

Abstract
Alcohol consumption is a leading cause of liver disease worldwide; thus, there is an urgent need to develop novel therapeutic interventions. Key events for the onset and progression of alcoholic liver disease result in part from the gut-to-liver interaction. Osteopontin is a cytokine present at high concentration in human milk, umbilical cord, and infants' plasma with beneficial potential. We hypothesized that dietary administration of milk osteopontin could prevent alcohol-induced liver injury perhaps by maintaining gut integrity and averting hepatic inflammation and steatosis. Wild-type mice were fed either the control or the ethanol Lieber-DeCarli diets alone or in combination with milk osteopontin for 3 wk, and parameters of gut and liver damage were measured. Milk osteopontin protected the stomach and the gut by increasing gland height, crypt cell plus enterocyte proliferation, and mucin content in addition to lowering macrophages, plasmacytes, lymphocytes, and neutrophils in the mucosa and submucosa in alcohol-fed mice. Milk osteopontin targeted the gut-liver axis, preserving the expression of tight-junction proteins in alcohol-fed mice thus maintaining intestinal integrity and permeability. There was protection from liver injury since transaminases, the activity scores, triglyceride levels, neutrophil infiltration, 3-nitrotyrosine residues, lipid peroxidation end products, translocation of gram-negative bacteria, lipopolysaccharide levels, and tumor necrosis factor-α were lower in cotreated than in ethanol-fed mice. Furthermore, milk osteopontin diminished ethanol-mediated liver injury in OPN knockout mice. Milk osteopontin could be a simple effective nutritional therapeutic strategy to prevent alcohol hepatotoxicity due, among others, to gut protective, anti-inflammatory, and anti-steatotic actions.
Keywords: gut, inflammation, osteopontin, steatosis
compelling evidence suggests that the gut-liver axis is associated with alcohol-induced liver injury, both in patients and in experimental animal models of alcoholic liver disease (ALD) (46). Indeed, gastrointestinal permeability is higher in alcoholics than in normal subjects (30, 31). Alcohol consumption disrupts the intestinal epithelial barrier function via direct effects of ethanol or acetaldehyde, its metabolite (21). Ethanol-induced leaky gut results in translocation of gram-negative bacteria from the intestinal lumen into the portal blood, elevating lipopolysaccharide (LPS) levels and triggering significant inflammation and liver injury (8, 29, 33, 46). Mice harboring a genetic deletion in the LPS signaling pathway are resistant to alcohol-induced liver injury, thus confirming the role of gut-derived LPS in eliciting alcohol-induced liver damage (26, 47, 52). Likewise, intestinal decontamination with nonabsorbable antibiotics reduces plasma LPS and prevents experimental ALD (2, 19, 20).
Anti-inflammatory treatment with corticosteroids is the only intervention demonstrated so far to improve outcome in alcoholic individuals. Patients with ALD often present chronic malnutrition with underlying liver disease worsening their prognosis. The most effective therapy for ALD is alcohol abstinence; however, for patients with severe forms of ALD such as alcoholic hepatitis and for those who do not achieve full alcohol abstinence, targeted therapies are urgently needed. Given the incidence of ALD and the increasing mortality rate, there is a pressing need to develop novel, inexpensive, and simple therapeutic interventions, perhaps using accessible nutritional approaches for slowing down and/or preventing disease progression.
Osteopontin (OPN) is a soluble cytokine and a matrix-associated protein present in the majority of tissues and body fluids (41). The concentration of OPN in human milk, umbilical cord, and infants' plasma is very high compared with that found in tissues or in adults, suggesting potential immunomodulatory and protective actions (36, 37). Milk OPN (m-OPN), which undergoes significant posttranslational modifications and proteolytic processing (9–11, 42), has been identified as gut-protective by maintaining the epithelial barrier function, providing mucosal defense in addition to opsonizing bacteria for phagocytosis, hence promoting host defense (37, 49). OPN knockout (Opn−/−) mice are impaired for clearing intracellular pathogens (3), and studies using an acute colitis model show exacerbated tissue destruction and reduced repair in Opn−/− compared with wild-type (WT) mice (15). Administration of exogenous OPN protected from the adverse effects of experimental colitis (15). Reports of increased OPN in plasma, as well as in the epithelial and submucosal layers of the intestine in inflammatory bowel disease and in short bowel syndrome, indicate that OPN plays an important function in protecting mucosal surfaces (25, 44). Thus, dietary supplementation with m-OPN could emerge as a possible nutritional strategy to protect from alcohol-induced liver injury.
We hypothesized that m-OPN could target the gut-liver axis protecting the gut by maintaining the integrity and the permeability of the epithelial barrier, blocking the translocation of enteric gram-negative bacteria into the liver, preventing sepsis and the LPS-mediated effects in the liver such as the increase in tumor necrosis factor-α (TNF-α), and as a consequence protecting from and/or delaying alcohol-induced liver injury. Thus, the overall goal of this work was to investigate how m-OPN averts alcohol hepatotoxicity and to determine whether administering m-OPN could be a useful strategy for preventing and/or slowing down ALD.
MATERIALS AND METHODS
General methods.
Alcohol, alanine aminotransferase (ALT), aspartate aminotransferase (AST), and triglycerides (TG) were analyzed using kits (Pointe Scientific, Canton, MI). LPS was measured using the chromogenic Limulus amebocyte lysate kit (BioWhittaker, Walkersville, MD). Western blot analysis for OPN was performed as previously (48) using the 2A1 OPN antibody (Ab) donated by Dr. D. T. Denhardt (Rutgers University) (28). The cytochrome P-450 2E1 Ab was provided by Dr. J. M. Lasker (Puracyp, Carlsbad, CA), and the Abs for alcohol dehydrogenase, cytokeratin-19, hedgehog (Hh), Gli-1, Gli-2, and calnexin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The Abs for zonnula occludens-1 (ZO-1), claudin-5, E-cadherin, and occludin were from Invitrogen (Carlsbad, CA). The enhanced chemiluminiscence reaction was developed using a Fujifilm LAS-4000 scanner and quantified with Image-J software. To measure bacterial growth, 25 mg of crushed livers, colons, jejunums, and ileums were plated onto LB-Agar dishes and incubated for 24 h at 37°C following which pictures were captured. Enteric bacteria were incubated with 100 μg/ml m-OPN for 15 min after which they were plated onto LB-Agar dishes and incubated for 24 h at 37°C. Details on general methodology can be found in previous publications (13, 28, 34, 35, 48). To measure gut permeability, a small molecule (FITC-Dextran, 3 kDa; Sigma, St. Louis, MO) was gavaged 3 h before the mice were killed, and fluorescence was measured in plasma. Human epithelial colorectal adenocarcinoma cells (Caco-2) were obtained from the American Type Culture Collection (ATCC, Manassas, VA).
