

Abstract
The field of stem-cell biology has been catapulted forward by the startling development of reprogramming technology. The ability to restore pluripotency to somatic cells through the ectopic co-expression of reprogramming factors has created powerful new opportunities for modelling human diseases and offers hope for personalized regenerative cell therapies. While the field is racing ahead, some researchers are pausing to evaluate whether induced pluripotent stem cells are indeed the true equivalents of embryonic stem cells and whether subtle differences between these cells might affect their research applications and therapeutic potential.
After a decade of constraints, pluripotent stem-cell biology is now a flourishing research area, following the achievement of a long-standing ambition — the successful derivation of pluripotent stem cells from a patient’s cells. In a momentous contribution, in 2006 Takahashi and Yamanaka illustrated how cell fates can be altered by the ectopic co-expression of transcription factors1. The manipulation of cell fates through reprogramming has altered fundamental ideas about the stability of cellular identity, stimulating major new directions in research into human disease modelling, tissue differentiation in vitro and cellular transdifferentiation. Despite heady progress, a major question remains: are the new induced pluripotent stem (iPS) cells equivalent to the classic embryonic stem (ES) cells and thus a suitable alternative for research and therapy? Whereas the initial wave of papers argued convincingly that the two cell types were functionally equivalent, a more refined analysis of how iPS cells behave in vitro, coupled with genome-wide genetic and epigenetic analysis, has revealed numerous subtle but substantial molecular differences, probably owing to technical limitations inherent in reprogramming. In this Review, we describe the derivation of iPS cells, outline the functional assessments of pluripotency, and then recount how global assessments of gene expression, gene copy number variation, DNA methylation and chromatin modification provide a more nuanced comparison of iPS cells and ES cells. Ultimately, we detail how these features influence the utility of each of these cell types for disease modelling and therapeutics, and we offer predictions for the evolution of the art of reprogramming somatic cells.
Pluripotent stem cells
The years since Takahashi and Yamanaka’s breakthrough have seen a flood of papers touting advances in reprogramming technology, including alternative methods for reprogramming and the successful derivation of iPS cells from various cell types. Although the field has advanced at a breathtaking pace, investigators have recently taken a step back to more critically evaluate iPS cells relative to ES cells and have endeavoured to fully understand how these cell populations differ from one another in the hope of closing the gap between the two populations. Taking clues from the data, it seems that researchers should attempt to define each cell type more accurately and to understand its inherent properties rather than ask whether these two classes of pluripotent cell are identical. Although ES cells and iPS cells are arguably equivalent in all of their functions, these cells are bound to harbour subtle differences and to have distinct but complementary roles in research because of their distinct origins and modes of derivation. To appreciate the differences between ES cells and iPS cells, we must first define what it means to be pluripotent.
The term pluripotency has been assigned to a variety of cell types with a wide range of functional capacities. In its loosest sense, pluripotent describes a cell that can generate cell types from each of the three embryonic germ layers: the endoderm, mesoderm and ectoderm. At the strict end of the range of definitions, however, pluripotent describes a cell that can give rise to an entire organism, generating every cell type within that organism. The property of cell pluripotency was first exposed by Driesch in 1891, when he separated the two cells of the early sea urchin blastocyst and observed the development of two complete sea urchins2. Many decades later, studies of embryo aggregation and blastocyst chimaerism by Mintz and colleagues3, Gardner4 and Brinster5, in the 1960s and 1970s, solidified the idea that the cells of the inner cell mass of the mouse blastocyst were pluripotent, and the isolation of mouse teratocarcinoma stem cells and native ES cells by Evans and Kaufman6 and Martin7, in 1981, ushered in the era of culturing pluripotent stem cells in a dish. The first successful isolation of human ES cells by Thomson and colleagues, in 1998, brought forth a surge of excitement in the scientific community and beyond8. The potential to understand early human development, tissue formation and differentiation in vitro through studying ES cells seemed to offer limitless possibilities. The opportunity to model diseases, discover disease mechanisms and, ultimately, use cell therapy for previously untreatable conditions was particularly alluring.
The derivation of ES cells from the human embryo, however, sparked controversy in the United States and led to a presidential executive order that restricted government funding9. The limited numbers of stem cell lines that were approved for research lacked the diversity necessary to address some of the most compelling questions, particularly those related to modelling and treating disease10. Most ES cells represented generic cells isolated from presumably normal embryos — except for those from embryos that had been tested by pre-implantation diagnostics and found to carry genetic diseases. The generic lines were not matched to a particular patient, so products derived from them for transplantation purposes would face rejection by the transplant recipient’s immune system or necessitate that the recipient receive lifelong therapy with toxic immunosuppressive medication. To compound these limitations, when human ES cells are cultivated on mouse feeder cells, the human cells can incorporate mouse components that render the ES cells subject to immune rejection.
To realize the full potential of ES cells, researchers foresaw that customized, personalized pluripotent stem cells specific to each patient would be generated by using somatic-cell nuclear transfer (SCNT) — the procedure that had been used successfully to clone Dolly the sheep from adult mammary cells. Nuclear-transfer-generated ES (ntES) cell lines would capture a patient’s complete genome in a cell that could be induced to become any tissue, thus allowing differentiation into disease-relevant cells for analysis or cell-replacement therapy. Despite successful proof of principle in mouse studies11, and the clear distinctions between generating ntES cells for medical research and creating cloned blastocysts for reproduction, the ethical controversy driven by widespread opposition to human cloning has severely curtailed research into human SCNT. Only this year, when investigators gained access to a large number of human oocytes, was the derivation of pluripotent stem cells through human SCNT accomplished12. However, the investigators in this study needed to leave the oocyte nucleus intact to derive pluripotent stem cells, so the resultant cells were triploid, thus affording research applications for these cells but limiting their suitability for therapeutic use12.
Despite the many hindrances to the study and derivation of human ES and ntES cells over the past decade, great strides were being made in understanding the pathways that regulate the maintenance and pluripotency of ES cells. This progress was not lost on those seeking an alternative source of personalized patient-specific stem cells, and in 2006 Takahashi and Yamanaka announced the successful derivation of iPS cells from adult mouse fibroblasts through the ectopic co-expression of only four genes1. In an elegant screen of 24 gene candidates selected for their links to ES-cell pluripotency, these researchers found four factors that were sufficient to reprogram adult fibroblasts into iPS cells: OCT4 (also known as POU5F1), SOX2, Krüppel-like factor 4 (KLF4) and c-MYC. This historic contribution inspired an astonishing flurry of follow-on studies, with successful reprogramming quickly translated to human fibroblasts13–15 and then to a wide variety of other cell types, including pancreatic β cells16, neural stem cells17,18, mature β cells19, stomach and liver cells20, melanocytes21, adipose stem cells22 and keratinocytes23, demonstrating the seemingly universal capacity to alter cellular identity.
Mouse and human iPS cells differ in appearance, however, with mouse iPS-cell colonies appearing more dome-like and refractile than human iPS-cell colonies. Human iPS-cell colonies are flatter than those of mice and are akin to a distinct type of pluripotent stem cell that is derived from the epiblast of the early mouse embryo24, a feature that indicates that mouse and human iPS cells, like mouse and human ES cells, probably reflect distinct developmental states (Fig. 1). The pluripotent state of mouse stem cells is called a ‘naive’ state because it closely resembles the most primitive state, or ground state, of the mouse inner cell mass; this is different from the more ‘primed’ state of human stem cells, which proliferate in response to different cytokines, reflecting the distinct developmental statuses of these populations25. Regardless of the method of derivation, iPS cells maintain the key features of ES cells, including the ability to propagate in culture indefinitely and the capacity to generate cells from each of the three embryonic germ layers (see ref. 26 for a review). Such broad similarities are not proof that iPS cells are molecularly or functionally equivalent to ES cells. Yamanaka’s intention was to derive an alternative source of pluripotent stem cells with the same range of functions as ES cells but offering even greater potential for clinical use. To determine the degree of success garnered by reprogramming, we must explore the set of assays that were developed to assess the key characteristic of ES cells: pluripotency.
Figure 1. Morphology of pluripotent stem cell types.
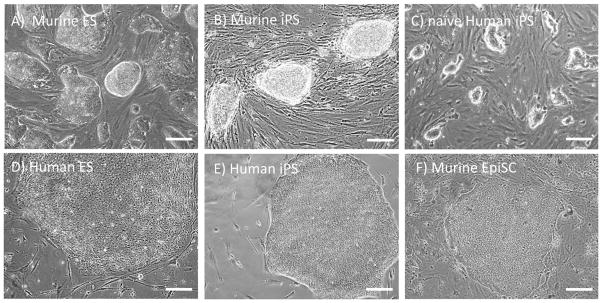
Mouse ES (a) and iPS (b) cells form dome-shaped, refractile colonies. These colonies are in contrast to the flat morphology of mouse epiblast-derived stem cells (f), which resemble human ES (d) and iPS (e) cells. Human iPS cells induced into a naive pluripotent state by treatment with chemical inhibitors82–85 (c) parallel the morphology of mouse ES and iPS cells. Scale bars, 50 μm.
