

Abstract
The ability to carry out simultaneous orthogonal exchange chemistries has opened new opportunities for increasing the numerical and structural diversity accessible to Dynamic Combinatorial Chemistry. We present proof-of-concept experiments demonstrating this concept is transferrable to resin-bound DCC, facilitating the generation and analysis of libraries with greater structural diversity.
From its beginnings little more than a decade ago, Dynamic Combinatorial (or Dynamic Covalent) Chemistry1 has developed into a useful method for the identification of novel binders for biological targets, new catalysts, new materials, and as a method for studying the rapidly emerging field of systems chemistry.2 The numerical diversity of a dynamic combinatorial library (DCL), and the range of chemical space it can access, increases dramatically with the number of bonds undergoing exchange. However, this increased potential dynamic complexity brings with it two major challenges. First, the formation of trimers (or higher order structures) is entropically disfavored relative to dimers, and therefore it is possible that libraries capable of forming higher-complexity structures will nonetheless consist primarily of dimers, unless the entropic penalty is overcome by compensating enthalpic factors. Second, if one does produce higher-diversity libraries, the analytical challenge presented by the library increases proportionately. Efforts to simulate DCLs by Ludlow and Otto have provided compelling evidence that“large” libraries can undergo efficient amplification processes,3 but to date the number of solution-phase libraries with > 1000 members has been vanishingly small.4
Building on a longstanding interest in our group in the use of phase segregation as a method for facilitating chemical synthesis,5 in 2006 we introduced the concept of Resin-Bound Dynamic Combinatorial Chemistry (RBDCC) in order to simplify the analytical challenges associated with DCC.6 In essence, this method inverts the affinity chromatography-like methods we7 and others8,9,10 have explored, in which the “target” molecule (a biopolymer or small molecule guest) was immobilized on resin. In RBDCC, one half of a dimer library is immobilized on synthesis resin beads (Figure 1a). Dynamic exchange with solution-phase monomers allows production of the full DCL; subsequent incubation of the mixture with a fluorophore-tagged target allows library evolution and separation of bead-bound library members graded based on fluorescence intensity. We have used this methodology to successfully identify novel compounds binding DNA,6 and two biomedically important RNA targets: one associated with the regulation of frameshifting in HIV,11 and a triplet repeat RNA implicated in Type 1 myotonic dystrophy.12
Fig. 1.
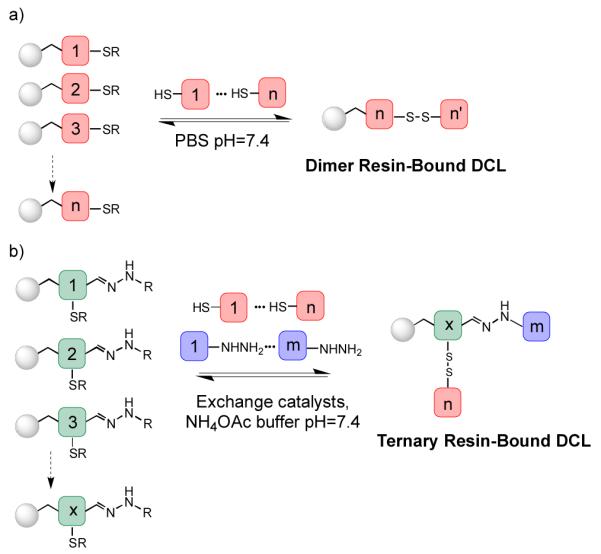
(a) Resin-bound dynamic combinatorial library and (b) Ternary resin-bound dynamic combinatorial library generation
To move beyond these early successes, it would be advantageous to extend the RBDCC technique to larger, nondimeric libraries. In many of the earliest DCL experiments, library monomers were synthesized carrying both partners of a chemically reactive pair (i.e. alcohol/ester, thiol/thiol, amine/aldehyde, etc.) allowing stoichiometric production of higher-order oligomers. In the context of expanding the range of RBDCC, although simple to implement, this would render identification of selected compounds difficult. An alternative and increasingly popular strategy involves the use of two orthogonally addressable exchange chemistries.13 In principle, this reduces the potential for entropic bias favoring dimers over trimers, since one can run the DCL selection as two successive processes, one for each type of exchange chemistry, as well as in a simultaneous exchange mode. The success of aniline catalysis as a method for accelerating acyl hydrazone exchange14 is particularly notable, as this potentially facilitates the use of simultaneous hydrazone and disulfide exchange at biopolymer-friendly pH values. To test the viability of this concept in a resin-bound format, we modified our generic nucleic acid-targeted library design as shown in Figure 1b.
