

Abstract
Hepcidin (HAMP) negatively regulates iron absorption, degrading the iron exporter ferroportin at the level of enterocytes and macrophages. We showed that mice with β-thalassemia intermedia (th3/+) have increased anemia and iron overload. However, their hepcidin expression is relatively low compared to their iron burden. We also showed that the iron metabolism gene Hfe is down-regulated in concert with hepcidin in th3/+ mice. These observations suggest that low hepcidin levels are responsible for abnormal iron absorption in thalassemic mice and that down-regulation of Hfe might be involved in the pathway that controls hepcidin synthesis in β-thalassemia. Therefore, these studies suggest that increasing hepcidin and/or Hfe expression could be a strategy to reduces iron overload in these animals. The goal of this paper is to review recent findings that correlate hepcidin, Hfe, and iron metabolism in β-thalassemia and to discuss potential novel therapeutic approaches based on these recent discoveries.
Keywords: β-thalassemia, iron overload, hepcidin, Hfe, lentiviral vectors
Introduction
The liver peptide hepcidin (HAMP)1,2 is the key regulator of iron absorption. Hepcidin triggers the degradation of ferroportin (FPN),3 the only known iron exporter expressed by duodenal enterocytes, hepatocytes, and macrophages. Although the mechanisms that regulate this peptide are not yet fully understood, a series of interrelated factors have been shown to affect hepcidin expression. Hepcidin is down-regulated by erythropoiesis,4 anemia, and hypoxia,5 whereas it is up-regulated by iron overload6 and inflammation.5,7–9
One of the major complications of β-thalassemia is iron overload due to the increased iron demand associated with efforts to satisfy an augmented erythropoietic activity. Iron accumulation in different organs can lead to several complications including heart and liver failure, diabetes mellitus, and hypopituitarism.10 For this reason, a major focus of clinical management has been on chelation therapy as a means of slowing the progression of iron overload.11 In β-thalassemia major, where no β-globin is produced, iron overload is largely due to the blood transfusions needed to sustain life. However, many studies have also indicated that iron overload in β-thalassemia is associated with increased iron uptake from the diet. Patients suffering from β-thalassemia intermedia, a milder form of the disease, do not usually require transfusion therapy but eventually develop iron overload due to increased iron absorption from the gastrointestinal tract.10,12 Therefore, better understanding the mechanisms of increased dietary iron absorption in β-thalassemia, especially in nontransfused patients, could lead to new therapies to manage this problem.
Hepcidin, HFE, and iron overload in β-thalassemia
β-Thalassemia is characterized by ineffective erythropoiesis (IE), anemia, and iron overload. Recent studies point to the key role of hepcidin in modulating iron absorption in β-thalassemia. Hepcidin is a peptide hormone synthesized in the liver,2,13 which has been shown to be the primary regulator of iron metabolism.14 Hepcidin exerts its effect via binding to the iron exporter ferroportin and inducing its internalization and degradation.3 Blocking iron export from cells allows this peptide to control iron flow to the blood stream, including uptake from the duodenum, recycling within macrophages, and release from the storage compartment in the liver. Regulation of hepcidin expression is a complex process requiring integration of several signals including iron, hypoxia, erythropoietic demand, and inflammatory status.14 Iron accumulation and inflammation drive hepcidin expression higher to limit iron release into the blood stream. On the other hand, anemia and hypoxia strongly down-regulate hepcidin, increasing iron availability. In β-thalassemia patients, anemia and iron overload coexist and exert two competing signals in modulating hepcidin expression. The first evidence of hepcidin disregulation in β-thalassemia was reported in 2004, with low hepcidin expression in mouse models of this disease.15 Using mouse models of β-thalassemia intermedia (th3/+) and major (th3/th3), it has been shown that the level of IE determines organ iron distribution and the expression level of hepcidin.16 In th3/+ mice, iron accumulates progressively in the spleen and, at a lower rate, in hepatic Kupffer cells, whereas in th3/th3 animals, iron overload occurs rapidly and involves predominantly the liver parenchymal cells. Several reports also showed that the liver hepcidin mRNA level is excessively low in th3/th3 mice,15–17 where extreme IE is able to override an expected increase in hepcidin due to the high liver iron concentration. On the other hand, hepcidin is initially decreased in young th3/+ mice,15,17,18 although it increases over time, reaching the same level as that of wild-type (wt) mice. However, the up-regulation of hepcidin was not proportional to the concurrent increase of iron overload observed in the th3/+ animals.16 These data suggested that low expression of hepcidin could be responsible for the high iron levels in these animals.