Assessment of pluripotency
In the past few years, consistent standards for the identification and evaluation of iPS cells and for the assessment of their functional equivalence to ES cells have become widely accepted27. A variety of reprogramming methods have been developed to derive iPS cells, and each has advantages and disadvantages (Table 1). Assessing reprogramming begins with identifying compact colonies that have distinct borders and well-defined edges and comprise cells with a large nucleus, large nucleoli and scant cytoplasm. A wide range of colony morphologies result from reprogramming, and although many colonies appear morphologically similar to ES cells, only some colonies have comparable molecular and functional features. To accurately distinguish reprogrammed, bona fide iPS cells from those that are only partially reprogrammed, investigators look for a series of molecular hallmarks. Fully reprogrammed cells express a network of pluripotency genes, including OCT4, SOX2 and NANOG, in levels comparable to ES cells, and they reactivate telomerase gene expression, downregulate THY1 and upregulate SSEA1 (ref. 28).
Table 1.
Methods for reprogramming somatic cells to iPS cells
| Vector type | Cell type | Factors* | Efficiency (%) | Advantages | Disadvantages | |
|---|---|---|---|---|---|---|
| Integrating | Retroviral1,14,86,87 | Fibroblasts, neural stem cells, stomach cells, liver cells, keratinocytes, amniotic cells, blood cells and adipose cells | OSKM, OSK, OSK + VPA, or OS + VPA | ~0.001–1 | Reasonably efficient | Genomic integration, incomplete proviral silencing and slow kinetics |
| Lentiviral15,16,88,89 | Fibroblasts and keratinocytes | OSKM or miR302/367 cluster + VPA | ~0.1–1.1 | Reasonably efficient and transduces dividing and non-dividing cells | Genomic integration and incomplete proviral silencing | |
| Inducible lentiviral23,28 | Fibroblasts, β cells, keratinocytes, blood cells and melanocytes | OSKM or OSKMN | ~0.1–2 | Reasonably efficient and allows controlled expression of factors | Genomic integration and requirement for transactivator expression | |
| Excisable | Transposon90 | Fibroblasts | OSKM | ~0.1 | Reasonably efficient and no genomic integration | Labour-intensive screening of excised lines |
| loxP-flanked lentiviral91 | Fibroblasts | OSK | ~0.1–1 | Reasonably efficient and no genomic integration | Labour-intensive screening of excised lines, and loxP sites retained in the genome | |
| Non-integrating | Adenoviral92,93 | Fibroblasts and liver cells | OSKM | ~0.001 | No genomic integration | Low efficiency |
| Plasmid94,95 | Fibroblasts | OSNL | ~0.001 | Only occasional genomic integration | Low efficiency and occasional vector genomic integration | |
| DNA free | Sendai virus96 | Fibroblasts | OSKM | ~1 | No genomic integration | Sequence-sensitive RNA replicase, and difficulty in purging cells of replicating virus |
| Protein97,98 | Fibroblasts | OS | ~0.001 | No genomic integration, direct delivery of transcription factors and no DNA-related complications | Low efficiency, short half-life, and requirement for large quantities of pure proteins and multiple applications of protein | |
| Modified mRNA99 | Fibroblasts | OSKM or OSKML + VPA | ~1–4.4 | No genomic integration, bypasses innate antiviral response, faster reprogramming kinetics, controllable and high efficiency | Requirement for multiple rounds of transfection | |
| MicroRNA100 | Adipose stromal cells and dermal fibroblasts | miR-200c, miR-302s or miR-369s | ~0.1 | Efficient, faster reprogramming kinetics than commonly used lentiviral or retroviral vectors, no exogenous transcription factors and no risk of integration | Lower efficiency than other commonly used methods |
OSKM and similar factor names represent combinations of reprogramming factors: K, KLF4; L, LIN28; M, c-MYC; N, NANOG; O, OCT4; S, SOX2; and VPA, valproic acid.
Markers of pluripotency
Positive staining for alkaline phosphatase activity has been widely used as a marker of pluripotency; however, recently published data have shown this enzyme to be insufficient as a marker of true iPS cells, because intermediately reprogrammed cells also stain positively29. The same report shows that iPS cells that are generated by virus-mediated reprogramming silence proviral genes when the endogenous pluripotency genes are activated and that this event is paired with the expression of the embryonic antigens SSEA3, TRA-1-60, TRA-1-81, DNA methyltransferase 3β (DNMT3β) and REX1 (ref. 29). Genome-wide epigenetic reprogramming is crucial for deriving fully reprogrammed cells, and the degree of success is measured, in part, by evaluating the methylation status at the promoters of the genes responsible for maintaining pluripotency, as well as at the genes important for driving differentiation30. A crucial event during epigenetic reprogramming is the reactivation of the silent X chromosome, which occurs late in reprogramming and represents a hallmark of ground-state pluripotency28,30,31. If iPS cells acquire all of these molecular features, they are expected to behave like ES cells and to demonstrate reprogramming-factor independence, which is marked by silencing of the proviral transgenes. Variations in epigenetic reprogramming, the extent of methylation, the persistence of expression of integrated proviruses and other known and unknown factors can alter the differentiation potential of iPS cells. Because of the potential for heterogeneity, it is essential to know as much as possible about the nature of a cell line before labelling it pluripotent.
Functional assays of pluripotency
When iPS cell lines are isolated and documented to carry the molecular features of fully reprogrammed cells, they are typically also assessed in functional assays. Characterization of the functional abilities of iPS cells begins with in vitro differentiation. The cells can be differentiated as embryoid bodies — compact balls of loosely organized tissues that resemble the gastrulating embryo — or through two-dimensional directed differentiation in a culture dish. Such cultures can then be assessed for markers of each of the three germ layers. Analysis of the pluripotency of mouse cells typically entails the development of a chimaera, which evaluates the potential of iPS cells to contribute to the normal development of adult tissues after injection into the blastocyst. Whether germline transmission occurs after blastocyst chimaerism is measured by the ability of chimaeras to produce all-iPS-cell mice in their offspring. These offspring have the genomic integrity of the injected iPS cell line, as well as the ability to form functional germ cells. The highest stringency test for mouse iPS cells — tetraploid complementation — entails the injection of iPS cells into tetraploid blastocysts to measure the ability of the iPS cells to direct the normal development of an entire organism. This test has been accomplished for only a limited subset of iPS cells32–34, although the successful cells had an efficiency that paralleled tetraploid complementation carried out with ES or ntES cells33,35,36.
The current functional gold standard for human iPS cells involves the evaluation of teratoma formation. In this assay, the in vivo differentiation potential of human iPS cells is measured after their injection subcutaneously or intramuscularly into immunodeficient mice37,38. If the cells are truly pluripotent, they will form well-differentiated tumours comprising elements from each of the three germ layers. This assay provides information about the spontaneous differentiation potential of the injected iPS cells. Although it is the most stringent assay available for human iPS cells, it is not powerful enough to assess whether iPS cells can produce all of the cell types of the human body, and it also cannot assess the contribution of iPS cells to the germ line. All of these functional assays have a caveat: the standards for iPS cells are still hotly debated, especially when anticipating the use of iPS cells for therapy39. Adopting a consistent set of standards that can be applied uniformly worldwide is essential as stem-cell research and applications move forward.
Functional differences between iPS cells and ES cells
Despite the multitude of assays used to evaluate pluripotency, and although many parallels have been found between iPS cells and ES cells, there is a wide range of evidence showing that there are subtle yet substantial differences between these cell types. Disparities were first observed in the differentiation abilities of iPS cells and ES cells in both teratoma-forming and in vitro differentiation assays. Some mouse iPS cells showed lower efficiencies of teratoma formation than ES cells, whereas some human iPS cells showed less propensity to differentiate along haematopoietic, neuroepithelial and neuronal lineages than human ES cells40–41,42. Some researchers interpreted these findings to mean that iPS cells have an intrinsically lower differentiation capacity than ES cells41, whereas other research groups have offered different explanations, including that the cell of origin might have a specific effect on the differentiation capacities of the derived iPS cells.
The results from cell-of-origin studies indicated that the parental cell can influence the differentiation capacity of the resultant iPS cells. In one study, mouse bone-marrow-derived and B-cell-derived iPS cells showed more efficient differentiation along haematopoietic lineages than did fibroblast-derived iPS cells or neural-progenitor-derived iPS cell lines. Interestingly, treatment of the neural-progenitor-derived iPS cells with trichostatin A, a potent histone-deacetylase inhibitor, plus 5-azacytidine, a methylation-resistant cytosine analogue, increased the blood-forming capacity of these cells, suggesting that their limitations were due to epigenetic modifications. Whereas the bone-marrow-derived and neural-progenitor-derived iPS cells contributed well to all tissues in the chimaera assay, including to the germ line, the fibroblast-derived iPS cells contributed only poorly43. This study laid some of the early groundwork for later lines of investigation that probed the molecular differences between iPS cells and ES cells and provided an explanation for the functional differences between these cells.
