

Abstract
Objective
Although the majority of sudden cardiac arrests (SCA) occur in patients with ischemic heart disease, the effect of therapeutic hypothermia (TH) on arrhythmia susceptibility during acute global ischemia is not well understood. While both ischemia and severe hypothermia are arrhythmogenic, patients undergoing TH do not have an increase in arrhythmias, despite the fact that most SCA occur in the setting of ischemia. We hypothesized that mild hypothermia induced prior to myocardial ischemia and reperfusion will have a beneficial effect on ischemia-related arrhythmia substrates.
Design
We developed a model of global ischemia and reperfusion in the canine wedge preparation in order to study the transmural electrophysiologic effects of ischemia at different temperatures.
Setting
Animal Study
Subjects
Male mongrel dogs.
Interventions
Canine left ventricle wedge preparations at 1) Control (36° C) or 2) Mild hypothermia (MH), to simulate temperatures used in TH (32° C), were subjected to 15 minutes of no-flow ischemia and subsequently reperfused.
Measurements
Optical action potentials were recorded spanning the transmural wall of left ventricle. Action potential duration (APD) for epicardial, mid-myocardial, and epicardial cells were measured. Transmural dispersion of repolarization (DOR) and conduction velocity (CV) were measured at baseline, during ischemia and reperfusion.
Main Results
No difference was seen at baseline for CV or DOR between groups. CV decreased from .46±.02 to .23 ±.07 m/s and DOR increased from 30±5 to 57±4 ms in the control group at 15 min of ischemia. Mild hypothermia attenuated both ischemia-induced CV slowing (decreasing from .44±.02 to .35±.03 m/s, p=.019), and the ischemia-induced DOR increase (25±3 to 37±7 ms, p=.037). Epicardial conduction block was observed in 6/7 preparations of the control group, but no preparations in the MH group developed conduction block (0/6).
Conclusions
Mild hypothermia attenuated ischemia-induced increase in DOR, conduction slowing and block, which are known mechanisms of arrhythmogenesis in ischemia. These data suggest that TH may decrease arrhythmogenesis during myocardial ischemia.
Keywords: hypothermia, dispersion of repolarization, arrhythmias, ventricular fibrillation, optical mapping, resuscitation
Introduction
Sudden cardiac arrest (SCA) related to ventricular arrhythmias is a major cause of mortality in the United States, accounting for over 500,000 deaths annually.(1) The vast majority of patients suffering SCA have evidence of coronary heart disease and up to 15-64% have evidence of recent coronary occlusion (2) Therapeutic hypothermia (TH) (cooling to 32-34°C) has now become the standard of care for patients who suffer cardiac arrest and have subsequent return of spontaneous circulation (ROSC).(3-5) The most recent Advanced Cardiac Life Support Guidelines (ACLS) recommends induction of TH after ROSC both after VF arrest (Class I, LOE B) and PEA/asystole (Class IIb, LOE C).(6)
However, hypothermia is arrhythmogenic. Arrhythmias are encountered in the majority of patients with severe hypothermia (less than 30° C), including VF that can be refractory to standard therapy.(7,8) Hypothermia prolongs action potential duration (APD), resulting in prolongation of the QT interval as currents governing repolarization are particularly susceptible to hypothermia.(9-13) Importantly, hypothermia increases transmural dispersion of repolarization (DOR), a well-known prerequisite for reentrant excitation and arrhythmogenesis.(14-17) We recently demonstrated in the canine ventricular wedge preparation that cooling to 26° C profoundly increases DOR which could be directly linked to a mechanism of VF in severe hypothermia.(18) However, we also observed that mild hypothermia at temperatures typically used during TH, was not associated with increases in DOR or arrhythmia susceptibility under normal, non-ischemic conditions. The effects of hypothermia on DOR and conduction under global ischemic conditions, as seen is resuscitation from cardiac arrest, are unknown.