Administration of Hamp25 peptide to thalassemic mice
Rivera and colleagues have shown that single 50-μg doses of HAMP25 are sufficient to prevent iron absorption in wt animals for up to 48 h.19 Using a similar approach, we administered a synthetic murine version (Hamp25) of the peptide to th3/+ and wt mice, and evaluated the effect on hematological parameters and organ iron content in vivo. The synthetic Hamp25 peptide was injected intravenously (i.v.). Injection of PBS was used as a negative control.20 All studies were done with age- and weight-matched 2-month-old female mice, kept on an iron sufficient diet containing 35-ppm of iron.20 Because th3/+ animals might require higher doses than wt mice to prevent abnormal iron absorption, we first tested the effects of single 50- and 100-μg doses delivered i.v., and found that 100 μg decreased serum iron levels in wt mice more than 50 μg.20 We also evaluated the effect of multiple doses of Hamp25 (50 μg) to maximize the effect on iron absorption. The Hamp25 was injected daily for 14 days, after which we monitored serum iron levels by a colorimetric assay,19 using serum from normal mice as a standard. We also measured organ iron contents and hematological parameters.20
After the last injection we observed decreased serum iron levels in both wt and th3/+ mice, compared to animals injected with PBS (N = 3–5 per group, Fig. 1A). However, measurement of organ iron contents did not show significant differences between Hamp25- and PBS-injected mice.20 However, hematological parameters such as Hb, RBC levels, and reticulocyte counts were not changed in wt and th3/+ mice treated with the peptide compared to PBS controls.20 Interestingly, quantitative real-time PCR analysis of the liver of these mice revealed downregulation of endogenous hepcidin expression after injection of the peptide (Fig. 1B). This observation suggests that a feedback mechanism is in place to adjust the concentration of hepcidin in the circulation (exogenous + endogenous). A possible mechanism might be that administration of Hamp25, by lowering the level of serum iron and the Tf saturation, triggers a physiological response to decrease expression of endogenous hepcidin. Alternatively, but less likely, the Hamp25 peptide itself might play a role in the regulation of hepcidin expression.
Figure 1.

Administration of a synthetic Hamp25 peptide reduces serum iron levels and down-regulates endogenous hepcidin expression. Wt and th3/+ mice (N = 3–5 animals/group) were injected once daily with 50 μg of Hamp25 for 14 days. Control mice were injected with PBS. (A) Serum was isolated from peripheral blood on days 0 and 14, and its iron levels measured by colorimetric assay (Thermo Electron Corporation, Melbourne, Australia), according to the manufacturer’s instructions. Both wt and th3/+ mice treated with Hamp25 showed a dramatic reduction of serum iron after 14 days of treatment, while no significant changes were detected in control mice. (B) Quantitative real-time PCR (Q-PCR) analysis on the livers from mice treated with Hamp25 and PBS showed down-regulation of endogenous Hamp expression after injection of the peptide. Error bars represent SDs calculated using at least three animals per group. The P values (*P <0.05) were calculated using Student’s t-test.
There are at least two explanations for these results. First, the dose of Hamp25 used may have been insufficient to reduce iron absorption to a level that would also reduce tissue iron levels in 14 days. Second, having observed that the level of endogenous hepcidin was reduced in treated mice, we speculate that the dose and the serum half-life4 of the Hamp25 may have been inadequate to compensate for the decreased synthesis of endogenous hepcidin. Therefore, future studies will focus on administering increased doses of Hamp25, to determine the optimal concentration capable of reducing iron overload without impairing erythropoiesis.
