

1. Introduction
Biologically-formed hard tissues, referred to as biominerals, have intrigued the materials engineering community for years because of the high degree of crystallographic control that is exerted during the precipitation of the bioinorganic crystals. In recent years, there has been a shift in attention, from prior studies that focused on specific organic-inorganic interactions that modulate the crystal morphology via the conventional crystallization pathway, to recent studies that find that many biominerals are formed via an amorphous precursor pathway. It has become clear that the things we thought we had learned about biominerals before, may or may not be relevant to truly understanding the mechanisms involved in biomineralization. Having witnessed this paradigm shift first hand, I am inclined to provide a review from this historical perspective, where I hope to belay some ideas about where we were, where we are, and where we are going, with respect to understanding how these shells and other biominerals are formed. Therefore, one goal of this review is to try and provide a link between the prior literature and the new literature, which might be useful to newcomers in the field, whom I suspect may find it confusing and difficult to integrate the findings in these different types of studies across this time period. A second goal is to try and integrate some of the knowledge obtained from in vitro model systems, which can be more amenable to obtaining mechanistic information, with the in vivo and ex vivo observational studies on biominerals. A third goal is to demonstrate that there may be certain unifying principles found in biomineral systems that seem widely diverse, such as from diatoms, to mollusk shells, to vertebrate bones and teeth. A final goal (the not so hidden agenda), is to demonstrate that not only is there as strong likelihood that many biominerals are formed by an amorphous precursor, but that the amorphous phase may possess fluidic properties that impart new processing capabilities to the system. Of course those who know my work will readily assess that I am referring to the polymer-induced liquid-precursor (PILP) process, which has been a primary focus in my lab. Along these lines, some new hypotheses are presented regarding the morphogenesis of certain biominerals, such as mollusk nacre, kidney stones, and bones and teeth, along with a review of the literature that provides support to these new ideas. The intent is to stimulate thoughtful discussions in this rapidly emerging area, which seemingly provides a unifying principle in biomineralization.
1.1. Biomineral Overview
Biominerals that have intrigued the materials engineer for years. The diversity of biominerals became particularly evident by the variety of books on biominerals that emerged in the late 80’s to 90’s,1–11 which has continued with a few more recent additions to this nice collection.12–16 Many people are probably not even aware of the diversity of biologically formed minerals, so a sampling is provided in Table 1 to illustrate some of the different chemistries that evolved in the formation of various biological hard tissues. The calcium carbonate (CaCO3) biominerals of invertebrates, such as mollusk shells and sea urchin spines, have been particularly well studied due to their accessibility, and because of the high degree of crystallographic control that is achieved in these biologically formed crystals. The calcium phosphate (CaP) biominerals of the vertebrates, such as bones and teeth, have also been extensively investigated because of their remarkable structure and mechanical properties, and for the more obvious reasons of health related issues.14 Calcium oxalate (CaOx) biominerals can be found in plants, but the majority of studies have focused on the pathological form of CaOx that is found in kidney stones, which often occurs in conjunction with CaP deposits.8,14
Table 1.
Examples of the diversity of biominerals. This list is by no means comprehensive, where some 70 different mineral phases have been identified to date.1–11
| Biogenic Minerals | Formula | Organism | Biological Location | Biological Function |
|---|---|---|---|---|
| Calcium Carbonates (calcite, vaterite, aragonite, Mg-calcite, amorphous) |
CaCO3 (Mg,Ca)CO3 CaCO • nH2O |
Many marine organisms, Aves,
Plants Mammals |
shell, test, eye lens, crab cuticle, eggshells, leaves inner ear | exoskeleton, optical, mechanical strength, protection, gravity receptor, buoyancy device, Ca store |
| Calcium Phospates (hydroxylapatite, dahllite, octacalcium phosphate) |
Ca10(PO4)6(OH)2 Ca5(PO4,CO3)3(OH) Ca8H2(PO4)6, ? |
Vertebrates Mammals Fish, Bivalves |
bone, teeth, scales, gizzard plates, gills mitochondria | endoskeleton, ion store, cutting/grinding, protection, precursor |
| Calcium Oxylates (whewellite, wheddellite) |
CaC2O4 •
H2O CaC2O4 • 2H2O |
Plants Fungi Mammals |
leaves hyphae renal stones |
protection/deterrent,
Ca storage/removal, pathological |
| Iron Oxides (magnetite, goethite, lepidocrocite, ferrihydrite) |
Fe3O4 α-FeOOH, γ-FeOOH 5Fe2O3 • 9H2O |
Bacteria,
Chitons Tuna/salmon Mammals |
intracellular, teeth, head, filaments, Ferritin protein | magnetotaxis, magnetic orientation, mechanical strength, iron storage |
| Sulfates (gypsum, celestite, barite) |
CaSO4 •
2H2O SrSO4 BaSO4 |
Jellyfish Acantharia Loxodes, Chara |
statoconia cellular intracellular statoliths |
gravity
receptor skeleton gravity device/receptor |
| Halides (flourite, hieratite) |
CaF2 | Mollusc, Crustacean | gizzard plate statocyst |
crushing gravity perception |
| Sulfides (pyrite, sphalerite, wurtzite, galena, greigite) |
FeS2 ZnS, PbS Fe3S4 |
Thiopneutes | cell wall | sulfate reduction/ion removal? |
| Silicon oxides (silica) |
SiO2 • nH2O | Diatoms Radiolaria Plants, etc. |
cell wall cellular leaves |
exoskeleton skeleton protection |
There are a variety of non-calcific biominerals as well, such as the biosilica found in diatoms, sponge spicules, and some plants; strontium sulfate lattices in acantharia (algae); iron oxides found in chiton and limpet teeth, as well as magnetotactic bacteria (including iron sulfides); and a most unusual copper hydroxide mineral found in the teeth-like jaws of a bloodworm.17 The diversity of biominerals and examples of their sophisticated structures has been presented in several excellent books and review articles, some of which were listed above. The focus of this review is to look for commonalities in the biomineralization process amongst these diverse organisms and their seemingly different biomineralized tissues, particularly with respect to the role of amorphous precursors in modulating the mineralization process. There are many figures in this review, but not with the intent to just show pretty biomineral pictures. Most were carefully selected to illustrate key morphological features that may be providing mechanistic information. By comparing the features observed from biominerals, and correlating them with the crystallochemical mechanisms deciphered from in vitro model systems, perhaps the materials chemist can begin to understand the physicochemical mechanisms involved in sculpting these beautiful biomineral structures.
