term sequel after repair of tetralogy of Fallot, causing progressive right ventricular (RV) dilation and failure. We describe the physiologic and molecular characteristics of the first murine model of RV volume overload. PI was created by entrapping the pulmonary valve leaflets with sutures. Imaging, catheterization, and exercise testing were performed at 1, 3, and 6 mo and compared with sham controls. RNA from the RV free wall was hybridized to Agilent whole genome oligonucleotide microarrays. Volume overload resulted in RV enlargement, decreased RV outflow tract shortening fraction at 1 mo followed by normalization at 3 and 6 mo (39 ± 2, 44 ± 2, and 41 ± 2 vs. 46 ± 3% in sham), early reversal of early and late diastolic filling velocities (E/A ratio) followed by pseudonormalization (0.87 ± 0.08, 0.82 ± 0.08, and 0.96 ± 0.08 vs. 1.04 ± 0.03; P < 0.05), elevated end-diastolic pressure (7.6 ± 0.7, 6.9 ± 0.8, and 7 ± 0.5 vs. 2.7 ± 0.2 mmHg; P < 0.05), and decreased exercise duration (26 ± 0.4, 26 ± 1, and 22 ± 1.3 vs. 30 ± 1.1 min; P < 0.05). Subendocardial RV fibrosis was evident by 1 mo. At 1 mo, 372 genes were significantly downregulated. Mitochondrial pathways and G protein-coupled receptor signaling were the most represented categories. At 3 mo, 434 genes were upregulated and 307 downregulated. While many of the same pathways continued to be downregulated, TNF-α, transforming growth factor-β1 (TGF-β1), p53-signaling, and extracellular matrix (ECM) remodeling transitioned from down- to upregulated. We describe a novel murine model of chronic RV volume overload recapitulating aspects of the clinical disease with gene expression changes suggesting early mitochondrial bioenergetic dysfunction, enhanced TGF-β signaling, ECM remodeling, and apoptosis.
Keywords: ventricles, remodeling, congenital, heart failure, right ventricle
although left ventricular dysfunction is by far the major cause of heart failure in adults, right ventricular (RV) dysfunction is an important etiology of heart failure in children. One of the more common causes of RV dysfunction is chronic pulmonary insufficiency (PI), a major long-term sequela after repair of tetralogy of Fallot. PI causes RV volume overload leading to progressive RV dilation in over 30% of patients (16). Although PI is usually well tolerated for many years, many patients more than 10 yr after repair have progressive exercise intolerance, RV diastolic dysfunction, atrial and ventricular arrhythmias, and sudden death (2, 5, 16). Systolic dysfunction is not an early feature of PI. It is characterized mostly by diastolic dysfunction (9, 14, 27). Thus chronic PI is an important determinant of late morbidity and mortality in these postoperative patients and often necessitates reintervention. However, options for replacement valves are less than perfect and often become insufficient shortly after implantation especially in the growing child. Identifying the optimal timing of intervention has been challenging, since the risk of reoperation and prosthetic valve degeneration must be balanced against the need to prevent irreversible RV dysfunction (39). Additionally, 44% of patients presenting with RV enlargement continue to have RV enlargement despite intervention (8, 10, 12, 18, 21). This may reflect irreversible molecular changes within the myocardium associated with chronic volume overload and may be a precursor to arrhythmias and sudden death even after pulmonary valve replacement. The lack of a small animal model of RV volume overload has been an impediment to understanding the molecular mechanisms of this disorder and its progression to RV failure.
In contrast, the mechanisms of left ventricular (LV) remodeling have been well described but predominantly in models of ischemia and pressure overload. Much less is known about the mechanisms of LV remodeling secondary to chronic volume overload (7, 17, 25, 29, 31, 38, 42). There are less data available in the volume overloaded RV (3), and only one prior study in a large animal model (1). Although the response of the RV and LV to afterload stress has many similarities, there are also important differences related to different embryologic precursors and differences in global structure, cell signaling, and calcium handling in response to hemodynamic loading (4, 19, 20, 22, 26, 36).
We describe the first murine model of RV volume overload, seeking to evaluate RV remodeling as it progresses from the subacute to the chronic state to determine its fidelity to human disease and to perform an initial genome-wide evaluation of gene expression changes as the RV progresses from early to late diastolic dysfunction. We hypothesize that chronic RV volume overload is associated with unique patterns of gene expression compared with subacute volume overload, providing further insight into the molecular mechanisms of RV remodeling under clinically relevant conditions. The development of this murine model will allow investigators to access the entire armamentarium of genetically altered murine strains to elucidate molecular mechanisms of RV diastolic dysfunction and potential biomarkers for clinical use.
METHODS
Model of RV Volume Overload
Twelve-week-old male, FVB mice were used for all studies. Twelve-week-old mice represent adult mice at which time the described surgical procedure can be performed without excess mortality and without concerns about the effects of developmental processes in younger (or older) mice. Following tracheal intubation, anesthesia was maintained with 1.5% isoflurane. Via a right thoracotomy, 10-0 silk sutures were placed through the pulmonary artery wall across the valve annulus to tether the two anterior-most pulmonary valve leaflets to the main pulmonary arterial wall to create PI (Fig. 1A). Postprocedure Doppler echocardiography showed reversal of flow into the branch pulmonary arteries, consistent with hemodynamically significant PI. PI mice were compared with sham-operated controls that underwent an identical surgical procedure with the exception of placement of the tethering sutures. Sham controls were a separate group of animals and had not failed insufficiency experiments. Following surgery, hearts were harvested at 1, 3, and 6 mo after physiologic assessment. The RV free wall was separated from the interventricular septum and the LV and weighed. Total RNA was isolated from the RV.
Fig. 1.

A: pulmonary insufficiency was created by entrapping the pulmonary valve leaflets. Echocardiographic functional assessment. Echocardiograms performed during the progression of volume overload demonstrated (B) increasing right ventricular (RV) dilation (i), sham, mild RV dilation at 2 wk (ii), moderate to severe RV dilation at 1, 3, and 6m (iii), increasing heart rate (C), early decrease in right ventricular outflow tract (D; RVOT) shortening fraction followed by normalization, an early reversal in the tricuspid valve early and late diastolic filling velocities (E/A ratio) followed by pseudonormalization (E), and no change in the RV free wall velocity (S' peak; F). PI, pulmonary insufficiency; LV, left ventricular. *P < 0.05 vs. sham.
Model of Pulmonary Artery Constriction
The model of pulmonary artery constriction (PAC) used has been previously described by our group and others (36). Anesthesia procedures used were similar between PI and PAC. Via a right thoracotomy, a 7-0 silk suture was tied around the main pulmonary artery over a 27-G needle to create pulmonary artery constriction with a reproducible band gradient of >35 mmHg. PAC mice were compared with sham-operated controls that underwent identical surgical procedures with the exception of placement of the constricting suture. We have previously reported on this model, which demonstrates many key components that are observed in clinical RV disease: right bundle branch block (RBBB) by electrocardiogram, elevated RV end-diastolic pressure (RVEDP) by invasive hemodynamics, and progression to RV contractile failure, manifested by decreased RV outflow tract shortening fraction and RV free wall velocity. Hearts were harvested at 10 days after PAC at which time these mice have overt signs of RV clinical failure with liver enlargement, ascites, and high mortality and total RNA isolated (36).
