

Abstract
Bone has long been known to be responsive to mechanical loading. For at least 25 years it has been known that osteocytes sense mechanical load, and because of their response to mechanical loading, osteocytes are believed to be the mechanosensory cell. The Wnt/β-catenin signaling pathway has been shown to be crucial in bone development. Mutations in LRP5 and SOST, which cause high bone mass, have increased interest in the Wnt pathway as a potential target for osteoporosis therapy and have helped link Wnt/β-catenin signaling to bone’s response to mechanical loading. Because of its specificity to osteocytes, the Wnt inhibitor sclerostin is a target for anabolic bone therapies. The response of bone to mechanical loading is critically regulated by osteocytes secreting sclerostin, which binds to Lrp5.
Introduction
Bone has long been known to be responsive to mechanical loading. The ability of bone to functional adapt to forces was discovered in the late 19th and early 20th centuries [1-5]. For at least 25 years it has been known that osteocytes sense mechanical load [6], and because of their varied response to mechanical loading [7-11], osteocytes are believed to be the cell that senses whether or not the bone is being loaded and signals osteoblasts and osteoclasts to respond accordingly [12, 13]. Osteocytes account for the vast majority of bone cells (90–95%) in the skeleton [12]. They are star-shaped, measure 9 μm by 20 μm in humans, and derive from mature osteoblasts that embed themselves into mineralized matrix and reside in the lacunae [14]. They communicate through their dendritic processes and have a cell spacing of about 25 μm. Each cell has some 50 dendritic processes that preferentially grow through the canaliculi toward the periosteal side of the bone [15].
Physiologically, mechanical forces are applied to bones through both muscle forces and ground reaction forces [16]. Forces on bone increase both the bone density and the geometrical properties of bone due to loading. Geometrically, the distribution of the bone material is more important than the cross-sectional area. Given the same amount of material, the bone with the higher moment of inertia (and related section modulus) is more resistant to bending, and the bone with the higher polar moment of inertia is more resistant to torsion. These moments of inertia are dependent on how the bone material is distributed [17]. Practically, this means that periosteal bone growth improves bone stiffness more than endosteal growth.
There are many examples of how bone adapts to loading. Athletes in high impact sports have higher bone mineral density and an improved section modulus than athletes in low impact sports and sedentary controls [18-20] and racquet sports athletes have higher bone density and section modulus in their dominant arm relative to their contralateral limb [21, 22]. Bed rest and spaceflight lead to decreased bone mineral density in humans [23-26] and in rodents, hindlimb suspension decreases the bone mineral content and moment of inertia of the unloaded bones [27, 28].
Recent work has causally linked alterations in Wnt signaling to changes in bone development and homeostasis. In this review, we introduce the cellular mechanisms associated with Wnt signaling, describe the key events that helped link Wnt signaling to bone disease, and discuss Wnt signaling in the osteocyte and the related anabolic bone therapies. We also describe specific experiments that have provided insights into the roles of Wnt signaling proteins produced by osteocytes (with an emphasis on sclerostin), which act in feedback mechanisms to control local response to mechanical loading.
Overview of Wnt signaling
Wnt signaling plays central roles in regulating the development of many tissues and organs, and alterations in the pathway are commonly associated with human disease. The first Wnt gene was identified by Nusse and Varmus in 1982, when they reported the presence of a common proviral insertion site found in tumors induced by mouse mammary tumor virus [29]. Viral insertion resulted in increased expression of this “integration site 1” gene (Int1). Studies of Int1 were hampered by the challenges associated with purifying the protein in biologically active form, so a central focus of early research on this gene focused on evaluating the genetic pathways associated with the homolog of Int1 in Drosophila, a gene known as wingless [30]. To provide clarity, researchers in the field then reorganized the nomenclature to reflect the contributions of studies focused on both Int1 and wingless, renaming the emerging protein family as “Wnt” (wingless + Int1) [31]. The clinical significance of this pathway came into sharper focus as downstream signaling components were identified. For example, one component, the adenomatous polyposis coli (APC) gene, is deleted in a significant majority of colorectal tumors [32]. This, combined with numerous other studies, identified regulation of the cytoplasmic and nuclear levels of β-catenin as a key point of activity for Wnts.
At the cellular level, Wnts activate several signaling cascades, including the most commonly studied (“canonical”) pathway, which results in stabilization of the β-catenin protein [33]. This pathway is initiated when a Wnt protein binds to a receptor complex that includes a member of the Frizzled family of seven-transmembrane receptors plus either Lrp5 (low-density lipoprotein-related receptor 5) or Lrp6 [34]. Formation of this receptor complex results in the phosphorylation of the cytoplasmic tail of Lrp5 or Lrp6, leading to the formation of a binding site for axin [35]. Axin is normally found in a multiprotein complex that also includes APC and glycogen synthase kinase 3 (GSK3). In the absence of an upstream Wnt signal, GSK3 phosphorylates residues near the amino terminus of β-catenin, targeting β-catenin for ubiquitin-dependent proteolysis. The recruitment of axin to the phosphorylated tail of Lrp5/6 inhibits the activity of GSK3 towards β-catenin (or perhaps the subsequent ubiquitination), leading to increased β-catenin levels in the cytoplasm. The increased cytoplasmic levels ultimately lead to β-catenin’s nuclear translocation, its binding to members of the LEF/TCF family of DNA binding proteins, and the transactivation of target-gene promoters. Recently, it has emerged that the stability and nuclear levels of the transcriptional activator TAZ are also regulated by the same process that controls β-catenin levels, because TAZ enters the nucleus as part of a β-catenin complex [36]. Thus, sites driven by TAZ transactivation, independent of TCF/LEF sites, may also be directly regulated by Wnt signaling (Figure 1).
Figure 1. Schematic of inactivated (left) and activated (right) Wnt/β-catenin signaling pathway.
