

Abstract
Lowe syndrome is an X-linked disorder characterized by cataracts at birth, mental retardation and progressive renal malfunction that results from loss of function of the OCRL1 (oculocerebrorenal syndrome of Lowe) protein. OCRL1 is a lipid phosphatase that converts phosphatidylinositol 4,5-bisphosphate to phosphatidylinositol 4-phosphate. The renal pathogenesis of Lowe syndrome patients has been suggested to result from alterations in membrane trafficking, but this cannot fully explain the disease progression. We found that knockdown of OCRL1 in zebrafish caused developmental defects consistent with disruption of ciliary function, including body axis curvature, pericardial edema, hydrocephaly and impaired renal clearance. In addition, cilia in the proximal tubule of the zebrafish pronephric kidney were longer in ocrl morphant embryos. We also found that knockdown of OCRL1 in polarized renal epithelial cells caused elongation of the primary cilium and disrupted formation of cysts in three-dimensional cultures. Calcium release in response to ATP was blunted in OCRL1 knockdown cells, suggesting changes in signaling that could lead to altered cell function. Our results suggest a new role for OCRL1 in renal epithelial cell function that could contribute to the pathogenesis of Lowe syndrome.
Keywords: kidney; Lowe syndrome; MDCK; oculocerebrorenal syndrome of Lowe; phosphatidylinositol 4,5-bisphosphate; zebrafish
Lowe syndrome is an X-linked disorder characterized by cataracts at birth, mental retardation and renal malfunction that results from mutations in the gene encoding oculocerebrorenal syndrome of Lowe (OCRL1) (1). OCRL1 is a lipid phosphatase that hydrolyzes the 5′ phosphate of phosphatidylinositol 4,5-bisphosphate (PIP2) and phosphatidylinositol 3,4,5-trisphosphate (PIP3), with a preference for the former substrate in vitro (2,3). OCRL1 is localized primarily at the trans-Golgi network (TGN), but is also present on a subset of endosomes and associated with clathrin-coated pits (4–8). Mutations that cause Lowe syndrome result in complete loss of OCRL1 catalytic activity (2,9). OCRL1 appears to be the major PIP2-hydrolyzing enzyme in human renal proximal tubule cells, and cells lacking OCRL1 have slightly elevated levels of PIP2 (3,10).
Lowe syndrome patients almost universally present with renal disease from birth, including acidosis, amino aciduria, phosphaturia and low molecular weight proteinuria (11). The defect in protein reabsorption may result from improper function or trafficking of the cell surface protein megalin, a large multiligand receptor that recycles at the apical domain of polarized epithelial cells (12). The demonstration that a subpopulation of OCRL1 binds to the endosomal adaptor APPL1 in clathrin-coated pits has fueled this speculation (6). OCRL1 also interacts with clathrin, α-adaptin and numerous Rab proteins, including Rab5 and Rab6, as well as Ses/IPIPs (4,8,13–16).
The potential link between loss of OCRL1 function and defective megalin trafficking has been reinforced by studies of Dent disease, originally described as resulting from defective function of the CLC-5 Cl−/H+ antiporter. Dent disease patients present with renal symptoms that are similar, though not identical to Lowe syndrome patients; however, Dent disease patients have none of the extra-renal symptoms characteristic of Lowe syndrome. There is strong evidence that loss of CLC-5 function leads to decreased uptake of fluid phase markers and megalin ligands in knockout mouse models (17–22). Surprisingly, a significant fraction of patients diagnosed with Dent disease were recently found to have mutations in OCRL1 rather than CLC-5; these patients are now classified as having Dent 2 (23). Mutations in OCRL1 that cause Dent 2 are limited to nonsense or splicing mutations within the first seven exons of the gene or to missense mutations in the catalytic domain. It is likely that these mutations result in expression of alternatively spliced or partially active variants of OCRL1 that are sufficient for function in extrarenal tissues, but not in the kidney (24).
In disagreement with the prevailing hypothesis for OCRL1 function in the kidney, we found that endocytic trafficking of megalin and a megalin ligand was unaffected by knockdown of OCRL1 in polarized canine and human renal epithelial cells (10). More recent studies have suggested that OCRL1 depletion alters endocytic recycling, possibly by modulating actin dynamics, in nonpolarized cells (14,25). The varying results obtained using these models could reflect the fact that apical endocytosis is considerably slower than internalization in nonpolarized cells and is more difficult to quantitate, or might suggest a differing role for actin dynamics in these processes. A common finding by several groups is that loss of OCRL1 results in altered trafficking of the mannose 6-phosphate receptor leading to enhanced secretion of lysosomal hydrolases (4,10,25). This finding is consistent with clinical observations that Lowe syndrome patients have elevated plasma levels of lysosomal hydrolases (4,26).
Given the lack of a clear effect of OCRL1 knockdown on megalin trafficking in polarized cells, we have revisited the question of how loss of OCRL1 disrupts organ function in Lowe syndrome patients. Interestingly, while OCRL1 is ubiquitously expressed in tissues, the organs most affected in Lowe syndrome (eye, brain and kidney) are also affected in diseases caused by disruption of ciliary function (collectively termed ciliopathies). Several features of OCRL1 are also consistent with a potential role in regulating ciliary biogenesis. The ASH domain in OCRL1 has been found in several proteins localized to cilia as well as to the Golgi complex (27). In addition to its selectivity for PI4,5P2, OCRL1 is able in vitro to hydrolyze PI3,4,5P3 to PI3,4P2, and the generation of this product may play an important role in delivery of cargo to cilia (28). Moreover, OCRL1 is known to bind Rab8, a protein implicated in ciliary biogenesis (16,29–31). Also of note is that another PIP2 phosphatase, INPP5E, has been implicated in the ciliopathy MORM syndrome (32). With this in mind, we asked whether OCRL1 might play a role in ciliary biogenesis or function.
In our studies, we found that knockdown of OCRL1 in zebrafish disrupted renal function and resulted in phenotypes consistent with ciliary defects. Moreover, we observed that primary cilia of Madin-Darby canine kidney (MDCK) cells depleted of OCRL1 were significantly longer than those of control cells. Similar to cells depleted of galectin-3, which also have elongated cilia, we found that MDCK cells grown in three-dimensional cultures formed aberrant cysts when OCRL1 was depleted. Additionally, cell signaling is apparently aberrant in OCRL1 depleted cells, as the intracellular release of calcium in response to ATP challenge was reduced. Our results suggest that loss of OCRL1 function affects multiple cellular processes that contribute to disease pathogenesis in the kidney.
Results
Knockdown of OCRL in zebrafish disrupts embryonic development and renal clearance
Knockout of the OCRL1 gene in mice does not recapitulate Lowe syndrome, likely due to expression of a homologous inositol polyphosphate-5-phosphatase called INPP5B. As an alternative approach to assess the role of OCRL1 in renal function, we examined the expression and function of ocrl in the zebrafish Danio rerio. Zebrafish have been widely used to model human disease, including many ciliopathies that disrupt kidney development and function (33,34). Zebrafish express a single copy of ocrl that is 59% identical at the amino acid level to human OCRL1, with the greatest identity found within the catalytic and RhoGAP domains of the protein. There is one INPP5B homolog in zebrafish (69). RT-PCR using primers specific for zebrafish ocrl revealed expression of ocrl mRNA in embryo extracts as early as the 13-somite stage and persisting for at least 48 hours postfertilization (hpf) (Figure 1A). In situ hybridization analysis using an antisense probe confirmed ocrl mRNA expression in the pronephric kidney, brain and eye at early stages and persisting through 48 hpf (Figure 1B, C). No expression was observed using a sense probe at 24 and 48 hpf (Figure 1D).