One investigation found that some iPS cells derived from human retinal-pigment epithelial cells show an increased propensity to differentiate back into these cells than do ES cells or iPS cells derived from other tissues44. More recently, Bar-Nur and colleagues showed that iPS cells generated from human pancreatic islet β cells retain open chromatin at the loci of key β-cell genes and that this correlates with a greater capacity to differentiate into insulin-producing cells both in vivo and in vitro than that of ES cells or isogenic non-β-cell-derived iPS cells45. These functional differences extend beyond differentiation and potency to disease modelling. For example, fragile X syndrome is caused by aberrant silencing of the FMR1 gene during human development; iPS cells that were reprogrammed from adult skin fibroblasts from an individual with fragile X syndrome failed to reactivate the FMR1 gene, whereas ES cells derived from embryos with this syndrome, as diagnosed by pre-implantation testing, expressed FMR1 (ref. 46). Consequently, the potential for epigenetic memory in the fragile-X-syndrome-derived iPS cells, and substantial differences between fragile-X-syndrome-derived iPS and ES cells, must be considered when studying this condition and potentially many other conditions. To determine whether the pluripotent cells being used are appropriate to address a particular question or to use in a given application, it is crucial to compare not only the in vivo and in vitro differentiation potentials but also the genetic and epigenetic disparities that underpin these functional differences.
iPS cells versus ES cells
Refined analyses, described in this section, have addressed whether iPS cells are suitable alternatives to classic ES cells for use in research and therapy.
Genetics and epigenetics
Global gene-expression analysis and bisulphite genomic sequencing accompanying early derivations of iPS cells provided the initial evidence for differences between iPS cells and ES cells at the epigenetic level14. Further exploration of these differences led to the identification of only a few, seemingly consistent, differences in global gene expression that were more pronounced in earlier passages of iPS cells47. Many of the differentially expressed genes were imprinted in ES cells48.
Looking beyond the expression patterns to the DNA sequence itself has revealed genetic variation between iPS cells and ES cells. A recent publication suggested that chromosomal aberrations are a common feature of stem-cell populations that are propagated in vitro, with each type of stem cell — whether ES cells, iPS cells or multipotent stem cells — being prone to distinct abnormalities49. Both human iPS and ES cells showed a tendency for gains at chromosomes 12 and 17. Whereas iPS cells had additional gains at chromosomes 1 and 9, ES cells had additional gains at chromosomes 3 and 20. Other work identified an accumulation of point mutations in reprogrammed cells, particularly occurring in oncogenic pathways50, whereas another study noted an increase in copy number variants (CNVs) in early-passage human iPS cells relative to intermediate-passage iPS cells or ES cells. The number and size of these CNVs were negatively correlated with the passage number in iPS cells, suggesting that a selective disadvantage is conferred by these aberrations. A comparison of iPS cells and their parental cell of origin showed that the majority of CNVs were created de novo in fragile regions of the genome50. A comprehensive study by Laurent and colleagues found a higher frequency of subchromosomal CNVs in pluripotent cell samples than in non-pluripotent cell samples51. This work uncovered variation between genomic regions enriched for CNVs in human ES cells and iPS cells. A small subset of samples of ES cells harboured a large number of duplications, whereas several iPS-cell samples contained moderate numbers of deletions. Reprogramming was associated with deletions in tumour-suppressor genes, whereas extended time in culture led to duplications of oncogenes in human iPS cells.
The finding that human iPS cells derived from a variety of tissues have residual, persistent donor-cell-specific gene-expression patterns sparks the question of whether the current measure of a fully reprogrammed cell is sufficient52 or whether iPS cells retain some type of ‘somatic memory’ from their past identity. To understand this observation better, a more detailed analysis at the epigenetic level is required.
Reprogramming cells to a pluripotent state entails global epigenetic remodelling and introduces epigenetic changes, some of which are necessary for reprogramming to occur and others of which are inadvertently introduced during the process. A failure to demethylate pluripotency genes is associated with partial reprogramming in iPS cells. Whole genome profiling of the DNA methylomes of multiple human iPS and ES cell lines, as well as somatic and progenitor cell lines, from different laboratories using different reprogramming techniques and with a variety of cells derived from distinct germ layers has shown that — although overall iPS-cell DNA methylomes closely resemble human ES-cell DNA methylomes — iPS cells have significant variability in their somatic memory, as well as aberrant iPS-cell-specific differential methylation. Some studies have suggested that this occurs in a passage-dependent manner, but others have shown that differentially methylated regions (DMRs) in iPS cells are transmitted to differentiated progeny at a high frequency and cannot be erased through passaging29,53–55. Overall, there are remarkable global similarities between the DNA methylomes of generic iPS and ES cells; however, a core set of DMRs that seems to represent hotspots of failed epigenetic reprogramming has been identified55. These DMRs are enriched for genes that are important for developmental processes58,55. The high incidence of unique DMRs in iPS cells compared with progenitor somatic cells or ES cells suggests that these patterns are stochastic and arise during reprogramming. In the most exhaustive comparison so far, Kim and colleagues reported that more DMRs were present in mouse iPS cells than in ntES or embryo-derived ES cells43. However, these DMRs did not pertain to specific loci and thus do not represent consistent differences between iPS cells and ES cells. This lack of consistency suggests that aberrant DMRs in mouse iPS cells reflect the technical limitations inherent in reprogramming, rather than indicating loci that can reliably distinguish ES cells from iPS cells43.
In addition, the residual iPS-cell-specific methylation in many iPS-cell isolates links these cells to their tissue of origin and, ultimately, affects their differentiation propensity43,55. Residual signatures can be distinct enough to enable the myeloid and lymphoid origins of blood-derived iPS cells to be discerned43. In iPS cells derived from non-haematopoietic cells, such as fibroblasts and neural progenitors, there can be residual repressive methylation at loci that are required for haematopoietic fates, reducing the blood-forming potential in vitro. Exogenous supplementation of neural-progenitor-derived iPS cells with the cytokine WNT3A can increase the blood-forming potential of these cells, supporting the idea that incomplete reprogramming owing to epigenetic marks can be overcome by manipulating the culture conditions. Treatment of cultures with demethylating agents or knockdown of DNMT1 expression has been shown to convert intermediately reprogrammed cells into fully pluripotent cells, further supporting this idea43.
When iPS cells are forced to differentiate along a particular lineage, they become more amenable to generating cells of that lineage after a further round of reprogramming. This finding shows that the differentiation propensity and DNA methylation profile can be reset, and it suggests that the ‘epigenetic memory’ of the donor cell can be exploited, especially in cases in which directed differentiation is particularly challenging43. Another important feature of epigenetic reprogramming is the reactivation of the inactivated X chromosome. During normal development in eutherian mammals (those with a placenta), one X chromosome is randomly inactivated in each cell in females. Whether this epigenetic silencing event is reset in iPS cells remains an area of controversy, in part because of the poor fidelity of X-inactivation markers in pluripotent cells56. Some studies have shown that the majority of female human iPS clones retain an inactivated X chromosome (which is transcriptionally silent)57, whereas others have indicated that some human iPS clones lose immunostaining for trimethylated H3K27 on the X chromosome (a marker of epigenetic silencing), indicating X reactivation58. In addition, some of the earliest studies of iPS cells showed X reactivation in reprogrammed female mouse fibroblasts31,58. However, recently published data support the finding that X reactivation does not occur in human iPS cells and, interestingly, reprogramming was found to favour expression of a particular X chromosome when induced from a mixed X-inactivated population of fibroblasts59. Finally, epigenetic reprogramming sometimes fails to properly restore bivalent domains, which mark developmental loci with active and repressive histone modifications60.
Although many of the studies cited here have generated data suggesting that there are epigenetic differences between iPS cells and ES cells, there are several limitations on extending these data to all iPS cells in a more general (and more useful) sense. The published comparisons were often made using iPS cells derived from a multitude of laboratories by a variety of methodologies, and reanalysis of the gene-expression microarray data using an unsupervised clustering algorithm shows a strong correlation between transcriptional signatures and specific laboratories for both iPS cells and ES cells. This finding indicates that specific culture protocols and laboratory environments can affect the transcriptional profile of iPS and ES cells. Therefore, the data produced in a particular laboratory might be specific to the cells derived there61.