m-OPN.
m-OPN was purified from bovine milk by anion-exchange chromatography as described elsewhere (43) (U.S. Patent 7,259,243). The m-OPN used is >97% pure OPN as analyzed by Edman sequencing, RP-HPLC, and MS analysis. Bovine m-OPN has been thoroughly characterized with respect to modifications and the fragmentation pattern (42). The preparation used in the current study is glycosylated and phosphorylated. It contains 28 phosphorylation sites, which are phosphorylated by 90–100%. Likewise, the five O-glycosylation sites are fully occupied (42). The form of bovine m-OPN used is significantly processed by the endogenous milk protease plasmin. A significant part of the OPN is present as the NH2-terminal fragment representing residues 1–145/147, which is in accordance with the way OPN is found in milk.
Induction of liver injury.
For the ethanol-feeding model, we used the commercially available control and ethanol Lieber-DeCarli diets (Bio-Serv, Frenchtown, NJ), which are equicaloric and have the same composition with respect to fat (42% of calories) and protein (16% of calories). The content of carbohydrates is 42% of total calories (dextrin-maltose) in the control diet and 12% of total calories in the ethanol diet, where up to 30% of carbohydrate calories are replaced by ethanol (32). Female C57BL/6J WT (10 wk old, ∼25 g of body wt; Jackson Laboratories, Bar Harbor, ME) were adapted to the liquid diet regime by feeding them the control diet for 3 days. The percentage of ethanol-derived calories was progressively increased from 10% (1 wk) to 20% (1 wk) and to 30% (1 wk). Mice were pair-fed, and body weight was monitored throughout the entire experiment. Mice consumed an average of 10 ml of diet/day. Water intake was similar in both groups as it was incorporated into the liquid diet. One-half of the mice received m-OPN in the diet. m-OPN was dissolved in water and incorporated into either the control or the ethanol diet at a concentration similar to the concentration in human milk (200 μg/ml) (14, 36). Female C57BL/6J Opn−/− mice (10 wk old, ∼25 g of body wt; Jackson Laboratories) were used to evaluate the protective effect of m-OPN under alcohol feeding. LPS-free bovine serum albumin was used to correct for exogenous protein intake in both control groups. All animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health. Protocols were approved by the Mount Sinai School of Medicine Institutional Animal Care and Use Committee.
Pathology.
The entire left liver lobe as well as the stomach, duodenum, jejunum, ileum, and colon were resected and fixed in 10% neutral-buffered formalin and processed into paraffin sections for hematoxylin and eosin (H&E) or immunohistochemical (IHC) staining. Blind analysis according to the Brunt classification (27) was used to determine the pathology scores. Ten ×100 fields were examined for necroinflammatory activity, which was scored as follows: 0 for none, 1 for <2 foci per ×100 field, 2 for 2–4 foci per ×100 field, 3 for 5–10 foci per ×100 field, 4 for >10 foci per ×100 field. The density of the necroinflammatory activity was also calculated per square millimeter over ×100 fields. Ductular reaction was evaluated according to Clouston et al. (12). Livers were also embedded in optimum cutting temperature compound, sectioned at 5 μM, and stained with oil red O followed by morphometric assessment of the positive area. Blind analysis according to Eaton et al. (18) was used to determine the inflammation score for the stomach and the gut segments. Image-Pro plus 7.0 Software (Media Cybernetics, Bethesda, MD) was used to measure villus height, crypt depth, and to calculate gland height. A picture of the stage micrometer was collected under ×100 magnification and imported into IPP 7.0 software; pictures from the tissues were collected at the same magnification and resolution as the stage micrometer. With the use of IPP software, the pixels from villus height and crypt depth were compared with the pixels from the stage micrometer, and the villus height, crypt depth, and gland height were calculated. Cell proliferation was assessed on Ki67 IHC followed by morphometric assessment.
Immunohistochemistry and special staining.
The 2A1 OPN Ab was used on IHC and was previously tested in livers from Opn−/− mice in C57BL/6J and 129sv genetic background to ensure specificity (data not shown) (28, 48). The Abs for 3-nitrotyrosine (3-NT) and 4-hydroxynonenal (4-HNE) were from Chemicon. The Abs for Ki67 and TNF-α were from Millipore (Billerica, MA) and Fitzgerald (Acton, MA), respectively. Immunochemical reactions were developed using the Histostain Plus detection system (Invitrogen). The Abs for ZO-1, claudin-2/5, and occludin (rabbit IgGs; Invitrogen) were used on immunofluorescences on frozen sections followed by Alexa-594-conjugated goat anti-rabbit IgG secondary Ab (Invitrogen). Naphtol AS-D chloroacetate esterase (NASDCA; Sigma) was used for in situ detection of neutrophils (22). Alcian blue periodic acid-Schiff (AB-PAS) staining was used to detect glycogen, mucin, and other polysaccharides. For computer-assisted morphometric assessment, the integrated optical density was calculated on 10 random fields/section containing similar size portal tracts or hepatic veins at ×100 and using Image-Pro 7.0 Software. Ki67, NASDCA, and TNF-α staining was quantified by counting the number of positively stained cells on 20 random fields/section at ×200.
Statistical analysis.
Data were analyzed by a two-factor analysis of variance, and results are expressed as means ± SE (at least n = 6).
RESULTS
m-OPN preserves the stomach and gut architecture under ethanol consumption.
In an effort to dissect whether the protective action of m-OPN from the noxious effects of alcohol involved structural adaptation, we determined the changes in the stomach and gut architecture induced by ethanol feeding and evaluated whether these effects were prevented by administration of m-OPN. First, samples from stomach were scored for villus length, crypt depth, and gastric gland height as indicators of increased absorptive area. H&E staining revealed increased villus height, crypt depth, and gastric gland height in cotreated mice compared with ethanol-fed mice (Fig. 1, A and B). Thus, m-OPN protected the stomach from alcohol-mediated structural changes.