Figure 1. OCRL1 is expressed in the zebrafish kidney, eye and brain.

A) RT-PCR was performed using ocrl-specific primers to detect expression of ocrl in embryo extracts collected at the 13-somite stage (13ss), 24 hpf and 48 hpf. B) In situ hybridization for ocrl expression was performed on embryos collected at the 6-somite stage (6ss), 13ss, 24 hpf and 48 hpf. No expression was observed at the 6ss. At the other stages, expression of ocrl in the brain (B), eye (E) and kidney (K) is marked by arrowheads. C) Higher magnification images of the pronephric kidney distal tubule and duct are shown to highlight ocrl expression in this region. D) Representative images of embryos at 24 hpf and 48 hpf incubated with ocrl sense probe are shown as a negative control.
We next assessed the effect of knocking down ocrl using a translation-blocking morpholino (MO). The ocrl-depleted embryos were developmentally impaired, and exhibited abnormal body curvature, hydrocephaly and pericardial edema. Morphants were classified at 48 hpf as wild type (class I), moderate (class II) and severe (or dead, class III) based on the extent of body curvature and edema (Figure 2A). The percentage of moderately and severely affected morphants increased with the dose of ocrl MO injected, with 6, 6.5 and 7 ng MO yielding approximately 70, 80 and 100% class II and class III phenotypes, respectively (Figure 2B).
Figure 2. Ocrl morphants develop pericardial edema and body curvature.

A) Following MO injection, 48 hpf zebrafish larvae were classified into three categories: class I, wild type; class II, moderate body curvature and mild pericardial edema; and class III, severe body curvature and severe pericardial edema. Representative images are shown. B) Embryos were injected with the indicated dose of control or ocrl MO or left uninjected and classified as above at 48 hpf. At 7.5 ng, all the embryos were dead at 24 hpf. For rescue studies, embryos were injected with mRNA encoding wild type ocrl or the phosphatase-dead R559G mutant, followed by injection with 6.0 ng of ocrl MO. Phenotypes were classified at 48 hpf. The graph shows quantitation of three independent injections with at least 50 injected embryos per dose in each experiment, except for R559G, which represents data from 64 embryos.
To confirm that the effects we observed were due to loss of ocrl, we attempted to rescue the phenotypes by expressing MO-resistant ocrl mRNA. Embryos were injected at the one-cell stage with 200 pg of synthetic wild-type zebrafish ocrl mRNA followed by injection of 6 ng ocrl or control MO before the eight cell stage. Phenotypes were then analyzed at 48 hpf. Injection of 200 pg ocrl mRNA without subsequent MO injection had no deleterious consequences on embryonic development (not shown). Importantly, expression of wild-type ocrl partially rescued the morphant phenotype, with only approximately 40% of embryos exhibiting moderate (class II) or severe (class III) phenotypes (compared with 70% induced by injection of 6 ng ocrl MO alone). In contrast, injection of the ocrl R559G, which contains a mutation in the conserved catalytic domain found in Lowe syndrome patients, failed to rescue the morphant phenotype (Figure 2B)
Notably, the defects in embryonic development that we observed are consistent with compromised kidney and ciliary function in zebrafish. We therefore asked whether renal function was impaired in ocrl morphant embryos. Morphant and control embryos were injected in the common cardinal vein with 10 kDa rhodamine dextran at 48 hpf and renal clearance was followed over time using fluorescence imaging. Only moderately affected morphants (class II) were selected for these experiments to minimize indirect effects due to severe developmental defects. Whereas embryos injected with the control MO efficiently cleared the dextran over a 24-h period, embryos injected with the ocrl MO retained significant amounts of dextran in the circulatory system, consistent with aberrant fluid clearance (Figure 3). The impaired clearance was not due to blockage of the pronephric duct, as there was no obstruction of the cloaca in these morphants (Figure S1).
Figure 3. Kidney function is disrupted after ocrl knockdown.

A) 1 ng of 10 kDa Rhodamine dextran was injected into the common cardinal vein of control embryos (n = 32) or moderately (class II) affected ocrl morphants (n = 25; all injected with 6 ng MO) at 24 hpf. Images were acquired at 1, 5 and 24 h post-injection. B) The progressive loss of fluorescence near the cardinal vein area (white box at 0 h) was quantified and plotted as described in Materials and Methods.
The pronephric kidney tubule contains both monociliated and multiciliated cells (35,36). To determine whether knockdown of ocrl altered cilia morphology, we visualized cilia in the pronephric kidney tubule at 24 hpf using anti-acetylated tubulin. We also included antibodies against IFT88, which localizes in part to basal bodies (37). We consistently observed that the cilia were more disorganized in the morphants, consistent with a widening of the tubule (Figure 4). We also quantified the length of the cilia in the pronephric kidney. The average length of cilia was 5.2 μm in embryos injected with control MO. Interestingly, cilia were elongated in ocrl morphants (average length of 6.7 μm; Figure 4), We also imaged cilia in the Kupffer’s vesicle at the 13-somite stage and found no difference in cilia abundance or length between morphants and controls (data not shown).
Figure 4. Cilia are elongated in the proximal tubule of ocrl morphants.

Zebrafish embryos injected with 6 ng control or ocrl MO were fixed and processed at 24 hpf for indirect immunofluorescence to detect acetylated α-tubulin (green) and IFT88 (red). Representative images of pronephric kidney proximal tubules in control and ocrl morphant larvae are shown and cilia length is plotted below (mean ± SEM from four embryos per condition; >100 cilia quantitated per condition). *p < 0.001 by Student’s t test. Scale bar: 5 μm.
Knockdown of OCRL1 in renal epithelial cells results in elongation of cilia
The phenotypes and defects in pronephric kidney function of zebrafish morphants led us to examine the effect of OCRL1 knockdown on cilia morphology and function in mammalian renal epithelial cells. To this end, MDCK cells were transfected with control siRNA or siRNA targeting OCRL1, cultured for 4 days on permeable supports, and processed for indirect immunofluorescence with anti-acetylated tubulin to visualize and quantitate cilia length. Western blotting confirmed that knockdown of OCRL1 was efficient (typically >80%, Figure 5A). Interestingly, we observed that cilia on cells transfected with OCRL1 siRNA were clearly longer than cilia on cells treated with control siRNA (Figure 5B). Cilia lengths from ≥100 cells for each condition were measured using VOLOCITY software, sorted by length, and plotted in rank order from shortest to longest (Figure 5C). Typically, approximately 90% of cilia were <5 μm in length in control cells, although a few very long cilia (up to ~20 μm) were also seen. In OCRL1 depleted cells, the shortest cilia observed were the same length as control, but the distribution of lengths rapidly diverged. Rank sum analysis confirmed that the median length (3.38 μm versus 1.72 μm for cells treated with OCRL1 versus control siRNA, respectively) was statistically different (p < 0.001) in cells treated with OCRL1 siRNA compared with control. This observation was extremely reproducible: cilia lengths in OCRL1-depleted cells were markedly longer than control in 19 of 23 experiments, and in no experiment was the mean cilia length in cells treated with OCRL1 siRNA shorter than control.
Figure 5. Knockdown of OCRL1 in renal epithelial cells causes elongation of cilia.

A) Filter-grown MDCK cells treated with siRNA targeting OCRL1 or control siRNA were solubilized and blotted to detect OCRL1 and β-actin (as a loading control). We typically observed 50–95% knockdown of OCRL1 in our experiments. B) Filter-grown MDCK cells treated with control or OCRL1 siRNA were fixed and processed for indirect immunofluorescence to detect acetylated tubulin. Cells were viewed by confocal microscopy to visualize cilia. Bar: 10 μm. C) Individual cilia lengths (at least 100 per sample) were measured from random fields of cells treated with control or OCRL1 siRNA, sorted in ascending order by length and plotted from shortest to longest. Each circle in the graph represents the length of a single cilium. The median cilia length is 3.38 μm versus 1.72 μm for cells treated with OCRL1 versus control siRNA, respectively, and is statistically different in the two samples (rank sum test p < 0.001). D) Scanning electron microscopy of MDCK cells treated with control or OCRL1 siRNA shows no obvious difference in cilia morphology. Two representative images for each condition are shown. Bar: 1 μm.
