

Abstract
Microglia, the brain's resident immune cells, are phagocytes of the macrophage lineage that have a key role in responding to inflammation and immune challenge in the brain. More recently, they have been shown to have a number of important roles beyond immune surveillance and response, including synaptic pruning during development and the support of adult neurogenesis. Microglial abnormalities have been found in several neuropsychiatric conditions, though in most cases it remains unclear whether these are causative or are a reaction to some other underlying pathophysiology. Here we summarize postmortem, animal, neuroimaging, and other evidence for microglial pathology in major depression, schizophrenia, autism, obsessive-compulsive disorder, and Tourette syndrome. We identify gaps in the existing literature and important areas for future research. If microglial pathology proves to be an important causative factor in these or other neuropsychiatric diseases, modulators of microglial function may represent a novel therapeutic strategy.
1. Introduction
Microglia are the brain's resident immune cells. Unlike neurons and other types of glia, which are of neuroectodermal origin, microglia are macrophage-lineage cells derived from hematopoietic progenitors. Convergent data suggests that microglial activation occurs in a number of neuropsychiatric conditions. This raises intriguing questions about the contribution of dysregulated brain-immune interactions to the pathogenesis of these conditions. Here we review the clinical and preclinical literature implicating microglia in the pathophysiology of several psychiatric disorders.
Historically, microglia have been presumed to be quiescent under physiological conditions and activated only upon immune challenge or in response to neuronal damage or debris. This is consistent with a role in neurodegeneration, in which microglial activation could be a consequence of a degenerative process, as these phagocytic cells participate in cleaning up cellular debris. Alternatively, dysregulated activation of cytotoxic microglial processes may contribute to neuronal damage and degeneration. A role for microglial activation has long been suggested in the pathophysiology of neurodegenerative conditions such as Alzheimer's disease, Parkinson's disease, and dementia associated with the human immunodeficiency virus.
More recently, a series of important findings have challenged the notion that microglia are dormant when not activated by inflammation or immune challenge. Microglia have been found to be required for the development of mature synapses during embryogenesis [1] and to regulate the number of functional synapses both in culture [2] and in vivo [3]. They also regulate adult neurogenesis [4]. This new appreciation that microglia regulate neuronal function and homeostasis under physiological conditions, in the absence of immune challenge or inflammation, raises the possibility that disruption of such processes may contribute to pathological conditions characterized by neuronal or synaptic dysfunction, rather than frank neurodegeneration. In particular, dysregulated synaptic physiology has been a major focus of recent interest in studies of the pathophysiology of mood [5] and psychotic disorders [6], and abnormalities of neurogenesis may similarly contribute to psychiatric disease [7].
The observation of microglial activation or other abnormalities in any particular psychiatric condition does not establish whether microglia are causal contributors to the pathophysiological process or, rather, are activated in response to cellular damage or other aspects of the core pathology. Experimental studies recapitulating pathophysiological processes in animal models provide the best avenue to explore this challenging question of causality. Such investigations are in their infancy, and a pathogenic role for dysregulated microglia in any neuropsychiatric condition remains largely hypothetical. This is an exciting area of ongoing research in neuropsychiatry.
2. Microglia
An exhaustive discussion of microglial physiology is beyond our scope here. Before summarizing evidence for a contribution of microglial dysregulation to several psychiatric conditions, we briefly introduce key points and molecular markers that are specifically relevant to that discussion.
Microglia are small cells of the macrophage lineage found throughout the brain. They are readily identified in brain tissue by their expression of a variety of macrophage markers; several of these have been widely used in the studies summarized below and merit specific mention (for a review see [8]). Microglia, like macrophages, can be identified by their expression of the marker CD11b (also known as complement receptor 3). The expression of the ionized calcium-binding adapter molecule 1 (Iba1) is restricted to microglial cells and is an excellent marker for the analysis of microglial ramifications (Figure 1). The activation of microglia, and of peripheral macrophages that infiltrate the brain under pathological conditions, can be monitored by their expression of CD45; resting microglia are CD45low, whereas macrophages are CD45high. These two populations can be readily distinguished and isolated by flow cytometry. Microglial activation also leads to upregulation of CD11b and Iba1. Microglia also express macrosialin (CD68), a molecule involved in phagocytosis.
Figure 1.
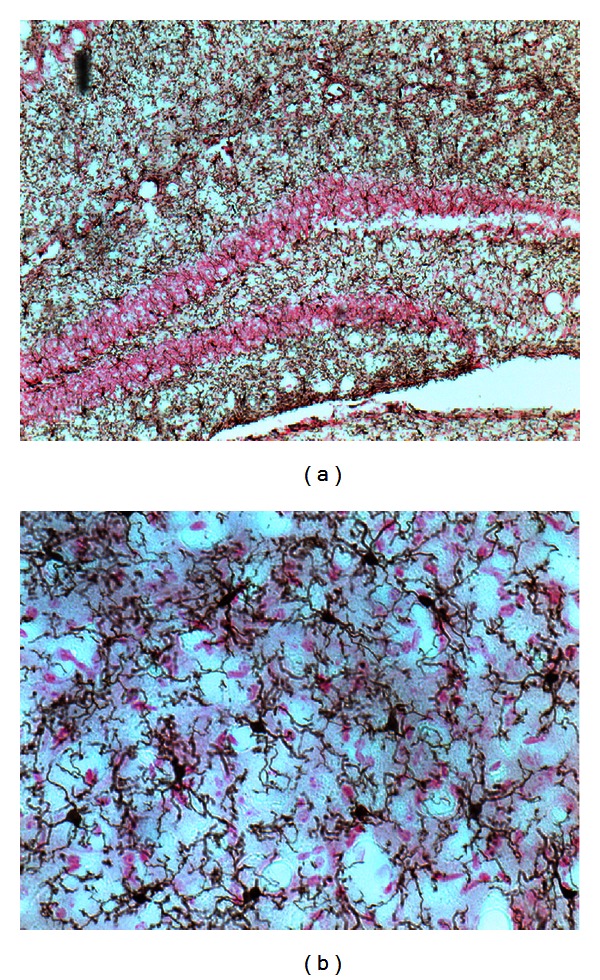
(a) Distribution of Iba1+ microglial cells in the mouse hippocampus. Total cells are stained with fast red. (b) High magnification of microglial staining in the striatum, showing cell bodies and ramifications.
Microglia take several morphological forms. Early in development they have an amoeboid appearance, similar to peripheral macrophages, while in the adult central nervous system they take on characteristic ramified morphology, with long, thin processes (Figure 1). Microglia are highly motile cells, extruding and retracting their processes every few minutes [9]. This has been interpreted as an active sampling of the environment; given the number of microglia in the brain and their motility, it has been calculated that they can explore the entire extracellular space in the brain in a few hours [10]. Such extensive sampling may allow them to search for signs of infection, cellular debris, or other inducing stimuli.
Exposure to bacterial antigens such as lipopolysaccharide (LPS) produces the classical cytotoxic activated microglial phenotype. Activated microglia may become hyperramified or amoeboid/phagocytic [11]. Hyperramified microglia exhibit increased arborization, with thick processes. During the transformation into the amoeboid form, microglia retract the processes and enlarge their cell bodies. Activated microglia produce the proinflammatory cytokines interleukin (IL)-1β, tumor necrosis factor (TNF)-α and IL-6, among others [12]. LPS-activated microglia also upregulate the inducible form of the nitric oxide synthase (iNOS) and produce nitric oxide. This activation of microglial cells is required for their effector immune function in the normal brain. However, dysregulation of this physiological process can lead to neurodegeneration [8, 13].