Use of lentiviral vectors expressing HAMP or HFE in a liver-specific manner
We showed that Hfe is down-regulated in the same way as hepcidin in thalassemic mice, suggesting a correlation between the two genes.16 Our data on wt mice have shown that the hepcidin mRNA level in the liver is 90 times higher than that of Hfe. In th3/+ mice, the hepcidin level is only 20 times higher than that of Hfe, because of the down-regulation of hepcidin.16 Therefore, a small increase in the expression of Hfe might up-regulate hepcidin to a larger extent. It is important to mention that by increasing hepcidin so as to reduce iron absorption, we could also impair erythropoiesis by reducing iron recycling through macrophages. Therefore, different strategies need to be developed to increase hepcidin levels so as to achieve a therapeutic dose of hepcidin that does not exacerbate anemia in th3/+ mice. To investigate a different approach to achieve the up-regulation of hepcidin in thalassemic mice, we produced lentiviral vectors designed to increase the expression of hepcidin in mice either directly or indirectly through the over-expression of Hfe. We generated three lentiviral vectors, named pCCL.TTR.Hamp.IRES.GFP.WPRE (THIGW), pCCL.TTR.Hamp.WPRE (THampW), and pCCL.TTR.Hfe.WPRE (THfeW), respectively, that carry either hepcidin or Hfe cDNAs and can express the proteins in a liver-specific fashion.20,21 A similar control vector, pCCL.TTR.GFP.WPRE (TGW), expressing the enhanced green fluorescent protein (eGFP) that allows for the identification of transduced cells, was generated as well. Tissue specificity was achieved using a promoter generated by fusion of the synthetic hepatocyte-specific enhancer with the murine transthyretin (TTR) promoter.21–23 All the vectors contain specific sequences that are able to improve lentiviral mediated gene transfer, and that are commonly inserted in HIV-derived vectors as two cis-acting elements. These are a polypurine tract sequence (cPPT) and a central termination sequence (CTS). They are present within the HIV polymerase (pol) gene, their function being to facilitate nuclear import of the viral preintegration complex.24 A second sequence element, termed woodchuck hepatitis virus post-transcriptional regulatory element (WPRE)25 was also inserted. WPRE is able to increase transgene expression in the context of plasmid DNAs or viral vectors.25 Viral stocks were generated by transient transfection into 293T cells as previously described.26–29 We generated two lentiviral vectors for the expression of Hamp, respectively named pCCL.TTR.Hamp.IRES.GFP.WPRE (THIGW) and pCCL.TTR.Hamp.WPRE (THampW). Because of the internal ribosome entry site (IRES) element, the THIGW vector expresses hepcidin and eGFP from a bicistronic mRNA, whereas eGFP allows for the identification of transduced cells.20 In addition, we produced a pCCL.TTR.Hfe.WPRE (THfeW) vector for the expression of Hfe, and a control vector pCCL.TTR.GFP.WPRE (TGW) expressing the green fluorescent protein (eGFP) (Fig. 2).20 All the vectors were used to transduce human and mouse hepatic cell lines, such as HepG2, Hepa1–6, and Hep3B, to determine whether they were suitable for expressing hepcidin or Hfe in a liver-specific fashion. Flow cytometric analysis (FACS) of HepG2 cells transduced with the TGW vector showed the percentage of GFP-positive cells to be 77%. GFP levels in HepG2 cells transduced with THIGW were slightly lower, showing 41% of GFP-positive cells (Fig. 3A).20
Figure 2.
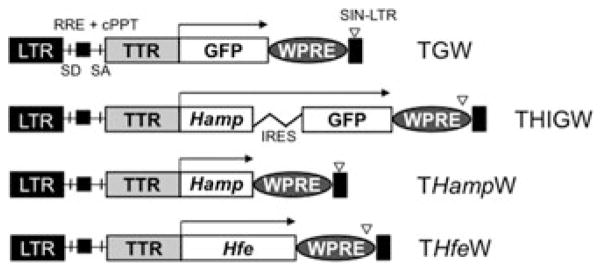
Schematic representation of the lentiviral vectors used during this study. The components shown are the long terminal repeat (LTR), the self-inactivating long terminal repeat (Sin-LTR), the HIV rev-response and central polypurine tract elements (RRE, cPPT), the splice donor (SD), the splice acceptor (SA), the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE), the liver-specific TTR promoter (TTR), the internal ribosome entry site (IRES), the enhanced green fluorescent protein (eGFP), and the mouse Hamp and Hfe cDNA sequences.