In addition, most iPS colonies are clones derived from a single reprogrammed cell, whereas ES cells used for analysis are typically non-clonal. The subcloning of ES cells has revealed genetic and epigenetic anomalies that would probably have otherwise gone undetected in the heterogeneous ES-cell population62. With regard to somatic memory, there is poor overlap between the gene sets that have been reported to be characteristic of a particular cell type of origin, suggesting that the retention of somatic memory is stochastic and is a reflection of the technical failure of reprogramming to fully erase the somatic epigenome. To exacerbate the issue, the iPS cells used for comparison often have different genetic backgrounds and have frequently been derived from fibroblasts that were already heterogeneous in their make-up, affecting both the gene-expression patterns and the functionality of the iPS cells.
Throughout the literature, there is also a lack of correlation between the gene-expression patterns and the epigenetic patterns observed. An additional consideration is the presence of different viral insertions in individual iPS cell lines, which can also affect the functionality of the derived cells1. Evidence to support this idea is provided by the reduced number of differences observed among iPS cells and between iPS and ES cells when transgenes are removed63. Many of the aforementioned studies have focused on differences in either transcriptional profiles or changes in epigenetic marks; however, the most recent studies have evaluated iPS cells and ES cells from both of these angles in parallel, together with their in vitro differentiation potential, generating the most comprehensive and compelling data that have been published so far.
Holistic analysis
Stadtfeld and colleagues explored the epigenetic and functional discrepancies between iPS cells and ES cells using a novel reprogramming strategy that allowed direct comparison of genetically matched cells derived from the same source32. These authors derived iPS and ES cells from mice carrying an integrated doxycycline-inducible reprogramming cassette in every cell, a strategy that sidesteps the confounding effects of variable genetic backgrounds and viral integration that have been observed in other studies. The overall messenger RNA and microRNA expression patterns of iPS cells and ES cells were indistinguishable except for the aberrant silencing of a few transcripts localized to the imprinted Dlk1–Dio3 gene cluster on chromosome 12qF1, a region that is important for development. A failure to reactivate this locus meant that iPS cells contributed poorly to chimaeras and were unable to generate all-iPS-cell mice. By contrast, iPS cells with normal Dlk1–Dio3 expression contributed to high-grade chimaeras and supported the development of viable all-iPS-cell mice. The treatment of iPS cells that failed to reactivate Dlk1–Dio3 with a histone-deacetylase inhibitor rescued the ability of these clones to support the development of all-iPS-cell mice by relieving this region of aberrant hypermethylation. However, recent data from iPS and ES cells derived from a mouse strain carrying a distinct reprogramming cassette suggest that different levels of expression of reprogramming factors, rather than aberrant silencing of Dlk1–Dio3, account for the different behaviour of the cell lines64. The disparate results from these studies highlight that iPS cells can behave differently based on subtle variations in the expression of only a few loci.
To systematically compare human iPS cells derived from different somatic cell types and ES cells, Ohi and colleagues compared ES cells with iPS cells reprogrammed from somatic cells representative of the three embryonic germ layers74. Transcriptional and epigenetic profiling of these cells showed transcriptional differences, owing, in part, to incomplete promoter methylation, which enabled iPS cells to be discerned on the basis of their cell of origin. The differential methylation between iPS cells and ES cells did not correlate with varying levels in DNA methyltransferases; however, the authors found a nonrandom pattern of incompletely silenced genes in genetic regions that are isolated from other genes that undergo silencing during reprogramming. This finding could be explained by inefficient or delayed recruitment of the silencing machinery and DNA methyltransferases to particular somatic genes because of the isolated location of these genes65.
In another comprehensive study, Polo and colleagues evaluated the effect of cellular origin on the gene-expression pattern, epigenetic properties and functional abilities of genetically matched mouse iPS cells66. Using the same ‘secondary’ reprogramming strategy used by Wernig and colleagues (whereby reprogramming is assessed using tissues from a mouse generated from iPS cells carrying integrated, doxycycline-inducible reprogramming factors), Polo and colleagues generated iPS cells from tail-tip fibroblasts, splenic B cells, bone-marrow-derived granulocytes and skeletal muscle precursors, and they showed that each iPS cell line retained a transcriptional memory of its cell of origin. This memory was evident in that markers for each respective cell of origin remained actively expressed and was supported by the finding that iPS cell lines derived from the same cell of origin clustered together on the basis of global transcriptional data. A similar correlation was found on evaluation of methylation patterns, which showed subtle but substantial differences, reflecting the consequences of different histone marks. The effects of somatic memory extended beyond the genetic and epigenetic levels to functional significance, affecting the autonomous differentiation potential of the different iPS cell lines after embryoid-body formation. A clear bias that reflected the cells of origin was observed in the iPS cell lines. Notably, the transcriptional, epigenetic and functional effects evaluated in early-passage iPS cell lines became less significant with continued passaging. This finding indicated that complete reprogramming is a gradual process that extends beyond the time frame necessary to observe the activation of endogenous pluripotency genes, transgene-free growth and differentiation into cell types from each of the three germ layers.
Exploring equivalence
Having considered data from a multitude of published studies, generated by the painstaking efforts of many research groups, we return to our earlier question: are iPS cells equivalent to ES cells? The answer is not straightforward. Rather, there is an emerging consensus that iPS cells and ES cells are neither identical nor distinct populations. Instead, they are overlapping, with greater variability inherent within each population than between the populations. The heterogeneity and behaviour of each class of cell line is more complex than has previously been thought. The two pluripotent stem-cell types are, in theory, functionally equivalent; however, in practice, they harbour genetic and epigenetic differences that reflect their different histories. It remains to be seen whether there are any consistent molecular distinctions between iPS cells and ES cells.
It is also important to consider that, in contrast to long-standing belief, ES cells themselves have considerable epigenetic heterogeneity and have differing propensities for differentiation — much like those found in iPS cells67,68. These observations, paired with those discussed in this Review, are a call for researchers to take a step back from the direct comparison of iPS cells and ES cells, and they highlight the need to redefine what it means to belong to either of these cell classes. Some researchers have already taken heed of this message and generated a bioinformatics assay for pluripotency72, whereas others have produced a ‘scorecard’ to evaluate the character of both iPS and ES cell lines and predict the quality and utility of any pluripotent cell line in a high-throughput manner47,69. Using DNA methylation mapping, gene-expression profiling and a quantitative differentiation assay, Bock and colleagues made a systematic comparison of 20 established ES cell lines and 12 iPS cell lines53. They confirmed that, despite overall similarities, transcriptional and epigenetic variation is common between iPS cell lines, between ES cell lines, and between iPS cell and ES cell populations. These data provide a reference for the variation among human pluripotent cell lines, which assists in predicting the functional consequences of these differences. We can conclude from these studies that any given iPS cell line generated by today’s technology might not be completely equivalent to the ideal ES cell.
The differences between iPS cells and ES cells, as well as those among iPS cells, clearly affect the utility of these cells in research, disease modelling and therapeutics, providing an impetus for investigators to evaluate their cell populations carefully and precisely. The differences do not diminish the potential of iPS cells, given that iPS cells have considerable advantages over ES cells. Rather than replacing ES cells with iPS cells, it is becoming clear that these two cell types complement one another. Researchers are still in the process of developing the necessary protocols to harness the potential of iPS cells; however, as it becomes clear how to evaluate the genetic, epigenetic and functional status of different iPS cell lines, further applications of these cells will be uncovered, and progress will be made in creating iPS cell lines and designing protocols to accomplish the ambitious goals of the field.
Medical applications of iPS cells
Generating patient-specific stem cells has been a long-standing goal in the field of regenerative medicine. Despite considerable challenges, generating disease-specific and patient-specific iPS cells through reprogramming has become almost routine. These cells provide a unique platform from which to gain mechanistic insight into a variety of diseases, to carry out in vitro drug screening, to evaluate potential therapeutics and to explore gene repair coupled with cell-replacement therapy (Fig. 2). In the past few years, the number of reports on applications of iPS cells has steadily increased, testifying to the broad influence of this breakthrough technology (Table 2). Despite the continued presence of substantial hurdles, the pace of this work is such that no review can capture the current state of the field; thus, we point to a few publications that highlight the promising medical applications of iPS cells but also indicate their key limitations.
Figure 2. Medical applications of iPS cells.

Reprogramming technology and iPS cells have the potential to be used to model and treat human disease. In this example, the patient has a neurodegenerative disorder. Patient-specific iPS cells — in this case derived by ectopic co-expression of transcription factors in cells isolated from a skin biopsy — can be used in one of two pathways. In cases in which the disease-causing mutation is known (for example, familial Parkinson’s disease), gene targeting could be used to repair the DNA sequence (right). The gene-corrected patient-specific iPS cells would then undergo directed differentiation into the affected neuronal subtype (for example, midbrain dopaminergic neurons) and be transplanted into the patient’s brain (to engraft the nigrostriatal axis). Alternatively, directed differentiation of the patient-specific iPS cells into the affected neuronal subtype (left) will allow the patient’s disease to be modelled in vitro, and potential drugs can be screened, aiding in the discovery of novel therapeutic compounds.