An initial set of monomers was designed (Figure 2) containing a thiol (A1, A2), an acyl hydrazine (C1, C2) or both a thiol and acyl hydrazone (B1, B2). We anticipated that use of glycine and proline as the two “variable” amino acids would provide sufficient structural differences to allow separation of library components by HPLC, without introducing the reactivity and protecting group complications that might occur through the use of amino acids with heteroatom-containing side chains. Monofunctional monomers A1, A2, C1, and C2 incorporated an aromatic moiety (here N-methyl indole carboxylic acid) to provide a readily observable chromophore for HPLC analysis and by analogy to the heterocyclic groups included in our previous RNA- and DNA-targeted libraries. Library components were synthesized on Wang resin using standard solid-phase Fmoc peptide synthesis protocols. Following cleavage, the structures of solution-phase monomers were confirmed bymass spectrometry.
Fig. 2.
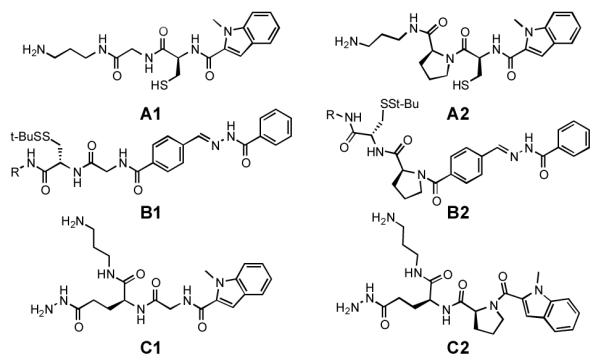
Library members. For solution phase experiments, R = (CH2)3NH2; for resin-bound experiments, R = (CH2)5CONH2
Before attempting experiments with immobilized components, the behavior of the library was studied in a fully solution-phase system. Both thiopropanol and glutathione15 were tested as facilitators of disulfide exchange, while aniline was employed as a catalyst for hydrazone exchange.14 As expected, equilibration of A1 and A2 with B1 and B2 (0.13 mM each, 1:1:1:1 ratio) in the presence of one equivalent of thiopropanol in 50mM ammonium acetate buffer (pH 7.4), containing 5.5% DMSO, for 3 days yielded several new product peaks in the HPLC trace (figure 3, green trace). MALDI data confirmed formation of several desired disulfides AB as well as AA, BB and thiopropanol adducts. Similarly, reaction of B1 and B2 with C1 and C2 (0.13 mM each, 1:1:1:1 ratio) in a pH 7.4 solution yielded the exchanged hydrazones in the presence of 37.5 equivalents of aniline (figure 3, red trace). This reaction occured much slower in the absence of aniline, consistent with the expected lack of reactivity of the uncatalyzed system at this pH.
Fig. 3.
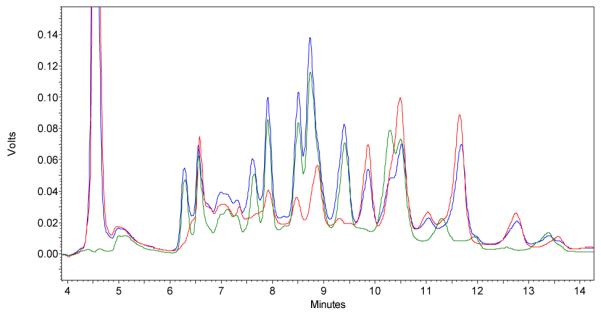
Solution phase library constructed from A1, A2, B1, B2 and thiopropanol (green trace); B1, B2, C1, C2 and aniline (red trace); A1, A2, B1, B2, C1, C2, thiopropanol and aniline (blue trace) after 3 days of equilibration in ammonium acetate buffer (pH 7.4).
Simultaneous disulfide and acyl hydrazone exchange was examined by mixing monomers A1 (thiol exchange), C1 (acyl hydrazone exchange), and B1 (bidirectional exchange) with thiopropanol and aniline and allowing them to equilibrate for 3 days. Formation of dimer B1C1 and trimer A1B1C1 were observed by MALDI. This complexity was increased further through the inclusion of all four monofunctional monomers (A1, A2, C1, C2), the two bifunctional monomers B1 and B2, and exchange catalysts thiopropanol and aniline. Following equilibration, two different peaks corresponding to mixed ABC structures were observed by MALDI-MS. A1B1C1 and A2B2C2, the ABC trimers expected to be in lowest concentration based on a statistical distribution of products, were not detected. Incorporating a 10-fold higher concentration of A and C monomers (building blocks A1, A2, B1, B2, C1, C2 in a 10:10:1:1:10:10 ratio) provided an overall increase in the formation of exchange products as measured by HPLC, although MALDI-MS did not change significantly.