Fig. 1.

Milk osteopontin (m-OPN) preserves the stomach, duodenum, and jejunum architecture under ethanol consumption. Wild-type (WT) mice were fed 3 wk either the control or the alcohol Lieber-DeCarli diet alone or in combination with 200 μg/ml m-OPN. Hematoxylin and eosin (H&E) staining from the stomach showed more erosion (arrow) in alcohol-treated than in cotreated mice (A). Structural changes were quantified as villus height, crypt depth, and gastric gland height (B). H&E staining showed more erosion (arrow) in the duodenum and jejunum in alcohol-treated than in m-OPN-cotreated mice (C and E). Structural changes in the duodenum and jejunum were quantified as villus height, crypt depth, and gland height. Crypt cell and enterocyte proliferation were quantified on Ki67 immunohistochemistry (IHC). The inflammation scores are also shown (D and F); n = 6 mice in each group. *P < 0.05, **P < 0.01, and ***P < 0.001 for any alcohol-treated group vs. its own control. ●P < 0.05, ●●P < 0.01, and ●●●P < 0.001 for any m-OPN treated vs. its own control.
The mucosal epithelium serves as a barrier between the microbiota and the gut lumen (16). Breakdown of the barrier function leads to infiltration of bacteria and luminal noxious agents triggering inflammation (1). Consequently, next we assessed the extent of ethanol-mediated injury in the intestine and the potential protective effects of m-OPN. H&E staining from the duodenum showed greater villus and gland height in cotreated vs. ethanol-treated mice (Fig. 1, C and D). This was accompanied by enhanced crypt cell and enterocyte proliferation as quantified on Ki67 IHC and by lower macrophages, plasmacytes, lymphocytes, and neutrophils in the mucosa and submucosa in alcohol-fed mice (Fig. 1D). Likewise, the jejunum presented enhanced crypt depth and enterocyte proliferation (Fig. 1, E and F) in cotreated vs. ethanol-treated mice along with a slight improvement of the inflammation score (Fig. 1F). Hence, the data indicate a protective role of m-OPN in preserving the gut architecture and in preventing inflammation.
m-OPN maintains tight-junction protein expression under ethanol consumption.
Although no significant differences were observed in the H&E staining from ileum and colon (data not shown), we speculated that perhaps functional adaptation might have occurred. Tight junctions are scaffolds of various transmembrane proteins and adaptor proteins that cross-link junctional membrane proteins to the actin cytoskeleton as well as to intracellular signaling components. When tight-junction proteins remain intact, they prevent both uptake of macromolecules and translocation of bacteria and enteric bacterial products to the mesenteric lymph nodes and to the liver via the portal vein. Chronic ethanol drinking increases microbial proliferation and acetaldehyde, a biproduct of ethanol metabolism, and opens intestinal tight junctions (4, 5, 38, 40), thus enhancing LPS levels in portal blood, with subsequent transport to the liver triggering hepatotoxic effects. Several studies indicate a central role for acetaldehyde in disrupting gut integrity; however, nothing is known on the potential protective role of m-OPN in preventing the increase in gut permeability by ethanol.
To dissect whether m-OPN secures tight junctions, we performed immunofluorescence for tight-junction proteins. Ethanol-treated mice showed loss of ZO-1, occludin, and claudin-5 expression in colon and of occludin and claudin-5 in ileum, whereas m-OPN helped maintain the expression of these proteins in mice cotreated with ethanol (Fig. 2, A and B). No differences were observed in claudin-2 expression in colon (Fig. 2C) and ileum (data not shown). To validate the protective role of m-OPN, Caco-2 cells were cotreated with ethanol in the presence of m-OPN. Coincubation with m-OPN preserved the expression of ZO-1, E-cadherin, and occludin (Fig. 2D) and enhanced cell proliferation (data not shown). Although the Hh signaling pathway has been involved in maintaining the expression of tight-junction proteins (51), coincubation of Caco-2 cells with m-OPN did not change the expression of Hh, Gli-1, and Gli-2 compared with controls (data not shown). These results suggest that m-OPN preserves junction structures that seal the paracellular space to preserve the integrity of the mucosal barrier perhaps via mechanisms independent from the Hh signaling pathway.
Fig. 2.

m-OPN maintains tight-junction protein expression in colon and ileum under ethanol consumption and preserves gut permeability. WT mice were fed 3 wk either the control or the alcohol Lieber-DeCarli diet alone or in combination with 200 μg/ml m-OPN. Immunofluorescence for tight-junction proteins was performed on frozen sections. Ethanol-treated mice showed loss of zonnula occludens-1 (ZO-1), occludin, and claudin-5 expression (yellow arrows) in colon (A) and occludin and claudin-5 in ileum (B), whereas m-OPN preserved the expression of all proteins in mice cotreated with ethanol. All groups of mice showed similar claudin-2 expression (yellow arrows) in the colon regardless of treatment (C). m-OPN preserves tight-junction protein expression in Caco-2 cells cotreated with ethanol. Caco-2 cells were coincubated with 0–150 mM ethanol in the presence of 0–50 nM m-OPN, and the expression of ZO-1, E-cadherin, and occludin was evaluated at 6 and 24 h by Western blot. Results are average values ± SE of arbitrary densitometry units over the control assigned a value of 1; n = 3. *P < 0.05 and **P < 0.01 for alcohol-treated vs. control. ●P < 0.05, ●●P < 0.01, and ●●●P < 0.001 for m-OPN cotreated vs. ethanol (D). m-OPN preserves gut permeability. Translocation of FITC-Dextran from the gut to the plasma was lower in m-OPN-cotreated than in ethanol-treated mice; n = 6. ●●●P < 0.001 for m-OPN treated vs. control (E).
Because changes in tight-junction protein expression could lead to effects on gut permeability, next we measured translocation or flux of a small molecule from the gut to the plasma. WT mice fed the ethanol Lieber-DeCarli diet were gavaged with FITC-Dextran 3 h before death. There was a significant decrease in gut permeability in m-OPN-cotreated mice compared with ethanol-treated mice (Fig. 2E), suggesting a protective role for m-OPN.