Another molecular signature of microglia activation is the expression of the major histocompatibility complex (MHC) class II, or human leukocyte antigen (HLA-DR, -DP, and -DQ), which serves as an antigen presenter to T helper cells (CD3+CD4+). CD25+ T helper cells (regulatory T cells, or Tregs) are particularly important to the biology of microglia, and the interaction between these cell types has been implicated in the pathophysiology of neurodegenerative diseases. For example, in mouse models of Parkinson disease and HIV infection-associated neurodegeneration, Tregs were found to have a neuroprotective effect, attenuating microglia-mediated inflammation [14, 15]. CD25− effector T cells (Teffs) were found to have the opposite effect [15]. It has been hypothesized that protective Tregs recognize self-antigens and mediate protective autoimmunity [16, 17].
Microglia can also adopt a neuroprotective phenotype upon activation by cytokines such as IL-4 or IL-25 [18, 19]. These neuroprotective microglia do not produce neurotoxic cytokines like TNF-α. Rather, they produce insulin-like growth factor (IGF)-I and transforming growth factor (TGF)-β, among others [18, 20].
As noted above, microglia are of the hematopoetic lineage, though they populate the brain early in development. Under conditions of inflammation, additional macrophage-lineage cells can be recruited into the central nervous system and differentiate into a microglia-like phenotype. Activated microglia produce high levels of the chemokine (C-C) motif ligand 2 (CCL-2), also known as monocyte chemotactic protein-1 (MCP-1). CCL2/MCP-1 triggers microglia proliferation and also serves as a signal for microglia-induced neurodegeneration [21, 22]. As suggested by its name, MCP-1 also acts a recruiter of other inflammatory cells to the brain. Unlike resident microglial cells, infiltrating macrophages express the CCL2 receptor (CCR2) at high levels.
Fractalkine (CX3CL1) and its receptor (CX3CR1) are also involved in immune cell trafficking to the central nervous system [23]. CX3CR1 expression, unlike that of CCR2, is restricted to microglia in the brain. Mice that lack the CX3CR1 have impaired cognitive function and synaptic plasticity [24]. CX3CR1 has also been implicated in the physiological synaptic pruning mediated by microglia, a process that is needed for normal development of neural circuits; knockout animals have increased dendritic spines and immature synapses [1]. CX3CR1+ microglial cells are also required to support hippocampal neurogenesis [25].
3. Microglial Dysregulation in Depression and Anxiety Disorders
A link between immune dysregulation and the pathophysiology of at least some forms of major affective disorder has long been hypothesized, on the basis of several observations. Many studies have shown abnormalities in peripheral cytokines in depressed patients; indeed, these data have led some investigators to propose a primary immunological etiology for major depressive disorder [7]. Certain core symptoms of major depression, especially the somatic symptoms, resemble the “sickness behavior” that is produced by systemic infectious or inflammatory conditions [26]. Furthermore, significant depressive symptoms are frequently seen following treatment with the cytokine interferon alpha in the context of hepatitis C [27].
As the primary resident immune cells in the brain, microglia are obvious candidate mediators of abnormal brain-immune dialogue in depression. However, clinical evidence implicating microglial dysregulation in affective disorders is limited. In a postmortem study of frontal cortex in several neuropsychiatric conditions, Bayer et al. [28] found increased hippocampal microglia activation (as visualized by HLA-DR expression) in only one of 6 patients with major affective disorders. Similarly, Steiner et al. [29] analyzed several brain regions (dorsolateral prefrontal cortex, anterior cingulate cortex, mediodorsal thalamus, and hippocampus) in postmortem samples from depressive patients and did not find alterations of microglial density. Another report examined CD11b mRNA expression and found no differences in patients with mood disorders (major depression and bipolar disorder) compared to controls [30]. However, significant microgliosis has been observed in patients with depression who completed suicide, compared to patients who died via other methods and healthy controls [29].
Furthermore, a study comparing depressed patients who completed suicide to healthy controls demonstrated an increased density of microglia positive for quinolinic acid, an N-methyl-D-aspartate (NMDA) glutamate receptor agonist produced and released by activated microglia and by no other cells in the brain [31]. Abnormalities in glutamatergic neurotransmission have been implicated in depression by recent studies [32, 33], and glutamate modulators have been proposed as novel antidepressant agents [33, 34]. At sufficient doses, quinolinic acid (QA) is a neurotoxin, a gliotoxin, a proinflammatory mediator, and an oxidant and can alter the integrity and cohesion of the blood-brain barrier [35]. All of these effects—inflammation, oxidative stress, impaired neurogenesis, reduced glial cell number, and neuronal damage—have been implicated in depression [36]. Whether QA produced by activated microglia contributes to these phenomena under physiological conditions in the pathogenesis of depression remains an open question.
Animal models of mood disorders have been used to further investigate the potential pathogenic role of microglia. Chronic psychological stress, which can contribute to the development of depression [37], increases microglia activation in the prefrontal cortex of rats; the antibiotic minocycline, an anti-inflammatory drug that blocks microglial activation, is able to reverse both microglial abnormalities and attendant cognitive dysfunction in stressed animals [38]. Interestingly, minocycline also produces antidepressant-like effects in rats subjected to learned helplessness, a model of depression [39]. Another chronic stress model, repeated social defeat, increased the presence of deramified Iba1+ microglia in the medial amygdala, prefrontal cortex, and hippocampus, with increased levels of cytokines associated with cytotoxic microglial activation—IL-1β, IL-6, TNF-α, and iNOS—in CD11b+ cells [40, 41].
Conversely, events that activate microglia can have long-lasting behavioral consequences. Neonatal exposure of rats to LPS produces significantly increased anxiety-like behavior and hippocampal microglial activation in adulthood [42, 43].
Further animal evidence for a pathogenic role for microglia derives from mice deficient in the fractalkine receptor, CX3CR1, whose expression in the brain is restricted to microglia. These mice displayed enhanced depression-like behavior after treatment with LPS, which also triggered a persistent activated microglial phenotype in the hippocampus and prefrontal cortex [44]. Purified CD11b+/CD45low microglia from knockout mice expressed higher levels of IL-1β than wild-type controls after LPS challenge [8]. These results suggest that CX3CR1/CX3CL1 negatively regulates depressogenic actions of activated microglia [44], perhaps by directing microglia towards a neuroprotective phenotype [18, 19]. Interestingly, two enzymes in the quinolinic acid biosynthesis pathway—indoleamine 2,3-dioxygenase (IDO) and kynurenine monooxygenase (KMO)—were also increased in microglia from CX3CR1 knockout mice [44]. Pharmacological inhibition of IDO counteracted the LPS-induced depressive-like state in CX3CR1 knockout mice [45], providing evidence for the functional importance of QA in microglia-mediated pathogenic effects.
Several antidepressants have been found to prevent the neurodegenerative activation of microglia induced by LPS and cytokines in vitro [46–52]. This effect has been seen with different classes of antidepressants, including selective serotonin reuptake inhibitors, selective norepinephrine reuptake inhibitors, tricyclic antidepressants, and even ketamine. An exception is the monoamine oxidase inhibitor phenelzine, which was found to synergize with LPS in activating microglia, albeit at high concentrations [53].
4. Microglial Abnormalities in Schizophrenia
Several lines of evidence suggest immune dysregulation in the pathogenesis of schizophrenia. For example, maternal infection during pregnancy has been associated with schizophrenia, at least at the epidemiological level (reviewed in [54, 55]).