Figure 3.

(A) FACS analysis of the level of GFP in HepG2 cells transduced with the TGW and THIGW vectors. (B) Hamp, Hfe, GFP, and WPRE mRNA levels expressed by the TGW, THIGW, and THfeW vectors in HepG2 cells. Because the Hamp and Hfe sequences expressed by the vectors are originated from mouse, they were easily discriminated from the endogenous Hamp and Hfe sequences of HepG2 cells that are, instead, of human origin.
To measure the VCN we used Q-PCR analysis of WPRE levels (since WPRE is present in all the vectors) on DNA extracted from transduced HepG2 cells. WPRE values were normalized to endogenous control values obtained from amplification of the human ID1 gene. We observed an average copy number of 1.2 for THIGW and THampW, and 1.7 for THfeW.20 The TGW vector showed a higher VCN (2.1 copies/cell). In addition, we performed Q-PCR analysis on the cDNA from transduced HepG2 cells (human), using specific primers to detect the mouse hepcidin or Hfe mRNA sequences expressed by the vectors. In this way we were able to discriminate between the mRNAs produced by the vectors and the endogenous mRNA synthesized by the cells. Our results confirmed the specific expression of Hamp, Hfe, GFP, and WPRE by the vectors (Fig. 3B).20 From these analyses, the vectors appear suitable for in vivo experimentation. We plan to administer these vectors to thalassemic mice as a therapeutic tool to limit iron overload.
Future prospects
Developing new strategies to reduce excessive iron absorption and tissue iron overload is one of the most important goals in the treatment of β-thalassemia. The hepatic peptide hepcidin has an acknowledged primary role in regulating iron absorption and metabolism. We showed that hepcidin mRNA expression is low compared to iron burden in th3/+ mouse models of β-thalassemia, thus being responsible for abnormal iron absorption. Our studies aim to develop possible therapeutic strategies to up-regulate hepcidin levels in th3/+ mice.
Administration of a synthetic Hamp25 peptide showed effects on iron distribution in th3/+ mice, reducing serum iron levels but also down-regulating the expression of endogenous hepcidin. However, we did not observe significant changes in tissue iron concentrations. We hypothesized that these results might be caused by an insufficient dosage and/or length of treatment, so that only the more immediate effects on serum iron concentration were evident. In particular, it might be necessary to adjust the treatment with Hamp25 to compensate for the decreased synthesis of endogenous hepcidin.
From this perspective, the lentiviral vectors we used to transduce cells were able to express hepcidin, Hfe, and eGFP respectively, and to do so in a liver-specific manner due to the presence of the TTR promoter. We transduced common hepatic cell lines with the vectors, to assure high-transducibility, a good copy number, and high levels of expression. Moving forward, we will study the vectors in vivo to determine the level and stability of expression, as well as organ iron concentrations and hematological parameters.
Acknowledgments
This work was supported by the Cooley’s Anemia Foundation-CAF, the Associazione Veneta Lotta alla Talassemia (AVLT) (S.G.), and by grants from the Carlo and Micol Schejola Foundation, the Children’s Cancer and Blood Foundation and NIH-R21DK065169 (S.R.), R01DK55463 (R.W.G.), the American Portuguese Biomedical Fund (APBRF, USA)/Inova grant (P.R.). S.G. is a fellow of the Cooley’s Anemia Foundation. P.R is a fellow from Fundação para a Ciência e Tecnologia, Portugal (SFRH/BD/24813/2005).