Echocardiography
Transthoracic echocardiograms were performed using a GE Vivid 7 ultrasound platform with a 13-MHz probe. Anesthesia was maintained with 1.5% isoflurane for the duration of the procedure. As previously described, mice with a peak band gradient >35 mmHg, RV dilation, interventricular septum shift with encroachment on the LV cavity, and tricuspid insufficiency at 10 days after surgery were characterized as having severe pulmonary stenosis (PS) and were also used for this study. PI mice (n = 8/group/time point) and sham-operated controls (n = 6/group/time point) underwent echocardiograms every 2 wk after surgery through 6 mo to quantify the severity and progression of RV volume overload and its effect on RV morphology and function, including LV shortening fraction (SF) and RV outflow tract (RVOT) SF, measurement of peak early and late diastolic filling velocities (E/A ratio), and tissue Doppler imaging to assess RV free wall velocity (peak S'; Ref. 36).
Electrocardiogram
Electrocardiograms (ECGs) were obtained at 1, 3, and 6 mo after surgery in PI and sham (n = 6/group/time point) to evaluate RV conduction using Chart for Windows v4.1 (AD Instruments, Colorado Springs, CO). This was performed while the mice were still under light anesthesia following the echocardiograms.
Cardiac Catheterization
We have previously described invasive hemodynamic evaluation using a 1.4-F transducer-tipped micromanometer catheter (Millar Instruments, Houston, TX) inserted via the right jugular vein to determine RVEDP and change in pressure over time (dP/dt). Steady-state data were measured over an average of 10 beats (36). Data were obtained at 1, 3, and 6 mo after PI and sham surgery (n = 6/group/time point).
Magnetic Resonance Imaging
Cardiac magnetic resonance imaging (MRI) was performed at the small animal imaging facility, Stanford Center for innovation in in vivo imaging using a Magnex/Varian self-shielded 30-cm bore 7 Tesla magnet, a Research Resonance Instruments 9-cm bore gradient insert and the GE Healthcare Micro-Signa software environment. Anesthesia was maintained with 1.5% isoflurane for the duration of the procedure. With temperature, respiratory, and heart rate monitoring, mice were placed in the magnet and short-axis images were obtained to evaluate RV volume and function at 1, 3, and 6 mo after PI and sham surgery (n = 4/group/time point). Cardiac gating was performed to minimize motion artifacts. Images were obtained with slice thickness of 7 mm, 10 slices per acquisition, TE: 1 ms (echo time), TR: 2.8 ms (repetition time), and FS: 7 (fast scanning) Offline analysis was performed using QMass MR software.
Graded Treadmill Exercise
Exercise testing and metabolic measurements were performed using an Oxymax metabolic chamber and software (Columbus Instruments, Columbus, OH) using our previously described ramped exercise protocol at 1, 3, and 6 mo after surgery (n = 6/group/time point; Ref. 11).
Histopathology
Cross sections of the heart from PI and sham-operated controls were stained with Masson's trichrome to evaluate for collagen deposition at 1, 3, and 6 mo after surgery (n = 4/group/time point). The extent of fibrosis was quantified using NIH ImageJ software. The DeadEnd calorimetric TdT-mediated dUTP nick-end label (TUNEL) system (Promega) was used to assay for apoptotic cell death per manufacturer's guidelines. Sections were fixed, permeabilized, labeled with TdT, and blocked with hydrogen peroxide. Binding was achieved with streptavidin horseradish peroxidase and stained with DAB. The number of apoptotic cells were counted in five high-power fields/heart (n = 4/group) and averaged. Frozen tissue sections were labeled with wheat germ agglutinin conjugate in Hank's balanced salt solution and analyzed under a fluorescence microscope to determine myocyte cross-sectional area. Sixty cardiomyocytes were measured/heart (n = 3/group), and the data were averaged.
Gene Expression Profiling
Total RNA was isolated from the RV free wall of PI and sham-operated mice (miRNeasy Mini Kit; Qiagen). Following quantification, purity was confirmed by an A260/A280 ratio of 1.8 - 2.1 (NanoDrop 1000 spectrophotometer). Ten nanograms total RNA from each sample were used to synthesize cDNA following which cDNA labeled with cyanine-3 was synthesized, amplified, and purified. This was hybridized to Agilent mouse whole genome oligonucleotide microarrays representing ∼41,000 probes and scanned, and probe features were extracted using Agilent feature extraction software (Agilent One-Color Microarray Low Input Quick Amp Labeling Protocol). Mice with PI were evaluated at two time points after surgery: subacute RV volume overload at 1 mo and chronic RV volume overload at 3 mo (n = 4/group/time point). We next compared significantly dysregulated genes in the PI microarray data at 3 mo (volume overload induced diastolic heart failure) to PAC microarray data at 10 days (pressure overload induced systolic heart failure).
RT-PCR.
In brief, 200 ng of total RNA were reverse transcribed to cDNA and amplified over 40 cycles using the CFX384 Bio-Rad thermocycler. Primers were designed using the Primer 3 Output program. Table 1 shows the primer sequences used. Expression of “hypertrophy/heart failure” and “fetal program” genes were confirmed in a one-step quantitative (q)RT-PCR using SYBR green technology (Qiagen) at 1 and 3 mo. qRT-PCR was also used to validate selected genes obtained from the microarray data. Fold change in expression was compared between PI and sham using the 2 power (−ΔΔCt) method (30).
Table 1.
Primer sequences
| Gene | Primer Sequence |
|---|---|
| α-MHC+ | GCAGGCCCTGGCTCTTCAGC |
| α-MHC− | GCCTGCCTCCTCCAGCCTCT |
| β-MHC+ | GCCCTTTGACCTCAAGAAAG |
| β-MHC− | CTTCACAGTCACCGTCTTGC |
| FZD4+ | AGCAGGTCACAGCTTGGAGT |
| FZD4− | CAGTTGAAATCCCACCCAGT |
| SFRP2+ | CACGAGACCATGAAGGAGGT |
| SFRP2− | GAAGAGCGAGCACAGGAACT |
| CJUN+ | CAATGGGCACATCACCACTA |
| CJUN− | GACACTGGGAAGCGTGTTCT |
| CAMK2a+ | TCTCTGAAAGTGCCAAAGACCT |
| CAMK2a− | TCTGCTGCTAAGTCCTGAG |
| SERCA2+ | TGGAGAACGCTCACACAAAGA |
| SERCA2− | ATTCGTTGGAGCCCCATCT |
| ANP+ | TCGTCTTGGCCTTTTGGCT |
| ANP− | TCCAGGTGGTCTAGCAGGTTCT |
Primer sequences used for quantitative RT-PCR to validate microarray data.
Western Blotting
Protein expression of select p53 downstream genes, Ca2+ handling genes, and Akt was assessed. Proteins were separated by gel electrophoresis, transferred onto a nitrocellulose membrane, and detected using the following antibodies: CaMKII and Bcl2 (sc-5306 and sc-7382; Santa Cruz Biotechnology) and Akt and Bax (Cell Signaling; no. 4691 and 2772). GAPDH was used as the housekeeping gene.