A small early postmortem study found aberrant activation of microglial cells characterized by HLA-DR expression in a subset of individuals with schizophrenia [28], though other small early studies did not replicate this finding [56]. More recently, morphological analysis in postmortem tissue found evidence both of microglial activation and of microglial degeneration. A pair of larger postmortem studies found evidence of degeneration in HLA+ microglia cells from schizophrenic patients, such as cytoplasm shrinkage, damaged mitochondria, thinning, and shortening and fragmentation of their processes [57, 58]. Ultrastructural analysis has revealed activation of pericapillary microglia with enlarged and vacuolated cytoplasms, irregular nuclear contours, and increased lysosomes [59]. In another recent study, the density of cells positive for the β subunit of the MHC-II, which is common to HLA-DP/DR/DQ and is expressed on activated microglia, correlated with IL-1β expression in the brains of schizophrenic patients [60]. Although activated microglia are not the only possible source of IL-1β, the significant statistical correlation with the microglia-specific marker MHC-II/HLA suggests that they are likely to be the source in these brains.
Several other postmortem studies have provided further evidence of microglial activation, and of brain infiltration by other immune cells, in schizophrenia [29, 61–64]. One of these found differential microglial activation in different clinical subtypes of schizophrenia; HLA-DR+ microglia were increased in the posterior hippocampus of individuals with paranoid schizophrenia relative to those with residual schizophrenia. In contrast, patients with residual schizophrenia had a greater density of CD3+ and CD20+ lymphocytes in the same region [63].
Recently it has become possible to quantify microglial activation in vivo using a positron emission tomography (PET) ligand that recognizes the translocator protein (TSPO), a receptor found on activated microglial cells (as well as on other peripheral cell types). Binding of one such agent, (R)-[11C]PK11195, was increased, suggesting differential microglial activation, in gray matter [65] and in hippocampus [66] of patients with schizophrenia.
Animal models of schizophrenia face vexing challenges to their validity; with that caveat, findings in a few models support a possible role for microglial dysregulation. An animal model based on a cryolesion in the parietal cortex of juvenile mice, which produces later cortical atrophy and cognitive decline reminiscent of that observed in schizophrenia, induces a lasting increase in the number of microglia in cingulate cortex and hippocampus, with accompanying neurodegeneration [67]. An increased number of microglial cells and reduced arborization, which suggests activation, were also found in the hippocampus and the striatum of young rodents following embryonic polyriboinosinic-polyribocytidylic acid (Poly I:C) exposure [68, 69], which is proposed to recapitulate disrupted brain-immune interactions associated with schizophrenia and autism. Similar microglial abnormalities were observed in an experimental model of schizophrenia associated with hyperbilirubinemia [70].
Other findings are consistent with increased microglial activation in schizophrenia having a pathogenic role, and with its modulation having a role in treatment. The gene DISC-1 (disrupted-in-schizophrenia-1), in which mutations have been associated with schizophrenia and other serious mental illnesses in a large pedigree, is expressed in CD11b+ microglia, as well as in neurons [71]. In vitro studies have shown that several antipsychotics, including olanzapine, risperidone, aripiprazole, spiperone, perospirone, and ziprasidone, can inhibit microglia activation [72–77].
If microglial activation contributes to the pathophysiology of schizophrenia, then direct modulators of microglia function may be effective in the treatment of psychotic disease. The antibiotic and anti-inflammatory drug minocycline reduces microglial activation. A few years ago, uncontrolled case series began to appear reporting therapeutic benefit from the addition of minocycline to antipsychotic treatment in schizophrenia [78, 79]. In one subject treated with eight weeks of minocycline, added to stable antipsychotic treatment, perfusion of the posterior cingulate cortex was reduced after the minocycline augmentation [80]. In open-label studies, Miyaoka and colleagues found a significant decrease in both positive and negative symptoms after minocycline was added to an antipsychotic in subjects with schizophrenia [81], and when it was added to an antipsychotic and antidepressant in individuals with psychotic depression [82].
More recently, adjunctive minocycline, added to stable antipsychotic treatment, has been examined in controlled clinical trials, with promising early results. Two double-blind, placebo-controlled studies showed a beneficial effect of adjunctive minocycline therapy on the negative symptoms of schizophrenia [83, 84] and on cognitive function [83], compared to subjects receiving standard antipsychotic therapy. In all reports, the addition of minocycline to the treatment regimen was well tolerated. These reports provide a first therapeutic application of the theory that microglial activation may contribute to psychotic illnesses.
5. Microglia in Autism and Rett Syndrome
Several postmortem studies have suggested a role for microglial pathology in autism spectrum disorders. An initial study found marked microglial activation, measured by immunohistochemical quantification of HLA-DR expression, in the cerebellum, several cortical regions, and white matter in patients with autism [85]. A subsequent study described both increased density of microglial cells in the dorsolateral prefrontal cortex and a shift towards an amoeboid morphology, characterized by soma enlargement, process retraction and thickening, and extension of filopodia from processes, that is suggestive of activation and differentiation into the cytotoxic phenotype [86]. Similar results have been reported in the frontoinsular and visual cortices [87]. Interestingly, the organization of microglia-neuron interactions may be abnormal in autism; microglia are distributed closer to neurons of the dorsolateral prefrontal cortex [88]. The encirclement of neurons by microglial processes suggests an important role of this cell-cell interaction in the pathophysiology of autism.
These postmortem findings have been supported more recently by PET imaging with [11C]PK11195, the microglia-binding ligand described above. Increased [11C]PK11195 binding was observed in multiple brain regions (cerebellum, midbrain, pons, fusiform gyri, and the anterior cingulate and orbitofrontal cortices) in young adult subjects with autism spectrum disorder, suggesting increased microglial activation [89]. These PET findings must be interpreted with caution, as there is no microglia-free reference region to which binding can be normalized, but in conjunction with the postmortem work they build a consistent case for microglial excess in at least some cases of autism.
Similar abnormalities have been reported in several animal models that recapitulate aspects of the pathophysiology or symptomatology of autism. For instance, BTBR T+tf/J mice, which exhibit reduced social interaction and a restricted behavioral repertoire, recapitulating some of the core symptoms of autism, have increased MHC2-expressing microglia compared to control mice [90]. Terbutaline, a β2-adrenoceptor agonist used to arrest preterm labor, has been associated with increased concordance for autism in dizygotic twins [91]; postnatal administration of terbutaline to rat pups resulted in microglial activation and behavioral abnormalities that resemble apsects of autism [92]. Propionic acid-induced autistic-like behavior in laboratory animals is also accompanied by microglial activation, assayed as increased CD68 expression [93, 94].
Rett syndrome, an X-linked autism spectrum disorder characterized by the mutation of the methyl-CpG-binding protein-2 (MECP2) gene, has recently been associated with microglial dysfunction. MECP2-deficient microglia release excess glutamate, in vitro, via connexin 32 (Cx32) hemichannel-mediated release, as a consequence of enhanced glutaminase expression [95]. Interestingly, increased levels of glutamate and glutamine, measured using magnetic resonance spectroscopy (MRS), have been reported in young patients with Rett syndrome [96], suggesting that a similar glutamate dysregulation may occur in vivo in patients. Reductions in AMPA and NMDA glutamate receptor density in the putamen and in kainate (KA) glutamate receptor density in the caudate of Rett patients have also reported [97]. Similarly, glutamatergic neurotransmission is impaired in the animal model of this disease [98]. Immune-mediated dysregulation of glutamatergic neurotransmission has been proposed as a pathogenic mechanism in autism spectrum disorders more generally [99].