OPN expression in plasma, liver, stomach, and duodenum.
Western blot analysis demonstrated an increase in OPN in plasma and in feces (Fig. 3A) from m-OPN-treated mice. IHC analysis showed similar OPN protein expression in all livers, although there was an increase in the ethanol-fed mice (Fig. 3B, left). There were OPN+ cells in the stomach, colon, and duodenum from both groups of m-OPN-treated mice (Fig. 3B, center and right, and Fig. 3C, left), which could suggest OPN adhesion to gastric gland cells, enterocytes, or crypt cells that express OPN receptors such as CD44 and integrins (17, 39). The staining was specific as confirmed by omission of the primary Ab (data not shown).
Fig. 3.
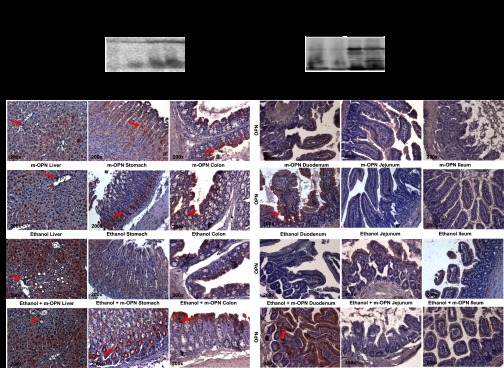
OPN expression enhances in plasma, feces, liver, stomach, colon, and duodenum. WT mice were fed 3 wk either the control or the alcohol Lieber-DeCarli diet alone or in combination with 200 μg/ml m-OPN. Western blot analysis from plasma (left) and feces (right) showed increased cleaved, full, and polymeric OPN expression in m-OPN-cotreated mice (A). There was an increase in endogenous hepatic OPN expression by ethanol treatment, with significant expression in cholangiocytes (red arrows) and hepatocytes (diffuse pattern) (B, left). There were abundant OPN+ cells (red arrows) in the stomach (B, center), colon (B, right), and duodenum (C, left) from both groups of m-OPN-treated mice. PV, portal vein. n = 6. ●P < 0.05 and ●●P < 0.01 for m-OPN treated vs. its own control.
Dietary supplementation with m-OPN protects from alcohol-induced liver injury.
Following evaluation of the stomach and the gut, next we examined whether m-OPN protected the liver from the noxious effects of alcohol. Ethanol feeding increased the liver-to-body weight ratio by approximately twofold; however, cotreatment with m-OPN partially blunted this increase (Fig. 4A). Serum alcohol levels and the expression of cytochrome P-450 2E1 and alcohol dehydrogenase, two enzymes that metabolize alcohol in the liver, were similar in both groups of ethanol-fed mice, hence suggesting that the protective effects of m-OPN were not because of differences in alcohol availability and/or metabolism (Fig. 4B). ALT and AST activities were elevated by three- and twofold, respectively, in ethanol-treated mice but were lowered to basal values by m-OPN (Fig. 4C). H&E staining and scoring as well as Western blot analysis for cytokeratin-19 revealed an increase in inflammation and a slight increase in ductular reaction in the ethanol-fed compared with the cotreated mice (Fig. 4, D and E). There was a twofold increase in the steatosis grade in ethanol-treated mice that was blunted in cotreated mice (Fig. 4F). Serum and liver TG were elevated in ethanol-treated mice but were lowered by m-OPN (Fig. 4G). In addition, Opn−/− mice presented significant liver injury when fed ethanol compared with WT mice, and this effect was partially prevented by coadministration of m-OPN (Fig. 4H). Indeed, the steatosis score dropped from 2.2 to 1. Collectively, these findings indicate that m-OPN prevents alcohol-induced liver injury and steatosis as well as other modes of liver damage.
Fig. 4.

Dietary supplementation with m-OPN protects from alcohol-induced liver injury and steatosis. WT mice were fed 3 wk either the control or the alcohol Lieber-DeCarli diet alone or in combination with 200 μg/ml m-OPN. A: liver-to-body weight ratio. B: serum alcohol concentration and liver cytochrome P-450 2E1 (CYP2E1) and alcohol dehydrogenase (ADH1) protein expression. C: serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities. D: H&E and oil red O staining showed more micro (green arrows)- and macrovesicular (blue arrows) steatosis in livers from alcohol-treated than in m-OPN-cotreated mice. E: the scores for inflammation and ductular reaction along with Western blot analysis for cytokeratin-19 are shown. F: the steatosis grade and neutral fat quantified by oil red O morphometric assessment. G: serum and liver triglyceride (TG) decreased in both groups of mice treated with m-OPN. n = 6; *P < 0.05, **P < 0.01, and ***P < 0.001 for any alcohol-treated group vs. its own control. ●P < 0.05, ●●P < 0.01, and ●●●P < 0.001 for any m-OPN treated vs. its own control. H: dietary supplementation with m-OPN lowers alcohol-induced liver injury in OPN knockout (Opn−/−) mice. Opn−/− mice fed the ethanol Lieber-DeCarli diet in combination with m-OPN showed less liver injury and steatosis than Opn−/− mice fed ethanol alone as shown by H&E (left) and oil red O staining (right); n = 6.
m-OPN lowers alcohol-induced hepatic lipid peroxidation and 3-NT residues.
Because of the significant role of lipid peroxidation (LPO) end products in the development of alcohol-induced liver injury, next we performed IHC to evaluate for 4-HNE, an α,β-unsaturated hydroxylalkenal produced by LPO in many cells. IHC (data not shown) and morphometric analysis showed greater 4-HNE staining in ethanol-treated compared with cotreated mice (Fig. 5A). This was also confirmed by measuring LPO end products by thiobarbituric acid reactive substances (data not shown). Hence, m-OPN could protect by preventing alcohol-induced LPO.
Fig. 5.