Statistical Analysis
Data other than microarray datasets are represented as means ± SE. Unpaired Student's t-test for two-group comparisons and ANOVA for three or more group comparisons was performed on continuous, normally distributed data. A P value of ≤0.05 was considered significant. Gene expression analysis was first performed using GeneSpring GX 11.5 software comparing PI vs. sham at each time point. Normalized data between the 20th to 100th percentiles with detected probes were used for further analysis. Quality control was performed following which unpaired t-test with Benjimini-Hochberg multiple testing correction was applied to the data. Significantly altered gene transcripts with a corrected P value of <0.05 and with a fold change ≥2 up- or downregulated were considered for further analysis. Database for Annotation, Visualization and Integrated Discovery (DAVID) bioinformatics database version 6.7 was queried to identify significant KEGG pathways enriched in the above gene sets with a false detection rate <5% (33).
Animal Care
All procedures were performed in accordance with National Institutes of Health standards and were approved by the Administrative Panel on Laboratory Animal Care at Stanford University.
RESULTS
Model of RV Volume Overload and Failure
There was no significant variability in the response to the creation of PI. After gaining experience with this surgical technique, we see no mortality.
Echocardiographic evidence of diastolic dysfunction.
As early as 2 wk after the creation of PI, there was qualitatively mild RV dilation by two-dimensional imaging. At 1 mo, there was qualitatively moderate RV dilation with no further increase in dilation at 3 and 6 mo (Fig. 1B). There was no tricuspid regurgitation at any time point. M-mode evaluation demonstrated normal LV SF between sham and PI at 1, 3, and 6 mo (40 ± 1 vs. 39 ± 2, 38 ± 2, and 40 ± 2%, P = ns). Heart rate increased by 3 mo after PI (Fig. 1C) while the RVOT SF decreased early in the course at 1 mo followed by normalization (46 ± 3 vs. 39 ± 2, 44 ± 2, and 41 ± 2%; Fig. 1D). Tricuspid valve inflow pulsed-wave Doppler (Fig. 1E) showed a reversal in peak E/A ratio compared with sham at 1 and 3 mo after PI representing impaired relaxation with normalization by 6 mo (1.04 ± 0.03 vs. 0.87 ± 0.08, 0.82 ± 0.08, and 0.96 ± 0.08). This may represent pseudonormalization where the trans-tricuspid profile appears normal but the relaxation pattern is abnormal. This phase occurs before the development of restrictive filling. RV free wall velocity (S'), representing RV systolic function was unchanged at all time points (Fig. 1F). In contrast to our model of PAC, none of the mice with PI developed overt signs of RV failure such as peripheral edema, and no deaths were seen over a 6-mo followup.
RBBB.
RV dilation resulted in a RBBB pattern with a prolonged QRS duration (25 ± 2 vs. 15 ± 1 ms; P < 0.01) at all time points in PI vs. sham (Fig. 2A). There was no further QRS prolongation over time.
Fig. 2.

Electrocardiography and invasive hemodynamics. A: right bundle branch block (RBBB) pattern with QRS prolongation was seen in at all time points in severe PI vs. a narrow QRS in sham. RV end-diastolic pressure (B; RVEDP) and RV dP/dt (C) was elevated in animals with severe pulmonary insufficiency vs. sham -perated, age-matched controls. *P < 0.01 vs. sham.
Elevated RVEDP.
Invasive hemodynamics demonstrated significantly elevated RVEDP at all time points compared with sham. dP/dt, a preejection-phase index of systolic function, was increased. However, neither the RVEDP nor the dP/dt progressively changed during 6-mo of followup (Fig. 2, B and C).
Quantification of RV volume and systolic function by MRI.
With PI, RV end-diastolic and end-systolic volumes increased significantly compared with sham at all time points (Fig. 3), providing quantification of our subjective echocardiographic data. RV ejection fraction and stroke volume increased at all times points. The increase in RVEDV did not compromise LV volume and LV ejection fraction, which were similar to sham-operated controls.
Fig. 3.

Cardiac MRI. A: 2-dimensional short axis images demonstrate a normal sized RV in the sham-operated animals while animals with PI demonstrated significant RV dilation. RV end diastolic volume (B), RV ejection fraction (C), RV end systolic volume (D), and RV stroke volume (E) were significantly increased in PI (n = 4/time point) compared with sham-operated controls (n = 12) at all time points. *P < 0.05, **P < 0.01 vs. sham.
Decreased exercise endurance.
RV volume overload resulted in a decrease in exercise duration compared with sham at all time points (Fig. 4). There was no progressive decline over time. There was no change in peak V̇o2, V̇co2, or respiratory exchange ratio compared with sham at any time point.
Fig. 4.

Graded treadmill exercise testing. There was a decrease in exercise capacity with PI compared with sham-operated controls at all points which remained stable during follow up without evidence of progressive decline. (n = 6/group/time point) *P < 0.01 vs. age-matched sham-operated controls.
RV morphometrics.
RV free wall weight, heart weight and liver weight (a sign of right-sided heart failure) were significantly increased at all time points after PI, although there was no incremental increase from 1 to 6 mo (Fig. 5, A–C). As expected, lung weight (a sign of left-sided heart failure) and LV weight were not increased compared with sham (Fig. 5D) and none of the animals showed evidence of ascites.
Fig. 5.

Morphometrics. RV (A), heart (B), and liver wt to body wt (C) were increased at all time points after the creation of PI. D: there was no difference in lung weight. *P < 0.05 vs. sham.
Fibrosis, Apoptosis, and Myocyte Hypertrophy
We quantified fibrosis in trichrome-stained tissue sections. There was minimal fibrosis at 1 mo but an increase at 3 and 6 mo following PI (Fig. 6, A and B), most prominent in the subendocardium. Apoptotic nuclei by TUNEL staining were noted in endothelial cells, interstitial cells and fibroblasts. There was rare cardiomyocyte apoptosis. This was significantly increased in PI at 3 mo (Fig. 6, C and D). Myocyte cross-sectional area as assessed by wheat germ agglutinin staining was also significantly increased in PI at 3 mo compared with sham (Fig. 6, E and F).
Fig. 6.

Histopathology. A: cross-sectional views of the heart at ×10 magnification with Masson's trichrome staining demonstrating lack of fibrosis in sham-operated mice, minimal subendocardial fibrosis at 1 mo after PI, and a significant increase in fibrosis at 3 and 6 mo after PI. B: quantification of RV fibrosis showed a significant increase in fibrosis at all time points compared with sham (*P < 0.01); significant increase in fibrosis from 1 mo to 3 and 6 mo (**P < 0.01); there was no difference in the fibrosis between the 3- and 6-mo time points. C: apoptotic nuclei (arrow) were seen in sham and PI in endothelial cells and fibroblasts with minimal cardiomyocyte apoptosis. D: number of TdT-mediated dUTP nick-end label (TUNEL) -positive cells in 5 high-power fields/heart were counted and averaged. This was significantly increased in PI compared with sham (*P < 0.05). E: wheat germ agglutinin demonstrated an increase in myocyte cross-sectional area in PI. F: myocyte area was significantly increased in PI compared with sham (*P < 0.001).