Most of the associations catalogued above between microglial dysregulation and psychopathology are correlational. A groundbreaking recent study in an animal model of Rett syndrome provides one of the few clear pieces of evidence for the causal importance of such an association. Restoration of wild-type microglia by bone marrow transplantation or genetic rescue attenuated the Rett syndrome-like symptomatology in MECP2-null mice [100]. This immediately suggests therapeutic possibilities in this devastating disease.
6. Microglia in Obsessive-Compulsive Disorder and Tourette Syndrome
Several lines of evidence have long suggested an association between immune dysregulation and obsessive-compulsive disorder (OCD) (e.g., [101, 102]). A specific role for microglia in the pathophysiology of OCD, or of the related compulsive grooming disorder, trichotillomania, has been suggested by a recent study in a mouse model. Ten years ago, mice with a knockout of the HoxB8 gene, a homeobox developmental patterning gene expressed prominently in macrophage-lineage hematopoietic cells, were observed to exhibit excessive grooming behavior; this excessive grooming has been proposed to model OCD symptomatology [103] More recently, HoxB8 mutant microglia were found to be necessary and sufficient for this excessive grooming phenotype. The phenotype can be rescued by transplantation of wild-type bone marrow into mutant Hoxb8 mice, which leads to repopulation of the brain with wild-type microglia. Conversely, transplant of Hoxb8 mutant bone marrow into wild-type mice can induce the pathological grooming behavior [104]. The mechanisms of this fascinating effect remain unclear. In the nervous system, Hoxb8 plays an important role in the formation of the spinal cord, sensory responses, and development of noradrenergic autonomic neurons [105, 106]. In the immune system, Hoxb8 appears to be involved in the maintenance of monocyte precursors by blocking differentiation of myeloid progenitors from primary marrow into macrophages, dendritic cells, and probably microglia [107–109]. In the brain, a subset of microglia—not all—express HoxB8. The specific physiological role of this particular subset of Hoxb8+ microglia has yet to be described.
There have been few postmortem studies in OCD, and none described to date have investigated microglial activation. Therefore, while these observations in HoxB8 knockout mice are fascinating and suggest exciting new directions for research, their direct applicability to the pathophysiology of OCD remains to be established. No mutations in the HoxB8 gene have yet been described in association with OCD, grooming disorders, or related conditions.
Tourette syndrome is a developmental neuropsychiatric disorder characterized by involuntary motor and phonic tics; it has a high comorbidity with OCD. There is as yet no direct postmortem evidence for microglial dysregulation in Tourette syndrome, but several molecular findings suggest a possible relationship. A nonsignificant two-fold increase in the expression of the surface marker CD45, which is higher in activated than resting microglia, was reported in postmortem basal ganglia [110]. Additionally, elevated expression of CCL2/MCP-1 was observed in these patients. Elevation of this chemokine may promote microglial activation, particularly of the subtype that express its receptor CCR2 [111]. The involvement of microglial dysregulation in Tourette syndrome is an intriguing area for future study.
7. Conclusion: Open Questions and New Directions
The data summarized above provide intriguing evidence for microglial dysregulation in a number of psychiatric conditions. However, they also highlight several important areas for ongoing research before these associations can be considered conclusive.
First, the direct data for associations between microglia and psychopathology rest in many cases on small postmortem studies; in the case of OCD, direct human data are entirely lacking. Ongoing high-quality postmortem work is essential to strengthen the evidence that microglial abnormalities are seen in these conditions. Findings in animal model systems are intriguing and can provide important supportive data and mechanistic insight, but because of the difficulties in fully modeling the pathophysiology of psychiatric disease in an animal, they cannot substitute for direct observations in human tissue. The recent development of PET ligands that can measure microglial activation, such as [11C]PK11195, provides an important new source of parallel data for microglial activation in patients.
Second, in many cases, postmortem investigations have revealed abnormal microglial activation in only a subset of individuals. For example, in one early investigation across several disorders [28], excessive microgliosis was seen in one of six subjects with major affective disorders and three of 14 subjects with schizophrenia. This suggests that, to the extent that abnormal microglial activation contributes to disease, it does so in a heterogeneous fashion. The distinction between patterns of immune dysregulation in paranoid versus residual schizophrenia described above [63] provides one candidate hypothesis to explain this heterogeneity. It will be important to better characterize which patients within each heterogeneous clinical population exhibit microglial abnormalities. PET imaging of microglial activation is likely to be an essential tool for this project. A related question is how microglial activation differs between phenotypically distinct disorders. If microglia are excessively activated in both depression and schizophrenia, what determines the difference between these conditions?
Third—and perhaps most importantly—the causal role of microglial activation in the pathophysiology of these conditions remains to be established. Most of the data described above are purely correlational and leave open the question of whether microglial pathology is a cause of neuronal dysfunction and damage and of behavioral symptomatology or, rather, whether neurons (and/or glia) are damaged by independent pathological processes and microglial activation develops as a consequence. In this regard, the recent demonstration that bone marrow transplant (and thus microglial replacement) can mitigate core phenotypes in a Rett model mouse is of immense importance [100]. This represents the best evidence for a causal role, rather than a reactive one, for microglial pathology in the development of behavioral symptomatology. Similar evidence in the HoxB8 knockout mouse [104] establishes a parallel causal role for microglia in their grooming phenotype, but its clinical importance is lessened by the still-tenuous connections between excessive grooming in this mouse model and the pathophysiology of OCD in human patients. The apparent ability of minocycline augmentation to improve the symptoms of schizophrenia [83, 84] provides some early clinical evidence for a causal role for microglial activation in this condition, though this conclusion is limited by minocycline's multiple mechanisms of action.
Finally, as has been emphasized at several points above, microglia can take on distinct phenotypes, and the separate contribution of these different subsets of microglial to psychiatry pathophysiology remains almost entirely obscure. For example, different cytokines can lead microglia to differentiate into cytotoxic or neuroprotective phenotypes; how these are differently dysregulated in various psychiatric conditions remains to be demonstrated. Similarly, only a subset of microglia in a wild-type mouse express the HoxB8 gene; the functional importance of this subset is emphasized by the phenotype of the HoxB8 knockout mouse, but the mechanistic details remain unclear. Finally, there is growing appreciation of the distinction between brain resident microglia and macrophage-lineage cells that enter the brain later during development, or even in adulthood [111, 112]. Functional distinctions between these microglial subpopulations are not yet well understood.
Our understanding of the role of microglia in normal brain function is rapidly evolving. Until recently, these resident immune cells were thought to be entirely passive in the absence of an immune challenge and to become activated only in the context of inflammation. Exciting recent data have revealed important role for microglia even in the absence of any inflammation or immune challenge. This is particularly clear in the context of brain development; the role for microglia in normal adult brain function and homeostasis is less well established.
These recently appreciated noninflammatory functions of microglia create a rich new field for the understanding of pathophysiological processes. The evidence for microglial dysregulation in the pathophysiology of several psychiatric conditions, which we have summarized here, is intriguing. While these associations remain inconclusive in most cases, this is an exciting area of ongoing research. To the extent that microglial dysregulation proves to be causally important in the development of neuropsychiatric disease, it may provide a fruitful new area for therapeutic intervention.