1.2. Why are Materials Scientists Interested in Biominerals?
First, I will discuss in more detail some specific features found in biominerals that demonstrate why they are deemed worth studying. I am often asked by colleagues, why is there so much literature on CaCO3, when this material has a rather limited number of commercial applications? One answer to this question is that CaCO3 is a relatively easy model system to work with, and an endless variety of morphologies can be formed no matter what additive is thrown in the mix (hence, a large body of literature). But more seriously, biomimetic engineers hope to be able to extrapolate the knowledge gained from such model systems and apply it to other inorganic systems, including non-biological materials, to regulate crystallographic properties for advanced materials applications. For example, it would be useful to know how biomineralization processes provide for chemical, spatial, structural, morphological and constructional control of their inorganic constituents.13 In addition, in an economy that is increasingly becoming environmentally and energy conscious, there is great interest in the development of new approaches to ‘green chemistry’.
The biomimetic approach has been particularly successful in terms of mimicking some of the biomechanical attributes of biomineral tissues, such as nanolaminated composites mimicking nacre.18–22 On the other hand, these hybrid composites were not made using biomimetic processing techniques. While biomimetic synthetic strategies are finding some success,23–27 we have a ways to go before the full synthetic attributes, such as how biomineral tissues are molded and formed into hierarchical structures, can also be capitalized on for commercial products. For example, there is a desire to develop functional ceramics (such as ferroelectrics, piezoelectrics, magnetics, optics, dielectrics, etc.) with controlled structures (nano- and hierarchical) that can be built from the bottom-up approach. Some progress is being made in this area, particularly with the development of biopanning techniques that “evolve” inorganic binding peptides through the combinatorial approach to genetic engineering.28–34 These types of systems involve specific molecular recognition between the peptide and inorganic crystal, which can be used to nucleate or assemble inorganic crystals. Of course molecular recognition is a primary tenet of biological systems, at least with respect to the organic interactions found in proteins and DNA. However, the role of molecular recognition is less understood in the inorganic systems, and furthermore, if the crystal forms from an amorphous precursor, the role of such specific binding events in biomineral formation is not entirely clear. Before getting to this, I will present a short historical perspective on the traditional views of biomineral formation, which were initially focused on the conventional crystallization pathway and the specificity of molecular recognition events at the organic-inorganic interface. Then I will proceed into a discussion on new theories based on the presence of an amorphous precursor, returning to the same biominerals discussed in this historical perspective, but from this new found perspective. Given that this is a very lengthy paper, those who are already familiar with the older biomineral literature may want to jump ahead to Section 2, which provides a discussion on the amorphous precursor pathways. Those who are already familiar with crystallization via amorphous phases may want to jump ahead to Section 4, where new hypotheses are presented with respect to the crystallochemical mechanisms responsible for the distinctive morphological features found in biominerals.
1.2.1. The Exemplary Calcium Carbonate Biominerals of Invertebrates
The mollusk shell is a well-studied biomineral that nicely illustrates the high degree of control exerted in biological systems (Figure 1). For example, the shell contains several types of microstructural layers that are composed of different phases of CaCO3,35 where for example prismatic layers contain elongated prisms of calcite,36–40 while nacreous layers are composed of flat tablets of aragonite (Figure 1b–d).41–45 Then, within these layers, the crystallographic orientation is also regulated, with a preferred [001] orientation of the aragonite tablets in the nacreous layer. The shapes of the biocrystals have long been an enigma because neither the elongated prisms of calcite nor the flat tablets of nacre are comparable to the inorganically grown habits of calcite or aragonite, which typically form rhombohedral- and needle-shaped crystals (in the form of polycrystalline spherulites), respectively (Figure 2).
Figure 1.