The arrhythmia substrates which occur during acute ischemia have been extensively studied.(15-17) Acute ischemia induces profound and complex electrophysiologic effects resulting in increased transmural DOR. Ischemia also induces conduction slowing and conduction block. Transmural electrophysiologic heterogeneities and conduction block are fundamental to the mechanism of arrhythmias in ischemia. (16, 17)
As TH is more widely employed, it is crucial to understand the interaction of a potentially arrhythmogenic therapy used in an arrhythmogenic situation. All patients with ROSC from a VF arrest undergo a period of global cardiac ischemia, but there is limited data on the electrophysiologic sequellae of TH in the post-resuscitated heart or in ischemia-induced arrhythmias. This interaction is particularly important as the vast majority of VF arrests are caused by myocardial ischemia and infarction. Importantly, clinical and laboratory observations suggest that mild hypothermia after cardiac arrest is not associated with an increase in arrhythmias.(19-21). It is possible that TH attenuates the substrate for ischemia-induced arrhythmias; specifically transmural DOR and transmural decreases in conduction velocity. Better understanding of the mechanisms by which TH might be antiarrhythmic in the setting of acute ischemia may lead to 1) Novel therapeutic targets to decrease ventricular arrhythmias both during ischemia and the post-arrest patient, 2) Refinement of TH protocols in order to optimize the potential cardioprotective effect of TH, and 3) the potential utilitization of TH as a therapy in patients prior to cardiac arrest.
Our overall hypothesis is that mild hypothermia induced during myocardial ischemia would be antiarrhythmic. Based on our previous studies in the non-ischemic heart, we predicted that mild hypothermia will attenuate the underlying electrophysiologic derangements that promote reentrant arrhythmias during ischemia: transmural conduction slowing and increased DOR. We used a model of global ischemia in the canine ventricular wedge preparation which allows the study of transmural electrophysiologic effects of mild hypothermia during ischemia and reperfusion.
MATERIALS AND METHODS
Study Design
Experiments were carried out in accordance with Public Health Service guidelines for the care and use of laboratory animals. 13 male mongrel dogs (approx. 15-20 kg) were used in this study. Organ harvest by right lateral thoracotomy was performed under pentobarbital (50mg/ml) I.V. general anesthesia and the intact heart was rapidly excised. The details of our system for high resolution transmural optical mapping of the arterial perfused canine wedge preparation were described previously.(16,18,22-24) Briefly, wedges of myocardium surrounding secondary branches of the LAD and circumflex were harvested from the left ventricle (approximately 3 × 2 × 1.5 cm) were isolated. The corresponding branch of coronary artery was cannulated and Langendorff perfused with Tyrode's solution (140 mM NaCl 4.03 mM KCl, 1.8 mM CaCl2, 5.5 mM dextrose, 0.5 mM MgSO4, 0.9 mM NaH2PO4, 10mM HEPES, NaOH titrated to a pH 7.41, oxygenated with 100% O2). The wedge was then placed in the chamber with the transmural surface against the glass imaging plate and perfused with an approximate perfusion pressure of 40 mmHg and a perfusion rate of 12-20 ml/min. Pressure and rate were monitored throughout the experiment. Wedges were then perfused the above Tyrodes solution and a voltage sensitive dye (di-4-ANNEPS, 15 μM). Action potentials were recorded from 256 sites simultaneously with high spatial (.89 mm), temporal (.5 ms) and voltage (.5mv) resolution. Optical magnification of 1 to 1.2x was used. Blebbistatin (5 μM) was used to eliminate motion artifact from the optical signals. We have previously validated a method for precise homogeneous temperature manipulation in the canine wedge preparation. (18) The imaging chamber was encased in a water insulated circuit, and temperature was measured using a digital temperature probe (Omega) in the water bath, allowing for temperature precision of 0.1°C. Temperature was controlled at 36°C (Control group) or 32°C (MH group) prior to inducing ischemia so as to achieve and maintain specific temperature during the ischemia protocol.