References
- 1.Paolicelli RC, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 2.Ji K, Akgul G, Wollmuth LP, Tsirka SE. Microglia actively regulate the number of functional synapses. PLOS ONE. 2013;8(2) doi: 10.1371/journal.pone.0056293.e56293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sierra A, Encinas JM, Deudero JJP, et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7(4):483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duman R, Aghajanian G. Synaptic dysfunction in depression: potential therapeutic targets. Science. 2012;338(6103):68–72. doi: 10.1126/science.1222939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coyle JT, Basu A, Benneyworth M, Balu D, Konopaske G. Glutamatergic synaptic dysregulation in schizophrenia: therapeutic implications. Handbook of Experimental Pharmacology. 2012;(213):267–295. doi: 10.1007/978-3-642-25758-2_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eyre H, Baune BT. Neuroplastic changes in depression: a role for the immune system. Psychoneuroendocrinology. 2012;37(9):1397–1416. doi: 10.1016/j.psyneuen.2012.03.019. [DOI] [PubMed] [Google Scholar]
- 8.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiology Review. 2011;91(2):461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 9.Trenblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. The Journal of Neuroscience. 2011;31(45):16064–16069. doi: 10.1523/JNEUROSCI.4158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nimmerjahn A, Kirchhoff F, Helmchen F. Neuroscience: resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo . Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 11.Stence N, Waite M, Dailey ME. Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia. 2001;33(3):256–266. [PubMed] [Google Scholar]
- 12.Bilbo SD, Schwarz JM. Early-life programming of later-life brain and behavior: a critical role for the immune system. Frontiers in Behavioral Neuroscience. 2009;3:p. 14. doi: 10.3389/neuro.08.014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Czeh M, Gressens P, Kaindl AM. The yin and yang of microglia. Developmental Neuroscience. 2011;33(3-4):199–209. doi: 10.1159/000328989. [DOI] [PubMed] [Google Scholar]
- 14.Reynolds AD, Banerjee R, Liu J, Gendelman HE, Mosley RL. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson’s disease. Journal of Leukocyte Biology. 2007;82(5):1083–1094. doi: 10.1189/jlb.0507296. [DOI] [PubMed] [Google Scholar]
- 15.Liu J, Gong N, Huang X, Reynolds AD, Mosley RL, Gendelman HE. Neuromodulatory activities of CD4+CD25+ regulatory T cells in a murine model of HIV-1-associated neurodegeneration. Journal of Immunology. 2009;182(6):3855–3865. doi: 10.4049/jimmunol.0803330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kipnis J, Schwartz M. Controlled autoimmunity in CNS maintenance and repair: naturally occurring CD4+CD25+ regulatory T-cells at the crossroads of health and disease. NeuroMolecular Medicine. 2005;7(3):197–206. doi: 10.1385/NMM:7:3:197. [DOI] [PubMed] [Google Scholar]
- 17.Avidan H, Kipnis J, Butovsky O, Caspi RR, Schwartz M. Vaccination with autoantigen protects against aggregated β-amyloid and glutamate toxicity by controlling microglia: effect of CD4+CD25+ T cells. European Journal of Immunology. 2004;34(12):3434–3445. doi: 10.1002/eji.200424883. [DOI] [PubMed] [Google Scholar]
- 18.Butovsky O, Talpalar AE, Ben-Yaakov K, Schwartz M. Activation of microglia by aggregated β-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-γ and IL-4 render them protective. Molecular and Cellular Neuroscience. 2005;29(3):381–393. doi: 10.1016/j.mcn.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 19.Maiorino C, Khorooshi R, Ruffini F, et al. Lentiviral-mediated administration of IL-25 in the CNS induces alternative activation of microglia. Gene Therapy. 2012 doi: 10.1038/gt.2012.58. [DOI] [PubMed] [Google Scholar]
- 20.Zhou X, Spittau B, Krieglstein K. TGFβ signalling plays an important role in IL4-induced alternative activation of microglia. Journal of Neuroinflammation. 2012;9:p. 210. doi: 10.1186/1742-2094-9-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hinojosa AE, Garcia-Bueno B, Leza JC, Madrigal JLM. CCL2/MCP-1 modulation of microglial activation and proliferation. Journal of Neuroinflammation. 2011;8, article 77 doi: 10.1186/1742-2094-8-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang G, Meng Y, Li W, et al. Neuronal MCP-1 mediates microglia recruitment and neurodegeneration induced by the mild impairment of oxidative metabolism. Brain Pathology. 2011;21(3):279–297. doi: 10.1111/j.1750-3639.2010.00445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prinz M, Priller J. Tickets to the brain: role of CCR2 and CX3CR1 in myeloid cell entry in the CNS. Journal of Neuroimmunology. 2010;224(1-2):80–84. doi: 10.1016/j.jneuroim.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 24.Rogers JT, Morganti JM, Bachstetter AD, et al. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. Journal of Neuroscience. 2011;31(45):16241–16250. doi: 10.1523/JNEUROSCI.3667-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bachstetter AD, Morganti JM, Jernberg J, et al. Fractalkine and CX3CR1 regulate hippocampal neurogenesis in adult and aged rats. Neurobiology of Aging. 2011;32(11):2030–2044. doi: 10.1016/j.neurobiolaging.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dantzer R. Cytokine, sickness behavior, and depression. Immunology and Allergy Clinics of North America. 2009;29(2):247–264. doi: 10.1016/j.iac.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Udina M, Castellvi P, Moreno-España J, et al. Interferon-induced depression in chronic hepatitis C: a systematic review and meta-analysis. Journal of Clinical Psychiatry. 2012;73(8):1128–1138. doi: 10.4088/JCP.12r07694. [DOI] [PubMed] [Google Scholar]
- 28.Bayer TA, Buslei R, Havas L, Falkai P. Evidence for activation of microglia in patients with psychiatric illnesses. Neuroscience Letters. 1999;271(2):126–128. doi: 10.1016/s0304-3940(99)00545-5. [DOI] [PubMed] [Google Scholar]
- 29.Steiner J, Bielau H, Brisch R, et al. Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. Journal of Psychiatric Research. 2008;42(2):151–157. doi: 10.1016/j.jpsychires.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 30.Dean B, Gibbons AS, Tawadros N, Brooks L, Everall IP, Scarr E. Different changes in cortical tumor necrosis factor-α-related pathways in schizophrenia and mood disorders. Molecular Psychiatry. 2012 doi: 10.1038/mp.2012.95. [DOI] [PubMed] [Google Scholar]
- 31.Steiner J, Walter M, Gos T, et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? Journal of Neuroinflammation. 2011;8:p. 94. doi: 10.1186/1742-2094-8-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hashimoto K. Emerging role of glutamate in the pathophysiology of major depressive disorder. Brain Research Reviews. 2009;61(2):105–123. doi: 10.1016/j.brainresrev.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 33.Sanacora G, Treccani G, Popoli M. Towars a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology. 2012;62(1):63–77. doi: 10.1016/j.neuropharm.2011.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathews DC, Henter ID, Zarate CA. Targeting the glutamatergic system to treat major depressive disorder: rationale and progress to date. Drugs. 2012;72(10):1313–1333. doi: 10.2165/11633130-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guillemin GJ. Quinolinic acid, the inescapable neurotoxin. FEBS Journal. 2012;279(8):1356–1365. doi: 10.1111/j.1742-4658.2012.08485.x. [DOI] [PubMed] [Google Scholar]