With a successful demonstration of simultaneous bidirectional disulfide and hydrazone exchange in the model solution library, we next turned to evaluating the system in a solid-phase context. Building blocks B1 and B2 were synthesized on TentaGel Macrobead resin, linked via aminohexanoic acid (R) to a photocleavable linker (2-nitrophenyl glycine) using standard solid-phase Fmoc peptide synthesis protocols. Incorporation of the aminohexanoic acid and photocleavable linker attachment was done in order to structure the test compounds as closely as possible to what would be used in a more diverse library, which would require this sort of attachment to allow for acid-mediated deprotection of amino acid sidechains without cleavage from the resin. After cleaving portions of the resin for each monomer and verifying identity by mass spectrometry, several exchange experiments were set up similar to the solution phase experiments described above.
First, control experiments were carried out in which only either hydrazone or disulfide exchange was allowed to occur. For hydrazone exchange, resin-bound building block B1 was allowed to equilibrate with the solution phase building blocks C1 and C2 in the presence of aniline for 6 days. For disulfide exchange, resin-bound building block B1 was first pretreated with 20 equivalents of thiopropanol for 24h, then allowed to equilibrate with solution phase building block A1 for 6 days. Following equilibration, solution phase library components were drained from the resin, which was then washed with water (x3), DMF (x3), THF (x3), and CH3CN (x3), and resuspended in a 4:1 mixture of acetonitrile:methanol. Photocleavage using a compact UV lamp was allowed to proceed for 6–8 hours, after which samples were analyzed by HPLC and MALDI-MS. In both cases we observed formation of either AB or BC dimer products.
Next, a simultaneous exchange experiment was attempted using 3 building blocks, one of each kind (A1 and C1 in solution, B1 on resin) in a 1:1:1 ratio. However, only dimers A1B1 and B1C1 were observed following as much as 6 days of equilibration. This result can be attributed to slower kinetics for the resin-bound exchange. In order to facilitate the exchange reactions, the concentration of solution phase building blocks was increased 10 fold (10:1:10 ratio). In this case, after 6 days of equilibration we observed formation of the desired trimer A1B1C1 (Figure 4).
Fig. 4.
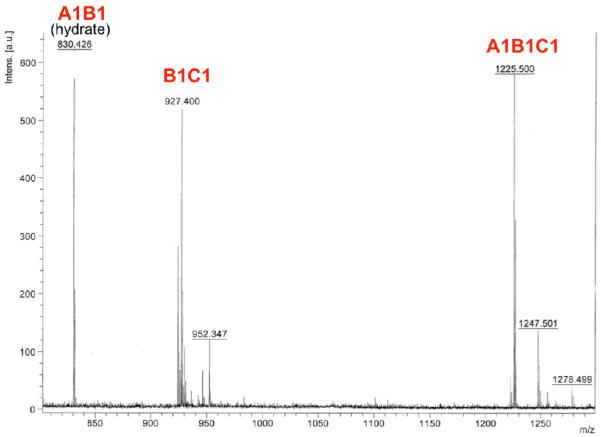
Resin bound library constructed from A1 and C1 in solution and resin bound B1, after 6 days of equilibration in the presence of aniline at pH 7.4
Finally, we set up the full library equilibration experiment in quadruplicate (2 experiments for 1 week of equilibration, and 2 experiments for 2 weeks). Two resin-bound, thiopropanol modified, bi-functional building blocks B1 and B2 were allowed to equilibrate for 1 or 2 weeks with solution phase thiol building blocks A1 and A2 and hydrazone building blocks C1 and C2 (1:1:10:10:10:10) in ammonium acetate buffer (pH=7.4) in the presence of aniline. After equilibration, resin beads were washed, and products were cleaved from the resin. After 1 week of equilibration, 3 different ABC products were detected as well as several dimers AB and BC (Fig. 5). Trimer A1B1C1 (M+H = 1225.5, all 3 varied amino acids were glycine), could not be detected. Comparison of HPLC traces along with MALDI data for duplicate experiments equilibrated for 1 week with those equilibrated for 2 weeks showed that additional reaction time does not significantly alter library composition.
Fig. 5.
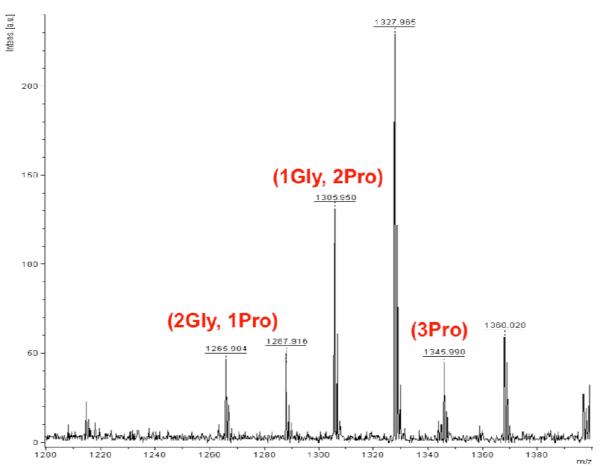
Resin bound library constructed from A1, A2, C1, C2 in solution and resin bound B1 and B2, after 1 week of equilibration in the presence of aniline at pH 7.4.