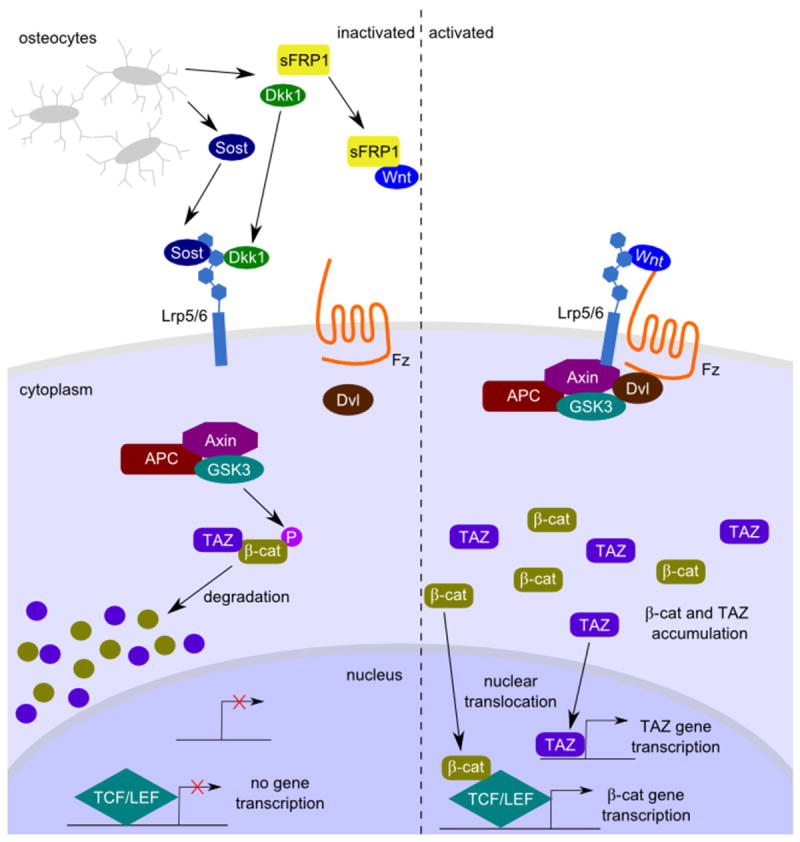
Wnt ligands bind to a receptor complex that includes a member of the Frizzled family and either Lrp5 or Lrp6 (right). In the absence of a Wnt signal (left), a “destruction” complex that includes Axin, APC, and GSK3 facilitates GSK3-mediated phosphorylation of β-catenin, targeting it for ubiquitin-dependent proteolytic degradation. In the presence of a Wnt signal, the cytoplasmic domain of Lrp5/6 is phosphorylated, which serves as a binding site for Axin, recruiting the destruction complex to the membrane and inhibiting its activity. This results in increased cytoplasmic levels of B-catenin, which can enter the nucleus and interact with members of the LEF/TCF family of DNA binding proteins to activate target gene promoters. Recently, regulation of the stability of the transcriptional transactivator, TAZ, has also been linked to B-catenin degradation, allowing GSK3-mediated phosphorylation of B-catenin to also regulate the targets of TAZ transactivation. A complex regulatory network has evolved to inhibit Wnt signaling at the level of the plasma membrane. For example, SOST and Dkk1 both bind to Lrp5/6 to inhibit the ability of Wnt to bind and activate signaling through its receptor complex.
Wnt signaling and bone disease
Studies of the molecular mechanisms of Wnt signaling as related to osteoblast function were stimulated by three seminal studies published in 2001 and 2002. The first was a long-term study focused on identifying the underlying genetic cause of the pediatric syndrome osteoporosis pseudoglioma (OPPG) [37]. OPPG is characterized by severe, early-onset osteoporosis and is also associated with abnormal eye vasculature [38]. In 2001, the underlying genetic mutation for this autosomal recessive disorder was found to be inactivating mutations in the gene encoding LRP5 [39]. This report was followed shortly by two manuscripts showing that some patients with an inherited predisposition to high bone mass carry a point mutation in LRP5 (G171V) that is causally associated with the increased bone mass [40, 41]. Subsequent generation of mice carrying germline inactivating mutations in Lrp5 further confirmed the importance of this gene by accurately modeling phenotypes observed in OPPG syndrome [42-44]. In addition, a strain of mice expressing the G171V version of Lrp5 specifically in osteoblasts developed high bone mass, further confirming role of Lrp5 in skeletal homeostasis [45].
While the mechanisms underlying the effect of LRP5 mutations on bone mass are still being debated in the literature, an important advance came from studies on two other disorders associated with increased bone mass: sclerosteosis and van Buchem disease [46]. Both disorders are caused by loss of expression of the gene SOST, which encodes the protein sclerostin [47, 48]. In sclerosteosis, this loss is due to inactivating mutations in the coding region, while the underlying defect in van Buchem disease is a 52-kilobase deletion in a putative regulatory element necessary for expression of SOST [49]. Subsequent studies found that SOST, which is specifically secreted from osteocytes [50-52] and some types of chondrocytes [53-55], is normally bound to the LRP5 protein to inhibit its signaling [56-58]. In patients with the high bone mass associated mutation in LRP5, the ability of SOST to bind and down-regulate LRP5 function is lost, leading to increased bone growth [56, 57, 59, 60]. Other proteins such as dickkopf 1 (DKK1) and mesoderm development (MESD) also bind to wild-type LRP5 [61-63], but not to mutant forms of LRP5 linked to high bone mass [64]. This evidence, combined with several mouse models in which LRP5 (and the related LRP6 protein) function is specifically altered within the osteoblast and osteocyte lineage [65-67], has led to a model proposing that Lrp5 and Lrp6 function within osteoblasts to regulate osteoblast function. It should be noted that another model has been proposed, in which Lrp5 is involved in the regulation of serotonin secretion from the enterrochromaffin cells of the intestine [68]. Alterations in serum serotonin then lead to changes in osteoblast function. The relative contributions of these two models are still being assessed. For a more thorough discussion of the current status of therapies targeting serotonin, we refer readers to a recent review on this topic [69].
Wnt signaling in osteocytes and related bone therapies
Osteocytes express several known inhibitors of the Wnt/β-catenin pathway. Of these inhibitors, sclerostin is most commonly linked to osteocytes because its expression is generally considered to be relatively specific to those cells [50-52]. Osteocytes secrete sclerostin along their dendrites in the canaliculi after the cells became embedded in mineralized matrix [70]. Consistent with the high bone mass phenotype of sclerosteosis and van Buchem disease patients, mice with a deletion of Sost had dramatically increased bone mineral density that was due to increased bone formation rather than to decreased osteoclast activity [71, 72], while overexpression of Sost decreased bone mass and strength due to decreased bone formation [50].