- 36.Maes M, Fisar Z, Medina M, Scapagnini G, Nowak G, Berk M. New drug targets in depression: inflammatory, cell-mediated immune, oxidative and nitrosative stress, mitochondrial, antioxidant, and neuroprogressive pathways. And new drug candidates-Nrf2 activators and GSK-3 inhibitors. Inflammopharmacology. 2012;20(3):127–150. doi: 10.1007/s10787-011-0111-7. [DOI] [PubMed] [Google Scholar]
- 37.Pittenger C, Duman RS. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology. 2008;33(1):88–109. doi: 10.1038/sj.npp.1301574. [DOI] [PubMed] [Google Scholar]
- 38.Hinwood M, Morandini J, Day TA, Walker FR. Evidence that microglia mediate the neurobiological effects of chronic psychological stress on the medial prefrontal cortex. Cerebral Cortex. 2012;22(6):1442–1454. doi: 10.1093/cercor/bhr229. [DOI] [PubMed] [Google Scholar]
- 39.Arakawa S, Shirayama Y, Fujita Y, et al. Minocycline produced antidepressant-like effects on the learned helplessness rats with alterations in levels of monoamine in the amygdala and no changes in BDNF levels in the hippocampus at baseline. Pharmacology Biochemistry and Behavior. 2011;100(3):601–606. doi: 10.1016/j.pbb.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 40.Wohleb ES, Hanke ML, Corona AW, et al. β-adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. Journal of Neuroscience. 2011;31(17):6277–6288. doi: 10.1523/JNEUROSCI.0450-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wohleb ES, Fenn AM, Pacenta AM, Powell ND, Sheridan JF, Godbout JP. Peripheral innate immune challenge exaggerated microglia activation, increased the number of inflammatory CNS macrophages, and prolonged social withdrawal in socially defeated mice. Psychoneuroendocrinology. 2012;37(9):1491–1505. doi: 10.1016/j.psyneuen.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sominsky L, Walker AK, Ong LK, Tynan RJ, Walker FR, Hodgson DM. Increased microglial activation in the rat brain following neonatal exposure to a bacterial mimetic. Behavioral Brain Research. 2012;226(1):351–356. doi: 10.1016/j.bbr.2011.08.038. [DOI] [PubMed] [Google Scholar]
- 43.Wang KC, Fan LW, Kaizaki A, Pang Y, Cai Z, Tien LT. Neonatal lipopolysaccharide exposure induces long-lasting learning impairment, less anxiety-like response and hippocampal injury in adult rats. Neuroscience. 2013;234:146–157. doi: 10.1016/j.neuroscience.2012.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Corona AW, Huang Y, O’Connor JC, et al. Fractalkine receptor (CX3CR1) deficiency sensitizes mice to the behavioral changes induced by lipopolysaccharide. Journal of Neuroinflammation. 2010;7, article 93 doi: 10.1186/1742-2094-7-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Corona AW, Norden DM, Skendelas JP, et al. Indoleamine 2,3-dioxygenase inhibition attenuates lipopolysaccharide induced persistent microglial activation and depressive-like complications in fractalkine receptor (CX3CR1)-deficient mice. Brain Behavior and Immunity. 2012 doi: 10.1016/j.bbi.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang Y, Lee JJ, Hsieh CY, Hsiao G, Chou DS, Sheu JR. Inhibitory effects of ketamine on lipopolysaccharide-induced microglial activation. Mediators of Inflammation. 2009;2009:7 pages. doi: 10.1155/2009/705379.705379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu D, Wang Z, Liu S, Wang F, Zhao S, Hao A. Anti-inflammatory effects of fluoxetine in lipopolysaccharide(LPS)-stimulated microglial cells. Neuropharmacology. 2011;61(4):592–599. doi: 10.1016/j.neuropharm.2011.04.033. [DOI] [PubMed] [Google Scholar]
- 48.Obuchowicz E, Kowalski J, Labuzek K, Krysiak R, Pendzich J, Herman ZS. Amitriptyline and nortriptyline inhibit interleukin-1β and tumour necrosis factor-α release by rat mixed glial and microglial cell cultures. International Journal of Neuropsychopharmacology. 2006;9(1):27–35. doi: 10.1017/S146114570500547X. [DOI] [PubMed] [Google Scholar]
- 49.Hashioka S, Klegeris A, Monji A, et al. Antidepressants inhibit interferon-γ-induced microglial production of IL-6 and nitric oxide. Experimental Neurology. 2007;206(1):33–42. doi: 10.1016/j.expneurol.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 50.Horikawa H, Kato TA, Mizoguchi Y, et al. Inhibitory effects of SSRIs on IFN-γ induced microglial activation through the regulation of intracellular calcium. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2010;34(7):1306–1316. doi: 10.1016/j.pnpbp.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 51.Tynan RJ, Weidenhofer J, Hinwood M, Cairns MJ, Day TA, Walker FR. A comparative examination of the anti-inflammatory effects of SSRI and SNRI antidepressants on LPS stimulated microglia. Brain Behavior and Immunity. 2012;26(3):469–479. doi: 10.1016/j.bbi.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 52.Zhang F, Zhou H, Wilson BC, Shi JS, Hong JS, Gao HM. Fluoxetine protects neurons against microglial activation-mediated neurotoxicity. Parkinsonism and Related Disorders. 2012;18(supplement 1):213–217. doi: 10.1016/S1353-8020(11)70066-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chung HS, Kim H, Bae H. Phenelzine (monoamine oxidase inhibitor) increases production of nitric oxide and proinflammatory cytokines via the NF-κB pathway in lipopolysaccharide-activated microglia cells. Neurochemical Research. 2012;37(10):2117–2124. doi: 10.1007/s11064-012-0833-y. [DOI] [PubMed] [Google Scholar]
- 54.Anderson G, Maes M. Schizophrenia: linking prenatal infection to cytokines, the tryptophan catabolite (TRYCAT) pathway, NMDA receptor hypofunction, neurodevelopment and neuroprogression. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2013;42:5–19. doi: 10.1016/j.pnpbp.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 55.Hagberg H, Gressens P, Mallard C. Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Annals of Neurology. 2012;71(4):444–457. doi: 10.1002/ana.22620. [DOI] [PubMed] [Google Scholar]
- 56.Togo T, Akiyama H, Kondo H, et al. Expression of CD40 in the brain of Alzheimer’s disease and other neurological diseases. Brain Research. 2000;885(1):117–121. doi: 10.1016/s0006-8993(00)02984-x. [DOI] [PubMed] [Google Scholar]
- 57.Wierzba-Bobrowicz T, Lewandowska E, Kosno-Kruszewska E, Lechowicz W, Pasennik E, Schmidt-Sidor B. Degeneration of microglial cells in frontal and temporal lobes of chronic schizophrenics. Folia Neuropathologica. 2004;42(3):157–165. [PubMed] [Google Scholar]
- 58.Wierzba-Bobrowicz T, Lewandowska E, Lechowicz W, Stepień T, Pasennik E. Quantitative analysis of activated microglia, ramified and damage of processes in the frontal and temporal lobes of chronic schizophrenics. Folia Neuropathologica. 2005;43(2):81–89. [PubMed] [Google Scholar]