Dynamic Gene Expression Changes Associated with RV Volume Overload and Diastolic Dysfunction from Early to Chronic Stages
RV gene expression was evaluated using Agilent mouse whole genome oligonucleotide microarrays at 1 and 3 mo after PI and compared with age matched sham-operated controls (Fig. 7A). Microarrays were not performed at 6 mo since the physiologic data were totally similar at the 3- and 6-mo time points representing a stable, compensated state. Genes were considered to be significantly regulated if Benjimini-Hochberg multiple testing corrected P value was <0.05 with a fold change ≥2. Significant genes were further subject to DAVID functional annotation analysis.
Fig. 7.
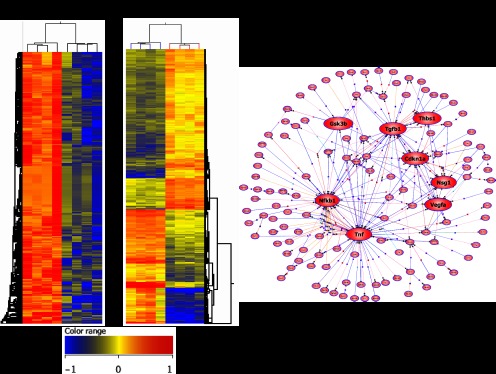
Heatmap and pathway analysis. A: heatmap at 1 and 3 mo comparing PI with sham-operated controls. 372 genes were downregulated by >2-fold at 1 mo. Corrected P value <0.05. At 3 mo 434 genes were upregulated and 307 were downregulated by >2-fold. Corrected P value <0.05. B: pathway analysis at 3 mo after PI demonstrated key nodal points highlighted here in red including upregulation in transforming growth factor-β1 (TGF-β1), thrombospondin, and TNF signaling and downregulation in NF-κB1 signaling.
Transcriptional changes during early volume overload.
At 1 mo after creation of PI, no genes were significantly upregulated and 372 genes were downregulated based on the above criteria. Metabolic pathways, Wnt, Ca2+, MAPK, and G protein-coupled receptor (GPCR) signaling were the most downregulated pathways (Table 2). Phosphofructokinase, a key rate-limiting enzyme in glycolysis, and aconitase, an upstream TCA cycle enzyme, were among the downregulated metabolic enzymes important for ATP production. Multiple genes encoding transport of nutrients across the cell membrane were also downregulated. Both canonical and noncanonical Wnt signaling were downregulated as were cell adhesion molecules such as glycoproteins and contactin. Transforming growth factor-β (TGF-β) pathway genes, genes related to actin, O-glycan, and chondroitin sulfate biosynthesis were downregulated. ATP-binding transporters, CREB-binding protein, and insulin-like growth factor binding protein emerged as key hubs in the network analysis. The most downregulated genes at 1 mo, Abca7, Tpte, Mmp19, Ubap1, and Olfr685 (3.5- to 4.5-fold change) are known to be involved in transmembrane transport, breakdown of the extracellular matrix (ECM), ubiquitin homeostasis, and GPCR.
Table 2.
KEGG pathway analysis of 372 downregulated genes at 1 mo after PI
| KEGG Pathway | Related Genes |
|---|---|
| Glycolysis, TCA cycle, amino acid, and linoleic acid metabolism | Abat, Pfkfb2, Aoc2, Nubpl, Mov10, Akr1e1, Igfbp3 |
| Purine, amino/nucleotide sugar metabolism | Adcy1, Cmah |
| Respiratory chain | Ndufs7, Nufa1 (Complex I); Uqcr11 (Complex III, ATP5k (Complex V) |
| ATP-binding transporters; mitochondrial solute carriers | Abca13, Abca7, Abcc2, Nubpl, Nlrp4b, Oas1a, Stard9, Mtpap, Mov10, Ddx4; Slc25a4 |
| Cell cycle, folate biosynthesis | Crebbp, Alppl2, Cd160, Ly6d, Tnfrsf23, Relt, Sprn, Negr1, Mmp19, Zdhhc24 |
| Calcium signaling pathway | Adcy1, Chran7, Grik1, Ptger1 |
| GPCR protein signaling pathway; Neuroactive ligand receptors | Olfactory receptors, Adora1, Rgs17, Ntsr1, Ptger1, Ccr6, Grm, Gabr |
| Wnt signaling pathway | Wnt1, fzd5/10, Dkk1, Ctnna, Tcf12/20/23, Tcfe4, Tcfap4, Bcl9, Crebbp |
| Cell adhesion | Cadm3, Cntnap1, Gpa33, Cd7, Sectm1a, Cntn4, Itgav |
| MAPK signaling pathway | Rasa2, Rasal2, Gmip, Flnb, Tpte, Map3k2, Bcr |
| Endocytosis, glycosaminoglycan degradation, lysosomal function | Agap2, ArsB, Ctsg, Klk1b27, C1rl, Cela1, Neu4, Mmp19, Ubap1 |
| TGF-ß signaling pathway | Crebbp, Gdf5/7 |
| O-glycan/chondroitin sulfate biosynthesis, Actin cytoskeleton regulation | Galntl5, Caln1, Zdhhc24, Tmem59, Il17rc, Tmem150b, BC021785, Derl3, EvI2b, Sectm1a, Enah |
| p53 signaling pathway | Rrm2b, SESN2, SHISA5, Igfbp3 |
Genes enriched in pathways regulating metabolism, transmembrane transport, cell adhesion, degradation, and transforming growth factor-β (TGF-β) signaling were downregulated. Genes representing hubs are in bold, and the most downregulated genes are underlined. PI, pulmonary insufficiency. GPCR, G protein-coupled receptor.
Transcriptional changes during chronic volume overload.
At 3 mo after PI, 434 genes were upregulated (Table 3) and 307 were downregulated (Table 4) by a fold change >2 and corrected P value <0.05. Pathways related to metabolism, cell cycle, Ca2+ signaling, and GPCR, which were downregulated at 1 mo, continued to remain downregulated at 3 mo after PI, while TGF-β, ECM, and p53 signaling, which were downregulated at 1 mo, transitioned to being upregulated by 3 mo (Fig. 8A). Protein expression of select p53 downstream genes demonstrated a decrease in Bcl2 expression and an increase in Bax expression at 3 mo after PI compared with sham (P < 0.01) (Fig. 9, A and B). CaMKII protein expression was also downregulated at 1 and 3 mo after PI (P < 0.05; Fig. 9C). Interestingly, pathways related to Wnt signaling, cell adhesion, MAPK signaling, and proteosomal degradation demonstrated distinct components of each pathway that were either up or downregulated. Novel pathways were also noted at 3 mo involving regulation of transcription, insulin, mammalian target of rapamycin (mTOR), and adipocytokine signaling (Table 5).
Table 3.