Since the Wnt signaling pathway has been shown to be crucial in bone development, it has received much interest as a potential target for osteoporosis therapy [73]. Specifically, the genetic linkage of the high bone mass diseases sclerosteosis and van Buchem disease to the SOST gene plus the specificity of sclerostin in osteocytes point to sclerostin’s potential use as an anabolic bone agent. The only currently available anabolic drug for treating osteoporosis is teriparatide (Forteo®; Eli Lilly and Company, Indianapolis, IN) [74]. Teriparatide is the human recombinant form of parathyroid hormone (PTH) and acts through the PTH receptor. Patients receiving intermittent teriparatide treatment had higher bone mineral density than those treated with bisphosphonates [75]. Treatment with PTH drives bone formation by decreasing sclerostin expression [76]. In wild-type and estrogen-deprived rats, PTH treatment directly regulated Sost transcription, decreased Sost/sclerostin expression, and increased bone mineral density [77]. When the PTH receptor was constitutively activated in osteocytes, mice had reduced sclerostin and increased bone mass. After the deletion of Lrp5 in these mice, the high bone mass phenotype was no longer apparent [78]. An alternative, but not mutually exclusive model, is that PTH signals directly through LRP6 to activate β-catenin. Taken together, PTH functions as an anabolic bone agent through the osteocytes to decrease sclerostin expression and activate the Wnt/β-catenin pathway through Lrp5.
Sclerostin antibodies are being developed to target the protein directly in order to improve bone mineral density. In preclinical studies, the administration of the sclerostin antibody AMG 785 (Amgen Inc., Thousand Oaks, CA) increased the formation of trabecular, periosteal, endosteal, and intractorical bone of postmenopausal osteoporotic rats [79] and cynomolgus monkeys [80]. In a phase I study in humans, a single dose of the sclerostin antibody increased bone mineral density in the hip and spine after 85 days relative to placebo controls [81]. In a phase II trial on postmenopausal osteoporotic women with femoral neck T-scores of −3.5 to −2, sclerostin antibody treatment increased bone mineral density in the hip and spine significantly more than bisphosphonate and teriparatide treatment after one year with more density increase in the spine than the hip. Bone density increased rapidly through the first six months but the rate of increase slowed in the second six months [82]. In both trials the drug was well-accepted with mild side effects. If the increases in density translate to functional increases in strength and decreases in fracture risk, and longer term trials demonstrate continued tolerability and safety, sclerostin antibody treatment will be an effective, bone-specific anabolic treatment for osteoporosis. The clinical success of PTH and the early successes of the sclerostin antibodies demonstrate the importance of the Wnt signaling pathway through osteocytes in bone formation.
In addition to sclerostin, osteocytes express the Wnt inhibitors Dkk1 and secreted frizzled-related protein 1 (sFRP1). Both play a role in regulating bone mass. Dkk1 inhibits osteoblast differentiation and bone formation by binding to Lrp5/6 [61, 62, 83], and Lrp5 high bone mass mutant mice have altered Dkk1-Lrp5 binding [64]. Deletion of a single allele of Dkk1 is enough to increase bone formation and improve structural characteristics but has no effect on bone resorption [84]. sFRP1 inhibits Wnt signaling either by binding to Wnts and preventing them from binding to the Lrp5/6 complex [85] or by binding directly to the Lrp5/6 complex to prevent Wnts from binding there [86]. Mice with sFRP1 deleted have increased trabecular bone mineral density, and in vitro, their osteoblasts show increased proliferation and differentiation into osteocytes [87]. sFRP1 expression is at peak levels in early osteocytes undergoing cell death and at decreased levels in mature osteocytes, which demonstrates that sFRP1 is involved in negative regulation of osteocyte survival [88].
Osteocyte-like MLO-Y4 cells have been used in fluid flow shear studies to demonstrate other pathways that are involved in cross talk with the Wnt/β-catenin pathway. One of the proposed mechanisms by which osteocytes sense mechanical load is through interstitial fluid flow through the lacunae-canaliculi network—for two mechanosensory reviews in this issue, see [89, 90]—which causes a shear stress on the cells [91]. Fluid flow shear stress in MLO-Y4 cells induces prostaglandin E2 (PGE2) and increases the number of gap junctions and the expression of the gap junction protein connexin 43 (Cx43) [92]. PGE2 in turn activates cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA) [93] and protects cells from dexamethasone-induced apoptosis by increasing the phosphorylation of GSK3, which causes nuclear translocation of β-catenin [94]. Osteoblasts and osteocytes not subjected to fluid flow but treated with PGE2 also show β-catenin nuclear translocation and activated Wnt signaling [95]. Under shear stress, increased expression of PGE2 activates the PI3K/Akt pathway, which in turn causes phosphorylation of GSK3, nuclear translocation of β-catenin, and the activation of Wnt signaling independent of Lrp5/6 [96]. β-catenin also induces expression of Cx43, which increases osteocyte communication through gap junctions [97]. Taken together, these results demonstrate that there is cross talk between PGE2, PI3K/Akt, and Wnt signaling and that PGE2 can activate Wnt signaling independent of Lrp5/6.
Studies in conditional knockout mice have demonstrated the importance of the Wnt/β-catenin pathway in regulating the osteoclast inhibitor osteoprotegerin (OPG). Increased OPG through β-catenin promotes osteoblast differentiation and prevents the differentiation of osteoclasts [98]. The conditional deletion of β-catenin in osteoblast precursors (using collagen I alpha I-; Col1a1-Cre) mature osteoblasts (osteocalcin-; Ocn-Cre), and osteocytes (dentin matrix acidic phosphoprotein 1-; DMP1-Cre) leads to a decreased level of OPG and an increased number of osteoclasts [98-100]. These conditional knockouts demonstrate the importance of β-catenin through the differentiation of osteoblast precursors (Col1a1+ cells) to osteoblasts (Ocn+ cells) to osteocytes (DMP1+ cells) in the regulation of OPG.
Role of Wnt signaling in response to mechanical loading
Shortly after the discovery of the link between Lrp5 and bone mass, Johnson hypothesized that Lrp5 is crucial in the sensation and response of bone to load [101]. Mice carrying germline mutations in Lrp5 have been made that model the high [45, 65] and low bone mass [42-44] phenotypes. Johnson’s hypothesis was confirmed when mice with a deletion of Lrp5 did not respond to mechanical loading [102]. Furthermore, mice with missense mutations of Lrp5 (A214V and G171V) that cause high bone mass had an altered response to mechanical loading. One of these mutations (A214V) increased periosteal bone formation compared with wild-type controls, while the other (G171V) improved endosteal bone formation compared with the wild-type [103]. The mechanosensitivity of Lrp5 acts at least in part through the osteocytes, because mice with an osteocyte-specific deletion of Lrp5 were less responsive to mechanical loading [104].