Table 2.
Diseases modelled with iPS cells
| Disease | Molecular defect of donor cell | Cell type differentiated from iPS cells | Disease phenocopied in differentiated cells | Drug or functional tests |
|---|---|---|---|---|
| Neurological | ||||
| Amyotrophic lateral sclerosis (ALS) | Heterozygous Leu144Phe mutation in SOD1 | Motor neurons and glial cells | ND | No |
| Spinal muscular atrophy (SMA) | Mutations in SMN1 | Neurons and astrocytes, and mature motor neurons | Yes | Yes |
| Parkinson’s disease | Multifactorial; mutations in LRRK2 and/or SNCA | Dopaminergic neurons | No | Yes |
| Huntington’s disease | 72 CAG repeats in the huntingtin gene | None | NA | No |
| Down’s syndrome | Trisomy 21 | Teratoma with tissue from each of the three germ layers | Yes | No |
| Fragile X syndrome | CGG triplet repeat expansion resulting in the silencing of FMR1 | None | NA | No |
| Familial dysautonomia | Mutation in IKBKAP | Central nervous-system lineage, peripheral neurons, haematopoietic cells, endothelial cells and endodermal cells | Yes | Yes |
| Rett’s syndrome | Heterozygous mutation in MECP2 | Neural progenitor cells | Yes | Yes |
| Mucopolysacchar idosis type IIIB (MPS IIIB) | Homozygous mutation in NAGLU | Neural stem cells and differentiated neurons | Partially | Yes |
| Schizophrenia | Complex trait | Neurons | Yes | Yes |
| X-linked adrenoleukodystr ophy (X-ALD), childhood cerebral ALD (CCALD) and adrenomyeloneur opathy (AMN) | Mutation in ABCD1 | Oligodendrocytes and neurons | Partially | Yes |
| Haematological | ||||
| ADA SCID | Mutation or deletion in ADA | None | ND | No |
| Fanconi’s anaemia | FAA and FAD2 corrected | Haematopoietic cells | No (corrected) | No |
| Schwachman–Bodian–Diamond Syndrome | Multifactorial | None | NA | No |
| Sickle-cell anaemia | Homozygous HbS mutation | None | NA | No |
| β-Thalassaemia | Homozygous deletion in the β-globin gene | Haematopoietic cells | ND | No |
| Polycythaemia vera | Heterozygous Val617Phe mutation in JAK2 | Haematopoietic progenitors (CD34+CD35+) | Partially | No |
| Primary myelofibrosis | Heterozygous mutation in JAK2 | None | NA | No |
| Metabolic | ||||
| Lesch–Nyhan syndrome (carrier) | Heterozygous mutation in HPRT1 | None | NA | No |
| Type 1 diabetes | Multifactorial; unknown | β-Cell-like cells (express somatostatin, glucagon and insulin; glucose-responsive) | ND | No |
| Gaucher’s disease, type III | Mutation in GBA | None | NA | No |
| α1-Antitrypsin deficiency (A1ATD) | Homozygous mutation in the α1-antitrypsin gene | Hepatocyte-like cells (fetal) | Yes | No |
| Glycogen storage disease Ia (GSD1a) | Defect in glucose-6-phosphate gene | Hepatocyte-like cells (fetal) | Yes | No |
| Familial hypercholesterol aemia | Autosomal dominant mutation in LDLR | Hepatocyte-like cells (fetal) | Yes | No |
| Crigler–Najjar syndrome | Deletion in UGT1A1 | Hepatocyte-like cells (fetal) | ND | No |
| Hereditary tyrosinaemia, type 1 | Mutation in FAHD1 | Hepatocyte-like cells (fetal) | ND | No |
| Pompe disease | Knockout of Gaa | Skeletal muscle cells | Yes | No |
| Progressive familial cholestasis | Multifactorial | Hepatocyte-like cells (fetal) | ND | No |
| Hurler syndrome (MPS IH) | Genetic defect in IDUA | Haematopoietic cells | No | No |
| Cardiovascular | ||||
| LEOPARD syndrome | Heterozygous mutation in PTPN11 | Cardiomyocytes | Yes | No |
| Type 1 long QT syndrome | Dominant mutation in KCNQ1 | Cardiomyocytes | Yes | No |
| Type 2 long QT syndrome | Missense mutation in KCNH2 | Cardiomyocytes | Yes | Yes |
| Primary immunodeficiency | ||||
| SCID or leaky SCID | Mutation in RAG1 | None | NA | No |
| Omenn syndrome (OS) | Mutation in RAG1 | None | NA | No |
| Cartilage-hair hypoplasia (CHH) | Mutation in RMRP | None | NA | No |
| Herpes simplex encephalitis (HSE) | Mutation in STAT1 or TLR3 | Mature cell types of the central nervous system | No | No |
| Other category | ||||
| Duchenne muscular dystrophy | Deletion in the dystrophin gene | None | NA | No |
| Becker muscular dystrophy | Unidentified mutation in dystrophin | None | NA | No |
| Dyskeratosis congenita (DC) | Deletion in DKC1 | None | NA | No |
| Cystic fibrosis | Homozygous deletion in CFTR | None | NA | No |
| Friedreich’s ataxia (FRDA) | Trinucleotide GAA repeat expansion in FXN | Sensory and peripheral neurons, and cardiomyocytes | Partially | No |
| Retinitis pigmentosa | Heterogeneity in causative genes and mutations: mutations in RP9, RP1, PRPH2 or RHO | Retinal progenitors, photoreceptor precursors, retinal-pigment epithelial cells and rod photoreceptor cells | Yes | Yes |
| Recessive dystrophic epidermolysis bullosa (RDEB) | Mutation in COL7A1 | Haematopoietic cells, and epidermis-like keratinocytes that differentiate into cells of all three germ layers in vivo | Partially | Yes |
| Scleroderma | Unknown | None | NA | No |
| Osteogenesis imperfecta | Mutation in COL1A2 | None | NA | No |
An extended version of this table includes references and more information about drug and functional tests (Supplementary Table 1). ABCD1, ATP-binding cassette, sub-family D, member 1; ADA, adenine deaminase; CFTR, cystic fibrosis transmembrane conductance regulator; COL1A2, α2-chain of type I collagen; COL7A1, α1-chain of type VII collagen; DKC1, dyskerin; FAA, Fanconi’s anaemia, complementation group A; FAD2, Fanconi’s anaemia, complementation group D2; FAHD1, fumarylacetoacetate hydrolase; FMR1, fragile X mental retardation 1; FXN, frataxin; Gaa, acid α-glucosidase; GBA, acid β-glucosidase; HbS, sickle haemoglobin; HPRT1, hypoxanthine phosphoribosyltransferase 1; IDUA, α-L-iduronidase; JAK2, Janus kinase 2; KCNH2, potassium voltage-gated channel, subfamily H (eag-related), member 2; KCNQ1, potassium voltage-gated channel, KQT-like subfamily, member 1; LDLR, low-density lipoprotein receptor; LRRK2, leucine-rich repeat kinase 2; MECP2, methyl CpG binding protein 2; NA, not applicable; NAGLU, α-N-acetylglucosaminidase; ND not determined; PRPH2, peripherin 2; PTPN11, protein tyrosine phosphatase, non-receptor type 11; RAG1, recombination activating gene 1; RHO, rhodopsin; RMRP, RNA component of mitochondrial-RNA-processing endoribonuclease; RP, retinitis pigmentosa; SCID, severe combined immunodeficiency; SMN1, survival of motor neuron 1; SNCA, α-synuclein; SOD1, superoxide dismutase 1; STAT1, signal transducer and activator of transcription 1; TLR3, Toll-like receptor 3; UGT1A1, UDP glucuronosyltransferase 1 family, polypeptide A1.
In 2009, Lee and colleagues harnessed iPS cells to demonstrate disease modelling and drug screening for familial dysautonomia, a rare genetic disorder of the peripheral nervous system70. In almost all cases, familial dysautonomia is caused by a single point mutation in the gene encoding the inhibitor of nuclear factor-κB (IκB)-kinase-complex-associated protein (IKBKAP) that manifests as an extensive autonomic nervous system deficit and dysfunction in small-fibre sensory neurons. Although many traditional cell-based models have been used to study the pathogenesis of familial dysautonomia and screen for candidate drugs, none has used symptom-relevant human cell types. With the successful derivation of iPS cells from patients with familial dysautonomia, investigators produced central and peripheral nervous system precursors and subsequently found three disease-related phenotypes, thus providing validation that disease-relevant cell types could accurately reflect disease pathogenesis in vitro. After screening with multiple compounds, the authors showed that the disease phenotype could be partially normalized by kinetin, a plant hormone. This initial report demonstrated how iPS cells can facilitate the discovery of therapeutic compounds and described how these cells provided a platform for modelling different severities of familial dysautonomia and for generating predictive tests to determine differences in the clinical manifestation of the disorder.