- 59.Uranova NA, Zimina IS, Vikhreva OV, Krukov NO, Rachmanova VI, Orlovskaya DD. Ultrastructural damage of capillaries in the neocortex in schizophrenia. The World Journal of Biological Psychiatry. 2010;11(3):567–578. doi: 10.3109/15622970903414188. [DOI] [PubMed] [Google Scholar]
- 60.Fillman SG, Cloonan N, Catts VS, et al. Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Molecular Psychiatry. 2013;18(2):206–214. doi: 10.1038/mp.2012.110. [DOI] [PubMed] [Google Scholar]
- 61.Radewicz K, Garey LJ, Gentleman SM, Reynolds R. Increase in HLA-DR immunoreactive microglia in frontal and temporal cortex of chronic schizophrenics. Journal of Neuropathology and Experimental Neurology. 2000;59(2):137–150. doi: 10.1093/jnen/59.2.137. [DOI] [PubMed] [Google Scholar]
- 62.Steiner J, Mawrin C, Ziegeler A, et al. Distribution of HLA-DR-positive microglia in schizophrenia reflects impaired cerebral lateralization. Acta Neuropathologica. 2006;112(3):305–316. doi: 10.1007/s00401-006-0090-8. [DOI] [PubMed] [Google Scholar]
- 63.Busse S, Busse M, Schiltz K, et al. Different distribution patterns of lymphocytes and microglia in the hippocampus of patients with residual versus paranoid schizophrenia: further evidence for disease course-related immune alterations? Brain Behavior and Immunity. 2012;26(8):1273–1279. doi: 10.1016/j.bbi.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 64.Drexhage RC, Hoogenboezem TA, Cohen D, et al. An activated set point of T-cell and monocyte inflammatory networks in recent-onset schizophrenia patients involves both pro- and anti-inflammatory forces. International Journal of Neuropsychopharmacology. 2011;14(6):746–755. doi: 10.1017/S1461145710001653. [DOI] [PubMed] [Google Scholar]
- 65.van Berckel BN, Bossong MG, Boellaard R, et al. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biological Psychiatry. 2008;64(9):820–822. doi: 10.1016/j.biopsych.2008.04.025. [DOI] [PubMed] [Google Scholar]
- 66.Doorduin J, de Vries EFJ, Willemsen ATM, de Groot JC, Dierckx RA, Klein HC. Neuroinflammation in schizophrenia-related psychosis: a PET study. Journal of Nuclear Medicine. 2009;50(11):1801–1807. doi: 10.2967/jnumed.109.066647. [DOI] [PubMed] [Google Scholar]
- 67.Sargin D, Hassouna I, Sperling S, Sirén AL, Ehrenreich H. Uncoupling of neurodegeneration and gliosis in a murine model of juvenile cortical lesion. Glia. 2009;57(7):693–702. doi: 10.1002/glia.20797. [DOI] [PubMed] [Google Scholar]
- 68.Juckel G, Manitz MP, Brüne M, Friebe A, Heneka MT, Wolf RJ. Microglial activation in a neuroinflammational animal model of schizophrenia—a pilot study. Schizophrenia Research. 2011;131(1–3):96–100. doi: 10.1016/j.schres.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 69.Ratnayake U, Quinn TA, Castillo-Melendez M, Dickinson H, Walker DW. Behaviour and hippocampus-specific changes in spiny mouse neonates after treatment of the mother with the viral-mimetic Poly I:C at mid-pregnancy. Brain Behavior and Immunity. 2012;26(8):1288–1299. doi: 10.1016/j.bbi.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 70.Liaury K, Miyaoka T, Tsumori T, et al. Morphological features of microglial cells in the hippocampal dentate gyrus of Gunn rat: a possible schizophrenia animal model. Journal of Neuroinflammation. 2012;9:p. 56. doi: 10.1186/1742-2094-9-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seshadri S, Kamiya A, Yokota Y, et al. Disrupted-in-Schizophrenia-1 expression is regulated by β-site amyloid precursor protein cleaving enzyme-1-neuregulin cascade. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(12):5622–5627. doi: 10.1073/pnas.0909284107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bian Q, Kato T, Monji A, et al. The effect of atypical antipsychotics, perospirone, ziprasidone and quetiapine on microglial activation induced by interferon-γ . Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2008;32(1):42–48. doi: 10.1016/j.pnpbp.2007.06.031. [DOI] [PubMed] [Google Scholar]
- 73.Hou Y, Wu CF, Yang JY, et al. Effects of clozapine, olanzapine and haloperidol on nitric oxide production by lipopolysaccharide-activated N9 cells. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2006;30(8):1523–1528. doi: 10.1016/j.pnpbp.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 74.Kato T, Monji A, Hashioka S, Kanba S. Risperidone significantly inhibits interferon-γ-induced microglial activation in vitro. Schizophrenia Research. 2007;92(1–3):108–115. doi: 10.1016/j.schres.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 75.Kato T, Mizoguchi Y, Monji A, et al. Inhibitory effects of aripiprazole on interferon-γ-induced microglial activation via intracellular Ca2+ regulation in vitro. Journal of Neurochemistry. 2008;106(2):815–825. doi: 10.1111/j.1471-4159.2008.05435.x. [DOI] [PubMed] [Google Scholar]
- 76.Kato TA, Monji A, Yasukawa K, et al. Aripiprazole inhibits superoxide generation from phorbol-myristate-acetate (PMA)-stimulated microglia in vitro: implication for antioxidative psychotropic actions via microglia. Schizophrenia Research. 2011;129(2-3):172–182. doi: 10.1016/j.schres.2011.03.019. [DOI] [PubMed] [Google Scholar]
- 77.Zheng LT, Hwang J, Ock J, Lee MG, Lee WH, Suk K. The antipsychotic spiperone attenuates inflammatory response in cultured microglia via the reduction of proinflammatory cytokine expression and nitric oxide production. Journal of Neurochemistry. 2008;107(5):1225–1235. doi: 10.1111/j.1471-4159.2008.05675.x. [DOI] [PubMed] [Google Scholar]
- 78.Miyaoka T, Yasukawa R, Yasuda H, Hayashida M, Inagaki T, Horiguchi J. Possible antipsychotic effects of minocycline in patients with schizophrenia. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2007;31(1):304–307. doi: 10.1016/j.pnpbp.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 79.Kelly DL, Vyas G, Richardson CM, et al. Adjunct minocycline to clozapine treated patients with persistent schizophrenia symptoms. Schizophrenia Research. 2011;133(1–3):257–258. doi: 10.1016/j.schres.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 80.Chaves C, de Marque CR, Wichert-Ana L, et al. Functional neuroimaging of minocycline’s effect in a patient with schizophrenia. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2010;34(3):550–552. doi: 10.1016/j.pnpbp.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 81.Miyaoka T, Yasukawa R, Yasuda H, Hayashida M, Inagaki T, Horiguchi J. Minocycline as adjunctive therapy for schizophrenia: an open-label study. Clinical Neuropharmacology. 2008;31(5):287–292. doi: 10.1097/WNF.0b013e3181593d45. [DOI] [PubMed] [Google Scholar]
- 82.Miyaoka T, Wake R, Furuya M, et al. Minocycline as adjunctive therapy for patients with unipolar psychotic depression: an open-label study. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2012;37(2):222–226. doi: 10.1016/j.pnpbp.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 83.Levkovitz Y, Mendlovich S, Riwkes S, et al. A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. Journal of Clinical Psychiatry. 2010;71(2):138–149. doi: 10.4088/JCP.08m04666yel. [DOI] [PubMed] [Google Scholar]