In the course of our studies, we used several different sub-clones of MDCK cells and noted some variation in mean cilia length between these. However, in all cases, depletion of OCRL1 resulted in elongation of cilia compared with cells treated with control siRNA (data not shown). Additionally, when differentiated, cilia-bearing cells were acutely deciliated using ammonium sulfate and then allowed to recover for 2 days, the restored cilia in OCRL1-depleted cells were again longer than control (Figure S2).
Loss of function of INPP5E, a phosphatidylinositol 5′-phospatase that localizes to cilia, has been shown to cause MORM syndrome, a cystic disease in which cilia are characteristically sparser than normal and have dilated ends (32). We therefore compared cilia morphology in control and OCRL1-depleted cells using scanning electron microscopy. While there was some variability in cilia length within each population, we found no obvious morphological differences between control and knockdown cells (Figure 5D).
We attempted to rescue the effect of OCRL1 knockdown on cilia length by heterologous expression of GFP-tagged siRNA-resistant wild type and mutant OCRL1. For these experiments, we generated stable cell lines expressing GFP-tagged wild type and catalytically inactive siRNA-resistant OCRL1 constructs, transfected the stably expressing and parental lines with control or OCRL1 siRNA and then quantitated cilia length in GFP-positive cells. These experiments proved difficult to interpret with confidence, as the stable cell lines that we isolated had variable cilia lengths compared with the parental controls. It is possible that overexpression of OCRL1 contributed to the variability in length and to the extent of rescue that we observed. Ultimately, these disparities made it difficult to rigorously assess whether overexpression of OCRL1 restored cilia length to control levels.
Although OCRL1 is ubiquitously expressed in tissues, many organs and cell types retain normal function in Lowe syndrome patients. We therefore asked whether OCRL1 depletion modulates cilia length in fibroblasts. We obtained essentially complete knockdown of OCRL1 in these cells (Figure 6A); however, this did not alter the cilia length profile of these cells measured after 3 days of serum starvation (Figure 6B) or under-fed conditions (data not shown). Upon shorter starvation periods, cilia in OCRL1-depleted fibroblasts were slightly longer than controls, consistent with our data in kidney cells (Figure 6C).
Figure 6. OCRL1 knockdown has no effect on cilia length in fibroblasts.

A) Western blot of OCRL1 and β-actin from human fibroblasts treated with control or OCRL1 siRNA. B) Cilia length was measured in human fibroblasts treated with control or OCRL1 siRNA after 72 h serum starvation and plotted as described in the legend to Figure 5. Median cilia length was identical (4.8 μm) in control and OCRL1-depleted cells. C) Cilia length was measured after 4 h of serum starvation. Median cilia length was 4.7 μm in control and 5.3 μm in OCRL-depleted cells.
Knockdown of OCRL1 disrupts lumen formation in cyst cultures
Increased cilia length has been linked with defective growth of MDCK cells in three-dimensional cyst cultures (38). We therefore examined the effect of OCRL1 knockdown on cilia and morphology of MDCK cysts. Cells transfected with control or OCRL1-specific siRNA were plated in Matrigel and fixed after 5 days in culture. Cells were processed for indirect immunofluorescence with anti-acetylated tubulin and anti-ZO-1 antibodies and cilia length was quantitated. As a positive control, we knocked down galectin-3, which has previously been demonstrated to result in elongated cilia (38). Cilia in cyst cultures of OCRL1-depleted cells were visibly longer compared with control, and quantitation confirmed that knockdown of either OCRL1 or galectin-3 caused elongation of cilia (Figure 7A, B). Moreover, we observed that there was a greater fraction of abnormal cysts (with either multiple lumens or filled lumen) in both OCRL1 and galectin-3 depleted cultures (Figure 7C). Interestingly, another recently published study has reported a similar effect of OCRL1 knockdown on cyst morphology (39).
Figure 7. OCRL1 knockdown disrupts cyst morphogenesis.

A) MDCK cells were transfected with control or OCRL1 siRNA and plated in Matrigel. Cells were fixed and processed for indirect immunofluorescence after 6 days to visualize nuclei (blue), acetylated tubu-lin (green) and ZO-1 (red). B) Cilia length was quantitated in MDCK cysts treated with control, OCRL1 or galectin-3 siRNA. C) Cyst morphology was quantitated in cells treated as in (B). Cysts with multiple or collapsed lumens were considered to be aberrant. Representative examples of a normal and abnormal cyst stained with anti-acetylated tubulin are shown. The average ± SD of 4–5 experiments for each condition is plotted (100 cysts counted per condition per experiment). *p < 0.001 versus control siRNA by Student t-test. Scale bars: 10 μm.
Calcium signaling in OCRL1-depleted cells
Mutations that result in shortened primary cilia are known to cause defects in cellular ATP release and calcium signaling that are thought to contribute to renal cyst formation (40–43). However, the effect of longer cilia on calcium signaling has not been examined. MDCK cells treated with control or OCRL1 siRNA were cultured on coverslips and loaded with Fura-2. Cells were then subjected to fluid shear and intracellular calcium was measured using ratiometric imaging. We did not detect any difference in calcium mobilization upon initiation of fluid shear in cells treated with control versus OCRL1 siRNA (data not shown). However, calcium mobilization in response to addition of extracellular ATP was significantly reduced in OCRL1-deficient cells compared with controls (Figure 8).
Figure 8. OCRL1 depletion reduces ATP-stimulated calcium mobilization.
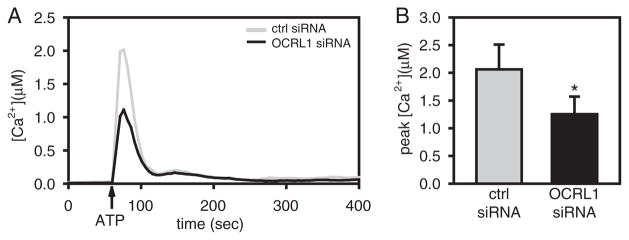
Confluent MDCK cells cultured on glass bottom dishes were transfected with control or OCRL1 siRNA. Ca2+ imaging was performed as described in Materials and Methods. A) ATP-stimulated calcium mobilization in control and OCRL1-depleted MDCK cells. Time course of change in [Ca2+]i in control and OCRL1 depleted cells. The time of addition of ATP (100 μM) is indicated with an arrow. Data represent the average from 9 to 10 independent experiments. B) Peak intracellular calcium following ATP addition in control and OCRL1 siRNA-treated cells. Data represent the mean ± SEM from 9 to 10 independent experiments. *p = 0.023 by Mann–Whitney.
Discussion
In the studies reported above we found that knockdown of OCRL1 in zebrafish elicited phenotypes consistent with a defect in cilia function and compromised pronephric kidney function. Additionally, loss of OCRL1 caused elongation of primary cilia in the zebrafish pronephric kidney as well as in mammalian renal epithelial cells and disrupted cyst formation in three-dimensional cell cultures. Calcium signaling in response to ATP challenge was also aberrant in renal cells depleted of OCRL1. Together, these results suggest that in addition to the known effects of OCRL1 depletion on membrane traffic and actin dynamics, altered ciliary biogenesis may also contribute to the pathogenesis of Lowe syndrome.