Upregulated pathways at 3 mo after PI
| KEGG Pathway | Genes |
|---|---|
| Wnt signaling | Lrp5 |
| Focal adhesion: Ctnnd1, Wisp1, Fzd 2, 4, 6, Daam2, Rock2 | |
| Wnt inhibitors: Sfrp2, Gsk3b, Pp2a | |
| Focal adhesion and adherens junction | Acta1, Col4a1/a2, Fn1, Flnc, Fgfr1, Pten, Thbs1, Vefga, ctnnd1, Map3k7, |
| MAPK signaling pathway | Mapk4, Mapk6, Map3k7, Rasl11a, Rab12, Rab31, Akt3 |
| Ubiquitin-mediated proteolysis | Ube2c/2d1/2m/4a, Uba1, Smurf2, Herc2, Prpf19, Pias4, Cbl |
| TGF-β signaling pathway | Tgfß1, Thbs1, Tnfα, Smad6, Smurf2, Rock2, Ltbp1, Ppp2r1a, Rps6kb2 |
| ECM-receptor interaction | Col4a1/2, Fn1, Fgfr1, Thbs1, Lama3, Lamc2, Sdc4, Mfap4, Loxl3, Pstn |
| Pathways in cancer, p53, and ErbB signaling | Proliferation: Cebpa, Ar, Cdkn1a, Mitf, Akt3, TGFα/ß1, Vegfa, Thbs1 |
| Apoptosis: Casp8, Dapk1, Gsk3b, Steap3 | |
| Insulin and mTOR signaling pathway | Glucose uptake: Akt3, Cbl; |
| Protein synthesis: Eif4e2, Rps6kb2; | |
| Glycogenolysis: Gsk3b, Pygm; | |
| Inhibition of fatty acid biosynthesis: Prkaa2, Prkag2, Prkar2a | |
| Adipocytokine signaling pathway | Pparα, Akt3, Tnfα, Prkaa2, Prkag2, Cst13, Clec2d |
| Inflammation | Tnfα, chemokines, Tgfß1 |
Pathways upregulated at 3m after creation of PI included extracellular matrix (ECM) formation, cell death, and degradation. Genes representing hubs are in bold, and genes most upregulated are underlined. mTOR, mammalian target of rapamycin.
Table 4.
Pathways downregulated at 3 m in PI
| KEGG Pathway | Related Genes |
|---|---|
| Pentose and glucuronate interconversions; fatty acid uptake | Ugt2b34, Akr1b8, Galk2; Fabp7 |
| Wnt signaling pathway | Wnt inhibitors: Dkk1, Axin2; Gbp3, Tcfap2a |
| Cell adhesion | Cadm1, Col2a1, Actb, Glg1 |
| Cell cycle and division | E2f4, Brca2, Mnd1, Plk5, Smc4, Vps24, Gadd45b, Krtap16-7 |
| MAPK signaling pathway | Gad45b, Nfκb1, Ppp5c, Hsp8, Spry4, Mdfi, Map3k15, Rab10/5c |
| Ubiquitin-mediated proteolysis, lysosomal function | Fbxo2, Ube2 s, Ube3a, Uckl1, Trim68, Proz, Klk1b16, Tmprss4, Tnfaip8, Ctso, Lamp3, Dnajb8 |
| Calcium signaling pathway | CamK2, Cacna2d1, Atp2b2, Mid1, Spsb2, |
| DNA damage and repair | Ogg1, Brca2, Rrm2b, Tdg |
| Regulation of transcription-DNA dependent, RNA metabolic processes | Nfκb1, Cebpd, E2f4, Evx2, Mdfi, Sox15/16, Creb3l3, forkhead box genes, Zinc finger proteins |
| Ion channel activity | Atp2b2, Atp7b, Rab10, Cacnad2, Fabp7, Kcnk7/2, Kcnh4 |
| GPCR protein signaling pathway; neuroactive ligand-receptors | Agtr1a, Agtrap, Npffr2, Grin2d, Mtnr1b, Mpffr2, P2x, Syn1 |
| Natural killer cell-mediated cytotoxicity | Shc2, Ifna9, killer cell lectin-like receptors, Pik3d |
These included ubiquitin-mediated proteolysis, Ca2+ signaling, transcription, and GPCR signaling. Genes representing hubs are in bold, and the most downregulated genes are underlined.
Fig. 8.

Quantitative RT-PCR to confirm microarray data. Select Ca2+ handling genes demonstrated a downregulation in CAMK2 and sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA; A), while (B) there was reactivation of the fetal gene program with increasing ANP and β-myosin heavy chain (MHC; B). *P < 0.05 vs. sham, **P < 0.05 vs. PI 1 mo. C: there was excellent correlation between microarray and RT-PCR data other than for Sfrp2, which did not show upregulation by quantitative (q)RT-PCR.
Fig. 9.

Protein expression. p53 downstream target Bcl2 expression was decreased at 3 mo after the creation of PI (A); Bax expression was increased at 3 mo (B); CaMKII expression was decreased at 1 and 3 mo after PI (C); and Akt expression was unaltered in PI compared with sham-operated controls (D). *P < 0.05, **P < 0.01.
Table 5.
Progression of dysregulated pathways in volume overload over time
| KEGG Pathways | Pulmonary Insufficiency at 1 mo | Pulmonary Insufficiency at 3 mo |
|---|---|---|
| Metabolism (glycolysis, TCA cycle, amino acid, ATP transporters) | ↓ | ↓ |
| Cell cycle | ↓ | ↓ |
| Calcium signaling | ↓ | ↓ |
| GPCR signaling | ↓ | ↓ |
| Wnt signaling | ↓ | ↓↑ |
| MAPK signaling | ↓ | ↓↑ |
| Degradation | ↓ | ↓↑ |
| Cell adhesion | ↓ | ↓↑ |
| TGF-β signaling | ↓ | ↑ |
| p53 signaling | ↓ | ↑ |
| Cytoskeleton | ↓ | ↑ |
| Repair of damaged DNA | — | ↓ |
| Regulation of transcription | — | ↓ |
| Ion channel activity | — | ↓ |
| NK cell-mediated cytotoxicity | — | ↓ |
| Insulin, mTOR signaling | — | ↑ |
| Adiopocytokine signaling | — | ↑ |
Many of the pathways downregulated at 1 mo were also downregulated at 3 mo. No pathways were significantly upregulated at 1 mo. TGF-β and p53 signaling and c pathways related to the cytoskeleton were downregulated at 1 mo but demonstrated upregulation by 3 mo. Pathways related to Wnt, MAPK, degradation, and cell adhesion were downregulated at 1 mo. Over time, these pathways demonstrated areas of up- and downregulation. Several novel pathways became significant at 3 mo including insulin and adipocytokine signaling.
Gene Expression Changes Associated with RV Volume Overload and Diastolic Dysfunction by Category (at 3 mo)
Inflammation, collagen deposition, and ECM reorganization.
Inflammation pathways via TNF-α, chemokine, and TGF-β signaling were upregulated. Cadherin, a target of canonical Wnt signaling, and other ECM genes including thrombospondin, laminin, fibronectins, and collagen were upregulated as were noncanonical Wnt pathway genes playing a role in focal adhesion and cytoskeletal changes. However, while some cell adhesion molecules were upregulated, many were also downregulated (Tables 3 and 4).
Metabolic shift away from β-oxidation and increased glycogenolysis.