Mechanical loading decreases Sost transcription and sclerostin protein expression while increasing bone formation [11, 105]. Mechanical loading also decreases the transcription of Dkk1, while sFRP1 transcription is unchanged [11]. When mice underwent unloading through hindlimb tail suspension, Sost transcription significantly increased in the tibia, while increases in Dkk1 and sFPR1 transcription approached significance [11], though a recent study has suggested that sclerostin response may be site-specific [106]. Local down-regulation of sclerostin in osteocytes is required for mechanotransduction-based bone formation [107], and mice with a deletion of Sost that underwent unloading through hindlimb tail suspension were resistant to bone loss [72]. Taken together, these reports suggest that the response of bone to mechanical loading is crucially regulated by osteocytes secreting sclerostin, which binds to Lrp5. When osteocytes sense a mechanical load, they reduce the expression of Wnt inhibitors, most prominently sclerostin. This down-regulation allows Lrp5 to be instead bound by Wnts, which may already be present or may have been up-regulated by the mechanical loading [108], and the result is activation of the Wnt/β-catenin signaling pathway.
Summary
The reports at the beginning of the last decade demonstrating that mutations in LRP5 are causally associated with changes in human bone mass stimulated extensive research into understanding the underlying mechanisms. This work demonstrated that components of this pathway, including LRP5, are required for osteocytes to respond to mechanical load. In addition, regulation of secretion of the Wnt inhibitor, SOST, from osteocytes plays a key role in coordinating the response to these mechanical signals. However, there are several outstanding questions remaining to be addressed. For example, what is the mechanism by which LRP5 is activated via mechanical loading? Does this involve a Wnt ligand? If so, which one(s)? Answers to these questions will further inform the development of therapies based on activating this pathway to treat osteoporosis and other bone diseases.
Acknowledgments
The authors thank David Nadziejka for technical editing of the manuscript and Michaela Kneissel for comments. Work in the Williams Laboratory is supported by National Institutes of Health grant AR053293 (BOW) and by Van Andel Research Institute.
Footnotes
The authors declare that they have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wolff J. Das gesetz der transformation der knochen. Berlin: Quarto; 1892. [Google Scholar]
- 2.Koch JC. The laws of bone architecture. Am J Anat. 1917;21:177–298. [Google Scholar]
- 3.Thompson DW. On growth and form. Cambridge University Press; 1942. [Google Scholar]
- 4.Cowin SC. Bone mechanics handbook. CRC Press; 2001. [Google Scholar]
- 5.van Rietbergen B, Huiskes R, Eckstein F, Ruegsegger P. Trabecular bone tissue strains in the healthy and osteoporotic human femur. J Bone Miner Res. 2003;18:1781–8. doi: 10.1359/jbmr.2003.18.10.1781. [DOI] [PubMed] [Google Scholar]
- 6.Pead MJ, Suswillo R, Skerry TM, Vedi S, Lanyon LE. Increased 3H-uridine levels in osteocytes following a single short period of dynamic bone loading in vivo. Calcif Tissue Int. 1988;43:92–6. doi: 10.1007/BF02555153. [DOI] [PubMed] [Google Scholar]
- 7.Skerry TM, Bitensky L, Chayen J, Lanyon LE. Early strain-related changes in enzyme activity in osteocytes following bone loading in vivo. J Bone Miner Res. 1989;4:783–8. doi: 10.1002/jbmr.5650040519. [DOI] [PubMed] [Google Scholar]
- 8.Klein-Nulend J, van der Plas A, Semeins CM, Ajubi NE, Frangos JA, Nijweide PJ, Burger EH. Sensitivity of osteocytes to biomechanical stress in vitro. FASEB J. 1995;9:441–5. doi: 10.1096/fasebj.9.5.7896017. [DOI] [PubMed] [Google Scholar]
- 9.Gluhak-Heinrich J, Ye L, Bonewald LF, Feng JQ, MacDougall M, Harris SE, Pavlin D. Mechanical loading stimulates dentin matrix protein 1 (DMP1) expression in osteocytes in vivo. J Bone Miner Res. 2003;18:807–17. doi: 10.1359/jbmr.2003.18.5.807. [DOI] [PubMed] [Google Scholar]
- 10.Gluhak-Heinrich J, Pavlin D, Yang W, MacDougall M, Harris SE. MEPE expression in osteocytes during orthodontic tooth movement. Arch Oral Biol. 2007;52:684–90. doi: 10.1016/j.archoralbio.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, Mantila SM, Gluhak-Heinrich J, Bellido TM, Harris SE, Turner CH. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283:5866–75. doi: 10.1074/jbc.M705092200. [DOI] [PubMed] [Google Scholar]
- 12.Bonewald LF. Osteocytes as dynamic multifunctional cells. Ann N Y Acad Sci. 2007;1116:281–90. doi: 10.1196/annals.1402.018. [DOI] [PubMed] [Google Scholar]
- 13.Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26:229–38. doi: 10.1002/jbmr.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noble BS. The osteocyte lineage. Arch Biochem Biophys. 2008;473:106–11. doi: 10.1016/j.abb.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 15.Sugawara Y, Kamioka H, Honjo T, Tezuka K, Takano-Yamamoto T. Three-dimensional reconstruction of chick calvarial osteocytes and their cell processes using confocal microscopy. Bone. 2005;36:877–83. doi: 10.1016/j.bone.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 16.Sharkey NA, Ferris L, Smith TS, Matthews DK. Strain and loading of the second metatarsal during heel-lift. J Bone Joint Surg Am. 1995;77:1050–7. doi: 10.2106/00004623-199507000-00011. [DOI] [PubMed] [Google Scholar]
- 17.van der Meulen MC, Jepsen KJ, Mikic B. Understanding bone strength: size isn’t everything. Bone. 2001;29:101–4. doi: 10.1016/s8756-3282(01)00491-4. [DOI] [PubMed] [Google Scholar]
- 18.Robinson TL, Snow-Harter C, Taaffe DR, Gillis D, Shaw J, Marcus R. Gymnasts exhibit higher bone mass than runners despite similar prevalence of amenorrhea and oligomenorrhea. J Bone Miner Res. 1995;10:26–35. doi: 10.1002/jbmr.5650100107. [DOI] [PubMed] [Google Scholar]