Ischemia protocol
We developed a model of global ischemia in the canine wedge preparations in order to evaluate the electrophysiologic effects of ischemia as seen in SCA. To induce global ischemia, flow of oxygenated Tyrode's solution was stopped. Measurements were made at 5, 10, and 15 minutes of no-flow ischemia. Flow was then resumed and measurements were made at 5 and 10 minutes of reperfusion.
Methods of Measurement
Action potential duration (APD) was measured by using an average of 5 epicardial (EPI), mid-myocardial (M), and endocardial (ENDO) cells, respectively. Transmural cell types were defined by previously validated anatomic and functional criterion. (22) EPI cells were defined as cells 1-2 mm from the epicardial surface. ENDO cells were defined as cells 1-2 mm from the ENDO surface. M cells were defined as the cells with the longest APD that were not EPI or ENDO cells. Dispersion of repolarization (DOR) was defined as the difference between the APD of the longest and shortest cell type. Conduction velocity (CV) was determined by a previously validated vector analysis technique.(25)
For arrhythmia induction, an identical programmed electrical stimulation (PES) protocol was performed at baseline, during ischemia, and during reperfusion in five preparations of the control group and four preparations of the MH group each group.(23). PES can induce conduction block, reentrant extra beats, monomorphic or polymorphic VT, and VF. VT was defined as 3 or more continuous extra beats. During ENDO pacing (S1), up to two premature beats were delivered from either the EPI or ENDO surface (S2 and S3), until failure to capture the preparation or an arrhythmia was induced. Typically, induced arrhythmias self-terminate in the wedge preparation allowing for serial comparison of arrhythmia susceptibility in any one preparation. PES was considered positive if there was a new inducible VT or VF from baseline or spontaneous VT or VF was observed. Baseline arrhythmias reproduced during ischemia were not considered positive.
Statistical analysis was performed using IBM SPSS Statistics 20 (IBM Corp, 2011). The primary analysis focused on the difference in electrophysiologic parameters between the MH and control group at baseline and during ischemia. Student's T-tests were used to analyze the differences in mean APD, CV, and DOR at baseline and at 15 min of ischemia. Furthermore, in order to analyze the group-time from baseline to ischemia and then reperfusion as well as accounting for possible baseline differences between groups, repeated measure analysis of variance (ANOVA) was performed with baseline covariate analysis and the EM algorithm (SAS Institute Inc.) for imputation of the minimal amount of missing data points through the trend (4 of 78) Group effects and differences between specific means were considered significant with a p≤0.05. All mean data is represented with the value and standard error of the mean. Statistical significance for the figures is represented by an asterisk (*) or as otherwise stated in the figure legends.
RESULTS
Mild hypothermia attenuates ischemia induced DOR and preserves AP morphology
The effects of global ischemia on APD, AP morphology, DOR, and conduction time (CT) are shown in a representative control experiment in Figure 1, Panel A. At baseline, DOR is 20 ms, and repolarization gradient is minimal. During ischemia, CT is markedly slowed and the repolarization gradient increases. APD shortens, specifically in the epicardium with significant morphological changes demonstrating ischemia (triangulation of the AP). DOR increases to 71 ms. Upon reperfusion, APD morphology returns to baseline, conduction time improves, and DOR returns closer to baseline.
Figure 1. Mild hypothermia attenuates ischemia-induced derangements in action potential duration, conduction time and repolarization gradients.


Panel A. Representative action potentials from each cell type during ischemia and reperfusion from the control group are shown. ECG is also shown. Left. Action potentials at baseline demonstrate a baseline dispersion of 20 ms. Middle. During ischemia, marked prolongation of dispersion is observed, secondary to significant epicardial action potential duration shortening. Right. After 10 min reperfusion, dispersion and action potential duration returns toward baseline. Panel B. Representative action potentials during ischemia and reperfusion from the mild hypothermia group are shown. ECG is also shown. Left. Action potentials at baseline. Middle. During ischemia epicardial shortening is attenuated. Right. After 10 min reperfusion, dispersion returns to baseline.