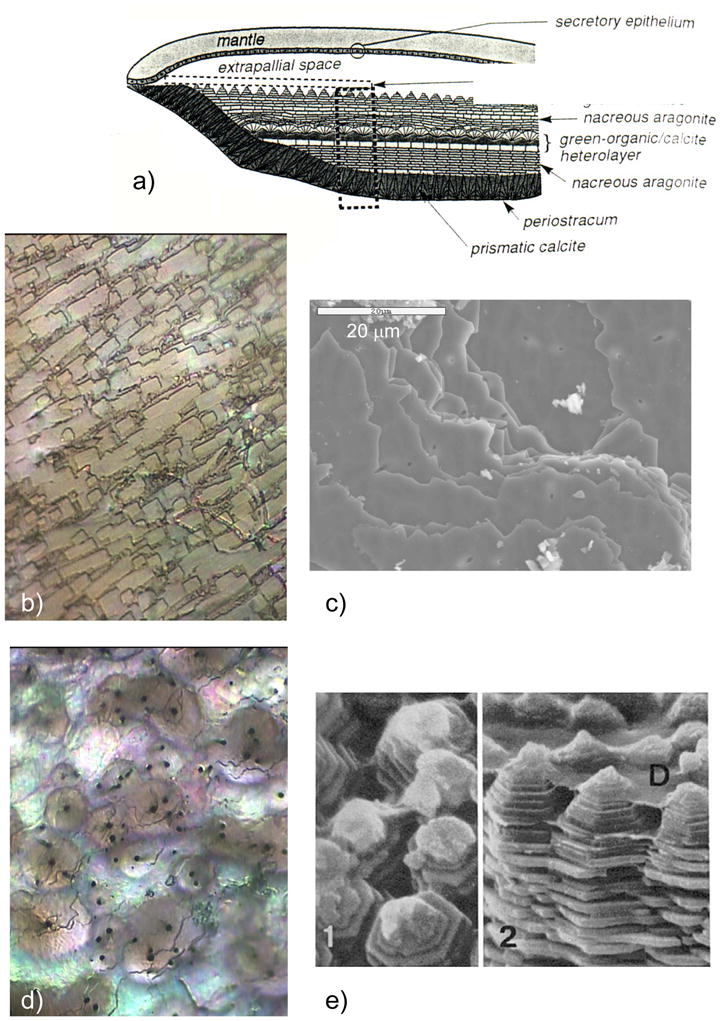
CaCO3 biominerals in mollusk shells, with comparison between sheet and columnar nacre. (a) A schematic provided by Zaremba et al.35 of the cross section of a shell illustrates the different layers within a mollusk shell, where the nacreous layers are composed of thin tablets of aragonite, while the prismatic layers are composed of elongated prisms of calcite. The epithelial cells in the mantel secrete the mineralizing reactants into the extrapallial space, which then deposit mineral layers modulated through interactions with the organic matrices or compartments. (b) Polarized light micrograph (reflection mode) of the upper growing surface of sheet nacre, which is most often found in bivalves, such as Atrina rigida shown here. (c) SEM of fractured sheet nacre also reveals smooth and continuous sheets of mineral, which are composed of aragonite crystals ~ 500 nm thick. (d) Polarized light micrograph (reflection mode) of the upper growing surface of columnar nacre, which is most often found in gastropods (although Geukensia demissa is pictured here). (e) SEM of the growing surface of columnar nacre from gastropods shows conical arrays of tablets. Tablet growth in the vertical direction is constrained by some type of compartment, but with further lateral expansion of the tablets, they impinge to form continuous aragonite layers (as seen at the bottom). Some groups suggest that mineral bridges span the layers keeping the crystallographic orientation within a column in registry. Pores, which could allow for such bridging, appear to present in the optical micrograph shown in (d). (a) (Reprinted with permission from ref 35. Copyright 1996 American Chemical Society.) (d) (Reprinted with permission from ref 325. Copyright 1979 Malacological Society of Japan.)
Figure 2.

Typical morphologies of CaCO3 crystals grown via the conventional crystallization pathway. (a) Faceted calcite crystals with rhombohedral habit. Bar = 100 μm. (b) Polycrystalline spherulites of aragonite formed with magnesium additive at a 50:10 mM concentration of Mg:Ca ion, simulating seawater concentrations. The partial spherulite on the right that has not branched out fully, and the morphology is referred to as a “sheath-of-wheat”. The needle-like morphology of the individual polycrystals is common for aragonite. Bar = 10 μm. (a) (Reprinted with permission from ref 203. Copyright 2000 Elsevier Science B.V..)
There are two main types of nacre, columnar and sheet nacre,3,42 both of which will be a focus of discussion in Section 4.1. In sheet nacre, found mostly in bivalves, flat tablets can be seen on the growing surface (Figure 1b), which grow laterally and fuse into a continuous mineral sheet (Figure 1c). Although the tablets are often described as having a hexagonal shape, images of the newly forming tablets on the surface show a diversity of shapes, ranging from round, to polygonal or hexagonal.41,46 In columnar nacre, found mainly in gastropods, there appears to be a high degree of locational control, where the aragonite tablets stack one upon each other to form columns as they grow (Figure 1d & e). In this case, the vertical growth proceeds faster than the lateral growth (by about 10 layers before the tablets come together46), so that the upper surface of the columns are conical until the tablets eventually expand laterally and fuse to then form continuous sheets of mineral (which can be seen at the bottom of the micrograph in Figure 1e).46
The layers of mineral in nacre are separated by an interlamellar organic matrix (referred to as a conchiolin membrane in some of the pioneering work on nacre),41,42,47–50 which according to Addadi et al.,51 consists of β-chitin with adsorbed acidic glycoproteins, which form a sandwich around a silk-like proteinaceous gel, within which the aragonite tablets grow. Some camps consider the uniform crystal orientation to result from epitaxial-like interactions with the organic matrix,52–55 while others suggest that simple growth kinetics can lead to the preferred orientation over substantial distances.56,57 It has also been suggested that the uniform crystal orientation within the columns of columnar nacre, or across layers of sheet nacre, could be carried over mineral bridges that span the conchiolin membrane through small perforations (which appear to be present in Figure 1d).44,46,58–60 This concept will be further discussed in Section 4.1.4. On the other hand, the originating [001] crystal orientation still needs to be regulated; and the evidence of neighboring tablets of uniform orientation in the a,b-plane over substantial distances has been considered to arise from the well-defined spatial relationship between the matrix (chitin fibrils) and crystallographic axes of aragonite.52–55 Rousseau’s recent work further supports the epitaxial hypothesis because the intracrystalline matrix was found to highly crystalline (see Figure 35 in Section 3.7), and when visualized with dark-field TEM using spots isolated for the organic matrix, the matrix appeared to be well connected (even with the interlamellar sheet), in a single-crystalline fashion within individual tablets.55
Figure 35.