- 19.Nikander R, Sievanen H, Heinonen A, Kannus P. Femoral neck structure in adult female athletes subjected to different loading modalities. J Bone Miner Res. 2005;20:520–8. doi: 10.1359/JBMR.041119. [DOI] [PubMed] [Google Scholar]
- 20.Bailey CA, Brooke-Wavell K. Exercise for optimising peak bone mass in women. Proc Nutr Soc. 2008;67:9–18. doi: 10.1017/S0029665108005971. [DOI] [PubMed] [Google Scholar]
- 21.Haapasalo H, Kontulainen S, Sievanen H, Kannus P, Jarvinen M, Vuori I. Exercise-induced bone gain is due to enlargement in bone size without a change in volumetric bone density: a peripheral quantitative computed tomography study of the upper arms of male tennis players. Bone. 2000;27:351–7. doi: 10.1016/s8756-3282(00)00331-8. [DOI] [PubMed] [Google Scholar]
- 22.Ducher G, Prouteau S, Courteix D, Benhamou CL. Cortical and trabecular bone at the forearm show different adaptation patterns in response to tennis playing. J Clin Densitom. 2004;7:399–405. doi: 10.1385/jcd:7:4:399. [DOI] [PubMed] [Google Scholar]
- 23.Leblanc AD, Schneider VS, Evans HJ, Engelbretson DA, Krebs JM. Bone mineral loss and recovery after 17 weeks of bed rest. J Bone Miner Res. 1990;5:843–50. doi: 10.1002/jbmr.5650050807. [DOI] [PubMed] [Google Scholar]
- 24.Collet P, Uebelhart D, Vico L, Moro L, Hartmann D, Roth M, Alexandre C. Effects of 1- and 6-month spaceflight on bone mass and biochemistry in two humans. Bone. 1997;20:547–51. doi: 10.1016/s8756-3282(97)00052-5. [DOI] [PubMed] [Google Scholar]
- 25.Miyamoto A, Shigematsu T, Fukunaga T, Kawakami K, Mukai C, Sekiguchi C. Medical baseline data collection on bone and muscle change with space flight. Bone. 1998;22:79S–82S. doi: 10.1016/s8756-3282(98)00020-9. [DOI] [PubMed] [Google Scholar]
- 26.Lang T, LeBlanc A, Evans H, Lu Y, Genant H, Yu A. Cortical and trabecular bone mineral loss from the spine and hip in long-duration spaceflight. J Bone Miner Res. 2004;19:1006–12. doi: 10.1359/JBMR.040307. [DOI] [PubMed] [Google Scholar]
- 27.Roer RD, Dillaman RM. Bone growth and calcium balance during simulated weightlessness in the rat. J Appl Physiol. 1990;68:13–20. doi: 10.1152/jappl.1990.68.1.13. [DOI] [PubMed] [Google Scholar]
- 28.Simske SJ, Guerra KM, Greenberg AR, Luttges MW. The physical and mechanical effects of suspension-induced osteopenia on mouse long bones. J Biomech. 1992;25:489–99. doi: 10.1016/0021-9290(92)90089-j. [DOI] [PubMed] [Google Scholar]
- 29.Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell. 1982;31:99–109. doi: 10.1016/0092-8674(82)90409-3. [DOI] [PubMed] [Google Scholar]
- 30.Rijsewijk F, Schuermann M, Wagenaar E, Parren P, Weigel D, Nusse R. The Drosophila homolog of the mouse mammary oncogene int-1 is identical to the segment polarity gene wingless. Cell. 1987;50:649–57. doi: 10.1016/0092-8674(87)90038-9. [DOI] [PubMed] [Google Scholar]
- 31.Nusse R, Varmus HE. Wnt genes. Cell. 1992;69:1073–87. doi: 10.1016/0092-8674(92)90630-u. [DOI] [PubMed] [Google Scholar]
- 32.Markowitz SD, Bertagnolli MM. Molecular origins of cancer: Molecular basis of colorectal cancer. N Engl J Med. 2009;361:2449–60. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 34.Joiner DM, Ke J, Zhong Z, Xu HE, Williams BO. LRP5 and LRP6 in development and disease. Trends Endocrinol Metab. 2013;24:31–9. doi: 10.1016/j.tem.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Azzolin L, Zanconato F, Bresolin S, Forcato M, Basso G, Bicciato S, Cordenonsi M, Piccolo S. Role of TAZ as mediator of Wnt signaling. Cell. 2012;151:1443–56. doi: 10.1016/j.cell.2012.11.027. [DOI] [PubMed] [Google Scholar]
- 37.Gong Y, Vikkula M, Boon L, Liu J, Beighton P, Ramesar R, Peltonen L, Somer H, Hirose T, Dallapiccola B, De Paepe A, Swoboda W, Zabel B, Superti-Furga A, Steinmann B, Brunner HG, Jans A, Boles RG, Adkins W, van den Boogaard MJ, Olsen BR, Warman ML. Osteoporosis-pseudoglioma syndrome, a disorder affecting skeletal strength and vision, is assigned to chromosome region 11q12-13. Am J Hum Genet. 1996;59:146–51. [PMC free article] [PubMed] [Google Scholar]
- 38.Beighton P. Osteoporosis-pseudoglioma syndrome. Clin Genet. 1986;29:263. doi: 10.1111/j.1399-0004.1986.tb00823.x. [DOI] [PubMed] [Google Scholar]
- 39.Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D, Zacharin M, Oexle K, Marcelino J, Suwairi W, Heeger S, Sabatakos G, Apte S, Adkins WN, Allgrove J, Arslan-Kirchner M, Batch JA, Beighton P, Black GC, Boles RG, Boon LM, Borrone C, Brunner HG, Carle GF, Dallapiccola B, De Paepe A, Floege B, Halfhide ML, Hall B, Hennekam RC, Hirose T, Jans A, Juppner H, Kim CA, Keppler-Noreuil K, Kohlschuetter A, LaCombe D, Lambert M, Lemyre E, Letteboer T, Peltonen L, Ramesar RS, Romanengo M, Somer H, Steichen-Gersdorf E, Steinmann B, Sullivan B, Superti-Furga A, Swoboda W, van den Boogaard MJ, Van Hul W, Vikkula M, Votruba M, Zabel B, Garcia T, Baron R, Olsen BR, Warman ML. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–23. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 40.Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, Wu D, Insogna K, Lifton RP. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346:1513–21. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 41.Little RD, Carulli JP, Del Mastro RG, Dupuis J, Osborne M, Folz C, Manning SP, Swain PM, Zhao SC, Eustace B, Lappe MM, Spitzer L, Zweier S, Braunschweiger K, Benchekroun Y, Hu X, Adair R, Chee L, FitzGerald MG, Tulig C, Caruso A, Tzellas N, Bawa A, Franklin B, McGuire S, Nogues X, Gong G, Allen KM, Anisowicz A, Morales AJ, Lomedico PT, Recker SM, Van Eerdewegh P, Recker RR, Johnson ML. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet. 2002;70:11–9. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holmen SL, Giambernardi TA, Zylstra CR, Buckner-Berghuis BD, Resau JH, Hess JF, Glatt V, Bouxsein ML, Ai M, Warman ML, Williams BO. Decreased BMD and limb deformities in mice carrying mutations in both Lrp5 and Lrp6. J Bone Miner Res. 2004;19:2033–40. doi: 10.1359/JBMR.040907. [DOI] [PubMed] [Google Scholar]