Indeed, notwithstanding the initial focus on a putative role of OCRL1 in modulating endocytosis, several recent studies have observed more global functions for this protein in cell polarity and division. Impaired cell migration and spreading have been demonstrated in Lowe syndrome patient cells (44). More recently, Grieve et al. (39) reported a dramatic effect of OCRL1 knockdown on renal cyst morphology and cell proliferation. Additionally, this study found that OCRL1 localized in part to tight junctions. Two other recent reports have implicated OCRL1 in actin and lipid dynamics required for cytokinesis (45,46). Given the relationship between cell division, actin dynamics and ciliogenesis (47,48), it is tempting to speculate that the effect we observed on ciliary length is related to this function of OCRL1. How the role of OCRL1 in cytokinesis contributes to the phenotypic defects in Lowe syndrome remains unclear.
Could Lowe syndrome be a ciliopathy?
The link between OCRL1 and cilia function suggests the possibility that Lowe syndrome might constitute a novel class of ciliopathy. While the analogy is certainly not perfect, there are interesting comparisons to make. Whereas all defects that lead to shortened cilia in renal cells are believed to result in cystic disease, some reported ciliopathy mutations instead cause elongation of cilia (49–51). Elongated cilia have also been observed in mouse renal tubules during the recovery from acute kidney injury and ureteral obstruction (52,53). Although ciliopathies frequently affect the same organs compromised in Lowe syndrome, many of the clinical features differ significantly. Patients with known ciliopathies do not typically have proteinuria, and instead present with cystic kidney disease and occasional situs inversus, neither of which is observed in Lowe syndrome. Moreover, while ocular problems in ciliopathy patients result from retinal degeneration, Lowe syndrome patients are born with cataracts resulting from necrosis of the lens epithelium. That said, many of the features commonly observed in Lowe syndrome, including cataracts, hypotonia and polyuria, high pitched voice and deep set eyes, have been reported in patients with Bardet-Biedl syndrome (BBS) (54). BBS patients can have mutations in any of the 12 protein subunits that comprise the BBSome, a complex that regulates cargo delivery to cilia, and some BBS knockout mice have elongated cilia (49). Moreover, similar to the variation in symptoms observed between mutations in OCRL1 that cause Lowe syndrome versus Dent 2 disease, mutations in the same ciliary gene can result in very different syndromes (54). For example, different mutations in the nephrocystin-3 protein that cause nephronophthisis can variably result in cystic kidney disease only, or in the broad range of clinical symptoms that include polydactyly, situs inversus and heart defects (55).
A new working model for OCRL1 function
On the basis of our own studies and the published work by others, we propose important functions for OCRL1 in both TGN and endosomal trafficking steps. First, localized OCRL1-mediated cleavage of PIP2 in the TGN and/or endosomes to produce phosphatidylinositol 4-phosphate (PI4P) is important for the efficient delivery of newly synthesized lysosomal hydrolases. These enzymes bind to the mannose 6-phosphate receptor (M6P-R) in the Golgi/TGN, and are ferried to early and ultimately late endosomes, where dissociation occurs. Subsequently, transient fusion of late endosomes with lysosomes enables delivery of the soluble hydrolases and recycling of the M6P-R. Importantly, the adaptor proteins that bind to M6P-R, including epsin R, AP-1, and the GGAs, all bind to phosphatidylinositol 4-phosphate (PI4P). While the abundant source of PI4P generated in the Golgi complex by phosphatidylinositol 4-kinases has been assumed to supply this lipid, the observations that lysosomal hydrolases are preferentially secreted and that M6P-R trafficking is disrupted in cells lacking OCRL1 suggests that this enzyme may also generate a functionally important pool of PI4P along the pathway.
How might OCRL1 modulate cilia length and how might this contribute to the kidney and/or extrarenal symptoms observed in Lowe syndrome? One possibility is that OCRL1 functions to generate a pool of PIP2 required for maintaining ciliary transport. Phosphoinositides are known to play roles in protein delivery to cilia (56,57). An intriguing target for these lipids is the exocyst, which plays a role in ciliary biogenesis and is known to bind to PIP2 (58–60). Alternatively, OCRL1 is able in vitro to hydrolyze PI3,4,5P3 to PI3,4P2, and the generation of this product may play an important role in delivery of cargo to cilia (28,61). Recent studies by Nachury and colleagues have found that the BBSome, a coat-forming complex that together with Rab8 directs transport carriers to cilia (28,31), binds selectively to PI3,4P2 (28). OCRL1 function in endocytic compartments might contribute to the pool of PI3,4P2 recognized by the BBSome. Notably, OCRL1 also interacts with Rab8 (16). While a detailed mechanism remains elusive, loss of OCRL1 might enhance delivery of some ciliary proteins by altering the localization or activity of the BBSome. The resulting elongated cilia may respond aberrantly to flow or other signaling cues in the renal tubule, resulting in dysfunctional downstream responses that could contribute to the renal pathology observed in Lowe syndrome patients.
While this manuscript was in preparation, two reports appeared in press that also document effects of OCRL1 depletion on cilia in zebrafish and in mammalian fibroblasts (62,63). Surprisingly, knockdown of OCRL1 in fibroblasts and retinal pigment epithelial cells was found to decrease cilia length in these studies. Although neither study examined cilia in renal cells, reduced cilia length in OCRL1-depleted cells is a surprising observation given the strong link between shortened cilia and formation of renal cysts, which are not a feature of Lowe syndrome. Clearly, additional experiments will be necessary to reconcile the disparate observations and to refine our understanding of the role of OCRL1 in ciliary biogenesis and function.
Materials and Methods
Zebrafish husbandry
Zebrafish were maintained under standard conditions and staged as described previously (64). Embryos were collected from group matings of wild-type adult zebrafish. All animal husbandry adheres to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
In situ hybridization
In situ hybridization was performed as previously described (64). ocrl full-length cDNA was purchased from Open Biosystems and the ocrl probe was made using the DIG labeling kit (Roche). T7 RNA polymerase was used for RNA synthesis.
Real-time PCR
Zebrafish embryos were homogenized with a plastic microcentrifuge pestle in 500 μL of TRI reagent (Ambion), and RNA was isolated using the RNeasy Micro Kit (Qiagen) according to the manufacturer’s protocol. Primer sets against ocrl were designed using PrimerQuest on the IDT Web site (forward primer: 5′-AGA CCA GTG ACC ACA AAC CTG TCA-3′; reverse primer: 5′-AGA ATG TCA TCC GGG CTT TCT CCA-3′). Synthesis of cDNA was performed using M-MLV reverse transcriptase (Ambion) according to the manufacturer’s recommendations. PCR was performed using the BioRad icycler and GeneAmp® High Fidelity PCR System (Applied Biosystems). The denaturing temperature was 95°C, the annealing temperature was 55 C and the extension temperature was 68°C with an amplification cycle of 30 min.
Microinjection
Zebrafish embryos were injected up to the eight-cell stage with the indicated doses of a translation blocking MO targeting ocrl (CGGAAATCCCAAATGAAGGTTCCAT) or a standard control MO. The same sequence targeting ocrl was recently also validated by Coon et al. (62). Embryos were allowed to develop in E3 culture medium (5 mM NaCl, 0.17 mM KCl, 0.4 mM CaCl2, 0.16 mM MgSO4) at 28.5°C. Images were taken and phenotypes classified at 48 hpf. For rescue experiments, MO-resistant wild type and R559G OCRL were prepared using the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies) and mRNAs were synthesized using mMESSAGE mMACHINE® SP6 kit (Invitrogen) according to the manufacturer’s recommendation. Zebrafish embryos were injected at the one-cell stage with 200 pg of synthetic wild type or mutated ocrl mRNA, and embryos were imaged and classified at 48 hpf.