Insulin signaling was upregulated via Akt, Cbl, and mTOR pathways, which regulate glucose uptake and protein synthesis. However, genes mediating glycogenolysis such as GSK3β and glycogen phosphorylase were also upregulated. Since Akt negatively regulates these genes, we evaluated Akt protein expression and found no difference between sham and PI (Fig. 9D). Fatty acid binding protein necessary for fatty acid synthesis was downregulated while AMP kinases, which inactivate key enzymes (acetyl-CoA carboxylase) involved in regulating de novo fatty acid biosynthesis, were upregulated. Adipocytokine signaling was also upregulated with an increase in TNF-α and AMP kinases. Glucoronidation, which is the formation of glucose and pyruvate from alternative sources via galatokinase and aldo-keto reductase, was downregulated.
Impaired Ca2+ and GPCR signaling and ion channel activity.
Calmodulin kinase, voltage-dependant Ca2+ channels, and plasma membrane calcium ATPase were downregulated. This was confirmed by qRT-PCR (Fig. 8A). Pathways regulating transcription, ion channel activity, and GPCRs such as angiotensin receptor 1, purinergic, and glutamate inotropic receptors were also downregulated.
Key hubs in the upregulated networks were TGF-β1, thrombospondin1, VEGFa, TNF-α and GSK3b, with Nfkb1 being a downregulated hub (Fig. 7B). These hubs reflect the key processes described above. Prpf19, Rsad1, Cst13, Clec2d, and Mapk4 were the most upregulated genes (ranging from 4.8- to 6.8-fold). These genes encode polyubiquitination, oxidoreductase activity, proteinase inhibitors, leptin receptors, and the classical MAPK pathway, respectively. Hsp40, Evx2, Zfp640, Klk1b16, and Krtap16–7 were the most downregulated genes (ranging from 10- to 16-fold). These genes encode degradation, transcription, peptidase activity, regulation of cell cycle, and proliferation.
Reactivation of the fetal gene program.
PI induced many components of the fetal gene program during the progression to diastolic dysfunction: a fourfold increase in ANP and β-myosin heavy chain, with no change in the adult type α-myosin heavy chain (Fig. 8B). qRT-PCR was performed on eight genes to confirm the microarray data (Fig. 8C). There was excellent correlation between qRT-PCR and the microarray data with the exception of Sfrp2.
Gene Expression Changes Associated with RV Volume vs. Pressure Overload
We compared the gene expression changes induced by RV volume overload with those secondary to RV pressure overload, using our previous model of RV pressure overload secondary to PAC (36, 41). Cell-cell adhesion molecules expressed in cardiac fibroblasts (PSTN, fold change 16) and ECM proteins (LOX, MFAP4, and FNBP4, fold change 8, 8, and 12, respectively) were not as highly expressed in RV volume overload (fold change 2–3). Clusterin, involved in ischemic protection, and the TGF-β-antagonist Nbl1 were upregulated in RVH (fold change 2.5 and 2.8, respectively) whereas TGF-β signaling was upregulated in RV volume overload (Table 3).
DISCUSSION
We have developed the first murine model of RV volume overload demonstrating stable, diastolic heart failure with preserved systolic function over a period of 6 mo. This model recapitulates key aspects of the clinical findings in human RV volume overload with RV enlargement, septal shift, RBBB, Doppler evidence of diastolic dysfunction, increased RVEDP, mild to moderately decreased exercise duration, subendocardial fibrosis, and a small increase in cardiomyocyte apoptosis. Patients with PI may do well for decades before RV systolic dysfunction supervenes, so our model accurately reproduces this compensated stage. This murine model therefore serves as a platform to evaluate the cellular and molecular mechanisms involved in the progression of RV remodeling by using a bioinformatics-based, systems biology approach. Our preliminary evaluation, using gene expression profiling, identified key hubs in affected signaling pathways, including TGF-β1, TNF-α, GSK3β, and NF-κB1 and delineated the complex interplay between pathways required to maintain a stable phenotype. Our specific findings include downregulation of gene expression suggestive of mitochondrial bioenergetic dysfunction, a key metabolic switch in energy production and ECM remodeling. There is an associated downregulation of pathways regulating Ca2+ and GPCR signaling.
Interestingly, many of the processes known to play a critical role in clinical heart failure and progression to a decompensated state are active even in this phase of stable diastolic heart failure with preserved RV systolic function. The metabolic pathway alterations in our model of RV volume overload are similar to those described in models of chronic LV volume overload (6, 23). There are, however, some important differences between the pathways activated in the RV vs. in the LV. Early in the process of RV volume overload, gene expression suggests that β-oxidation of fatty acids is maintained as the primary source of energy production. With progression of RV dilation, however, there is inhibition of components of β-oxidation with concomitant upregulation of glycogenolysis to maintain energy production. Complex 1, 3, and 5 of the respiratory chain are downregulated early in volume overload accompanied by upregulation in oxidoreductases. These changes have been reported in a rat model of aorto-caval fistula-induced LV volume overload and predispose the mitochondria to reactive oxygen species-induced injury, thereby contributing to progressive ventricular dilation (15). In this model of chronic RV volume overload, peroxisome proliferator-activated receptor-α (PPARα), a key regulator of fatty acid, glucose, and amino acid metabolism, remains upregulated whereas models of LV volume overload demonstrate a late downregulation in PPARα expression. PPARα may therefore represent a key difference between the adaptations to volume loading in the RV and LV (6). Less efficient energy utilization in Ca2+ handling processes along with downregulation of ATP-requiring Ca2+ handling genes may predispose the RV myocardium to contractile dysfunction with additional injury, such as early coronary artery insufficiency associated with aging or during cardiopulmonary bypass at the time of valve implantation (34).
Neurohormonal activation may act through a final common pathway via TGF-β to increase collagen production, fibronectin, and adhesion molecules to modulate ECM remodeling. While this is beneficial early in the disease process to maintain cardiac output, chronically it leads to progressive ventricular dilation and conduction abnormalities (6, 24, 28, 37). As RV fibrosis increases, there is an associated upregulation in not only TGF-β but also Wnt signaling mediators indicating their role in cytoskeletal changes. Similar to LV volume overload, Ca2+, and ion channel signaling pathways, GPCRs mediating neurotransmitter release and uptake are downregulated even early in the course of RV volume overload and in the absence of overt heart failure while adrenergic receptor expression is not altered at this stage (18, 35).
It is interesting to note that while the gene expression changes in the volume-loaded RV vs. LV are similar until the stage of stable heart failure, all models of LV volume overload progressed from diastolic to LV systolic heart failure whereas our model develops predominant diastolic heart failure as is seen in humans. The only other previously described animal model of chronic RV volume overload, in the pig, also does not develop RV systolic dysfunction (1, 6, 23, 28, 34, 37, 40). Whether this difference in the RV vs. LV in response to chronic volume overload phenotype is related to their different geometric structures, to markedly different afterloads, or to basic developmental differences in cardiomyocyte biology will be the subject of future research, aided by the development of this murine model of RV volume overload.
Finally, we can compare the gene expression changes induced by RV volume overload with those secondary to RV pressure overload, using our previous model of RV pressure overload secondary to PAC (36, 41). There were many similarities, representing pathways involved in matrix remodeling, actin cytoskeleton, and metabolism although they were not as highly expressed in RV volume overload. Interestingly, these differences are similar to previously described differences between pathways mediating LV volume and pressure overload (35).