Rousseau and coworker’s55 analysis of the intracrystalline matrix of nacre tablets. (a) Darkfield TEM image of nacre evidencing the crystalline structure of the organic matrix. Organic matter is in contrast when under Bragg conditions whilst the mineral phase remains systematically extinguished. (b) High-resolution TEM image of the square box region marked in (a), showing a ‘bridge’ in the interlaminar matrix between two tablets. The insets show the Fourier analyses of the two squared regions, evidencing only the highly crystallized organic matrix, and not the CaCO3 orthorhombic lattice of aragonite. Bar is 10 nm. (c) AFM topography(left) and phase contrast (right) images of the polished surface of nacre tablets. The phase contrast shows a foam-like structure of the intracrystalline organic matrix. (Reprinted with permission from ref 55. Copyright 2005 Elsevier Ltd..)
In summary, one can see that in the mollusks, control is exerted over crystal size, shape, texture, orientation, phase, and location. The formation of a crystal tablet may not seem extraordinary, but in the case of aragonite, it virtually never forms a single-crystalline, tabular morphology when grown synthetically. Instead, it is highly prone to twinning, and forms radiating needles in the form of a spherulite (Figure 2b). There are other biomineral examples with even more impressive morphologies that completely defy the symmetry of the underlying lattice.61 These biominerals often have smoothly curved surfaces, totally lacking the crystal facets that one would expect for a single crystal or lowest surface energy. A classic example is the spine of a sea urchin (Figure 3),62 which has a microporous and convoluted structure (Figure 3b), yet diffracts and behaves optically as single-crystalline calcite.63–66 Interestingly- the dimensions of the struts and pores in this bicontinuous structure are around 10 microns, orders of magnitude larger than synthetic bicontinuous structures generated in mesophase reactions (Figure 3c). When fractured, the spine exposes a conchoidal glassy fracture surface, rather than the well-defined cleavage planes of calcite (this is also seen in other echinoderm elements, sponge spicules, and foraminiferal shells67).54,68 The unusual properties are thought to arise from the presence of occluded intracrystalline proteins within the biocrystal,61,69–71 thus forming what I like to call, a single-crystalline composite (discussed further in Section 5.3). Synchrotron x-ray studies on sponge spicules, which also have symmetry-breaking morphologies, have shown (via coherence lengths and angular spreads) that there is a reduction in symmetry at the level of crystal domains, which is thought to be caused by selectively intercalated proteins.61,67,71
Figure 3.

Biominerals with ‘molded’ non-equilibrium morphologies are typical of the echinoderms, as demonstrated by the classic example- the sea urchin spine. (a) The urchin spine has a complex fenestrated structure, with smooth curved surfaces that lack facets, yet reportedly diffracts as single-crystalline calcite. The spines emanate from test plates of similar microporous structure, to provide exoskeletal protection to the organism. They are both composed of magnesium-bearing calcite. (b) A cross section of a spine from the urchin Arbacia tribuloides shows the typical microporous architecture. Bar = 1 mm. (c) Higher magnification of the fractured spine in (b) shows its internal bicontinuous morphology, along with a conchoidal, glassy fracture surface, which is a topic that has received considerable attention in the literature because calcite normally fractures along well-defined cleavage planes. Bar = 100 μm. (d) The structures throughout the spine are not necessarily homogeneous, which according to the prior hypothesized mechanism, would imply that many specific proteins would be required to modulate these different structures. Bar = 100 μm. (a) (Reprinted with permission from ref 67. Copyright 1993 American Associate for the Advancement of Science.)
The sea urchin is just one of the many examples of CaCO3 biominerals that exhibit such elaborate non-equilibrium morphologies (see for example coccoliths,72 foraminiferans,73 brittle stars74). The purpose of these elaborate morphologies is not always known, but at least in the case of the brittlestar, the array of calcitic mounds is thought to serve as a set of microlenses for gathering light.74 Lastly, it should be mentioned that these types of biominerals with non-equilibrium morphologies are usually formed within some type of membrane-bound vesicular compartment,75 highlighting the important role of compartmentalization during the ‘molding’ of the biomineral.72 In fact, in the case of larger biomineral elements, such as the urchin spine, a whole team of cells fuse their membranes to form a giant vesicle or syncitium,4 which expands the compartment as the biomineral grows to centimeters in length.
1.3. Biomineralization Mechanisms: The Historical Landscape
1.3.1. Calcium Carbonate Biominerals in the Invertebrates
Overall, one can see that the mechanism(s) involved in biomineralization enable control over crystal size, shape, orientation, phase, texture, and location (Figure 4). This is quite impressive considering the relatively limited set of reaction conditions that can be manipulated, because temperature and pressure are set by the physiological environment. Basically, the crystallographic control must be regulated through manipulation of the ionic reactants and/or the use of additives, which can be either inorganic dopants or organic additives (Figure 5). Magnesium ion, being present in large quantities in sea water (around 50 to 60 mM Mg2+, relative to the 12 mM Ca2+), can certainly be expected to influence the growth of CaCO3 crystals. Mg-ion has long been known to have an inhibitory effect on calcite growth.76 At low concentrations, Mg-ion alters the morphology of calcite to a prismatic form,77 while at high concentrations, it sufficiently inhibits the calcite phase to favor aragonite, typically in the form of spherulitic ‘dumbbells’. This inhibitory action is thought to arise from the increased solubility of the calcite phase, while the Mg-ion is not as effectively incorporated into the lattice of aragonite, so its solubility is less affected.76,78–80 On the other hand, there are many calcitic biominerals that contain quite high levels of magnesium (e.g., up to 30% in red coralline algae), at levels which generally don’t form during an inorganic precipitation (the relevance of this feature will be discussed in Section 5.1). Therefore, one can add composition, or impurity incorporation, to the already impressive list of crystallographic control.
Figure 4.