Rhodamine dextran clearance assay
Renal function was assayed as previously described (65,66). Briefly, 1 ng of 10 kDa Rhodamine dextran was injected into the common cardinal vein of embryos at 48 hpf. Embryos were imaged sequentially and under identical conditions at 1, 5 and 24 h post-injection. The loss of fluorescence near the common cardinal vein area over time was quantified using Adobe Photoshop.
Indirect immunofluorescence of cilia in zebrafish
Embryos at 48 hpf were incubated in Dent’s fixative (80% methanol and 20% DMSO) overnight at 4°C. After rehydration, embryos were incubated with block solution (PBS with 0.5% Tween 20 with 10% Normal Goat Serum) for 1 h at room temperature and then overnight at 4°C with mouse anti-acetylated tubulin (1:1000, 6-11B-1, Sigma) and rabbit anti-IFT88 (1:5000, gift from Brian Perkins; reviewed in 67). Alexa Fluor 568 goat anti-mouse (1:1000, Invitrogen) and Alexa Fluor 488 goat anti-rabbit (1:1000, Invitrogen) were used as secondary antibodies. Images of kidney tubules were acquired using a Leica DMI 6000 CS Trino microscope and processed and quantified using ImageJ.
Cell culture, siRNA knockdown and generation of stable cell lines
Type II MDCK cells were transfected with siRNA oligonucleotides targeting OCRL1 (10), galectin-3 (68) or luciferase (as a control) prior to plating on filter supports as described in Mo et al. (68) and used for experiments 4 days later. Duplicate filters were processed for western blotting with monoclonal anti-OCRL1 antibody (gift of Robert Nussbaum and Sharon Suchy) to monitor the efficiency of knockdown. Human fibroblast cultures were transfected with OCRL1 or control siRNA and incubated in serum-free medium the following day. Cells were processed for western blotting or indirect immunofluorescence 4 days after transfection.
Immunofluorescence and quantitation of cilia length
Filter- or coverslip-grown cells were fixed with 4% paraformaldehyde in 100 mM sodium cacodylate, 3 mM CaCl2, 3 mM MgCl2 and 3 mM KCl, pH 7.4 for 15 min (5 min at 37°C followed by 10 min at ambient temperature), quenched in PBS with 20 mM glycine and 75 mM NH4Cl for 5 min, permeabilized using 0.1% TritonX-100 in the quench solution for 10 min with gentle shaking, blocked in PBS with 1% fish skin gelatin (Sigma) and 0.1% saponin for 10 min at 37°C. This was followed by sequential incubation for 1 h with mouse monoclonal anti-acetylated tubulin antibody (1:400; Sigma) and fluorophore-coupled goat anti-mouse secondary antibody (1:500; Invitrogen) diluted in PBS with 0.5% gelatin and 0.025% saponin. After thorough washes between and after antibody incubations, squares of transwell filters (cut out from the transwells) or coverslips were mounted on glass slides using Prolong Gold antifade reagent with DAPI (Invitrogen). Mounted slides were dried overnight at room temperature. Random fields of cilia were acquired using a Leica DM6000B upright fluorescence microscope. Individual cilia lengths (at least 100 per sample) were measured using VOLOCITY software, sorted in ascending order by length and plotted from shortest to longest.
Scanning electron microscopy
MDCK cells were transfected with siRNA, plated on filters for 5 days, then fixed in 200 mM sodium cacodylate buffer, pH 7.4 containing 0.5% glutaraldehyde, 2% paraformaldehyde, 0.5 mM MgCl2 and 1 mM CaCl2 for 30–60 min at ambient temperature. Filters were washed with 100 mM sodium cacodylate buffer, pH 7.4, for 5 min. Subsequently, cells were fixed again with 1.5% OsO4 in 100 mM sodium cacodylate buffer, pH 7.4, for 1 h at 4°C with gentle shaking in a fume hood, rinsed repeatedly with deionized water, and washed for 5 min in deionized water with gentle shaking. Following fixation, cells were dehydrated by sequential soaking for 5 min each on ice in 40, 50, 75, 80, 90 and 95% ethanol. Cells were then incubated twice in 100% ethanol for 10 min at ambient temperature, critical point dried on a Samdri® -PVT-3D machine (Tousimis), and sputter coated using gold palladium in a Cressington Sputter Coater. Samples were imaged using a JEOL JSM-T300 scanning electron microscope.
Culture and staining of MDCK cysts
MDCK cells were detached from culture plates using trypsin and diluted to a final concentration of 2.5 × 104 cells/mL. Aliquots (0.2 mL) were pelleted, resuspended in 50 μL Matrigel Basement Membrane Matrix (BD Biosciences) and plated on cover slips. The solution was allowed to solidify at 37°C in 5% CO2 for 30 min prior to addition of growth medium. The cells were cultured and fed every 24–48 h for 5–7 days.
For immunofluorescence staining, cysts were fixed in 4% paraformaldehyde for 20 min, washed in PBS three times (5 min each), and permeabilized with 0.5% Triton X-100 in PBS for 10 min. After quenching with PBS-glycine three times (10 min each), samples were blocked in 1% BSA/0.1% saponin in PBS for 1 h, then incubated with antibodies in block overnight at 4°C. After washing three times with block buffer (10 min each), secondary antibodies were added for 1 h at ambient temperature. The samples were washed with block buffer once for 15 min and with PBS three times (10 min each), then were mounted with Prolong Gold antifade reagent with DAPI (Invitrogen). The slides were allowed to dry overnight and then sealed with nail polish.
Measurement of intracellular Ca2+
MDCK cells cultured on glass bottom dishes (MatTek Corporation) were transfected with control or OCRL1 siRNA. Ca2+ measurements were performed 2 days after transfection. Cells were loaded with Fura-2AM (5μM) (Invitrogen) in HEPES-buffered saline (HBS; 135 mM NaCl, 4.5 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 10 mM glucose, pH 7.4) for 2 h at room temperature. Fura-2AM loaded cells were washed with HBS and incubated for an additional 10 min to allow for further de-esterification of the dye. Glass bottom dishes were mounted in a QE-1 imaging chamber (Warner Instruments). The temperature of the chamber was maintained at 37°C with a dual channel bipolar temperature controller (model TC-344B, Warner Instruments). Ca2+ imaging was performed using an inverted Nikon Eclipse Ti microscope equipped with an ORCA-Flash 2.8 camera (Hamamatsu). For excitation we used a PhotoFluor II metal halide light source (89 North) connected to a Lambda 10–3 filter wheel system (Sutter Instruments) with 340 nm and 380 nm filters (Chroma Technology). Images were acquired and analyzed with NIS-elements software (Nikon). In situ calibration with ionomycin (5 μM) was performed to estimate Ca2+ calibration parameters under zero or saturating intracellular Ca2+ concentrations. The zero-free Ca2+ solution contained 135 mM NaCl, 4.5 mM KCl, 2 mM EGTA, 1 mM MgCl2, 10 mM HEPES and 10 mM glucose, pH 7.4. HBS was used to estimate Fura-2 ratio values at saturating Ca2+ concentrations. Ten cells were randomly selected for analysis per experiment.
Supplementary Material
Figure S1: Cloaca opening is normal in ocrl morphant zebrafish. Embryos injected with either control MO (left) or MO targeting ocrl (right) were examined for cloaca opening. Arrows indicate normal cloaca openings in both control and morphant larvae at 48 hpf.