Such applications of iPS cells in drug screening and discovery are destined to expand to encompass numerous disease conditions. Several research groups have generated models of long QT syndrome, a congenital disease with 12 types, each of which is associated with abnormal ion-channel function, a prolonged QT interval on an electrocardiogram and a high risk of sudden cardiac death owing to ventricular tachyarrhythmia. Much work has been carried out in animal models to probe the underlying mechanisms of this syndrome, but cardiomyocytes have distinct and complex electrophysiological properties that differ between species. In addition, the lack of in vitro sources of human cardiomyocytes and the inability to model patient-specific variations of this disease has impeded studies.
In a proof-of-principle study for using iPS cells to capture the physiological mechanisms of genetic variation, Moretti and colleagues differentiated iPS cells from individuals with type 1 long QT syndrome into cardiomyocytes and, as predicted, observed prolonged action potentials in the ventricular and atrial cells71. Using this model system, these investigators uncovered a dominant-negative trafficking defect associated with the particular mutation that causes this variant of long QT syndrome. Further investigation of long QT syndrome iPS-cell-derived cardiomyocytes showed that these cells had an increased susceptibility to catecholamine-induced tachyarrhythmia, and compounds that exacerbated the condition (including isoprenaline) were identified. Treatment of these cardiomyocytes with β-adrenergic receptor blockers attenuated the long QT phenotype.
Type 2 long QT syndrome has also been modelled in cardiomyocytes, by Itzhaki and colleagues72. The authors derived type 2 long QT syndrome iPS cells to evaluate the potency of existing and novel pharmacological agents that might exacerbate or ameliorate the condition. Their studies show that the long QT syndrome phenotype was aggravated by blockers of ERG-type potassium channels, whereas nifedipine, a calcium-channel blocker, and pinacidil, an agonist of ATP-sensitive potassium channels, both ameliorated the long QT syndrome phenotype, as shown by the decreased duration of action potentials in long QT syndrome cardiomyocytes, as well as the elimination of early after-depolarizations and the abolishment of all triggered arrhythmias. A possible limitation of these beneficial drugs is excessive shortening of the action-potential duration, leading to short QT syndrome.
Importantly, these studies established that the iPS-cell model can be used to identify complex cardiotoxic effects of drugs, as well as to define protective pharmacological agents, including optimal drug dosages. Given the number of drugs that have notoriously been withdrawn from the market owing to their tendency to induce arrhythmias, it is highly likely that the current inadequate approaches for assessing cardiotoxicity will be complemented by iPS-cell-based assessments of drug effects.
A study from our laboratory explored dyskeratosis congenita, a disorder of telomere maintenance, and provided an unanticipated insight into the basic biology of telomerase that has therapeutic implications73. In its most severe form, dyskeratosis congenita is caused by a mutation in the dyskerin gene (DKC1), which is X linked, leading to shortened telomeres and premature senescence in cells and ultimately manifesting as the degeneration of multiple tissues. Because the reprogramming of cells to an induced pluripotent state is accompanied by the induction of the gene encoding telomerase reverse transcriptase (TERT), we investigated whether the telomerase defect would limit the derivation and maintenance of iPS cells from individuals with dyskeratosis congenita. Although the efficiency of iPS-cell derivation was poor, we were able to successfully reprogram patient fibroblasts. Surprisingly, whereas the mean telomere length immediately after reprogramming was shorter than that of the parental fibroblast population, continued passage of some iPS cell lines led to telomere elongation over time. This process was accompanied by upregulation of the expression of TERC, which encodes the RNA subunit of telomerase.
Further analysis established that TERT and TERC, as well as DKC1, were expressed at higher levels in dyskeratosis-congenita-derived iPS cells than in the parental fibroblasts. We determined that the genes encoding these components of the telomerase pathway — including a cis element in the 3′ region of the TERC locus that is essential for a transcriptionally active chromatin structure — were direct binding targets of the pluripotency-associated transcription factors. Further analysis indicated that transcriptional silencing owing to a 3′ deletion in the TERC locus leads to the autosomal dominant form of dyskeratosis congenita by diminishing TERC transcription. Although telomere length is restored in dyskeratosis-congenita-derived iPS cells, differentiation into somatic cells is accompanied by a return to pathogenesis with low TERC expression and a decay in telomere length. This finding showed that TERC RNA levels are dynamically regulated and that the pluripotent state of the cells is reversible, suggesting that drugs that elevate or stabilize TERC expression might rescue defective telomerase activity and provide a therapeutic benefit. Although we set out to understand the pathogenesis of dyskeratosis congenita with this study, we showed that a high expression level of multiple telomerase components was characteristic of the pluripotent state more generally, illustrating how iPS cells can reveal fundamental aspects of cell biology.
An independent study of the reprogramming of cells from patients with dyskeratosis congenita confirmed the general transcriptional upregulation of multiple telomerase components and the maintenance of telomere lengths in many clones74; however, in this study, no clones with elongated telomeres were identified. The different outcomes of these studies highlight the limitations of iPS-cell-based disease models that are imposed by clonal variation as a result of the inherent technical infidelity of reprogramming75. This point also introduces an additional important consideration. Before a given iPS-cell disease model can be claimed to be truly representative of the disease, how many patients must be involved, and how many iPS cell lines must be derived from each patient? Although the answers to these questions are unclear, it is crucial to keep these issues in mind when generating disease models and making claims based on these models.
Although iPS cells are an invaluable tool for modelling diseases in vitro, the goal of developing patient-specific stem cells has also been motivated by the prospect of generating a ready supply of immune-compatible cells and tissues for autologous transplantation. At present, the clinical translation of iPS-cell-based cell therapies seems more futuristic than the in vitro use of iPS cells for research and drug development, but two groundbreaking studies have provided the proof of principle in mouse models that the dream might one day be realized. Hanna, Jaenisch and colleagues used homologous recombination to repair the genetic defect in iPS cells derived from a humanized mouse model of sickle-cell anaemia76. Directed differentiation of the repaired iPS cells into haematopoietic progenitors followed by transplantation of these cells into the affected mice led to the rescue of the disease phenotype. The gene-corrected iPS-cell-derived haematopoietic progenitors showed stable engraftment and correction of the disease phenotype — a significant finding as proof of principle for gene repair coupled to cell-replacement therapy.
In another landmark study from Jaenisch’s research group, Wernig and colleagues derived dopaminergic neurons from iPS cells that, when implanted into the brain, became functionally integrated and improved the condition of a rat model of Parkinson’s disease77. The successful implantation and functional recovery in this model is evidence of the therapeutic value of pluripotent stem cells for cell-replacement therapy in the brain — one of the most promising areas for the future of iPS-cell applications.
Together, these findings provide proof of principle for using reprogramming with gene repair and cell-replacement therapy for treating diseases. Using iPS cells in cell-replacement therapy offers the promise of therapeutic intervention that is not compounded by the use of immunosuppressive drugs to prevent tissue rejection, while harnessing targeted gene-repair strategies, such as homologous recombination and zinc-finger nucleases, to repair genetic defects. These strategies provide the opportunity for generating an unlimited population of stem cells that can be differentiated into the desired cell type for studying disease mechanisms, for screening and developing drugs or for developing a suitable cell-replacement therapy. There have been considerable advances and successes to this end; however, selecting an appropriate disease target, directing the differentiation of iPS cells into phenotype-relevant cell populations and identifying disease-relevant phenotypes remain major hurdles. It is unclear whether iPS cells used for cell-replacement therapy would completely evade an immune response when returned to the patient, because a recent study has shown the immune rejection of teratomas formed from iPS cells, even in syngeneic mice78. Nevertheless, iPS cells provide a promising model with which to study disease mechanisms, discover new therapies and develop truly personalized treatments.
Predictions for the evolution of the art
Few fields have enjoyed the remarkable upsurge in activity and excitement that followed the initial report of the reprogramming of somatic cells into iPS cells in 2006. Despite heady progress, crucial challenges must be met for the field to realize its full potential. There is as yet no consensus on the most consistent protocol or the optimal protocol for deriving the most reliable and, ultimately, the safest iPS cells. Increasing the reprogramming efficiency and effecting reprogramming without genetically modifying the cells are goals that have been achieved. However, using more-uniform protocols and more-rigorous controls would facilitate experimental consistency, and potentially therapeutic consistency, between laboratories and would yield standardized cell lines that could be used with confidence in both basic and applied studies.