Limitations
Although our model of chronic RV volume overload demonstrated RV diastolic dysfunction, the animals did not progress to systolic dysfunction. Our model may uncover biomarkers of early RV dysfunction especially those related to fibrosis but may not be capable of differentiating those related to overt RV failure. While the whole genome approach identifies key biological pathways thereby identifying targets for future research, these data need to be confirmed. However, creation of the model is the important first step in this process. There could be sex differences in the response to volume overload that need to be studied in the future (32).
In summary, we have developed the first murine model of RV volume overload progressing to diastolic dysfunction. This model recapitulates many of the clinical features in patients with volume overloaded RVs. Gene pathway analysis demonstrates a complex interaction of several major pathways required to maintain homeostasis at the molecular level, thereby maintaining a stable heart failure phenotype for a prolonged time period. The combination of the dynamic changes in bioenergetics and ECM remodeling may contribute to progressive RV dilation, fibrosis, and subsequently dysfunction. What characterizes the loss of this homeostasis with a subsequent shift to overt heart failure will be the subject of further investigation. The 3- to 6-mo time point representing 20–30 yr of a hemodynamic load in humans is the ideal time for the institution of therapy since patients are in a stable, compensated phase of diastolic dysfunction but have changes that render the myocardium vulnerable to injury thereby predisposing to systolic dysfunction (13). Improving energy efficiency and arresting cell death and fibrosis may be areas for potential therapeutics.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grant HL-061535 (to D. Bernstein), Children's Heart Center Research Fund, Lucile Packard Children's Hospital, and Stanford University. Support was also provided by a Children's Heart Foundation Grant (to D. Bernstein and S. Reddy).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: S.R., G.A.F., and D.B. conception and design of research; S.R., M.Z., D.-Q.H., E.K., R.P., and F.P.C. performed experiments; S.R. and J.M.S. analyzed data; S.R. interpreted results of experiments; S.R. and D.-Q.H. prepared figures; S.R. drafted manuscript; S.R., G.A.F., R.P., J.M.S., and D.B. edited and revised manuscript; S.R., M.Z., D.-Q.H., G.A.F., E.K., R.P., J.M.S., F.P.C., and D.B. approved final version of manuscript.
REFERENCES
- 1. Agger P, Hyldebrandt JA, Nielsen EA, Hjortdal V, Smerup M. A novel porcine model for right ventricular dilatation by external suture plication of the pulmonary valve leaflets–practical and reproducible. Interact Cardiovasc Thorac Surg 10: 962–966, 2010 [DOI] [PubMed] [Google Scholar]
- 2. Ammash NM, Dearani JA, Burkhart HM, Connolly HM. Pulmonary regurgitation after tetralogy of Fallot repair: clinical features, sequelae, and timing of pulmonary valve replacement. Congenit Heart Dis 2: 386–403, 2007 [DOI] [PubMed] [Google Scholar]
- 3. Bernstein D, Webber S. New directions in basic research in hypertrophy and heart failure: relevance for pediatric cardiology. Prog Pediatr Cardiol 32: 5–9, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Black BL. Transcriptional pathways in second heart field development. Semin Cell Dev Biol 18: 67–76, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chaturvedi RR, Redington AN. Pulmonary regurgitation in congenital heart disease. Heart 93: 880–889, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen YW, Pat B, Gladden JD, Zheng J, Powell P, Wei CC, Cui X, Husain A, Dell'italia LJ. Dynamic molecular and histopathological changes in the extracellular matrix and inflammation in the transition to heart failure in isolated volume overload. Am J Physiol Heart Circ Physiol 300: H2251–H2260, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cox MJ, Sood HS, Hunt MJ, Chandler D, Henegar JR, Aru GM, Tyagi SC. Apoptosis in the left ventricle of chronic volume overload causes endocardial endothelial dysfunction in rats. Am J Physiol Heart Circ Physiol 282: H1197–H1205, 2002 [DOI] [PubMed] [Google Scholar]
- 8. Davlouros PA, Karatza AA, Gatzoulis MA, Shore DF. Timing and type of surgery for severe pulmonary regurgitation after repair of tetralogy of Fallot. Int J Cardiol 97, Suppl 1: 91–101, 2004 [DOI] [PubMed] [Google Scholar]
- 9. Davlouros PA, Kilner PJ, Hornung TS, Li W, Francis JM, Moon JC, Smith GC, Tat T, Pennell DJ, Gatzoulis MA. Right ventricular function in adults with repaired tetralogy of Fallot assessed with cardiovascular magnetic resonance imaging: detrimental role of right ventricular outflow aneurysms or akinesia and adverse right-to-left ventricular interaction. J Am Coll Cardiol 40: 2044–2052, 2002 [DOI] [PubMed] [Google Scholar]
- 10. de Ruijter FT, Weenink I, Hitchcock FJ, Meijboom EJ, Bennink GB. Right ventricular dysfunction and pulmonary valve replacement after correction of tetralogy of Fallot. Ann Thorac Surg 73: 1794–1800; discussion 1800, 2002 [DOI] [PubMed] [Google Scholar]
- 11. Desai KH, Sato R, Schauble E, Barsh GS, Kobilka BK, Bernstein D. Cardiovascular indexes in the mouse at rest and with exercise: new tools to study models of cardiac disease. Am J Physiol Heart Circ Physiol 272: H1053–H1061, 1997 [DOI] [PubMed] [Google Scholar]
- 12. Discigil B, Dearani JA, Puga FJ, Schaff HV, Hagler DJ, Warnes CA, Danielson GK. Late pulmonary valve replacement after repair of tetralogy of Fallot. J Thorac Cardiovasc Surg 121: 344–351, 2001 [DOI] [PubMed] [Google Scholar]
- 13. Fox JG, Davisson M, Newcomer CE, Quimby FW, Smith A. The Mouse in Aging Research (2nd ed). The Mouse in Biomedical Research. New York: Academic, American College of Laboratory Animal Medicine, 2007, p. 637–672 [Google Scholar]
- 14. Frigiola A, Redington AN, Cullen S, Vogel M. Pulmonary regurgitation is an important determinant of right ventricular contractile dysfunction in patients with surgically repaired tetralogy of Fallot. Circulation 110: II153–157, 2004 [DOI] [PubMed] [Google Scholar]
- 15. Gladden JD, Zelickson BR, Wei CC, Ulasova E, Zheng J, Ahmed MI, Chen Y, Bamman M, Ballinger S, Darley-Usmar V, Dell'Italia LJ. Novel insights into interactions between mitochondria and xanthine oxidase in acute cardiac volume overload. Free Radic Biol Med 51: 1975–1984, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gregg D, Foster E. Pulmonary insufficiency is the nexus of late complications in tetralogy of Fallot. Curr Cardiol Rep 9: 315–322, 2007 [DOI] [PubMed] [Google Scholar]