Mild hypothermia attenuated the electrophysiological effects of global ischemia, as shown in a representative experiment in Panel B. At baseline, there is similar DOR, CT, and repolarization gradient to the control experiment. During ischemia, the APD shortening seen in the control group is attenuated. CT is not significantly slowed and the AP morphology remained similar to baseline, specifically in the epicardium. The large ischemia-induced increase in DOR is attenuated. Upon reperfusion, DOR returns to baseline.
Figure 2 summarizes the effect of mild hypothermia on DOR during ischemia. At baseline, mean APD is longer in the hypothermia group (230±6 ms vs. 287±7 ms, p<.001) but no significant difference is seen in DOR (30±5 ms vs. 24±3 ms, p=ns). At 15 min ischemia, the increase in DOR is significantly larger in the control vs. MH group (57±4 ms vs. 37±7 ms, p=.037). Upon reperfusion, DOR returns to baseline and no significant difference is seen (p=ns). Table 1 summarizes the difference in mean APD and each cell type from baseline to ischemia. The greatest change is seen in the epicardial cells (difference of 69 ms) in the control group which improved in the MH group (difference of 43 ms).
Figure 2. Mild hypothermia attenuates ischemia-induced dispersion of repolarization.

Summary data is shown in mild hypothermia and control groups during ischemia and reperfusion. No difference in transmural dispersion of repolarization was observed at baseline. Significant increase is observed at 15 min ischemia in the control group compared to therapeutic hypothermia group (denoted by *). Overall group by time difference was significant (p=.007)
Table 1.
Changes in APD from baseline and during ischemia in each group.
| Control (36°C) | MH (32°C) | |||
|---|---|---|---|---|
| Baseline | 15 min Ischemia | Baseline | 15 min Ischemia | |
| Mean APD (ms) | 230±6 | 178±13 | 287±7 | 250±15 |
| EPI (ms) | 211±7 | 142±15 | 272±8 | 229±12 |
| M (ms) | 238±7 | 197±12 | 292±8 | 260±18 |
| ENDO (ms) | 241±6 | 194±12 | 296±8 | 263±15 |
APD= action potential duration. EPI=epicardial action potential duration. M=mid myocardial action potential duration. ENDO=endocardial action potential duration. Inducible = induced with PES. Spontaneous= occurred during observation without PES. Temperature noted is where arrhythmia was first induced or observed during cooling or rewarming protocol. Block and reentry= conduction block and single reentrant beat mapped during PES. All VT was nonsustained polymorphic unless otherwise noted. All arrythmias resolved spontaneously. VF=sustained ventricular fibrillation.
Mild hypothermia attenuates ischemia-induced conduction slowing and epicardial conduction block
Conduction slowing was significantly attenuated during ischemia in the MH group. Figure 3 depicts isochrome maps of conduction time during ischemia and reperfusion in a representative example of both control and MH groups. In the control group, there is significant conduction slowing (crowding of isochrones) during ischemia with EPI conduction block (red line) that resolves during reperfusion. In the MH group, ischemia-induced conduction slowing is attenuated and no EPI block occurs. Figure 4 summarizes CV during ischemia reperfusion for both control and MH groups. At baseline, no significant difference in CV was appreciated (.46±.02 vs. .44±.02 m/s, p=ns). At 15 min of ischemia there was a significantly greater decrease in CV in the control group (.23 ±.07 m/sec vs. .35±.03 m/s, p=.019). Both groups returned to baseline upon rewarming.
Figure 3. Mild hypothermia attenuates ischemia- induced transmural conduction slowing.