Figure S2: Cilia in OCRL1-depleted MDCK cells are longer than control upon recovery from deciliation. A) MDCK cells were transfected with control or OCRL1 siRNA and plated on permeable supports. After 2 days, indicated samples were treated for 3 h with ammonium sulfate (NH4SO4) or left untreated, then allowed to recover for 2 days. Cells were processed for indirect immunofluorescence to detect acetylated tubulin 2 days after plating (top row), immediately after deciliation (middle row), and after recovery for 2 days (bottom row). B) Quantitation of cilia length in control or OCRL1-siRNA-treated cells grown on filters for 4 days (closed and open circles, respectively) or treated after 2 days with ammonium sulfate and allowed to recover for 2 days (closed and open triangles, respectively). Scale bar: 10 μm.
Acknowledgments
We thank Robert Nussbaum and Sharon Suchy for their generous gift of anti-OCRL1 antibody and Melanie Warnes for experimental support. This work was supported by grants from the NIH (DK064613) and the Lowe syndrome Association to O. A. W. H. F. was supported by NIH grant GM070736. We are grateful to the Urinary Tract Epithelial Imaging, Model Organisms, and Single Tubule Physiology Cores of the P30 Pittsburgh Center for Kidney Research (NIH DK079307) for technical support.
Footnotes
Additional Supporting Information may be found in the online version of this article:
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Charnas LR, Gahl WA. The oculocerebrorenal syndrome of Lowe. Adv Pediatr. 1991;38:75–107. [PubMed] [Google Scholar]
- 2.Suchy SF, Olivos-Glander IM, Nussabaum RL. Lowe syndrome, a deficiency of phosphatidylinositol 4,5-bisphosphate 5-phosphatase in the Golgi apparatus. Hum Mol Genet. 1995;4:2245–2250. doi: 10.1093/hmg/4.12.2245. [DOI] [PubMed] [Google Scholar]
- 3.Zhang X, Hartz PA, Philip E, Racusen LC, Majerus PW. Cell lines from kidney proximal tubules of a patient with Lowe syndrome lack OCRL inositol polyphosphate 5-phosphatase and accumulate phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 1998;273:1574–1582. doi: 10.1074/jbc.273.3.1574. [DOI] [PubMed] [Google Scholar]
- 4.Choudhury R, Diao A, Zhang F, Eisenberg E, Saint-Pol A, Williams C, Konstantakopoulos A, Lucocq J, Johannes L, Rabouille C, Greene LE, Lowe M. Lowe syndrome protein OCRL1 interacts with clathrin and regulates protein trafficking between endosomes and the trans-Golgi network. Mol Biol Cell. 2005;16:3467–3479. doi: 10.1091/mbc.E05-02-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choudhury R, Noakes CJ, McKenzie E, Kox C, Lowe M. Differential clathrin binding and subcellular localization of OCRL1 splice isoforms. J Biol Chem. 2009;284:9965–9973. doi: 10.1074/jbc.M807442200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erdmann KS, Mao Y, McCrea HJ, Zoncu R, Lee S, Paradise S, Modregger J, Biemesderfer D, Toomre D, De Camilli P. A role of the Lowe syndrome protein OCRL in early steps of the endocytic pathway. Dev Cell. 2007;13:377–390. doi: 10.1016/j.devcel.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dressman MA, Olivos-Glander IM, Nussbaum RL, Suchy SF. Ocrl1, a PtdIns(4,5)P(2) 5-phosphatase, is localized to the trans-Golgi network of fibroblasts and epithelial cells. J Histochem Cytochem. 2000;48:179–190. doi: 10.1177/002215540004800203. [DOI] [PubMed] [Google Scholar]
- 8.Ungewickell A, Ward ME, Ungewickell E, Majerus PW. The inositol polyphosphate 5-phosphatase Ocrl associates with endosomes that are partially coated with clathrin. Proc Natl Acad Sci USA. 2004;101:13501–13506. doi: 10.1073/pnas.0405664101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin T, Orrison BM, Leahey AM, Suchy SF, Bernard DJ, Lewis RA, Nussbaum RL. Spectrum of mutations in the OCRL1 gene in the Lowe oculocerebrorenal syndrome. Am J Hum Genet. 1997;60:1384–1388. doi: 10.1086/515471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cui S, Guerriero CJ, Szalinski CM, Kinlough CL, Hughey RP, Weisz OA. OCRL1 function in renal epithelial membrane traffic. Am J Physiol Renal Physiol. 2010;298:F335–345. doi: 10.1152/ajprenal.00453.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schurman SJ, Scheinman SJ. Inherited cerebrorenal syndromes. Nat Rev Nephrol. 2009;5:529–538. doi: 10.1038/nrneph.2009.124. [DOI] [PubMed] [Google Scholar]
- 12.Christensen EI, Birn H. Megalin and cubilin: multifunctional endocytic receptors. Nat Rev Mol Cell Biol. 2002;3:256–266. doi: 10.1038/nrm778. [DOI] [PubMed] [Google Scholar]
- 13.Hyvola N, Diao A, McKenzie E, Skippen A, Cockcroft S, Lowe M. Membrane targeting and activation of the Lowe syndrome protein OCRL1 by rab GTPases. EMBO J. 2006;25:3750–3761. doi: 10.1038/sj.emboj.7601274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noakes CJ, Lee G, Lowe M. The PH domain proteins IPIP27A and B link OCRL1 to receptor recycling in the endocytic pathway. Mol Biol Cell. 2011;22:606–623. doi: 10.1091/mbc.E10-08-0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swan LE, Tomasini L, Pirruccello M, Lunardi J, De Camilli P. Two closely related endocytic proteins that share a common OCRL-binding motif with APPL1. Proc Natl Acad Sci USA. 2010;107:3511–3516. doi: 10.1073/pnas.0914658107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukuda M, Kanno E, Ishibashi K, Itoh T. Large scale screening for novel rab effectors reveals unexpected broad Rab binding specificity. Mol Cell Proteomics. 2008;7:1031–1042. doi: 10.1074/mcp.M700569-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Piwon N, Gunther W, Schwake M, Bosl MR, Jentsch TJ. ClC-5 Cl-channel disruption impairs endocytosis in a mouse model for Dent’s disease. Nature. 2000;408:369–373. doi: 10.1038/35042597. [DOI] [PubMed] [Google Scholar]
- 18.Hryciw DH, Ekberg J, Pollock CA, Poronnik P. ClC-5: a chloride channel with multiple roles in renal tubular albumin uptake. Int J Biochem Cell Biol. 2006;38:1036–1042. doi: 10.1016/j.biocel.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Cai H, Cebotaru L, Hryciw DH, Weinman EJ, Donowitz M, Guggino SE, Guggino WB. ClC-5: role in endocytosis in the proximal tubule. Am J Physiol Renal Physiol. 2005;289:F850–F862. doi: 10.1152/ajprenal.00011.2005. [DOI] [PubMed] [Google Scholar]
- 20.Wang SS, Devuyst O, Courtoy PJ, Wang XT, Wang H, Wang Y, Thakker RV, Guggino S, Guggino WB. Mice lacking renal chloride channel, CLC-5, are a model for Dent’s disease, a nephrolithiasis disorder associated with defective receptor-mediated endocytosis. Hum Mol Genet. 2000;9:2937–2945. doi: 10.1093/hmg/9.20.2937. [DOI] [PubMed] [Google Scholar]
- 21.Christensen EI, Devuyst O, Dom G, Nielsen R, Van der Smissen P, Verroust P, Leruth M, Guggino WB, Courtoy PJ. Loss of chloride channel ClC-5 impairs endocytosis by defective trafficking of megalin and cubilin in kidney proximal tubules. Proc Natl Acad Sci USA. 2003;100:8472–8477. doi: 10.1073/pnas.1432873100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devuyst O, Jouret F, Auzanneau C, Courtoy PJ. Chloride channels and endocytosis: new insights from Dent’s disease and ClC-5 knockout mice. Nephron Physiol. 2005;99:69–73. doi: 10.1159/000083210. [DOI] [PubMed] [Google Scholar]