- 17. Herrmann KL, McCulloch AD, Omens JH. Glycated collagen cross-linking alters cardiac mechanics in volume-overload hypertrophy. Am J Physiol Heart Circ Physiol 284: H1277–H1284, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hutchinson KR, Guggilam A, Cismowski MJ, Galantowicz ML, West TA, Stewart JA, Jr, Zhang X, Lord KC, Lucchesi PA. Temporal pattern of left ventricular structural and functional remodeling following reversal of volume overload heart failure. J Appl Physiol 111: 1778–1788, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kaufman BD, Desai M, Reddy S, Osorio JC, Chen JM, Mosca RS, Ferrante AW, Mital S. Genomic profiling of left and right ventricular hypertrophy in congenital heart disease. J Card Fail 14: 760–767, 2008 [DOI] [PubMed] [Google Scholar]
- 20. Kondo RP, Dederko DA, Teutsch C, Chrast J, Catalucci D, Chien KR, Giles WR. Comparison of contraction and calcium handling between right and left ventricular myocytes from adult mouse heart: a role for repolarization waveform. J Physiol 571: 131–146, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lim C, Lee JY, Kim WH, Kim SC, Song JY, Kim SJ, Choh JH, Kim CW. Early replacement of pulmonary valve after repair of tetralogy: is it really beneficial? Eur J Cardiothorac Surg 25: 728–734, 2004 [DOI] [PubMed] [Google Scholar]
- 22. Lopez L, Cohen MS, Anderson RH, Redington AN, Nykanen DG, Penny DJ, Deanfield JE, Eidem BW. Unnatural history of the right ventricle in patients with congenitally malformed hearts. Cardiol Young 20, Suppl 3: 107–112, 2010 [DOI] [PubMed] [Google Scholar]
- 23. Melenovsky V, Benes J, Skaroupkova P, Sedmera D, Strnad H, Kolar M, Vlcek C, Petrak J, Benes J, Jr, Papousek F, Oliyarnyk O, Kazdova L, Cervenka L. Metabolic characterization of volume overload heart failure due to aorto-caval fistula in rats. Mol Cell Biochem 354: 83–96, 2011 [DOI] [PubMed] [Google Scholar]
- 24. Miner EC, Miller WL. A look between the cardiomyocytes: the extracellular matrix in heart failure. Mayo Clin Proc 81: 71–76, 2006 [DOI] [PubMed] [Google Scholar]
- 25. Namba T, Tsutsui H, Tagawa H, Takahashi M, Saito K, Kozai T, Usui M, Imanaka-Yoshida K, Imaizumi T, Takeshita A. Regulation of fibrillar collagen gene expression and protein accumulation in volume-overloaded cardiac hypertrophy. Circulation 95: 2448–2454, 1997 [DOI] [PubMed] [Google Scholar]
- 26. Reddy S, Zhao M, Hu DQ, Fajardo G, Hu S, Ghosh Z, Rajagopalan V, Wu JC, Bernstein D. Dynamic microRNA expression during the transition from right ventricular hypertrophy to failure. Physiol Genomics 44: 562–575, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Redington AN, Oldershaw PJ, Shinebourne EA, Rigby ML. A new technique for the assessment of pulmonary regurgitation and its application to the assessment of right ventricular function before and after repair of tetralogy of Fallot. Br Heart J 60: 57–65, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ryan TD, Rothstein EC, Aban I, Tallaj JA, Husain A, Lucchesi PA, Dell'Italia LJ. Left ventricular eccentric remodeling and matrix loss are mediated by bradykinin and precede cardiomyocyte elongation in rats with volume overload. J Am Coll Cardiol 49: 811–821, 2007 [DOI] [PubMed] [Google Scholar]
- 29. Rysa J, Leskinen H, Ilves M, Ruskoaho H. Distinct upregulation of extracellular matrix genes in transition from hypertrophy to hypertensive heart failure. Hypertension 45: 927–933, 2005 [DOI] [PubMed] [Google Scholar]
- 30. Schefe JH, Lehmann KE, Buschmann IR, Unger T, Funke-Kaiser H. Quantitative real-time RT-PCR data analysis: current concepts and the novel “gene expression's CT difference” formula. J Mol Med (Berl) 84: 901–910, 2006 [DOI] [PubMed] [Google Scholar]
- 31. Shah AS, Atkins BZ, Hata JA, Tai O, Kypson AP, Lilly RE, Koch WJ, Glower DD. Early effects of right ventricular volume overload on ventricular performance and beta-adrenergic signaling. J Thorac Cardiovasc Surg 120: 342–349, 2000 [DOI] [PubMed] [Google Scholar]
- 32. Shioura KM, Geenen DL, Goldspink PH. Sex-related changes in cardiac function following myocardial infarction in mice. Am J Physiol Regul Integr Comp Physiol 295: R528–R534, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Takewa Y, Chemaly ER, Takaki M, Liang LF, Jin H, Karakikes I, Morel C, Taenaka Y, Tatsumi E, Hajjar RJ. Mechanical work and energetic analysis of eccentric cardiac remodeling in a volume overload heart failure in rats. Am J Physiol Heart Circ Physiol 296: H1117–H1124, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Toischer K, Rokita AG, Unsold B, Zhu W, Kararigas G, Sossalla S, Reuter SP, Becker A, Teucher N, Seidler T, Grebe C, Preuss L, Gupta SN, Schmidt K, Lehnart SE, Kruger M, Linke WA, Backs J, Regitz-Zagrosek V, Schafer K, Field LJ, Maier LS, Hasenfuss G. Differential cardiac remodeling in preload vs. afterload. Circulation 122: 993–1003, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Urashima T, Zhao M, Wagner R, Fajardo G, Farahani S, Quertermous T, Bernstein D. Molecular and physiological characterization of RV remodeling in a murine model of pulmonary stenosis. Am J Physiol Heart Circ Physiol 295: H1351–H1368, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wei CC, Lucchesi PA, Tallaj J, Bradley WE, Powell PC, Dell'Italia LJ. Cardiac interstitial bradykinin and mast cells modulate pattern of LV remodeling in volume overload in rats. Am J Physiol Heart Circ Physiol 285: H784–H792, 2003 [DOI] [PubMed] [Google Scholar]
- 38. Wikman-Coffelt J, Walsh R, Fenner C, Kamiyama T, Salel A, Mason DT. Activity and molecular changes in right and left ventricular myosins during right ventricular volume overload. Biochem Med 14: 33–41, 1975 [DOI] [PubMed] [Google Scholar]
- 39. Yemets IM, Williams WG, Webb GD, Harrison DA, McLaughlin PR, Trusler GA, Coles JG, Rebeyka IM, Freedom RM. Pulmonary valve replacement late after repair of tetralogy of Fallot. Ann Thorac Surg 64: 526–530, 1997 [DOI] [PubMed] [Google Scholar]
- 40. Zendaoui A, Lachance D, Roussel E, Couet J, Arsenault M. Usefulness of carvedilol in the treatment of chronic aortic valve regurgitation. Circ Heart Fail 4: 207–213, 2011 [DOI] [PubMed] [Google Scholar]
- 41. Zhao M, Chow A, Powers J, Fajardo G, Bernstein D. Microarray analysis of gene expression after transverse aortic constriction in mice. Physiol Genomics 19: 93–105, 2004 [DOI] [PubMed] [Google Scholar]
- 42. Zile MR, Baicu CF, Gaasch WH. Diastolic heart failure–abnormalities in active relaxation and passive stiffness of the left ventricle. N Engl J Med 350: 1953–1959, 2004 [DOI] [PubMed] [Google Scholar]