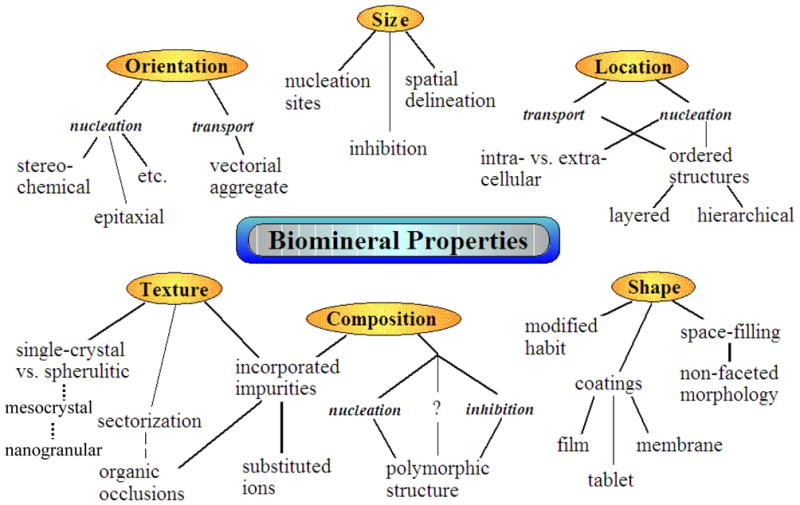
Crystallographic properties that are highly regulated during biomineralization, with some indication of potential processing strategies that provide for this crystallochemical control. Some of these features will form the basis of discussion in this review. (Reprinted with permission from ref 386. Copyright 2006 John Wiley & Sons, Ltd..)
Figure 5.
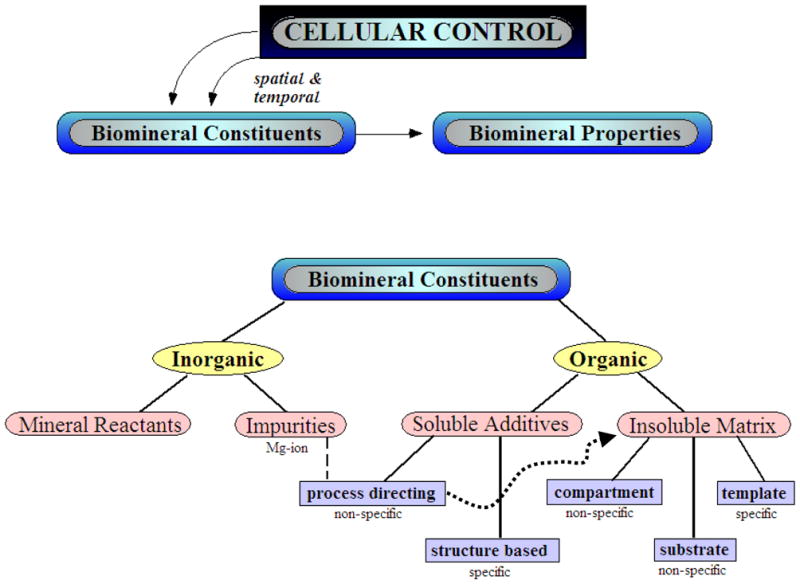
A simplistic view of the role of inorganic and organic constituents in regulating biomineral formation, keeping in mind that cellular processing provides for spatial and temporal control over the delivery of the species, and their subsequent organic-inorganic interactions. In addition to the obvious requirement of mineral reactants, the presence of inorganic impurities found in the physiological environment (such as Mg- and Sr- ions in sea water), can have a pronounced impact on the process, and particularly in combination with the soluble organic additives (such as acidic proteins), where both can synergistically promote the amorphous precursor pathway. The soluble additives, such as the acidic proteins, are considered as having either “process-directing” or “structure-based” interactions, where the former refers to stimulation of non-classical crystallization process (such as the amorphous precursor pathway), while the latter refers to the traditional view of additive interactions with specific crystallographic planes. Once an amorphous precursor is induced, the morphology can then be molded within vesicular compartments, or deposited on organic matrices as thin tablets, films, and coatings, where more specific structure-based interactions from a template may influence the crystallographic texture, phase and orientation. (Reprinted with permission from ref 386. Copyright 2006 John Wiley & Sons, Ltd..)