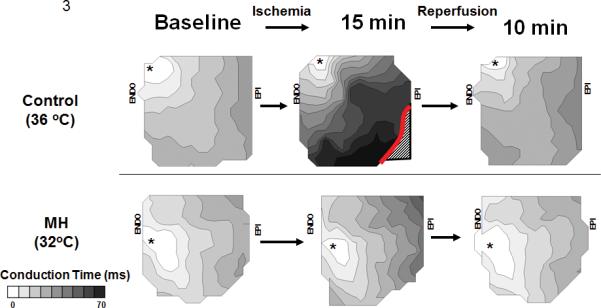
Activation maps of the transmural surface of the canine wedge preparation for conduction time are shown at baseline, 15 min ischemia, and 10 min reperfusion for control and therapeutic hypothermia groups Top. Control group. Ischemia increased conduction time and produced epicardial conduction block; both returned to baseline upon reperfusion. Bottom. Mild Hypothermia Group. Conduction slowing is attenuated during ischemia compared to the control group and returns to baseline upon reperfusion. No epicardial block is observed.
Figure 4. Mild hypothermia attenuates conduction slowing.

Summary data is shown for mild hypothermia and control groups during ischemia and reperfusion. No difference in conduction velocity was seen at baseline. Significant increase is seen at 15 min ischemia in the control group compared to mild hypothermia group (denoted by *). Overall group by time difference was significant (p=.027)
EPI conduction block was improved by mild hypothermia. In six of seven preparations in the control group, conduction block in the EPI was observed, while we did not observe EPI conduction block in any of the MH group (0/6 preparations).
Programmed Electrical Stimulation (PES)
Five preparations in the control group and four in the MH group underwent PES. In the control group, 3/5 had a positive PES during ischemia. VT was induced in two preparations at the area of conduction block, and one preparation had spontaneous VT at 15 min ischemia. In the MH group, 1 of 4 preparations had a new arrhythmia during ischemia and there was no evidence of epicardial block. In the preparation that was inducible, the VT origin was not related to transmural conduction slowing or block. Figure 5 depicts an arrhythmia induced during PES, resulting from EPI conduction block in the control group during ischemia.
Figure 5. Arrhythmia secondary to conduction block during ischemia.

Panel A. ECG and action potentials during programmed electrical stimulation are shown. Three endocardial paced beats (S1, S2, S3) are followed by induction of ventricular tachycardia. Panel B. Activation mapping before ischemia compared to 15 minutes of ischemia (S1 beat). Note that ischemia induces significant conduction slowing (crowding of isochrones). During ischemia, conduction fails in the epicardium (area with dashed line). Conduction block occurs during the S3 beat (block symbol in panel A ,red line in panel B) followed by the initiation of ventricular tachycardia, with the first beat reentering through the more electrically preserved subepicardium.
DISCUSSION
These data demonstrate that mild hypothermia, as induced during TH, attenuates the underlying electrophysiologic derangements that promote reentrant arrhythmias during ischemia. Mild hypothermia attenuates transmural conduction slowing and increased DOR, protecting against ischemia-induced arrhythmias and providing potential insights into the mechanisms underlying this protection.
Clinically, TH has not been shown to increase arrhythmias, despite a potentially arrhythmic treatment (hypothermia) being added to an already arrhythmogenic substrate (ischemia). Sagalyn et al reviewed 13 studies of patients undergoing TH after ROSC from cardiac arrest. Of the 656 patients treated with TH, (11%) had an arrhythmia vs. 30% (41/138) in the historical controls.(20)
Results of studies to assess the potential cardioprotective benefit of TH in MI prior to arrest have been mixed. Two clinical trials (COOL-MI and ICT-IT) did not demonstrate a reduction in infarct size or improvement of ejection fraction (26,27). However, subgroup analysis of patients undergoing TH before revascularization showed a possible benefit. Therefore, a recent feasibility study was performed to enroll patients in a clinical trial to assess this new question (28). Importantly, none of these studies demonstrated an increase in ventricular arrhythmias during hypothermia. Although the number of patients in these studies was small and the study aims addressed infarct size and cardiac function, the number of arrhythmias reported were less in the hypothermia groups.