- 43.Kato M, Patel MS, Levasseur R, Lobov I, Chang BH, Glass DA, 2nd, Hartmann C, Li L, Hwang TH, Brayton CF, Lang RA, Karsenty G, Chan L. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol. 2002;157:303–14. doi: 10.1083/jcb.200201089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iwaniec UT, Wronski TJ, Liu J, Rivera MF, Arzaga RR, Hansen G, Brommage R. PTH stimulates bone formation in mice deficient in Lrp5. J Bone Miner Res. 2007;22:394–402. doi: 10.1359/jbmr.061118. [DOI] [PubMed] [Google Scholar]
- 45.Babij P, Zhao W, Small C, Kharode Y, Yaworsky PJ, Bouxsein ML, Reddy PS, Bodine PV, Robinson JA, Bhat B, Marzolf J, Moran RA, Bex F. High bone mass in mice expressing a mutant LRP5 gene. J Bone Miner Res. 2003;18:960–74. doi: 10.1359/jbmr.2003.18.6.960. [DOI] [PubMed] [Google Scholar]
- 46.Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19:179–92. doi: 10.1038/nm.3074. [DOI] [PubMed] [Google Scholar]
- 47.van Bezooijen RL, Roelen BA, Visser A, van der Wee-Pals L, de Wilt E, Karperien M, Hamersma H, Papapoulos SE, ten Dijke P, Lowik CW. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199:805–14. doi: 10.1084/jem.20031454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Bezooijen RL, ten Dijke P, Papapoulos SE, Lowik CW. SOST/sclerostin, an osteocyte-derived negative regulator of bone formation. Cytokine Growth Factor Rev. 2005;16:319–27. doi: 10.1016/j.cytogfr.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 49.Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, Dioszegi M, Dikkers FG, Hildering P, Willems PJ, Verheij JB, Lindpaintner K, Vickery B, Foernzler D, Van Hul W. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39:91–7. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, Appleby M, Brunkow ME, Latham JA. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22:6267–76. doi: 10.1093/emboj/cdg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Turner CH, Warden SJ, Bellido T, Plotkin LI, Kumar N, Jasiuk I, Danzig J, Robling AG. Mechanobiology of the skeleton. Sci Signal. 2009;2:pt3. doi: 10.1126/scisignal.268pt3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weidauer SE, Schmieder P, Beerbaum M, Schmitz W, Oschkinat H, Mueller TD. NMR structure of the Wnt modulator protein Sclerostin. Biochem Biophys Res Commun. 2009;380:160–5. doi: 10.1016/j.bbrc.2009.01.062. [DOI] [PubMed] [Google Scholar]
- 53.van Bezooijen RL, Bronckers AL, Gortzak RA, Hogendoorn PC, van der Wee-Pals L, Balemans W, Oostenbroek HJ, Van Hul W, Hamersma H, Dikkers FG, Hamdy NA, Papapoulos SE, Lowik CW. Sclerostin in mineralized matrices and van Buchem disease. J Dent Res. 2009;88:569–74. doi: 10.1177/0022034509338340. [DOI] [PubMed] [Google Scholar]
- 54.Chan BY, Fuller ES, Russell AK, Smith SM, Smith MM, Jackson MT, Cake MA, Read RA, Bateman JF, Sambrook PN, Little CB. Increased chondrocyte sclerostin may protect against cartilage degradation in osteoarthritis. Osteoarthritis Cartilage. 2011;19:874–85. doi: 10.1016/j.joca.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 55.Roudier M, Li X, Niu QT, Pacheco E, Pretorius J, Graham K, Yoon BR, Gong J, Warmington K, Ke HZ, Black RA, Hulme J, Babij P. Sclerostin is expressed in articular cartilage but loss or inhibition does not affect cartilage remodeling during aging or following mechanical injury. Arthritis Rheum. 2012 doi: 10.1002/art.37802. in press. [DOI] [PubMed] [Google Scholar]
- 56.Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, Wu D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883–7. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 57.Semenov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005;280:26770–5. doi: 10.1074/jbc.M504308200. [DOI] [PubMed] [Google Scholar]
- 58.Veverka V, Henry AJ, Slocombe PM, Ventom A, Mulloy B, Muskett FW, Muzylak M, Greenslade K, Moore A, Zhang L, Gong J, Qian X, Paszty C, Taylor RJ, Robinson MK, Carr MD. Characterization of the structural features and interactions of sclerostin: molecular insight into a key regulator of Wnt-mediated bone formation. J Biol Chem. 2009;284:10890–900. doi: 10.1074/jbc.M807994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Semenov MV, He X. LRP5 mutations linked to high bone mass diseases cause reduced LRP5 binding and inhibition by SOST. J Biol Chem. 2006;281:38276–84. doi: 10.1074/jbc.M609509200. [DOI] [PubMed] [Google Scholar]
- 60.Ellies DL, Viviano B, McCarthy J, Rey JP, Itasaki N, Saunders S, Krumlauf R. Bone density ligand, Sclerostin, directly interacts with LRP5 but not LRP5G171V to modulate Wnt activity. J Bone Miner Res. 2006;21:1738–49. doi: 10.1359/jbmr.060810. [DOI] [PubMed] [Google Scholar]
- 61.Zhang Y, Wang Y, Li X, Zhang J, Mao J, Li Z, Zheng J, Li L, Harris S, Wu D. The LRP5 high-bone-mass G171V mutation disrupts LRP5 interaction with Mesd. Mol Cell Biol. 2004;24:4677–84. doi: 10.1128/MCB.24.11.4677-4684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li J, Sarosi I, Cattley RC, Pretorius J, Asuncion F, Grisanti M, Morony S, Adamu S, Geng Z, Qiu W, Kostenuik P, Lacey DL, Simonet WS, Bolon B, Qian X, Shalhoub V, Ominsky MS, Zhu Ke H, Li X, Richards WG. Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia. Bone. 2006;39:754–66. doi: 10.1016/j.bone.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 63.Ai M, Holmen SL, Van Hul W, Williams BO, Warman ML. Reduced affinity to and inhibition by DKK1 form a common mechanism by which high bone mass-associated missense mutations in LRP5 affect canonical Wnt signaling. Mol Cell Biol. 2005;25:4946–55. doi: 10.1128/MCB.25.12.4946-4955.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Balemans W, Piters E, Cleiren E, Ai M, Van Wesenbeeck L, Warman ML, Van Hul W. The binding between sclerostin and LRP5 is altered by DKK1 and by high-bone mass LRP5 mutations. Calcif Tissue Int. 2008;82:445–53. doi: 10.1007/s00223-008-9130-9. [DOI] [PubMed] [Google Scholar]