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.Krause A, et al. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000;480:147–150. doi: 10.1016/s0014-5793(00)01920-7. [DOI] [PubMed] [Google Scholar]
- 2.Park CH, et al. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276:7806–7810. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]
- 3.Nemeth E, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 4.Pak M, et al. Suppression of hepcidin during anemia requires erythropoietic activity. Blood. 2006;108:3730–3735. doi: 10.1182/blood-2006-06-028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicolas G, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. 2002;110:1037–1044. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pigeon C, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276:7811–7819. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 7.Nemeth E, et al. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101:2461–2463. doi: 10.1182/blood-2002-10-3235. [DOI] [PubMed] [Google Scholar]
- 8.Nemeth E, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113:1271–1276. doi: 10.1172/JCI20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood. 2006;108:3204–3209. doi: 10.1182/blood-2006-06-027631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bannerman RM, et al. Thalassemia intermedia, with iron overload, cardiac failure, diabetes mellitus, hypopituitarism and porphyrinuria. Am J Med. 1967;42:476–486. doi: 10.1016/0002-9343(67)90276-8. [DOI] [PubMed] [Google Scholar]
- 11.Giardina PJ, Grady RW. Chelation therapy in beta-thalassemia: an optimistic update. Semin Hematol. 2001;38:360–366. doi: 10.1016/s0037-1963(01)90030-7. [DOI] [PubMed] [Google Scholar]
- 12.Pippard MJ, et al. Iron absorption and loading in beta-thalassaemia intermedia. Lancet. 1979;2:819–821. doi: 10.1016/s0140-6736(79)92175-5. [DOI] [PubMed] [Google Scholar]
- 13.Nicolas G, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA. 2001;98:8780–8785. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nemeth E, Ganz T. The role of hepcidin in iron metabolism. Acta Haematol. 2009;122:78–86. doi: 10.1159/000243791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adamsky K, et al. Decreased hepcidin mRNA expression in thalassemic mice. Br J Haematol. 2004;124:123–124. doi: 10.1046/j.1365-2141.2003.04734.x. [DOI] [PubMed] [Google Scholar]
- 16.Gardenghi S, et al. Ineffective erythropoiesis in {beta}-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood. 2007;109:5027–5035. doi: 10.1182/blood-2006-09-048868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weizer-Stern O, et al. mRNA expression of iron regulatory genes in beta-thalassemia intermedia and beta-thalassemia major mouse models. Am J Hematol. 2006;81:479–483. doi: 10.1002/ajh.20549. [DOI] [PubMed] [Google Scholar]
- 18.De Franceschi L, et al. Liver expression of hepcidin and other iron genes in two mouse models of beta-thalassemia. Haematologica. 2006;91:1336–1342. [PubMed] [Google Scholar]
- 19.Rivera S, et al. Synthetic hepcidin causes rapid dose-dependent hypoferremia and is concentrated in ferroportin-containing organs. Blood. 2005;106:2196–2199. doi: 10.1182/blood-2005-04-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gardenghi S, et al. Increased hepcidin expression in Mice affected by β-thalassemia reduces iron overload with no effect on Anemia. Blood. 2008;112:53. [Google Scholar]
- 21.Follenzi A, et al. Efficient gene delivery and targeted expression to hepatocytes in vivo by improved lentiviral vectors. Hum Gene Ther. 2002;13:243–260. doi: 10.1089/10430340252769770. [DOI] [PubMed] [Google Scholar]
- 22.Costa RH, Grayson DR. Site-directed mutagenesis of hepatocyte nuclear factor (HNF) binding sites in the mouse transthyretin (TTR) promoter reveal synergistic interactions with its enhancer region. Nucleic Acids Res. 1991;19:4139–4145. doi: 10.1093/nar/19.15.4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vigna E, et al. Efficient Tet-dependent expression of human factor IX in vivo by a new self-regulating lentiviral vector. Mol Ther. 2005;11:763–775. doi: 10.1016/j.ymthe.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 24.Follenzi A, et al. Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV-1 pol sequences. Nat Genet. 2000;25:217–222. doi: 10.1038/76095. [DOI] [PubMed] [Google Scholar]
- 25.Zufferey R, et al. Woodchuck hepatitis virus post-transcriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J Virol. 1999;73:2886–2892. doi: 10.1128/jvi.73.4.2886-2892.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zufferey R, et al. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol. 1998;72:9873–9880. doi: 10.1128/jvi.72.12.9873-9880.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zufferey R, et al. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol. 1997;15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]
- 28.Naldini L, et al. in vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector [see comments] Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 29.Naldini L, et al. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci USA. 1996;93:11382–11388. doi: 10.1073/pnas.93.21.11382. [DOI] [PMC free article] [PubMed] [Google Scholar]