Ischemia-induced conduction slowing was improved by mild hypothermia. A possible explanation for this observation is that mild hypothermia maintained intercellular communication through gap junctions during ischemia, thereby maintaining conduction. Another possible mechanism includes temperature-related preservation of ischemia-sensitive conduction currents, specifically the fast Na current. We also observed that mild hypothermia attenuated ischemia-induced enhancement of DOR. It is likely that one or more temperature-sensitive repolarization currents are affected by hypothermia. IK,ATP is a temperature sensitive repolarization current that is activated by ischemia and attenuated by hypothermia. It is therefore possible that hypothermia prevented activation of this current, maintaining APD and DOR, and therefore playing a role in the beneficial effects of mild hypothermia observed in these studies. It is also possible that decreasing ATP utilization during hypothermia also had a beneficial effect of CV and repolarization time.
At this time, the only therapy directly treating the post-arrest patient is TH, based on studies demonstrating improved neurological outcome and survival benefit. Exploring potential mechanisms of benefit could provide insight not only into the improved morbidity and mortality observed with TH, but potentially the development of novel antiarrhythmic interventions during resuscitation as well as in patients suffering from ongoing ischemia.
These experiments have some limitations. First, electrophysiologic effects of hypothermia were evaluated only using optical mapping in the canine wedge preparation, and therefore in a two dimensional area of a small portion of the left ventricle. Therefore, we cannot account for changes in interventricular conduction or repolarization or other heterogeneities (such as apex to base heterogeneities) which could potentially play a role in arrhythmia susceptibility in a whole-heart model of global ischemia. However, two important mechanisms of arrhythmias, transmural conduction velocity and transmural dispersion or repolarization, are best studied using the transmural canine wedge model. Further studies in whole-heart models of resuscitation are warranted. Second, our protocols dictated that the MH group hypothermia was instituted at 32°C before the preparation was made ischemic, and does not directly simulate the clinical scenario where TH is induced soon after the onset of ischemia. We specifically chose this design to directly evaluate the effect of temperature on global ischemia ,and the time required to cool the canine wedge preparation to TH levels (longer than 15 minutes) exceeds the time that the canine wedge preparation remains viabile during global ischemia. Since there is likely ongoing ischemia during most cases of clinical resuscitation as well as near immediate cooling by EMS in many clinical TH protocols, we feel this model is clinically relevant. Future studies in models of low-flow or less severe ischemia and induction of TH are warranted. Although out of the scope of the current investigation, we also did not directly examine the potential mechanisms underlying the beneficial electrophysiological effect of mild hypothermia. Although these studies provide insight into potential antiarrhythmic effects of TH in ischemic myocardium, further studies will be required to determine the cellular and molecular basis for the protective effects observed.
CONCLUSIONS
Mild hypothermia, at temperatures similar to TH, may decrease arrhythmia susceptibility by preserving transmural CV while attenuating EPI conduction block and increases in DOR, suggesting that TH may be antiarrhythmic in the ischemic myocardium. With the greater adoption of TH, an improved understanding of the electrophysiologic effects of TH on arrhythmogenesis in the ischemic and post-resuscitated heart is of growing importance. These data suggest that improved understanding of the interaction between myocardial ischemia and TH may provide avenues for the development of new therapies in the post-arrest patient.