m-OPN lowers alcohol-induced lipid peroxidation, 3-NT residues and neutrophil infiltration. WT mice were fed 3 wk either the control or the alcohol Lieber-DeCarli diet alone or in combination with 200 μg/ml m-OPN. 4-Hydroxynonenal (4-HNE, A), 3-nitrotyrosine (3-NT, B), and naphthol AS-D chloroacetate esterase (NASDCA, C) morphometric analysis; n = 6. *P < 0.05, **P < 0.01, and ***P < 0.001 for any alcohol-treated group vs. its own control; ●P < 0.05, ●●P < 0.01, and ●●●P < 0.001 for any m-OPN treated vs. its own control.
Increased expression of inducible nitric oxide synthase contributes to alcohol-induced liver damage.
OPN has been identified as a negative feedback regulator of inducible nitric oxide synthase (NOS2) (53). Increased NOS2, NO·, and O2·− correlate with elevated nitration and oxidation of proteins, a posttranslational modification that causes liver injury. However, whether m-OPN contributes to prevent alcohol-induced liver injury by lowering protein nitration is unknown. Thus, we carried out IHC for 3-NT residues, the footprint of protein nitration (data not shown). The morphometric assessment indicates that m-OPN lowers alcohol-induced 3-NT protein adducts, thus contributing to prevent liver injury (Fig. 5B).
Ethanol-induced hepatic neutrophil infiltration is reduced by m-OPN.
Because alcohol increases neutrophil extravasation contributing to liver damage, we performed NASDCA staining followed by quantitative morphometric assessment. The number of NASDCA+ cells (i.e., neutrophils) was enhanced by alcohol feeding, yet it was significantly reduced by m-OPN treatment (Fig. 5C), hence reinforcing the protective role of m-OPN from alcohol-induced liver injury.
m-OPN preserves liver glycogen along with gastric and intestinal mucin content in cotreated mice.
Gluconeogenesis from glycogen is impaired by ethanol consumption (45). Innate immune responses include increased mucin secretion by goblet cells, which are key components of the first line of host defense against intestinal pathogens. AB-PAS staining and histological evaluation revealed that cotreatment with m-OPN conferred liver, gastric, and intestinal protection compared with ethanol-fed mice by enhancing liver glycogen along with gastric and intestinal mucin secretion (data not shown).
Hepatic bacterial growth, LPS, and TNF-α expression are decreased by m-OPN in cotreated mice.
Because a hallmark of alcohol-induced liver injury is bacterial translocation, which conditions LPS availability and TNF-α production (19), and our data indicated that m-OPN targeted the gut-liver axis preserving tight-junction integrity, next we studied whether bacterial translocation occurred under m-OPN treatment. Bacterial growth was present in crushed livers from ethanol-fed mice, but it was reduced by m-OPN (Fig. 6A). Moreover, bacterial growth in colon, jejunum, and ileum mucosa, likely a reflection of bacterial adhesion to the mucosa, was also prevented by cotreatment with m-OPN (Fig. 6B). Enteric bacteria were also incubated with m-OPN before plating them on LB agar, yet bacterial growth was not affected, suggesting that m-OPN does not kill enteric bacteria (Fig. 6C).
Fig. 6.

Hepatic and gut bacterial growth and tumor necrosis factor-α (TNF-α) expression decrease by m-OPN in cotreated mice. WT mice were fed 3 wk either the control or the alcohol Lieber-DeCarli diet alone or in combination with 200 μg/ml m-OPN. A: bacterial growth in crushed livers cultured in LB-Agar plates. B: bacterial growth in crushed colons, ileums and jejunums cultured in LB-Agar plates. EtOH, ethanol. C: enteric bacteria cultured in the presence or absence of m-OPN. D: liver lipopolysaccharide (LPS). E: TNF-α IHC depicts greater TNF-α+ sinusoidal cells (black arrows) in alcohol-treated than in m-OPN co-treated mice. CV, central vein. F: TNF-α morphometric analysis. n = 6; *P < 0.05 for any alcohol-treated group vs. its own control. ●●P < 0.01 for any m-OPN treated vs. its own control.
Because bacterial translocation conditions hepatic LPS availability (19), next we measured LPS levels. Ethanol feeding elevated LPS, and cotreatment with m-OPN significantly blunted the increase in LPS levels (Fig. 6D). This was also confirmed by measuring bacterial 16S rRNA (data not shown). Because elevated LPS increases TNF-α expression contributing to liver injury (5, 7, 50), to examine whether the protective role of m-OPN involved a decrease in TNF-α, alcohol-induced hepatic TNF-α positive staining was quantified on IHC. More TNF-α positive sinusoidal staining was observed in mice treated with ethanol compared with cotreated mice (Fig. 6, E and F). Moreover, ethanol increased Tnfα and Il-1 mRNA by two- and threefold, respectively, compared with controls, whereas m-OPN lowered these mRNAs to basal levels; however, no major differences were observed in Il-6 mRNA (data not shown). Overall, m-OPN prevented bacterial translocation, LPS availability, and TNF-α increase, thus blunting alcohol hepatotoxicity.
DISCUSSION
Key events for the onset and progression of ALD result from the gut-to-liver interaction (46). Reports of increased OPN in plasma and in the epithelial and submucosal layers of the intestine in inflammatory bowel disease and in short bowel syndrome indicate that OPN plays an important role in protecting mucosal surfaces (25, 44). Because the concentration of OPN in human milk, umbilical cord, and infants' plasma is very high compared with that found in adults, we hypothesized that dietary supplementation with m-OPN, which is highly posttranslationally modified compared with endogenous OPN (6), could protect from alcohol-induced liver injury resulting from gut protective and possibly anti-inflammatory and anti-steatotic effects in the liver.
The present study proved that oral intake of m-OPN increased plasma, liver, stomach, gut, and fecal OPN levels, preserved gut integrity, and lowered hepatic inflammation and steatosis, the latter three key events in the pathogenesis of ALD (23). Overall, the data suggest that m-OPN could prevent alcohol-induced injury. We demonstrated that m-OPN blocked the ethanol-mediated effects in the stomach and the upper segments of the intestine since there was improvement in the histopathology scores, increased mucin secretion, and greater gland height. In addition, there was significant enterocyte proliferation and an overall decrease in inflammation in the duodenum and jejunum. Furthermore, m-OPN prevented the increase in gut permeability by ethanol and preserved tight-junction integrity. Last, m-OPN preserved tight-junction protein expression in Caco-2 cells cotreated with ethanol. The likely mechanisms whereby m-OPN could preserve tight-junction proteins could include the following: 1) m-OPN may bind noxious substances that damage tight junctions and alter the expression of tight-junction proteins; 2) m-OPN could bind the intestinal lining, protect the integrity of tight junctions under alcohol consumption, and maintain tight-junction protein expression; 3) binding of m-OPN to bacteria may prevent their passage through the intestinal lining; and 4) m-OPN could promote epithelial regeneration and/or repair by enhancing enterocyte proliferation. Indeed, studies in an acute colitis model showed exacerbated tissue destruction and reduced repair in Opn−/− compared with WT mice (3, 15). In a follow-up study, the same authors showed that oral administration of a physiological concentration of m-OPN in the drinking water ameliorated the destructive host response in acute colitis (14).