- 23.Hoopes RR, Jr, Shrimpton AE, Knohl SJ, Hueber P, Hoppe B, Matyus J, Simckes A, Tasic V, Toenshoff B, Suchy SF, Nussbaum RL, Scheinman SJ. Dent Disease with mutations in OCRL1. Am J Hum Genet. 2005;76:260–267. doi: 10.1086/427887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shrimpton AE, Hoopes RR, Jr, Knohl SJ, Hueber P, Reed AA, Christie PT, Igarashi T, Lee P, Lehman A, White C, Milford DV, Sanchez MR, Unwin R, Wrong OM, Thakker RV, et al. OCRL1 mutations in Dent 2 patients suggest a mechanism for phenotypic variability. Nephron Physiol. 2009;112:27–36. doi: 10.1159/000213506. [DOI] [PubMed] [Google Scholar]
- 25.Vicinanza M, Di Campli A, Polishchuk E, Santoro M, Di Tullio G, Godi A, Levtchenko E, De Leo MG, Polishchuk R, Sandoval L, Marzolo M, De Matteis MA. OCRL controls trafficking through early endosomes via PtdIns4,5P(2)-dependent regulation of endosomal actin. EMBO J. 2011;30:4970–4985. doi: 10.1038/emboj.2011.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ungewickell AJ, Majerus PW. Increased levels of plasma lysosomal enzymes in patients with Lowe syndrome. Proc Natl Acad Sci USA. 1999;96:13342–13344. doi: 10.1073/pnas.96.23.13342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ponting CP. A novel domain suggests a ciliary function for ASPM, a brain size determining gene. Bioinformatics. 2006;22:1031–1035. doi: 10.1093/bioinformatics/btl022. [DOI] [PubMed] [Google Scholar]
- 28.Jin H, White SR, Shida T, Schulz S, Aguiar M, Gygi SP, Bazan JF, Nachury MV. The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell. 2010;141:1208–1219. doi: 10.1016/j.cell.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hou X, Hagemann N, Schoebel S, Blankenfeldt W, Goody RS, Erdmann KS, Itzen A. A structural basis for Lowe syndrome caused by mutations in the Rab-binding domain of OCRL1. EMBO J. 2011;30:1659–1670. doi: 10.1038/emboj.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knodler A, Feng S, Zhang J, Zhang X, Das A, Peranen J, Guo W. Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc Natl Acad Sci USA. 2010;107:6346–6351. doi: 10.1073/pnas.1002401107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, Jackson PK. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- 32.Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, Pernot E, Kisseleva MV, Compere P, Schiffmann SN, Gergely F, Riley JH, Perez-Morga D, Woods CG, Schurmans S. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet. 2009;41:1027–1031. doi: 10.1038/ng.427. [DOI] [PubMed] [Google Scholar]
- 33.Wessely O, Obara T. Fish and frogs: models for vertebrate cilia signaling. Front Biosci. 2008;13:1866–1880. doi: 10.2741/2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swanhart LM, Cosentino CC, Diep CQ, Davidson AJ, de Caestecker M, Hukriede NA. Zebrafish kidney development: basic science to translational research. Birth Defects Res Part C, Embryo Today: Rev. 2011;93:141–156. doi: 10.1002/bdrc.20209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kramer-Zucker AG, Olale F, Haycraft CJ, Yoder BK, Schier AF, Drummond IA. Cilia-driven fluid flow in the zebrafish pronephros, brain and Kupffer’s vesicle is required for normal organogenesis. Development. 2005;132:1907–1921. doi: 10.1242/dev.01772. [DOI] [PubMed] [Google Scholar]
- 36.Ma M, Jiang YJ. Jagged2a-notch signaling mediates cell fate choice in the zebrafish pronephric duct. PLoS Genetics. 2007;3:e18. doi: 10.1371/journal.pgen.0030018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robert A, Margall-Ducos G, Guidotti JE, Bregerie O, Celati C, Brechot C, Desdouets C. The intraflagellar transport component IFT88/polaris is a centrosomal protein regulating G1-S transition in non-ciliated cells. J Cell Sci. 2007;120(Pt 4):628–637. doi: 10.1242/jcs.03366. [DOI] [PubMed] [Google Scholar]
- 38.Koch A, Poirier F, Jacob R, Delacour D. Galectin-3, a novel centrosome-associated protein, required for epithelial morphogenesis. Mol Biol Cell. 2010;21:219–231. doi: 10.1091/mbc.E09-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grieve AG, Daniels RD, Sanchez-Heras E, Hayes MJ, Moss SE, Matter K, Lowe M, Levine TP. Lowe syndrome protein OCRL1 supports maturation of polarized epithelial cells. PLoS One. 2011;6:e24044. doi: 10.1371/journal.pone.0024044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu W, Murcia NS, Duan Y, Weinbaum S, Yoder BK, Schwiebert E, Satlin LM. Mechanoregulation of intracellular Ca2+ concentration is attenuated in collecting duct of monocilium-impaired orpk mice. Am J Physiol Renal Physiol. 2005;289:F978–F988. doi: 10.1152/ajprenal.00260.2004. [DOI] [PubMed] [Google Scholar]
- 41.Hovater MB, Olteanu D, Hanson EL, Cheng NL, Siroky B, Fintha A, Komlosi P, Liu W, Satlin LM, Bell PD, Yoder BK, Schwiebert EM. Loss of apical monocilia on collecting duct principal cells impairs ATP secretion across the apical cell surface and ATP-dependent and flow-induced calcium signals. Purinerg Signal. 2008;4:155–170. doi: 10.1007/s11302-007-9072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siroky BJ, Ferguson WB, Fuson AL, Xie Y, Fintha A, Komlosi P, Yoder BK, Schwiebert EM, Guay-Woodford LM, Bell PD. Loss of primary cilia results in deregulated and unabated apical calcium entry in ARPKD collecting duct cells. Am J Physiol Renal Physiol. 2006;290:F1320–F1328. doi: 10.1152/ajprenal.00463.2005. [DOI] [PubMed] [Google Scholar]
- 43.Rohatgi R, Battini L, Kim P, Israeli S, Wilson PD, Gusella GL, Satlin LM. Mechanoregulation of intracellular Ca2+ in human autosomal recessive polycystic kidney disease cyst-lining renal epithelial cells. Am J Physiol Renal Physiol. 2008;294:F890–F899. doi: 10.1152/ajprenal.00341.2007. [DOI] [PubMed] [Google Scholar]
- 44.Coon BG, Mukherjee D, Hanna CB, Riese DJ, 2nd, Lowe M, Aguilar RC. Lowe syndrome patient fibroblasts display Ocrl1-specific cell migration defects that cannot be rescued by the homologous Inpp5b phosphatase. Hum Mol Genet. 2009;18:4478–4491. doi: 10.1093/hmg/ddp407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ben El Kadhi K, Roubinet C, Solinet S, Emery G, Carreno S. The inositol 5-phosphatase dOCRL controls PI(4,5)P2 homeostasis and is necessary for cytokinesis. Curr Biol. 2011;21:1074–1079. doi: 10.1016/j.cub.2011.05.030. [DOI] [PubMed] [Google Scholar]
- 46.Dambournet D, Machicoane M, Chesneau L, Sachse M, Rocancourt M, El Marjou A, Formstecher E, Salomon R, Goud B, Echard A. Rab35 GTPase and OCRL phosphatase remodel lipids and F-actin for successful cytokinesis. Nat Cell Biol. 2011 doi: 10.1038/ncb2279. [DOI] [PubMed] [Google Scholar]