In addition to inorganic dopants, it has long been known that most biominerals contain some amount of organic material. This can range from the very small quantity of proteins occluded within the urchin spines (~0.02 wt%; about 10 proteins per 106 calcite unit cells68), to an interconnected “foam-like” network within individual tablets of sheet nacre,55 to a predominant matrix of collagen in bone (roughly 20% by weight, or 40% by volume).81 Two general categories of organics are described for biominerals, the insoluble matrix and the soluble additives. The insoluble matrix is composed of macromolecules that provide a framework or scaffold for the deposition of the mineral, such as chitin in the case of CaCO3 biominerals in some invertebrates, or collagen in the case of vertebrate bone and dentin. When this insoluble matrix is removed (such as by bleach), and then the remaining crystals dissolved (such as by weak acid), there is usually a small quantity of organic matter that remains, and this is composed predominately of water soluble, polyanionic proteins (or more commonly referred to as acidic proteins). These proteins tend to be highly negatively charged, far more so than any other proteins known (e.g. dentin phosphophoryn contains up to 70% charged amino acids), because they contain high quantities of aspartic and glutamic acid, as well as phosphorylated serine residues.82–87
The organic matrix provides unique mechanical properties to biomineralized tissues, which are optimized for both high strength and toughness, which is needed for load-bearing skeletal applications, or for impact and wear resistance in dental tissues or invertebrate exoskeletons. One important contributor is the nanostructural arrangement of the components, which is often organized into hierarchical levels of structure that make biominerals distinctly different than traditional engineering composites.45,81,88,89 In recent years, computational approaches have helped in deciphering the contribution of the organic phase towards toughening the brittle ceramic,90–92 and scanning force microscopy has also provided new insights into the nanomechanics of biomineral tissues.93–100
A feature that I find particularly fascinating about biominerals is that the organic matrix not only contributes to the biomechanical properties, but it is also contributes to the biosynthesis of the composite in the first place. It is generally considered that the insoluble matrix acts as a scaffold for establishing where the mineral will be deposited, as well as regulating the nucleation event in order to control crystal phase and orientation. Many biomimetic studies have used this approach in vitro to regulate the nucleation of a variety of inorganics grown on organic templates. Because of the complexity of the macromolecular matrices found in biominerals, many in vitro studies use simpler templates with well-defined chemistries and structures. For example, self-assembled monolayers (SAMs), which can be patterned on substrates using microcontact printing, provide templates of well-defined chemistry and spacing of functional groups.101 Aizenberg elegantly demonstrated the application of this technique for examining crystallization processes, by showing that patterned SAMs could serve as templates for locational control of the nucleation event,102 as well as modulate crystal orientation.102,103 In a similar approach, amphiphilic molecules can be spread at the air-water interface to form a Langmuir monolayer of well-defined chemistry, and this method provides some control over the spacing between functional groups through applied surface pressure. These monolayers have been used for patterning crystal location,104 but most commonly are used for examining the influence of organics on crystal orientation105–115 and crystal phase;116–119 but this approach (alone) generally has little to no impact on the crystal morphology (aside from partial habits expressed from controlled orientation)120 because the crystals grow outward from the template into the solution, generally taking on the traditional faceted equilibrium morphology. While Pokroy and Aizenberg121 have shown some shape modulation in calcite due to lattice mismatch between the monolayer and nucleating crystal face, the overall morphology is still one of well-faceted rhombohedral calcite.
While the insoluble matrix in biominerals can form a compartment, which plays an important role in delineating the shape of the forming crystal, an insoluble matrix in the form of a substrate has a relatively minor influence on crystal morphology, and is therefore often considered responsible for templating the nucleation event, thereby controlling crystal location, orientation and phase. Therefore, it has generally been assumed that crystal morphology in biominerals is regulated through interaction with soluble additives. The soluble polyanionic proteins, being found ubiquitously in nearly all calcific biominerals examined (including both vertebrate and invertebrate biominerals), have long been thought to play this type of role in regulating biomineral morphology. One prevalent hypothesis as to how this could be accomplished is through selective interactions of the additive with specific crystallographic faces, which alters the growth kinetics in those specific directions, thereby altering the crystal shape (Figure 6, adapted from these papers).54,122,123 The morphological influence of additives had been described in general crystal growth theory for some time,124 but to my knowledge, Addadi and Weiner were the first to suggest its potential applicability to biomineralization.85 It became a primary focus of this group’s research for some years, along with many others, who have demonstrated that an assortment of additives can alter the morphology of calcite crystals and other biologically relevant minerals.77,123,125–133 This was suggested to occur by adsorption to specific crystallographic faces, where the literature often represented some type of stereochemical or geometrical matching between the crystal lattice and the shape and/or spacing of charge groups on the additive.
Figure 6.

Modulation of crystal morphology through selective adsorption of additive onto specific crystallographic planes (adapted from various papers of Addadi et al.85). Some type of preferred interaction, either with lattice planes or with growth steps, alters the kinetics of growth in that direction. The stabilizing influence of an additive can even lead to the expression of new crystal faces that normally grow too fast to be expressed, as depicted in the bottom illustration for the C-faces.
More recent AFM studies indicate that this specificity in binding may actually be occurring at the growing step edges, and not the flat crystal faces.134,135 In any case, it is clear that additives can alter the growth kinetics and thermodynamics, where a stabilizing influence can lead to larger crystal faces; or an additive can even promote the expression of new crystal faces, which normally aren’t expressed due to high surface energy. Thus, it was considered that expression of the (001) planes in nacre tablets, which doesn’t occur in synthetic calcite or aragonite due to the high charge density of the (001) planes, could be due to selective interactions with soluble additives, such as the acidic proteins found associated with these biominerals.136 Likewise, the ‘sculpted’ non-equilibrium morphologies seen in various calcitic biominerals was also perceived as resulting from stereospecific interactions with soluble additives. The evidence to support this proposed mechanism was based in part on in vitro growth of crystals in the presence of additives, including proteins extracted from the biomineral.68,123,130,136,137 Although the elaborate biomineral morphology has not been duplicated with this approach, one can certainly see that the proteins influence crystal growth, and often produce the expression of new crystal faces, which although rough, have been correlated to the crystallographic planes expressed on the corresponding biomineral.85,138,139 This argument was further supported by the analysis of biomineral crystallographic textures, which as mentioned above, was suggested to arise from anisotropic intercalation of proteins after they had adsorbed to those specific crystallographic faces.61,67,71,130,140,141
The point I wish to make in this historical overview is that the additive interactions described for this mechanism of morphological modification are “specific” to a particular set of crystal faces/growth steps, and will be referred to here as ‘structure-directing’ additives. In contrast to this, I will be discussing the influence of ‘process-directing’ additives (Figure 5), which can modify the crystallization process by transforming the conventional crystal growth into an amorphous precursor process. Notably, the organic-inorganic interactions that lead to shape regulation in this case occur prior to the formation of any crystal structure, and therefore do not require interactions specific to crystal lattice arrangements. These process-directing agents can have a pronounced effect on crystal morphology, as well as other crystal properties, and thus provide an alternative explanation for the morphogenesis of biominerals. Because several of the biomineral systems that were originally thought to modified via specific structure-directing additives have now been found to form from an amorphous precursor (such as the examples mentioned above), the relevance of the once popular selective adsorption hypothesis has now come into question. We have suggested an alternative explanation for the anisotropic crystal textures which lay at the foundation of this hypothesis,142 as will be discussed in Section 5.2.3.