Moreover, cotreated mice were significantly protected from alcohol hepatotoxicity, and m-OPN protected Opn−/− mice that were fed ethanol. Because of the many factors that contribute to the pathogenesis of ALD, alcohol-mediated microbial proliferation, disturbance of gut permeability, and the increase in circulating gut-derived LPS play a central role in the induction of hepatic steatosis and inflammation (5, 46), and the data clearly indicated protective effects of m-OPN in the gut; we analyzed if the positive effects in the liver could involve a decrease in bacterial translocation. Indeed, bacterial growth and bacterial 16S rRNA were present in livers from ethanol-fed mice, yet they were significantly lowered by coadministration of m-OPN. This occurred along with a decrease in hepatic LPS levels and TNF-α+ cells in the sinusoids, certainly contributing to prevent liver injury.
The notion that m-OPN could protect from ALD is novel and may prevent alcohol-related effects in other organs as well. This work is original because it suggests an unrecognized, yet effective, therapeutic approach that could protect from alcohol hepatotoxicity. The novelty of the proposed work and the interrelated gaps that were filled in are as follows. First, the elucidation that m-OPN targets the gut-liver axis, protecting the mucosal barrier (preserving tight-junction protein expression and gut permeability) and blocking the translocation of gram-negative bacteria from the gut to the liver thus preventing sepsis, lowering LPS levels, and the LPS-mediated downstream effects in the liver, which could also be informative for other inflammatory diseases. Second is the recognition that m-OPN prevents liver injury by decreasing the LPS-mediated TNF-α increase, which could have implications for obesity, diabetes, liver regeneration, and the metabolic syndrome as well. We believe that part of the mechanism whereby m-OPN lowers steatosis is precisely by preventing bacterial translocation and thus the LPS increase in portal blood. This, in turn, prevents Kupffer cell activation and TNF-α production, which can benefit the liver preventing the well-documented prosteatotic action of TNF-α (24, 54).
In aggregate, these results suggest a protective role of m-OPN from the hepatotoxic effects of ethanol by targeting, among others, the gut-liver axis and key interconnected events driving ALD, hence addressing important features for disease progression. The results from this study provide novel insight for treating liver diseases associated with alcohol consumption. This effective nutritional therapeutic strategy could be very useful in developing countries and would have significant socioeconomic impact because it addresses an important health-related issue with a very simple and affordable approach provided by Mother Nature.
GRANTS
This work was supported by National Institute on Alcohol Abuse and Alcoholism Grants 5-R01-AA-017733, 5-R01-AA-017733-01S1, 5-P20-AA-017067, 5-P20-AA-017067-01S1, 5-P20-AA-017067-03S1, and 1-U01-AA-021887-01 (N. Nieto) and by The Danish Council for Independent Research (E. S. Sørensen).
DISCLOSURES
The authors have no conflict of interest to disclose.
AUTHOR CONTRIBUTIONS
Author contributions: X.G., Y.L., and T.-M.L. performed experiments; X.G., Y.L., T.-M.L., and N.N. analyzed data; X.G., Y.L., T.-M.L., and N.N. interpreted results of experiments; X.G., Y.L., T.-M.L., and N.N. prepared figures; X.G., Y.L., T.-M.L., E.S.S., and N.N. edited and revised manuscript; X.G., Y.L., T.-M.L., E.S.S., and N.N. approved final version of manuscript; N.N. conception and design of research; N.N. drafted manuscript.
REFERENCES
- 1. Acheson DW, Luccioli S. Microbial-gut interactions in health and disease. Mucosal immune responses. Best Pract Res Clin Gastroenterol 18: 387–404, 2004 [DOI] [PubMed] [Google Scholar]
- 2. Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 108: 218–224, 1995 [DOI] [PubMed] [Google Scholar]
- 3. Ashkar S, Weber GF, Panoutsakopoulou V, Sanchirico ME, Jansson M, Zawaideh S, Rittling SR, Denhardt DT, Glimcher MJ, Cantor H. Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Science 287: 860–864, 2000 [DOI] [PubMed] [Google Scholar]
- 4. Atkinson KJ, Rao RK. Role of protein tyrosine phosphorylation in acetaldehyde-induced disruption of epithelial tight junctions. Am J Physiol Gastrointest Liver Physiol 280: G1280–G1288, 2001 [DOI] [PubMed] [Google Scholar]
- 5. Bala S, Marcos M, Kodys K, Csak T, Catalano D, Mandrekar P, Szabo G. Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor α (TNFα) production via increased mRNA half-life in alcoholic liver disease. J Biol Chem 286: 1436–1444, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bissonnette N, Dudemaine PL, Thibault C, Robitaille G. Proteomic analysis and immunodetection of the bovine milk osteopontin isoforms. J Dairy Sci 95: 567–579, 2012 [DOI] [PubMed] [Google Scholar]
- 7. Cederbaum AI, Yang L, Wang X, Wu D. CYP2E1 sensitizes the liver to LPS- and TNF alpha-induced toxicity via elevated oxidative and nitrosative stress and activation of ASK-1 and JNK mitogen-activated kinases (Abstract). Int J Hepatol 2012: 582790, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Choudhry MA, Fazal N, Goto M, Gamelli RL, Sayeed MM. Gut-associated lymphoid T cell suppression enhances bacterial translocation in alcohol and burn injury. Am J Physiol Gastrointest Liver Physiol 282: G937–G947, 2002 [DOI] [PubMed] [Google Scholar]