Barring that, researchers must agree on standards of molecular analysis to ensure that the reprogrammed cells that most closely approximate the generic state of the naive genome can be identified. Because iPS cells are subject to the same type of culture adaptations that affect karyotypic integrity as human ES cells79, it is important to define protocols that minimize the time in culture. In addition, cell lines used in clinical applications will need to be evaluated frequently for aberrant culture-induced changes at all stages: from the somatic cells to the reprogrammed and differentiated cells80. Understanding the genomic alterations that take place during the reprogramming, culture and differentiation of iPS cells will be crucial for designing experiments and ensuring that the derived cells are functional, pure and appropriate for use in research and therapy. Minimizing any aberrations is important, but as long as researchers understand that aberrations will arise — and can describe and control their effects — even the imperfect cells can be used, and preferential differentiation can be taken advantage of whenever possible. Characteristics of iPS cells that were initially perceived as flaws, including varying differentiation propensities, might prove useful in clinical settings, to generate cell types that have been difficult to obtain thus far.
Generating more stringent markers of pluripotency and assays to distinguish the abilities of a given iPS cell line are key priorities. Building on the progress that has already been made using ES cells81, researchers must continue to improve the understanding of directed differentiation and to develop new protocols. With refined differentiation protocols, researchers will be able to investigate the pathophysiological basis of genetic diseases and carry out drug screening on affected cell types. In addition, these protocols will bring the field a step closer to patient-matched cells and tissues for clinical transplantation, a long-standing ambition of the stem-cell field that might be its ultimate measure of success.
Acknowledgments
G.Q.D. is supported by grants from the National Institutes of Health (RO1-DK70055, RO1-DK59279 and UO1-HL100001, as well as special funds from the American Recovery and Reinvestment Act of 2009 (RC2-HL102815), the Roche Foundation for Anemia Research, Alex’s Lemonade Stand Foundation and the Harvard Stem Cell Institute. G.Q.D. is an affiliate member of the Broad Institute, a recipient of Clinical Scientist Awards in Translational Research from the Burroughs Wellcome Fund and the Leukemia and Lymphoma Society, and an investigator of the Howard Hughes Medical Institute and the Manton Center for Orphan Disease Research. We gratefully acknowledge A. de Los Angeles for providing the images for Fig. 1a, d, f; T. Onder, for Fig. 1b; and A. Cherry, for Fig. 1c, e.
G.Q.D. is a member of the scientific advisory boards and holds stock in, or receives consulting fees from, the following companies: Johnson & Johnson, Verastem, Epizyme, iPierian, Solasia Pharma and MPM Capital.
References
- 1.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Driesch H. Entwicklungsmechanische Studien I. Der Wert der ersten beiden Furchungszellen in der Echinodermenentwickelung Experimentelle Erzeugung von Teil und Doppelbildungen. Ztschr f Wiss Zool. 1891;53:160–183. [Google Scholar]
- 3.Dewey MJ, Martin DW, Jr, Martin GR, Mintz B. Mosaic mice with teratocarcinoma-derived mutant cells deficient in hypoxanthine phosphoribosyltransferase. Proc Natl Acad Sci U S A. 1977;74:5564–5568. doi: 10.1073/pnas.74.12.5564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gardner RL. Mouse chimeras obtained by the injection of cells into the blastocyst. Nature. 1968;220:596–597. doi: 10.1038/220596a0. [DOI] [PubMed] [Google Scholar]
- 5.Brinster RL. The effect of cells transferred into the mouse blastocyst on subsequent development. J Exp Med. 1974;140:1049–1056. doi: 10.1084/jem.140.4.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 7.Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci U S A. 1981;78:7634–7638. doi: 10.1073/pnas.78.12.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomson JA, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 9.Vogel G, Holden C. Stem cells. Ethics questions add to concerns about NIH lines. Science. 2008;321:756–757. doi: 10.1126/science.321.5890.756b. [DOI] [PubMed] [Google Scholar]
- 10.Mosher JT, et al. Lack of population diversity in commonly used human embryonic stem-cell lines. N Engl J Med. 2010;362:183–185. doi: 10.1056/NEJMc0910371. [DOI] [PubMed] [Google Scholar]
- 11.Tabar V, et al. Therapeutic cloning in individual parkinsonian mice. Nat Med. 2008;14:379–381. doi: 10.1038/nm1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noggle S, et al. Human oocytes reprogram somatic cells to a pluripotent state. Nature. 2011;478:70–75. doi: 10.1038/nature10397. [DOI] [PubMed] [Google Scholar]
- 13.Park IH, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 15.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 16.Stadtfeld M, Brennand K, Hochedlinger K. Reprogramming of pancreatic beta cells into induced pluripotent stem cells. Curr Biol. 2008;18:890–894. doi: 10.1016/j.cub.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eminli S, Utikal J, Arnold K, Jaenisch R, Hochedlinger K. Reprogramming of neural progenitor cells into induced pluripotent stem cells in the absence of exogenous Sox2 expression. Stem cells. 2008;26:2467–2474. doi: 10.1634/stemcells.2008-0317. [DOI] [PubMed] [Google Scholar]
- 18.Kim JB, et al. Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature. 2008;454:646–650. doi: 10.1038/nature07061. [DOI] [PubMed] [Google Scholar]
- 19.Hanna J, et al. Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell. 2008;133:250–264. doi: 10.1016/j.cell.2008.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aoi T, et al. Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science. 2008;321:699–702. doi: 10.1126/science.1154884. [DOI] [PubMed] [Google Scholar]
- 21.Utikal J, Maherali N, Kulalert W, Hochedlinger K. Sox2 is dispensable for the reprogramming of melanocytes and melanoma cells into induced pluripotent stem cells. J Cell Sci. 2009;122:3502–3510. doi: 10.1242/jcs.054783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun N, et al. Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells. Proc Natl Acad Sci U S A. 2009;106:15720–15725. doi: 10.1073/pnas.0908450106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maherali N, et al. A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell. 2008;3:340–345. doi: 10.1016/j.stem.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tesar PJ, et al. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature. 2007;448:196–199. doi: 10.1038/nature05972. [DOI] [PubMed] [Google Scholar]
- 25.Wray J, Kalkan T, Smith AG. The ground state of pluripotency. Biochem Soc Trans. 2010;38:1027–1032. doi: 10.1042/BST0381027. [DOI] [PubMed] [Google Scholar]
- 26.Hanna JH, Saha K, Jaenisch R. Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell. 2010;143:508–525. doi: 10.1016/j.cell.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maherali N, Hochedlinger K. Guidelines and techniques for the generation of induced pluripotent stem cells. Cell Stem Cell. 2008;3:595–605. doi: 10.1016/j.stem.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 28.Stadtfeld M, Maherali N, Breault DT, Hochedlinger K. Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell. 2008;2:230–240. doi: 10.1016/j.stem.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan EM, et al. Live cell imaging distinguishes bona fide human iPS cells from partially reprogrammed cells. Nature biotechnology. 2009;27:1033–1037. doi: 10.1038/nbt.1580. [DOI] [PubMed] [Google Scholar]
- 30.Payer BLJ, Namekawa SH. X-inactivation and X-reactivation: epigenetic hallmarks of mammalian reproduction and pluripotent stem cells. Hum Genet. 2011 doi: 10.1007/s00439-011-1024-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maherali N, et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1:55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 32.Stadtfeld M, et al. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature. 2010;465:175–181. doi: 10.1038/nature09017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boland MJ, et al. Adult mice generated from induced pluripotent stem cells. Nature. 2009;461:91–94. doi: 10.1038/nature08310. [DOI] [PubMed] [Google Scholar]
- 34.Zhao XY, et al. iPS cells produce viable mice through tetraploid complementation. Nature. 2009;461:86–90. doi: 10.1038/nature08267. [DOI] [PubMed] [Google Scholar]
- 35.Eakin GS, Hadjantonakis AK, Papaioannou VE, Behringer RR. Developmental potential and behavior of tetraploid cells in the mouse embryo. Dev Biol. 2005;288:150–159. doi: 10.1016/j.ydbio.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 36.Eggan K, Jaenisch R. Differentiation of F1 embryonic stem cells into viable male and female mice by tetraploid embryo complementation. Methods Enzymol. 2003;365:25–39. doi: 10.1016/s0076-6879(03)65002-0. [DOI] [PubMed] [Google Scholar]
- 37.Lensch MW, Schlaeger TM, Zon LI, Daley GQ. Teratoma formation assays with human embryonic stem cells: a rationale for one type of human-animal chimera. Cell Stem Cell. 2007;1:253–258. doi: 10.1016/j.stem.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 38.Park IH, et al. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daley GQ, et al. Broader implications of defining standards for the pluripotency of iPSCs. Cell Stem Cell. 2009;4:200–201. doi: 10.1016/j.stem.2009.02.009. author reply 202. [DOI] [PubMed] [Google Scholar]
- 40.Miura K, et al. Variation in the safety of induced pluripotent stem cell lines. Nature biotechnology. 2009;27:743–745. doi: 10.1038/nbt.1554. [DOI] [PubMed] [Google Scholar]
- 41.Feng Q, et al. Hemangioblastic derivatives from human induced pluripotent stem cells exhibit limited expansion and early senescence. Stem cells. 2010;28:704–712. doi: 10.1002/stem.321. [DOI] [PubMed] [Google Scholar]