1.3.2. Calcium Phosphate Biominerals in the Vertebrates
The vertebrates primarily use CaP in their hard tissues, such as bones and teeth (with the notable exception of eggshells and otoliths of the inner ear). Bones and teeth have distinctly different biomineral features, probably because these tissues are created by cells from different lineages. Bones and dentin (the inner part of the tooth) are formed by osteoblast and dentinoblast cells, respectively, which are both derived from connective tissue; while dental enamel is formed by ameloblast cells, which are derived from epithelial tissue.2 There are interesting features in both with respect to the materials chemistry of their formation.
1.3.2.1. Vertebrate Bone (and Dentin)
In contrast to the beautifully sculpted crystals found in CaCO3 biominerals, the calcium phosphate (CaP) crystals found in bones are less interesting from a morphological perspective. For example, the hydroxyapatite (HA) crystals of bone (and dentin) are very small platelets, with rather irregular and ill-defined shapes, which are not readily identifiable at the micron scale (Figure 7a). The more exciting aspect of the vertebrate biominerals is the structural arrangement of the crystals. The structure is hierarchical,81 in which the nanoscopic platelets of HA are embedded within the fibrils of collagen (Figure 7b), which in turn are aligned roughly parallel within lamellae that are arranged concentrically around the vasculature, referred to as osteonal bone (Figure 7a). At the macroscopic level, the bone is then organized into trabecular (spongy) bone surrounded by more compact cortical bone. At the fundamental level of bone structure, which I will refer to as bone nanostructure, the HA crystals are crystallographically oriented roughly parallel to the long axis of the collagen fibrils (Figure 7b-inset), and given that they occupy roughly half the volume of the scaffold, one could consider bone to be an interpenetrating organic-inorganic composite. On a per weight basis, the composition of secondary bone is roughly 65:25:10 wt% (45/45/10 vol%) of Mineral:Organic:Water.81,143,144 Most of the organic component is fibrillar collagen, although there is a small amount (~3%) of non-collagenous proteins (NCPs) and polysaccharides present as well, which are thought to play an important role in the biomineralization process.82,83,86,145–149 There has been some ex vivo evidence from 3D-tomographic TEM imaging of naturally mineralizing turkey tendon, which has been used as a model of secondary bone formation (which is collagen directed), to suggest that the hydroxyapatite crystals may nucleate within the hole zones of collagen fibrils, as illustrated by the schematic put forth by Landis et al. (Figure 8).150 The intrafibrillar crystals then seem to outgrow the space of the hole zones, and spread throughout the interstices of the fibrils, eventually fusing to form an interpenetrating collagen-hydroxyapatite composite.
Figure 7.
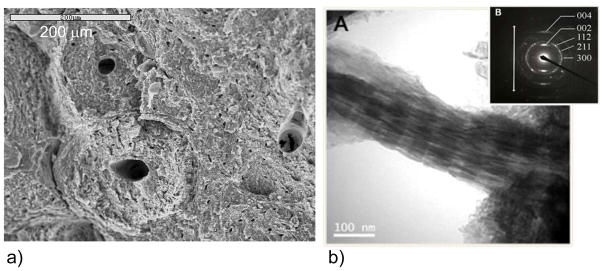
Bone is a hierarchically structure composite, composed of roughly equal volume fractions of organic and inorganic phases, along with around 10% water from the hydrated collagen matrix (or ~65:25:10 wt% of HA:collagen:H2O). (a) The microstructure of bone consists of concentric lamellae of mineralized collagen which wrap around the vasculature and nerves, referred to as osteons. (b) The focus of the discussion in this report is on bone’s nanostructured architecture, which can be seen via TEM to consist of nanocrystals of hydroxyapatite embedded within the collagen fibril. (inset) Selected area electron diffraction shows that the HA crystals are crystallographically oriented with their [001] axes roughly parallel to the long axis of the collagen fibril (indicated with arrow), and provide the dark striated contrast seen in this mineralized fibril extracted from equine cortical bone. The native banding pattern of type-I collagen can be observed due to the infiltration of the electron dense mineral, and staining was not used. Bar = 100 nm. (Reprinted with permission from ref 160. Copyright 2007 Elsevier B.V..)
Figure 8.

Schematic provided by Landis et al.150 depicting the early stages of mineralization in naturally mineralizing turkey tendon (a model of secondary bone formation), based on ex vivo TEM analysis with tomographic 3D reconstructions to determine the location and morphology of the newly forming crystals. The tropocollagen (triple-helical) molecules are depicted as cylinders, 300 nm in length by 1.23 nm in diameter, which self-assemble into a quarter-stagger array, leading to a periodicity of hole and overlap zones that is responsible for the 67 nm repetitive banding exhibited by type-I collagen. As described by Landis “Widthwise growth of crystals is not limited to single hole zones, but it may proceed into and beyond unoccupied hole zones in neighboring molecules. The (001) planes of developed crystals remain generally parallel. If two crystals form in adjacent zones, they apparently fuse to create a larger unit; development of crystals in this manner would lead to fused bands of mineral, irregular in c-axial length, along collagen hole zones in register and ultimately to thin parallel sheets of mineral throughout the assemblage of fibrils.” (Reprinted with permission from ref 150. Copyright 1993 Academic Press, Inc..)