- 84.Chaudhry B, Hallak J, Husain N, et al. Minocycline benefits negative symptoms in early schizophrenia: a randomised double-blind placebo-controlled clinical trial in patients on standard treatment. Journal of Psychopharmacology. 2012;26(9):1185–1193. doi: 10.1177/0269881112444941. [DOI] [PubMed] [Google Scholar]
- 85.Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Annals of Neurology. 2005;57(1):67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- 86.Morgan JT, Chana G, Pardo CA, et al. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biological Psychiatry. 2010;68(4):368–376. doi: 10.1016/j.biopsych.2010.05.024. [DOI] [PubMed] [Google Scholar]
- 87.Tetreault NA, Hakeem AY, Jiang S, et al. Microglia in the cerebral cortex in autism. Journal of Autism and Developmental Disorders. 2012;42(12):2569–2584. doi: 10.1007/s10803-012-1513-0. [DOI] [PubMed] [Google Scholar]
- 88.Morgan JT, Chana G, Abramson I, Semendeferi K, Courchesne E, Everall IP. Abnormal microglial-neuronal spatial organization in the dorsolateral prefrontal cortex in autism. Brain Research. 2012;1456:72–81. doi: 10.1016/j.brainres.2012.03.036. [DOI] [PubMed] [Google Scholar]
- 89.Suzuki K, Sugihara G, Ouchi Y, et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry. 2013;70(1):49–58. doi: 10.1001/jamapsychiatry.2013.272. [DOI] [PubMed] [Google Scholar]
- 90.Heo Y, Zhang Y, Gao D, Miller VM, Lawrence DA. Aberrant immune responses in a mouse with behavioral disorders. PLoS ONE. 2011;6(7) doi: 10.1371/journal.pone.0020912.e20912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Connors SL, Crowell DE, Eberhart CG, et al. β 2-adrenergic receptor activation and genetic polymorphisms in autism: data from dizygotic twins. Journal of Child Neurology. 2005;20(11):876–884. doi: 10.1177/08830738050200110401. [DOI] [PubMed] [Google Scholar]
- 92.Zerrate MC, Pletnikov M, Connors SL, et al. Neuroinflammation and behavioral abnormalities after neonatal terbutaline treatment in rats: implications for autism. Journal of Pharmacology and Experimental Therapeutics. 2007;322(1):16–22. doi: 10.1124/jpet.107.121483. [DOI] [PubMed] [Google Scholar]
- 93.MacFabe DF, Cain DP, Rodriguez-Capote K, et al. Neurobiological effects of intraventricular propionic acid in rats: possible role of short chain fatty acids on the pathogenesis and characteristics of autism spectrum disorders. Behavioural Brain Research. 2007;176(1):149–169. doi: 10.1016/j.bbr.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 94.MacFabe DF, Cain NE, Boon F, Ossenkopp KP, Cain DP. Effects of the enteric bacterial metabolic product propionic acid on object-directed behavior, social behavior, cognition, and neuroinflammation in adolescent rats: relevance to autism spectrum disorder. Behavioural Brain Research. 2011;217(1):47–54. doi: 10.1016/j.bbr.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 95.Maezawa I, Jin LW. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. Journal of Neuroscience. 2010;30(15):5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Horská A, Farage L, Bibat G, et al. Brain metabolism in rett syndrome: age, clinical, and genotype correlations. Annals of Neurology. 2009;65(1):90–97. doi: 10.1002/ana.21562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Blue ME, Naidu S, Johnston MV. Altered development of glutamate and GABA receptors in the basal ganglia of girls with Rett syndrome. Experimental Neurology. 1999;156(2):345–352. doi: 10.1006/exnr.1999.7030. [DOI] [PubMed] [Google Scholar]
- 98.Wood L, Shepherd GMG. Synaptic circuit abnormalities of motor-frontal layer 2/3 pyramidal neurons in a mutant mouse model of Rett syndrome. Neurobiology of Disease. 2010;38(2):281–287. doi: 10.1016/j.nbd.2010.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Blaylock RL, Strunecka A. Immune-glutamatergic dysfunction as a central mechanism of the autism spectrum disorders. Current Medicinal Chemistry. 2009;16(2):157–170. doi: 10.2174/092986709787002745. [DOI] [PubMed] [Google Scholar]
- 100.Derecki NC, Cronk JC, Lu Z, et al. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484(7392):105–109. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Swedo SE, Schrag A, Gilbert R, et al. Streptococcal infection, Tourette syndrome, and OCD: is there a connection? PANDAS: horse or zebra? Neurology. 74(17):1397–1398. doi: 10.1212/WNL.0b013e3181d8a638. [DOI] [PubMed] [Google Scholar]
- 102.Bhattacharyya S, Khanna S, Chakrabarty K, Mahadevan A, Christopher R, Shankar SK. Anti-brain autoantibodies and altered excitatory neurotransmitters in obsessive-compulsive disorder. Neuropsychopharmacology. 2009;34(12):2489–2496. doi: 10.1038/npp.2009.77. [DOI] [PubMed] [Google Scholar]
- 103.Greer JM, Capecchi MR. Hoxb8 is required for normal grooming behavior in mice. Neuron. 2002;33(1):23–34. doi: 10.1016/s0896-6273(01)00564-5. [DOI] [PubMed] [Google Scholar]
- 104.Chen SK, Tvrdik P, Peden E, et al. Hematopoietic origin of pathological grooming in Hoxb8 mutant mice. Cell. 2010;141(5):775–785. doi: 10.1016/j.cell.2010.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Holstege JC, De Graaff W, Hossaini M, et al. Loss of Hoxb8 alters spinal dorsal laminae and sensory responses in mice. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(17):6338–6343. doi: 10.1073/pnas.0802176105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Huber L, Ferdin M, Holzmann J, Stubbusch J, Rohrer H. HoxB8 in noradrenergic specification and differentiation of the autonomic nervous system. Developmental Biology. 2012;363(1):219–233. doi: 10.1016/j.ydbio.2011.12.026. [DOI] [PubMed] [Google Scholar]
- 107.Knoepfler PS, Sykes DB, Pasillas M, Kamps MP. HoxB8 requires its Pbx-interaction motif to block differentiation of primary myeloid progenitors and of most cell line models of myeloid differentiation. Oncogene. 2001;20(39):5440–5448. doi: 10.1038/sj.onc.1204710. [DOI] [PubMed] [Google Scholar]
- 108.Wang GG, Calvo KR, Pasillas MP, Sykes DB, Häcker H, Kamps MP. Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nature Methods. 2006;3(4):287–293. doi: 10.1038/nmeth865. [DOI] [PubMed] [Google Scholar]
- 109.Rosas M, Osorio F, Robinson MJ, et al. Hoxb8 conditionally immortalised macrophage lines model inflammatory monocytic cells with important similarity to dendritic cells. European Journal of Immunology. 2011;41(2):356–365. doi: 10.1002/eji.201040962. [DOI] [PubMed] [Google Scholar]
- 110.Morer A, Chae W, Henegariu O, Bothwell ALM, Leckman JF, Kawikova I. Elevated expression of MCP-1, IL-2 and PTPR-N in basal ganglia of Tourette syndrome cases. Brain, Behavior, and Immunity. 2010;24(7):1069–1073. doi: 10.1016/j.bbi.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 111.Mizutani M, Pino PA, Saederup N, Charo IF, Ransohoff RM, Cardona AE. The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. Journal of Immunology. 2012;188(1):29–36. doi: 10.4049/jimmunol.1100421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Saederup N, Cardona AE, Croft K, et al. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS ONE. 2010;5(10) doi: 10.1371/journal.pone.0013693.e13693 [DOI] [PMC free article] [PubMed] [Google Scholar]