- 42.Hu BY, et al. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc Natl Acad Sci U S A. 2010;107:4335–4340. doi: 10.1073/pnas.0910012107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim K, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu Q, Friedrich AM, Johnson LV, Clegg DO. Memory in induced pluripotent stem cells: reprogrammed human retinal-pigmented epithelial cells show tendency for spontaneous redifferentiation. Stem cells. 2010;28:1981–1991. doi: 10.1002/stem.531. [DOI] [PubMed] [Google Scholar]
- 45.Bar-Nur O, Russ HA, Efrat S, Benvenisty N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet Beta cells. Cell Stem Cell. 2011;9:17–23. doi: 10.1016/j.stem.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 46.Urbach A, Bar-Nur O, Daley GQ, Benvenisty N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell. 2010;6:407–411. doi: 10.1016/j.stem.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bock C, et al. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 2011;144:439–452. doi: 10.1016/j.cell.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pick M, et al. Clone- and gene-specific aberrations of parental imprinting in human induced pluripotent stem cells. Stem cells. 2009;27:2686–2690. doi: 10.1002/stem.205. [DOI] [PubMed] [Google Scholar]
- 49.Ben-David U, Mayshar Y, Benvenisty N. Large-scale analysis reveals acquisition of lineage-specific chromosomal aberrations in human adult stem cells. Cell Stem Cell. 2011;9:97–102. doi: 10.1016/j.stem.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 50.Hussein SM, et al. Copy number variation and selection during reprogramming to pluripotency. Nature. 2011;471:58–62. doi: 10.1038/nature09871. [DOI] [PubMed] [Google Scholar]
- 51.Laurent LC, et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell. 2011;8:106–118. doi: 10.1016/j.stem.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ghosh Z, et al. Persistent donor cell gene expression among human induced pluripotent stem cells contributes to differences with human embryonic stem cells. PLoS One. 2010;5:e8975. doi: 10.1371/journal.pone.0008975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wernig M, et al. A drug-inducible transgenic system for direct reprogramming of multiple somatic cell types. Nature biotechnology. 2008;26:916–924. doi: 10.1038/nbt1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mikkelsen TS, et al. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lister R, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shen Y, et al. X-inactivation in female human embryonic stem cells is in a nonrandom pattern and prone to epigenetic alterations. Proc Natl Acad Sci U S A. 2008;105:4709–4714. doi: 10.1073/pnas.0712018105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tchieu J, et al. Female human iPSCs retain an inactive X chromosome. Cell Stem Cell. 2010;7:329–342. doi: 10.1016/j.stem.2010.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marchetto MC, et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pomp O, et al. Unexpected X Chromosome Skewing during Culture and Reprogramming of Human Somatic Cells Can Be Alleviated by Exogenous Telomerase. Cell Stem Cell. 2011;9:156–165. doi: 10.1016/j.stem.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 60.Bernstein BE, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 61.Newman AM, Cooper JB. Lab-specific gene expression signatures in pluripotent stem cells. Cell Stem Cell. 2010;7:258–262. doi: 10.1016/j.stem.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 62.Humpherys D, et al. Epigenetic instability in ES cells and cloned mice. Science. 2001;293:95–97. doi: 10.1126/science.1061402. [DOI] [PubMed] [Google Scholar]
- 63.Soldner F, et al. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964–977. doi: 10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carey BW, et al. Reprogramming factor stoichiometry influences the epigenetic state and biological properties of induced pluripotent stem cells. Cell Stem Cell. 2011;9:588–598. doi: 10.1016/j.stem.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 65.Ohi Y, et al. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nature cell biology. 2011;13:541–549. doi: 10.1038/ncb2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Polo JM, et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nature biotechnology. 2010;28:848–855. doi: 10.1038/nbt.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martinez Y, et al. Cellular diversity within embryonic stem cells: pluripotent clonal sublines show distinct differentiation potential. J Cell Mol Med. 2011 doi: 10.1111/j.1582-4934.2011.01334.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Osafune K, et al. Marked differences in differentiation propensity among human embryonic stem cell lines. Nature biotechnology. 2008;26:313–315. doi: 10.1038/nbt1383. [DOI] [PubMed] [Google Scholar]
- 69.Muller FJ, et al. A bioinformatic assay for pluripotency in human cells. Nat Methods. 2011;8:315–317. doi: 10.1038/nmeth.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee G, et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature. 2009;461:402–406. doi: 10.1038/nature08320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moretti A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363:1397–1409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- 72.Itzhaki I, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–229. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- 73.Agarwal S, et al. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature. 2010;464:292–296. doi: 10.1038/nature08792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Batista LF, et al. Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature. 2011;474:399–402. doi: 10.1038/nature10084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Agarwal S, Daley GQ. Telomere dynamics in dyskeratosis congenita: the long and the short of iPS. Cell Res. 2011 doi: 10.1038/cr.2011.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hanna J, et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318:1920–1923. doi: 10.1126/science.1152092. [DOI] [PubMed] [Google Scholar]
- 77.Wernig M, et al. Neurons derived from reprogrammed fibroblasts functionally integrate into the fetal brain and improve symptoms of rats with Parkinson’s disease. Proc Natl Acad Sci U S A. 2008;105:5856–5861. doi: 10.1073/pnas.0801677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhao T, Zhang ZN, Rong Z, Xu Y. Immunogenicity of induced pluripotent stem cells. Nature. 2011;474:212–215. doi: 10.1038/nature10135. [DOI] [PubMed] [Google Scholar]
- 79.Harrison NJ, Baker D, Andrews PW. Culture adaptation of embryonic stem cells echoes germ cell malignancy. Int J Androl. 2007;30:275–281. doi: 10.1111/j.1365-2605.2007.00762.x. discussion 281. [DOI] [PubMed] [Google Scholar]
- 80.Mayshar Y, et al. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell. 2010;7:521–531. doi: 10.1016/j.stem.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 81.Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132:661–680. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 82.Hanna J, et al. Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc Natl Acad Sci U S A. 2010;107:9222–9227. doi: 10.1073/pnas.1004584107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Buecker C, et al. A murine ESC-like state facilitates transgenesis and homologous recombination in human pluripotent stem cells. Cell Stem Cell. 2010;6:535–546. doi: 10.1016/j.stem.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li W, et al. Generation of rat and human induced pluripotent stem cells by combining genetic reprogramming and chemical inhibitors. Cell Stem Cell. 2009;4:16–19. doi: 10.1016/j.stem.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 85.Nichols J, Smith A. Naive and primed pluripotent states. Cell Stem Cell. 2009;4:487–492. doi: 10.1016/j.stem.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 86.Lowry WE, et al. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc Natl Acad Sci U S A. 2008;105:2883–2888. doi: 10.1073/pnas.0711983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huangfu D, et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol. 2008;26:1269–1275. doi: 10.1038/nbt.1502. [DOI] [PubMed] [Google Scholar]
- 88.Sommer CA, et al. Induced pluripotent stem cell generation using a single lentiviral stem cell cassette. Stem cells. 2009;27:543–549. doi: 10.1634/stemcells.2008-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Anokye-Danso F, et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell. 2011;8:376–388. doi: 10.1016/j.stem.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Woltjen K, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458:766–770. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Somers A, et al. Generation of transgene-free lung disease-specific human induced pluripotent stem cells using a single excisable lentiviral stem cell cassette. Stem Cells. 2010;28:1728–1740. doi: 10.1002/stem.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhou W, Freed CR. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem cells. 2009;27:2667–2674. doi: 10.1002/stem.201. [DOI] [PubMed] [Google Scholar]
- 93.Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008;322:945–949. doi: 10.1126/science.1162494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Okita K, Nakagawa M, Hyenjong H, Ichisaka T, Yamanaka S. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322:949–953. doi: 10.1126/science.1164270. [DOI] [PubMed] [Google Scholar]
- 95.Si-Tayeb K, et al. Generation of human induced pluripotent stem cells by simple transient transfection of plasmid DNA encoding reprogramming factors. BMC Dev Biol. 2010;10:81. doi: 10.1186/1471-213X-10-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fusaki N, Ban H, Nishiyama A, Saeki K, Hasegawa M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:348–362. doi: 10.2183/pjab.85.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim D, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009;4:472–476. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhou H, et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell. 2009;4:381–384. doi: 10.1016/j.stem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Warren L, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–630. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Miyoshi N, et al. Reprogramming of Mouse and Human Cells to Pluripotency Using Mature MicroRNAs. Cell Stem Cell. 2011;8:633–638. doi: 10.1016/j.stem.2011.05.001. [DOI] [PubMed] [Google Scholar]