- 9. Christensen B, Klaning E, Nielsen MS, Andersen MH, Sorensen ES. C-terminal modification of osteopontin inhibits interaction with the alphaVbeta3-integrin. J Biol Chem 287: 3788–3797, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Christensen B, Nielsen MS, Haselmann KF, Petersen TE, Sorensen ES. Post-translationally modified residues of native human osteopontin are located in clusters: identification of 36 phosphorylation and five O-glycosylation sites and their biological implications. Biochem J 390: 285–292, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Christensen B, Schack L, Klaning E, Sorensen ES. Osteopontin is cleaved at multiple sites close to its integrin-binding motifs in milk and is a novel substrate for plasmin and cathepsin D. J Biol Chem 285: 7929–7937, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clouston AD, Powell EE, Walsh MJ, Richardson MM, Demetris AJ, Jonsson JR. Fibrosis correlates with a ductular reaction in hepatitis C: roles of impaired replication, progenitor cells and steatosis. Hepatology 41: 809–818, 2005 [DOI] [PubMed] [Google Scholar]
- 13. Cubero FJ, Nieto N. Ethanol and arachidonic acid synergize to activate Kupffer cells and modulate the fibrogenic response via tumor necrosis factor alpha, reduced glutathione, and transforming growth factor beta-dependent mechanisms. Hepatology 48: 2027–2039, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. da Silva AP, Ellen RP, Sorensen ES, Goldberg HA, Zohar R, Sodek J. Osteopontin attenuation of dextran sulfate sodium-induced colitis in mice. Lab Invest 89: 1169–1181, 2009 [DOI] [PubMed] [Google Scholar]
- 15. Da Silva AP, Pollett A, Rittling SR, Denhardt DT, Sodek J, Zohar R. Exacerbated tissue destruction in DSS-induced acute colitis of OPN-null mice is associated with downregulation of TNF-alpha expression and non-programmed cell death. J Cell Physiol 208: 629–639, 2006 [DOI] [PubMed] [Google Scholar]
- 16. Duerkop BA, Vaishnava S, Hooper LV. Immune responses to the microbiota at the intestinal mucosal surface. Immunity 31: 368–376, 2009 [DOI] [PubMed] [Google Scholar]
- 17. Dydensborg AB, Teller IC, Basora N, Groulx JF, Auclair J, Francoeur C, Escaffit F, Pare F, Herring E, Menard D, Beaulieu JF. Differential expression of the integrins alpha6Abeta4 and alpha6Bbeta4 along the crypt-villus axis in the human small intestine. Histochem Cell Biol 131: 531–536, 2009 [DOI] [PubMed] [Google Scholar]
- 18. Eaton KA, Danon SJ, Krakowka S, Weisbrode SE. A reproducible scoring system for quantification of histologic lesions of inflammatory disease in mouse gastric epithelium. Comp Med 57: 57–65, 2007 [PubMed] [Google Scholar]
- 19. Enomoto N, Ikejima K, Yamashina S, Hirose M, Shimizu H, Kitamura T, Takei Y, Sato, Thurman RG. Kupffer cell sensitization by alcohol involves increased permeability to gut-derived endotoxin. Alcohol Clin Exp Res 25: 51S–54S, 2001 [DOI] [PubMed] [Google Scholar]
- 20. Enomoto N, Yamashina S, Kono H, Schemmer P, Rivera CA, Enomoto A, Nishiura T, Nishimura T, Brenner DA, Thurman RG. Development of a new, simple rat model of early alcohol-induced liver injury based on sensitization of Kupffer cells. Hepatology 29: 1680–1689, 1999 [DOI] [PubMed] [Google Scholar]
- 21. Ferrier L, Berard F, Debrauwer L, Chabo C, Langella P, Bueno L, Fioramonti J. Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am J Pathol 168: 1148–1154, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fischer R, Hennekeuser HH, Kaufer C. Cytochemical demonstration of naphtol-AS-d-chloroacetate-esterase Auer's bodies. Klin Wochenschr 44: 1401–1402, 1966 [DOI] [PubMed] [Google Scholar]
- 23. Frazier TH, DiBaise JK, McClain CJ. Gut microbiota, intestinal permeability, obesity-induced inflammation, and liver injury. J Parenter Enteral Nutr 35: 14S–20S, 2011 [DOI] [PubMed] [Google Scholar]
- 24. Gao B. Hepatoprotective and anti-inflammatory cytokines in alcoholic liver disease. J Gastroenterol Hepatol 27, Suppl 2: 89–93, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gassler N, Autschbach F, Gauer S, Bohn J, Sido B, Otto HF, Geiger H, Obermuller N. Expression of osteopontin (Eta-1) in Crohn disease of the terminal ileum. Scand J Gastroenterol 37: 1286–1295, 2002 [DOI] [PubMed] [Google Scholar]
- 26. Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, Kurt-Jones E, Szabo G. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 48: 1224–1231, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hubscher SG. Histological assessment of non-alcoholic fatty liver disease. Histopathology 49: 450–465, 2006 [DOI] [PubMed] [Google Scholar]
- 28. Kazanecki CC, Kowalski AJ, Ding T, Rittling SR, Denhardt DT. Characterization of anti-osteopontin monoclonal antibodies: binding sensitivity to post-translational modifications. J Cell Biochem 102: 925–935, 2007 [DOI] [PubMed] [Google Scholar]
- 29. Keshavarzian A, Choudhary S, Holmes EW, Yong S, Banan A, Jakate S, Fields JZ. Preventing gut leakiness by oats supplementation ameliorates alcohol-induced liver damage in rats. J Pharmacol Exp Ther 299: 442–448, 2001 [PubMed] [Google Scholar]
- 30. Keshavarzian A, Fields JZ, Vaeth J, Holmes EW. The differing effects of acute and chronic alcohol on gastric and intestinal permeability. Am J Gastroenterol 89: 2205–2211, 1994 [PubMed] [Google Scholar]
- 31. Keshavarzian A, Holmes EW, Patel M, Iber F, Fields JZ, Pethkar S. Leaky gut in alcoholic cirrhosis: a possible mechanism for alcohol-induced liver damage. Am J Gastroenterol 94: 200–207, 1999 [DOI] [PubMed] [Google Scholar]