Acknowledgments
SOURCES OF FUNDING
This study was supported by Departmental MetroHealth Foundation Grant (JSP), Emergency Medicine Foundation Career Development Grant (LDW) and by National Institutes of Health Grant RO1- HL54807 (DSR)
Footnotes
The Authors have no conflicts of interest. All experiments were carried out in accordance with Public Health Service guidelines for the care and use of laboratory animals
Presented in part at the ReSS program, Scientific Sessions of the American Heart Association, Orlando, FL, 2009
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Hu D, Viskin S, Oliva A, et al. Genetic predisposition and cellular basis for ischemia-induced ST-segment changes and arrhythmias. J Electrocardiol. 2007;40:S26–S29. doi: 10.1016/j.jelectrocard.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zipes DP, Wellens HJ. Sudden cardiac death. Circulation. 1998;98:2334–2351. doi: 10.1161/01.cir.98.21.2334. [DOI] [PubMed] [Google Scholar]
- 3.Nolan JP, Neumar RW, Adrie C, et al. Post-cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A Scientific Statement from the International Liaison Committee on Resuscitation; the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; the Council on Stroke. Resuscitation. 2008;79:350–379. doi: 10.1016/j.resuscitation.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 4.Bernard SA, Gray TW, Buist MD, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557–563. doi: 10.1056/NEJMoa003289. [DOI] [PubMed] [Google Scholar]
- 5.HACA Investigators Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:5549–56. doi: 10.1056/NEJMoa012689. [DOI] [PubMed] [Google Scholar]
- 6.Peberdy MA, Callaway CW, Neumar RW, et al. Part 9: post-cardiac arrest care: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122:S768–786. doi: 10.1161/CIRCULATIONAHA.110.971002. [DOI] [PubMed] [Google Scholar]
- 7.White JD. Hypothermia: the Bellevue Experience. Ann Emerg Med. 1982;11:417–424. doi: 10.1016/s0196-0644(82)80038-3. [DOI] [PubMed] [Google Scholar]
- 8.Hauty MG, Esrig BC, Hill JG, et al. Prognostic factors in severe accidental hypothermia: experience from the Mt. Hood tragedy. J Trauma. 1987;27:1107–1112. [PubMed] [Google Scholar]
- 9.Yan GX. Antzelevitch C: Cellular basis for the electrocardiographic J wave. Circulation. 1996;93:372–379. doi: 10.1161/01.cir.93.2.372. [DOI] [PubMed] [Google Scholar]
- 10.Vassal T, Benoit-Gonin B, Carrat F, et al. Severe accidental hypothermia treated in an ICU: prognosis and outcome. Chest. 2001;120:1998–2003. doi: 10.1378/chest.120.6.1998. [DOI] [PubMed] [Google Scholar]
- 11.Gambassi G, Cerbai E, Pahor M, et al. Temperature modulates calcium homeostasis and ventricular arrhythmias in myocardial preparations. Cardiovasc Res. 1994;28:391–399. doi: 10.1093/cvr/28.3.391. [DOI] [PubMed] [Google Scholar]
- 12.Liu B, Arlock P, Wohlfart B, et al. Temperature effects on the Na and Ca currents in rat and hedgehog ventricular muscle. Cryobiology. 1991;28:96–104. doi: 10.1016/0011-2240(91)90011-c. [DOI] [PubMed] [Google Scholar]
- 13.Sprung J, Laszlo A, Turner LA, et al. Effects of hypothermia, potassium , and verapamil on the action potential characteristics of canine cardiac Purkinje fibers. Anesthesiology. 1995;82:713–722. doi: 10.1097/00000542-199503000-00013. [DOI] [PubMed] [Google Scholar]
- 14.Salama G, Kanai AJ, Huang D, et al. Hypoxia and hypothermia enhance spatial heterogeneities of repolarization in guinea pig hearts: analysis of spatial autocorrelation of optically recorded action potential durations. J Cardiovasc Electrophysiol. 1998;9:164–183. doi: 10.1111/j.1540-8167.1998.tb00897.x. [DOI] [PubMed] [Google Scholar]