Conclusions
We have demonstrated successful proof-of-concept experiments for orthogonal exchange chemistries operating individually and simultaneously across a three-component phase-segregated dynamic combinatorial library, or “ternary RBDCC”. Use of resin-bound monomers was found to require increases in the concentration of solution monomers in order to observe the formation of trimers. Further studies will be needed in order to ascertain how this affects affinity-based library selection directed by either a biopolymeric or small molecule target. As library equilibration was conducted near neutral pH in buffered solution, we anticipate that ternary RBDCC will prove useful for screening libraries intended to yield novel protein and nucleic acid binding molecules.
Supplementary Material
Acknowledgments
We thank the NIH (NINDS and NIAMS Grant #1R21NS071023-01) for support of this work.
Footnotes
Electronic Supplementary Information (ESI) available: synthetic procedures and full mass spectral analysis of all library experiments. see DOI: 10.1039/b000000x/
References
- 1.Corbett PT, Leclaire J, Vial L, West KR, Wietor J-L, Sanders JKM, Otto S. Chem. Rev. 2006;106:3652. doi: 10.1021/cr020452p. J. N. H. Reek and S. Otto, Dynamic Combinatorial Chemistry, Wiley VCH, Weinheim, 2010; B. L. Miller, Dynamic Combinatorial Chemistry in Drug Discovery, Bioorganic Chemistry, and Materials Science, Wiley, New Jersey, 2010. [DOI] [PubMed] [Google Scholar]
- 2.Ludlow RF, Otto S. Chem. Soc. Rev. 2008;37:101. doi: 10.1039/b611921m. [DOI] [PubMed] [Google Scholar]
- 3.Ludlow RF, Otto S. J. Am. Chem. Soc. 2010;132:5984. doi: 10.1021/ja1013689. [DOI] [PubMed] [Google Scholar]
- 4.Ludlow RF, Otto S. J. Am. Chem. Soc. 2008;130:12218. doi: 10.1021/ja803317k. [DOI] [PubMed] [Google Scholar]
- 5.Hari A, Miller BL. Angew. Chem. Int. Ed. 1999;38:2777. doi: 10.1002/(sici)1521-3773(19990917)38:18<2777::aid-anie2777>3.0.co;2-x. A. Hari and B. L. Miller, Org. Lett. 2000, 2, 691. [DOI] [PubMed] [Google Scholar]
- 6.McNaughton BR, Miller BL. Org. Lett. 2006;8:1803. doi: 10.1021/ol060330+. [DOI] [PubMed] [Google Scholar]
- 7.Klekota B, Hammond MH, Miller BL. Tetrahedron Lett. 1997;38:8639. [Google Scholar]
- 8.Eliseev AV, Nelen MI. J. Am. Chem. Soc. 1997;119:1147. [Google Scholar]
- 9.Hioki H, Still WC. J. Org. Chem. 1998;63:904. [Google Scholar]
- 10.Besenius P, Cormack PAG, Ludlow RF, Otto A, Sherrington DC. Org. Biomol. Chem. 2010;8:2414. doi: 10.1039/c000333f. [DOI] [PubMed] [Google Scholar]
- 11.McNaughton BR, Gareiss PC, Miller BL. J. Am. Chem. Soc. 2007;129:11306. doi: 10.1021/ja072114h. [DOI] [PubMed] [Google Scholar]
- 12.Gareiss PC, Sobczak K, McNaughton BR, Palde PB, Thornton CA, Miller BL. J. Am. Chem. Soc. 2008;130:16254. doi: 10.1021/ja804398y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Escalante AM, Orrillo AG, Furlan RLE. J. Comb. Chem. 2010;12:410. doi: 10.1021/cc100046r. A. Rodriguez-Docampo and S. Otto, Chem. Commun. 2008, 5301. [DOI] [PubMed] [Google Scholar]
- 14.Dirksen A, Dirksen S, Hackeng TM, Dawson PE. J. Am. Chem. Soc. 2006;128:15602. doi: 10.1021/ja067189k. V. T. Bhat, A. M. Caniard, T. Luksch, R. Brenk, D. J. Campopiano, and M. F. Greaney, Nature Chem. 2010, 2, 490. [DOI] [PubMed] [Google Scholar]
- 15.Whitney AM, Ladame S, Balasubramanian S. Angew. Chem. Int. Ed. 2004;43:1143. doi: 10.1002/anie.200353069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.