- 65.Cui Y, Niziolek PJ, MacDonald BT, Zylstra CR, Alenina N, Robinson DR, Zhong Z, Matthes S, Jacobsen CM, Conlon RA, Brommage R, Liu Q, Mseeh F, Powell DR, Yang QM, Zambrowicz B, Gerrits H, Gossen JA, He X, Bader M, Williams BO, Warman ML, Robling AG. Lrp5 functions in bone to regulate bone mass. Nat Med. 2011;17:684–91. doi: 10.1038/nm.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Joeng KS, Schumacher CA, Zylstra-Diegel CR, Long F, Williams BO. Lrp5 and Lrp6 redundantly control skeletal development in the mouse embryo. Dev Biol. 2011;359:222–9. doi: 10.1016/j.ydbio.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao L, Shim JW, Dodge TR, Robling AG, Yokota H. Inactivation of Lrp5 in osteocytes reduces Young’s modulus and responsiveness to the mechanical loading. Bone. 2013 doi: 10.1016/j.bone.2013.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yadav VK, Ryu JH, Suda N, Tanaka KF, Gingrich JA, Schutz G, Glorieux FH, Chiang CY, Zajac JD, Insogna KL, Mann JJ, Hen R, Ducy P, Karsenty G. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell. 2008;135:825–37. doi: 10.1016/j.cell.2008.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang W, Drake MT. Potential role for therapies targeting DKK1, LRP5, and serotonin in the treatment of osteoporosis. Curr Osteoporos Rep. 2012;10:93–100. doi: 10.1007/s11914-011-0086-8. [DOI] [PubMed] [Google Scholar]
- 70.Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Lowik CW, Reeve J. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19:1842–4. doi: 10.1096/fj.05-4221fje. [DOI] [PubMed] [Google Scholar]
- 71.Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, Kurahara C, Gao Y, Cao J, Gong J, Asuncion F, Barrero M, Warmington K, Dwyer D, Stolina M, Morony S, Sarosi I, Kostenuik PJ, Lacey DL, Simonet WS, Ke HZ, Paszty C. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23:860–9. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- 72.Lin C, Jiang X, Dai Z, Guo X, Weng T, Wang J, Li Y, Feng G, Gao X, He L. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J Bone Miner Res. 2009;24:1651–61. doi: 10.1359/jbmr.090411. [DOI] [PubMed] [Google Scholar]
- 73.Marie PJ, Kassem M. Osteoblasts in osteoporosis: past, emerging, and future anabolic targets. Eur J Endocrinol. 2011;165:1–10. doi: 10.1530/EJE-11-0132. [DOI] [PubMed] [Google Scholar]
- 74.Cusano NE, Bilezikian JP. Combination anabolic and antiresorptive therapy for osteoporosis. Endocrinol Metab Clin North Am. 2012;41:643–54. doi: 10.1016/j.ecl.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 75.Saag KG, Shane E, Boonen S, Marin F, Donley DW, Taylor KA, Dalsky GP, Marcus R. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med. 2007;357:2028–39. doi: 10.1056/NEJMoa071408. [DOI] [PubMed] [Google Scholar]
- 76.Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, O’Brien CA, Manolagas SC, Jilka RL. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology. 2005;146:4577–83. doi: 10.1210/en.2005-0239. [DOI] [PubMed] [Google Scholar]
- 77.Keller H, Kneissel M. SOST is a target gene for PTH in bone. Bone. 2005;37:148–58. doi: 10.1016/j.bone.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 78.O’Brien CA, Plotkin LI, Galli C, Goellner JJ, Gortazar AR, Allen MR, Robling AG, Bouxsein M, Schipani E, Turner CH, Jilka RL, Weinstein RS, Manolagas SC, Bellido T. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS One. 2008;3:e2942. doi: 10.1371/journal.pone.0002942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li X, Ominsky MS, Warmington KS, Morony S, Gong J, Cao J, Gao Y, Shalhoub V, Tipton B, Haldankar R, Chen Q, Winters A, Boone T, Geng Z, Niu QT, Ke HZ, Kostenuik PJ, Simonet WS, Lacey DL, Paszty C. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res. 2009;24:578–88. doi: 10.1359/jbmr.081206. [DOI] [PubMed] [Google Scholar]
- 80.Ominsky MS, Vlasseros F, Jolette J, Smith SY, Stouch B, Doellgast G, Gong J, Gao Y, Cao J, Graham K, Tipton B, Cai J, Deshpande R, Zhou L, Hale MD, Lightwood DJ, Henry AJ, Popplewell AG, Moore AR, Robinson MK, Lacey DL, Simonet WS, Paszty C. Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J Bone Miner Res. 2010;25:948–59. doi: 10.1002/jbmr.14. [DOI] [PubMed] [Google Scholar]
- 81.Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26:19–26. doi: 10.1002/jbmr.173. [DOI] [PubMed] [Google Scholar]
- 82.Reid IR. Osteoporosis treatment at ASBMR 2012. IBMS BoneKEy. 2012;9 [Google Scholar]
- 83.Heiland GR, Zwerina K, Baum W, Kireva T, Distler JH, Grisanti M, Asuncion F, Li X, Ominsky M, Richards W, Schett G, Zwerina J. Neutralisation of Dkk-1 protects from systemic bone loss during inflammation and reduces sclerostin expression. Ann Rheum Dis. 2010;69:2152–9. doi: 10.1136/ard.2010.132852. [DOI] [PubMed] [Google Scholar]
- 84.Morvan F, Boulukos K, Clement-Lacroix P, Roman Roman S, Suc-Royer I, Vayssiere B, Ammann P, Martin P, Pinho S, Pognonec P, Mollat P, Niehrs C, Baron R, Rawadi G. Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res. 2006;21:934–45. doi: 10.1359/jbmr.060311. [DOI] [PubMed] [Google Scholar]
- 85.Jones SE, Jomary C. Secreted Frizzled-related proteins: searching for relationships and patterns. Bioessays. 2002;24:811–20. doi: 10.1002/bies.10136. [DOI] [PubMed] [Google Scholar]
- 86.Bafico A, Gazit A, Pramila T, Finch PW, Yaniv A, Aaronson SA. Interaction of frizzled related protein (FRP) with Wnt ligands and the frizzled receptor suggests alternative mechanisms for FRP inhibition of Wnt signaling. J Biol Chem. 1999;274:16180–7. doi: 10.1074/jbc.274.23.16180. [DOI] [PubMed] [Google Scholar]