- 15.Carmeleit D. Cardiac Ionic Currents and Acute ischemia: From Channels to Arrhythmias. Physiological Reviews. 1999;79:917–1017. doi: 10.1152/physrev.1999.79.3.917. [DOI] [PubMed] [Google Scholar]
- 16.Akar FG, Laurita KR, Rosenbaum DS. Cellular basis for dispersion of repolarization underlying reentrant arrhythmias. J Electrocardiol. 2000;33(Suppl):23–31. doi: 10.1054/jelc.2000.20313. [DOI] [PubMed] [Google Scholar]
- 17.Kuo CS, Reddy CP, Munakata K, et al. Mechanism of ventricular arrhythmias caused by increased dispersion of repolarization. Eur Heart J. 1985;6(Suppl D):63–70. doi: 10.1093/eurheartj/6.suppl_d.63. [DOI] [PubMed] [Google Scholar]
- 18.Piktel JS, Jeyaraj D, Said TH, et al. Enhanced dispersion of repolarization explains increased arrhythmogenesis in severe versus therapeutic hypothermia. Circ Arrhythm Electrophysiol. 2011;4:79–86. doi: 10.1161/CIRCEP.110.958355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tiainen M, Parikka HJ, Makijarvi MA, et al. Arrhythmias and heart rate variability during and after therapeutic hypothermia for cardiac arrest. Crit Care Med. 2009;37:403–409. doi: 10.1097/CCM.0b013e31819572c4. [DOI] [PubMed] [Google Scholar]
- 20.Sagalyn E, Band RA, Gaieski D, et al. Therapeutic hypothermia after cardiac arrest in clinical practice: review and compilation of recent experiences. Crit Care Med. 2009;37:S223–S226. doi: 10.1097/CCM.0b013e3181aa5c7c. [DOI] [PubMed] [Google Scholar]
- 21.Kelly FE, Nolan JP. The effects of mild induced hypothermia on the myocardium: a systematic review. Anaesthesia. 2010;65:505–515. doi: 10.1111/j.1365-2044.2009.06237.x. [DOI] [PubMed] [Google Scholar]
- 22.Girouard SD, Laurita KR, Rosenbaum DS. Unique properties of cardiac action potentials recorded with voltage-sensitive dyes. J Cardiovasc Electrophysiol. 1996;7:1024–1038. doi: 10.1111/j.1540-8167.1996.tb00478.x. [DOI] [PubMed] [Google Scholar]
- 23.Pajouh M, Wilson LD, Poelzing S, et al. IKs blockade reduces dispersion of repolarization in heart failure. Heart Rhythm. 2005;2:731–738. doi: 10.1016/j.hrthm.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 24.Rosenbaum DS, Akar FG. Quantitative Cardiac Electrophysiology. First edition Marcel Dekker, Inc.; New York: 2002. The electrophysiological substrate for reentry: unique insight from optical mapping with voltage-sensitive dyes. [Google Scholar]
- 25.Fouts K, Fernandes B, Mal N, et al. Electrophysiological consequence of skeletal myoblast transplantation in normal and infarcted canine myocardium. Heart Rhythm. 2006;3:452–461. doi: 10.1016/j.hrthm.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 26.Dixon SR, Whitbourn RJ, Dae MW, et al. Induction of mild systemic hypothermia with endovascular cooling during primary percutaneous coronary intervention for acute myocardial infarction. J Am Coll Cardiol. 2002;40:1928–1934. doi: 10.1016/s0735-1097(02)02567-6. [DOI] [PubMed] [Google Scholar]
- 27.Hale SL, Kloner RA. Mild hypothermia as a cardioprotective approach for acute myocardial infarction: laboratory to clinical application. J Cardiovasc Pharmacol Ther. 2011;16:131–139. doi: 10.1177/1074248410387280. [DOI] [PubMed] [Google Scholar]
- 28.Götberg M, Olivecrona GK kaul S. A pilot study of rapid cooling by cold saline and endovascular cooling before reperfusion in patients with ST-elevation myocardial infarction. Circ Cardiovasc Interv. 2010;3:400–40. doi: 10.1161/CIRCINTERVENTIONS.110.957902. [DOI] [PubMed] [Google Scholar]