- 47.Kim S, Zaghloul NA, Bubenshchikova E, Oh EC, Rankin S, Katsanis N, Obara T, Tsiokas L. Nde1-mediated inhibition of ciliogenesis affects cell cycle re-entry. Nat Cell Biol. 2011;13:351–360. doi: 10.1038/ncb2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Avasthi P, Marshall WF. Stages of ciliogenesis and regulation of ciliary length. Differentiation; Res in Biol Divers. 2012;83:S30–S42. doi: 10.1016/j.diff.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mokrzan EM, Lewis JS, Mykytyn K. Differences in renal tubule primary cilia length in a mouse model of Bardet-Biedl syndrome. Nephron Exp Nephrol. 2007;106:e88–e96. doi: 10.1159/000103021. [DOI] [PubMed] [Google Scholar]
- 50.Tammachote R, Hommerding CJ, Sinders RM, Miller CA, Czarnecki PG, Leightner AC, Salisbury JL, Ward CJ, Torres VE, Gattone VH, 2nd, Harris PC. Ciliary and centrosomal defects associated with mutation and depletion of the Meckel syndrome genes MKS1 and MKS3. Hum Mol Genet. 2009;18:3311–3323. doi: 10.1093/hmg/ddp272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sohara E, Luo Y, Zhang J, Manning DK, Beier DR, Zhou J. Nek8 regulates the expression and localization of polycystin-1 and polycystin-2. J Am Soc Nephrol. 2008;19:469–476. doi: 10.1681/ASN.2006090985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang L, Weidenfeld R, Verghese E, Ricardo SD, Deane JA. Alterations in renal cilium length during transient complete ureteral obstruction in the mouse. J Anat. 2008;213:79–85. doi: 10.1111/j.1469-7580.2008.00918.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Verghese E, Weidenfeld R, Bertram JF, Ricardo SD, Deane JA. Renal cilia display length alterations following tubular injury and are present early in epithelial repair. Nephrol Dial Transplant. 2008;23:834–841. doi: 10.1093/ndt/gfm743. [DOI] [PubMed] [Google Scholar]
- 54.Baker K, Beales PL. Making sense of cilia in disease: the human ciliopathies. Am J Med Genet C Semin Med Genet. 2009;151C:281–295. doi: 10.1002/ajmg.c.30231. [DOI] [PubMed] [Google Scholar]
- 55.Bergmann C, Fliegauf M, Bruchle NO, Frank V, Olbrich H, Kirschner J, Schermer B, Schmedding I, Kispert A, Kranzlin B, Nurnberg G, Becker C, Grimm T, Girschick G, Lynch SA, et al. Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am J Hum Genet. 2008;82:959–970. doi: 10.1016/j.ajhg.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bae YK, Kim E, L’Hernault SW, Barr MM. The CIL-1 PI 5-phosphatase localizes TRP Polycystins to cilia and activates sperm in C. elegans. Curr Biol. 2009;19:1599–1607. doi: 10.1016/j.cub.2009.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mukhopadhyay S, Wen X, Chih B, Nelson CD, Lane WS, Scales SJ, Jackson PK. TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev. 2010;24:2180–2193. doi: 10.1101/gad.1966210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zuo XF, Guo W, Lipschutz JH. The exocyst protein Sec10 is necessary for primary ciliogenesis and cystogenesis in vitro. Mol Biol Cell. 2009;20:2522–2529. doi: 10.1091/mbc.E08-07-0772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu JL, Zuo XF, Yue P, Guo W. Phosphatidylinositol 4,5-bisphosphate mediates the targeting of the exocyst to the plasma membrane for exocytosis in mammalian cells. Mol Biol Cell. 2007;18:4483–4492. doi: 10.1091/mbc.E07-05-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He B, Xi FG, Zhang XY, Zhang J, Guo W. Exo70 interacts with phospholipids and mediates the targeting of the exocyst to the plasma membrane. Embo J. 2007;26:4053–4065. doi: 10.1038/sj.emboj.7601834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fields IC, King SM, Shteyn E, Kang RS, Folsch H. Phosphatidylinositol 3,4,5-trisphosphate localization in recycling endosomes is necessary for AP-1B-dependent sorting in polarized epithelial cells. Mol Biol Cell. 2010;21:95–105. doi: 10.1091/mbc.E09-01-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Coon BG, Hernandez V, Madhivanan K, Mukherjee D, Hanna CB, Barinaga-Rementeria Ramirez I, Lowe M, Beales PL, Aguilar RC. The Lowe syndrome protein OCRL1 is involved in primary cilia assembly. Hum Mol Genet. 2012;21:1835–1847. doi: 10.1093/hmg/ddr615. [DOI] [PubMed] [Google Scholar]
- 63.Luo N, West C, Murga-Zamalloa C, Sun L, Anderson RM, Wells C, Weinreb RN, Travers JB, Khanna H, Sun Y. OCRL localizes to the primary cilium: a new role for cilia in Lowe syndrome. Hum Mol Genet. 2012 doi: 10.1093/hmg/dds163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Groh ED, Swanhart LM, Cosentino CC, Jackson RL, Dai W, Kitchens CA, Day BW, Smithgall TE, Hukriede NA. Inhibition of histone deacetylase expands the renal progenitor cell population. J Am Soc Nephrol. 2010;21:794–802. doi: 10.1681/ASN.2009080851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cianciolo Cosentino C, Roman BL, Drummond IA, Hukriede NA. Intravenous microinjections of zebrafish larvae to study acute kidney injury. J Vis Exp. 2010 Aug;4(42):2079. doi: 10.3791/2079. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hentschel DM, Park KM, Cilenti L, Zervos AS, Drummond I, Bonventre JV. Acute renal failure in zebrafish: a novel system to study a complex disease. Am J Physiol Renal Physiol. 2005;288:F923–F929. doi: 10.1152/ajprenal.00386.2004. [DOI] [PubMed] [Google Scholar]
- 67.Krock BL, Perkins BD. The intraflagellar transport protein IFT57 is required for cilia maintenance and regulates IFT-particle-kinesin-II dissociation in vertebrate photoreceptors. J Cell Science. 2008;121(Pt 11):1907–1915. doi: 10.1242/jcs.029397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mo D, Potter BA, Bertrand CA, Hildebrand JD, Bruns JR, Weisz OA. Nucleofection disrupts tight junction fence function to alter membrane polarity of renal epithelial cells. Am J Physiol Renal Physiol. 2010;299:F1178–F1184. doi: 10.1152/ajprenal.00152.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ramirez IB-R, Pietka G, Jones DR, Divecha N, Alia A, Baraban SC, Hurlstone AFL, Lowe M. Impaired neural development in a zebrafish model for Lowe syndrome. Hum Mol Genet. 2012;21:1744–1759. doi: 10.1093/hmg/ddr608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Cloaca opening is normal in ocrl morphant zebrafish. Embryos injected with either control MO (left) or MO targeting ocrl (right) were examined for cloaca opening. Arrows indicate normal cloaca openings in both control and morphant larvae at 48 hpf.
Figure S2: Cilia in OCRL1-depleted MDCK cells are longer than control upon recovery from deciliation. A) MDCK cells were transfected with control or OCRL1 siRNA and plated on permeable supports. After 2 days, indicated samples were treated for 3 h with ammonium sulfate (NH4SO4) or left untreated, then allowed to recover for 2 days. Cells were processed for indirect immunofluorescence to detect acetylated tubulin 2 days after plating (top row), immediately after deciliation (middle row), and after recovery for 2 days (bottom row). B) Quantitation of cilia length in control or OCRL1-siRNA-treated cells grown on filters for 4 days (closed and open circles, respectively) or treated after 2 days with ammonium sulfate and allowed to recover for 2 days (closed and open triangles, respectively). Scale bar: 10 μm.