- 87.Bodine PV, Zhao W, Kharode YP, Bex FJ, Lambert AJ, Goad MB, Gaur T, Stein GS, Lian JB, Komm BS. The Wnt antagonist secreted frizzled-related protein-1 is a negative regulator of trabecular bone formation in adult mice. Mol Endocrinol. 2004;18:1222–37. doi: 10.1210/me.2003-0498. [DOI] [PubMed] [Google Scholar]
- 88.Bodine PV, Billiard J, Moran RA, Ponce-de-Leon H, McLarney S, Mangine A, Scrimo MJ, Bhat RA, Stauffer B, Green J, Stein GS, Lian JB, Komm BS. The Wnt antagonist secreted frizzled-related protein-1 controls osteoblast and osteocyte apoptosis. J Cell Biochem. 2005;96:1212–30. doi: 10.1002/jcb.20599. [DOI] [PubMed] [Google Scholar]
- 89.Klein-Nulend J, Bakker AD, Bacabac RG, Vatsa A, Weinbaum S. Mechanosensation and transduction in osteocytes. Bone. 2012 doi: 10.1016/j.bone.2012.10.013. in press, this issue. [DOI] [PubMed] [Google Scholar]
- 90.Nguyen AM, Jacobs CR. Emerging role of primary cilia as mechanosensors in osteocytes. Bone. 2012 doi: 10.1016/j.bone.2012.11.016. in press, this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Letechipia JE, Alessi A, Rodriguez G, Asbun J. Would increased interstitial fluid flow through in situ mechanical stimulation enhance bone remodeling? Med Hypotheses. 2010;75:196–8. doi: 10.1016/j.mehy.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 92.Cheng B, Kato Y, Zhao S, Luo J, Sprague E, Bonewald LF, Jiang JX. PGE(2) is essential for gap junction-mediated intercellular communication between osteocyte-like MLO-Y4 cells in response to mechanical strain. Endocrinology. 2001;142:3464–73. doi: 10.1210/endo.142.8.8338. [DOI] [PubMed] [Google Scholar]
- 93.Cherian PP, Cheng B, Gu S, Sprague E, Bonewald LF, Jiang JX. Effects of mechanical strain on the function of Gap junctions in osteocytes are mediated through the prostaglandin EP2 receptor. J Biol Chem. 2003;278:43146–56. doi: 10.1074/jbc.M302993200. [DOI] [PubMed] [Google Scholar]
- 94.Kitase Y, Barragan L, Qing H, Kondoh S, Jiang JX, Johnson ML, Bonewald LF. Mechanical induction of PGE2 in osteocytes blocks glucocorticoid-induced apoptosis through both the beta-catenin and PKA pathways. J Bone Miner Res. 2010;25:2657–68. doi: 10.1002/jbmr.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kamel MA, Picconi JL, Lara-Castillo N, Johnson ML. Activation of beta-catenin signaling in MLO-Y4 osteocytic cells versus 2T3 osteoblastic cells by fluid flow shear stress and PGE2: Implications for the study of mechanosensation in bone. Bone. 2010;47:872–81. doi: 10.1016/j.bone.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310:1504–10. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- 97.Xia X, Batra N, Shi Q, Bonewald LF, Sprague E, Jiang JX. Prostaglandin promotion of osteocyte gap junction function through transcriptional regulation of connexin 43 by glycogen synthase kinase 3/beta-catenin signaling. Mol Cell Biol. 2010;30:206–19. doi: 10.1128/MCB.01844-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Glass DA, 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751–64. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 99.Holmen SL, Zylstra CR, Mukherjee A, Sigler RE, Faugere MC, Bouxsein ML, Deng L, Clemens TL, Williams BO. Essential role of beta-catenin in postnatal bone acquisition. J Biol Chem. 2005;280:21162–8. doi: 10.1074/jbc.M501900200. [DOI] [PubMed] [Google Scholar]
- 100.Kramer I, Halleux C, Keller H, Pegurri M, Gooi JH, Weber PB, Feng JQ, Bonewald LF, Kneissel M. Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol Cell Biol. 2010;30:3071–85. doi: 10.1128/MCB.01428-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Johnson ML, Picconi JL, Recker RR. The gene for high bone mass. The Endocrinologist. 2002;12:445–453. [Google Scholar]
- 102.Sawakami K, Robling AG, Ai M, Pitner ND, Liu D, Warden SJ, Li J, Maye P, Rowe DW, Duncan RL, Warman ML, Turner CH. The Wnt co-receptor LRP5 is essential for skeletal mechanotransduction but not for the anabolic bone response to parathyroid hormone treatment. J Biol Chem. 2006;281:23698–711. doi: 10.1074/jbc.M601000200. [DOI] [PubMed] [Google Scholar]
- 103.Niziolek PJ, Warman ML, Robling AG. Mechanotransduction in bone tissue: The A214V and G171V mutations in Lrp5 enhance load-induced osteogenesis in a surface-selective manner. Bone. 2012;51:459–65. doi: 10.1016/j.bone.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhao L, Shim JW, Dodge TR, Robling AG, Yokota H. Inactivation of Lrp5 in osteocytes reduces Young’s Modulus and responsiveness to the mechanical loading. Bone. 2013 doi: 10.1016/j.bone.2013.01.033. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Moustafa A, Sugiyama T, Saxon LK, Zaman G, Sunters A, Armstrong VJ, Javaheri B, Lanyon LE, Price JS. The mouse fibula as a suitable bone for the study of functional adaptation to mechanical loading. Bone. 2009;44:930–5. doi: 10.1016/j.bone.2008.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Macias BR, Aspenberg P, Agholme F. Paradoxical Sost gene expression response to mechanical unloading in metaphyseal bone. Bone. 2013;53:515–9. doi: 10.1016/j.bone.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 107.Tu X, Rhee Y, Condon KW, Bivi N, Allen MR, Dwyer D, Stolina M, Turner CH, Robling AG, Plotkin LI, Bellido T. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone. 2012;50:209–17. doi: 10.1016/j.bone.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Robinson JA, Chatterjee-Kishore M, Yaworsky PJ, Cullen DM, Zhao W, Li C, Kharode Y, Sauter L, Babij P, Brown EL, Hill AA, Akhter MP, Johnson ML, Recker RR, Komm BS, Bex FJ. Wnt/beta-catenin signaling is a normal physiological response to mechanical loading in bone. J Biol Chem. 2006;281:31720–8. doi: 10.1074/jbc.M602308200. [DOI] [PubMed] [Google Scholar]