- 32. Lieber CS, DeCarli LM. The feeding of alcohol in liquid diets: two decades of applications and 1982 update. Alcohol Clin Exp Res 6: 523–531, 1982 [DOI] [PubMed] [Google Scholar]
- 33. Mathurin P, Deng QG, Keshavarzian A, Choudhary S, Holmes EW, Tsukamoto H. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology 32: 1008–1017, 2000 [DOI] [PubMed] [Google Scholar]
- 34. Ming Leung T, Lu Y, Yan W, Moron-Concepcion JA, Ward SC, Ge X, Conde de la Rosa L, Nieto N. Argininosuccinate synthase conditions the response to acute and chronic ethanol-induced liver injury in mice. Hepatology 55: 1596–1609, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nieto N, Friedman SL, Cederbaum AI. Cytochrome P450 2E1-derived reactive oxygen species mediate paracrine stimulation of collagen I protein synthesis by hepatic stellate cells. J Biol Chem 277: 9853–9864, 2002 [DOI] [PubMed] [Google Scholar]
- 36. Schack L, Lange A, Kelsen J, Agnholt J, Christensen B, Petersen TE, Sorensen ES. Considerable variation in the concentration of osteopontin in human milk, bovine milk, and infant formulas. J Dairy Sci 92: 5378–5385, 2009 [DOI] [PubMed] [Google Scholar]
- 37. Schack L, Stapulionis R, Christensen B, Kofod-Olsen E, Skov Sorensen UB, Vorup-Jensen T, Sorensen ES, Hollsberg P. Osteopontin enhances phagocytosis through a novel osteopontin receptor, the alphaXbeta2 integrin. J Immunol 182: 6943–6950, 2009 [DOI] [PubMed] [Google Scholar]
- 38. Seth A, Basuroy S, Sheth P, Rao RK. l-Glutamine ameliorates acetaldehyde-induced increase in paracellular permeability in Caco-2 cell monolayer. Am J Physiol Gastrointest Liver Physiol 287: G510–G517, 2004 [DOI] [PubMed] [Google Scholar]
- 39. Sheen-Chen SM, Ho HT, Chen WJ, Eng HL, Wu CH. Obstructive jaundice alters CD44 expression in rat small intestine. Digestion 65: 112–117, 2002 [DOI] [PubMed] [Google Scholar]
- 40. Sheth P, Seth A, Atkinson KJ, Gheyi T, Kale G, Giorgianni F, Desiderio DM, Li C, Naren A, Rao R. Acetaldehyde dissociates the PTP1B-E-cadherin-beta-catenin complex in Caco-2 cell monolayers by a phosphorylation-dependent mechanism. Biochem J 402: 291–300, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sodek J, Ganss B, McKee MD. Osteopontin. Crit Rev Oral Biol Med 11: 279–303, 2000 [DOI] [PubMed] [Google Scholar]
- 42. Sorensen ES, Hojrup P, Petersen TE. Posttranslational modifications of bovine osteopontin: identification of twenty-eight phosphorylation and three O-glycosylation sites. Protein Sci 4: 2040–2049, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sorensen ES, Petersen TE. Purification and characterization of three proteins isolated from the proteose peptone fraction of bovine milk. J Dairy Res 60: 189–197, 1993 [DOI] [PubMed] [Google Scholar]
- 44. Stoidis CN, Misiakos EP, Patapis P, Fotiadis CI, Spyropoulos BG. Potential benefits of pro- and prebiotics on intestinal mucosal immunity and intestinal barrier in short bowel syndrome. Nutr Res Rev 21: 1–9, 2010 [DOI] [PubMed] [Google Scholar]
- 45. Sumida KD, Cogger AA, Matveyenko AV. Alcohol-induced suppression of gluconeogenesis is greater in ethanol fed female rat hepatocytes than males. Alcohol 41: 67–75, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Szabo G, Bala S. Alcoholic liver disease and the gut-liver axis. World J Gastroenterol 16: 1321–1329, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Uesugi T, Froh M, Arteel GE, Bradford BU, Thurman RG. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology 34: 101–108, 2001 [DOI] [PubMed] [Google Scholar]
- 48. Urtasun R, Lopategi A, George J, Leung TM, Lu Y, Wang X, Ge X, Fiel MI, Nieto N. Osteopontin, an oxidant stress sensitive cytokine, up-regulates collagen-I via integrin alpha(V) beta(3) engagement and PI3K/pAkt/NFkappaB signaling. Hepatology 55: 594–608, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. van der Windt GJ, Hoogerwerf JJ, de Vos AF, Florquin S, van der Poll T. Osteopontin promotes host defense during Klebsiella pneumoniae-induced pneumonia. Eur Respir J 36: 1337–1345, 2010 [DOI] [PubMed] [Google Scholar]
- 50. Wang X, Lu Y, Xie B, Cederbaum AI. Chronic ethanol feeding potentiates Fas Jo2-induced hepatotoxicity: role of CYP2E1 and TNF-alpha and activation of JNK and P38 MAP kinase. Free Radic Biol Med 47: 518–528, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xiao C, Ogle SA, Schumacher MA, Schilling N, Tokhunts RA, Orr-Asman MA, Miller ML, Robbins DJ, Hollande F, Zavros Y. Hedgehog signaling regulates E-cadherin expression for the maintenance of the actin cytoskeleton and tight junctions. Am J Physiol Gastrointest Liver Physiol 299: G1252–G1265, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yin M, Bradford BU, Wheeler MD, Uesugi T, Froh M, Goyert SM, Thurman RG. Reduced early alcohol-induced liver injury in CD14-deficient mice. J Immunol 166: 4737–4742, 2001 [DOI] [PubMed] [Google Scholar]
- 53. Zhang XF, Liu S, Zhou YJ, Zhu GF, Foda HD. Osteopontin protects against hyperoxia-induced lung injury by inhibiting nitric oxide synthases. Chin Med J (Engl) 123: 929–935, 2010 [PubMed] [Google Scholar]
- 54. Zhao XJ, Dong Q, Bindas J, Piganelli JD, Magill A, Reiser J, Kolls JK. TRIF and IRF-3 binding to the TNF promoter results in macrophage TNF dysregulation and steatosis induced by chronic ethanol. J Immunol 181: 3049–3056, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]