The high degree of mineral loading imparts bone with high strength and rigidity, which is needed for providing skeletal support; and by surrounding the brittle ceramic particles with a ductile collagen matrix, bone also has remarkable toughness. The hierarchical structuring provides a means for varying the orientation of the fibrils, which in turn varies the orientation of the bioceramic-reinforcement nanocrystals, enabling the composite’s mechanical properties to be optimized depending on the directionality of the mechanical loading (which is sensed by the entrapped bone forming cells, called osteocytes). There are many excellent reviews on bone’s structure and mechanical properties,45,81,88,89 but the focus of this report is on the materials chemistry involved in the formation of bones and teeth, so a full list of citations on this nice body of work is not included here.
As was the case with CaCO3 biominerals, the CaP biominerals have historically been considered to arise from the conventional crystallization process (and still are by many). For example, because collagen itself does not seem to stimulate epitaxial growth of hydroxyapatite, it has been considered that some of the non-collagenous phosphoproteins might bind to the collagen fibrils to direct the nucleation event.145,151–154 Along these lines, Hoang et al.149 determined the x-ray structure of osteocalcin, which is the most abundant NCP, and found that the negatively charged surface coordinates five calcium ions in a spatial arrangement that is complementary to calcium ions in the hydroxyapatite lattice. However, an in vitro model demonstrating that an ‘epitaxial’ mechanism can lead to intrafibrillar mineralization has never been realized. On the other hand, there was some discussion in the older literature of an amorphous precursor in bone formation, and while some researchers found spectroscopic and microscopic evidence of what appeared to be an amorphous calcium phosphate phase;2,155–158 the issue was hotly debated, and around 25 years ago the debate was quelled when another group examined the early stages of bone formation in chick embryos and found no evidence to support the claims of an amorphous phase.159 We have reviewed this work in our recent MSER paper,160 and suggest that this issue needs to be revisited, as will be discussed further in Section 4.4. In addition, Crane et al.161 have recently found Raman spectroscopic evidence for an octacalcium phosphate precursor during intramembranous bone formation. It was suggested by them, and in a commentary by Weiner,162 that a transient amorphous precursor might also exist. In fact, in 2005 review paper discussing the prevalence of the amorphous phase in the invertebrates, Weiner et al.73 suggested that “It may be time to reconsider” the possibility of a transient precursor phase in bone. Indeed, this team has very recently (since my first draft of this review) found evidence of an abundant amorphous calcium phosphate phase in the continuously forming fin bones of zebrafish.163 Thus, my argument can now reallocated from suggesting this is a possibility to look for, to proposing a mechanism of how this might be accomplished (see Section 4.4).
1.3.2.2. Vertebrate Dental Enamel
The vertebrate tooth is composed of two types of biomineralized tissue, dentin (on the interior) and enamel (as an external abrasion resistant coating). Dentin is very similar to bone at the nanostructural level, in that it consists of collagen associated mineral, although the collagen is not organized into the higher level structures of osteons. Enamel, on the other hand, does not contain a collagen matrix, and is much more highly mineralized, thus providing a very hard and durable coating for the task of chewing. As mentioned above, ameloblasts are derived from an epithelial-based cell line, and therefore secrete a different set of proteins, which consists primarily of amelogenins and enamelins.2,4 The amelogenins are thought to self-assemble into spherical subunits which adsorb to and elongate the hydroxyapatite crystals,164,165 where they become arranged into long rope-like bundles, called rods or prisms, which can span across the entire thickness of the enamel. The hydroxyapatite rods are organized into higher level structures as they are “secreted” by the ameloblasts, as exemplified by the woven triple-ply architecture seen in the enamel of rodents (Figure 9a & b).4 During the maturation stage, nearly all the organic matrix is enzymatically degraded and removed, thereby leaving behind the most highly mineralized tissue of the vertebrates. The fascinating structure of enamel, in addition to highlighting the rather unusual ‘fibrous’ morphology of the constituent hydroxyapatite crystals (discussed further in Section 4.3.4), provides another remarkable example of the hierarchical structuring found in the vertebrate hard tissues.166
Figure 9.

‘Fibrous’ biominerals found in the teeth of both vertebrates and invertebrates. (a) Hierarchical structuring is seen in the dental enamel of vertebrates, which consists of polycrystalline “rods” or “prisms” of hydroxyapatite. In the rat enamel shown here, the structure resembles a ‘triple-ply weave’, where the ‘woven fibers’ are composed of hydroxyapatite. Other species display different 3-dimensional structures, but the fundamental prismatic foundation is the same. Bars are 10 μm (top) and 1 μm (bottom). (b) At higher magnification of a more mature tooth, one can see that the vertically-oriented prisms bend and wrap around each other to form the dense woven microstructure. (c) The tooth of a sea urchin is composed of “rods” or “needles” of calcite that emanate from large plates of calcite, and are embedded in amorphous (or poorly crystalline) CaCO3. (d) The “rods” can bend and curve, even though they are single-crystalline Mg-bearing calcite (~6 μm in diameter, but this can change throughout), and as in the urchin spine, represent non-equilibrium morphologies that expose unstable curved surfaces. (e) SEM of the longitudinal fracture plane of a chiton tooth, showing elongated ‘fibrous’ units of magnetite near the top of the tooth. x 6750. (f) Mineralized fibers in the tip of a Glycera (bloodworm) jaw composed of a copper- and chlorine-containing mineral called atacamite, (Cu2(OH)3Cl). Bar = 1 μm. (a & b) (Reprinted with permission from ref 438. Copyright 1984 Academic Press, Inc..) (c) (Reprinted with permission from ref 223. Copyright 2004 American Chemical Society.) (e) (Reprinted with permission from ref 169. Copyright 1990 with kind permission of Springer Science and Business Media.) (f) (Reprinted with permission from ref 17. Copyright 2002 American Associate for the Advancement of Science.)
1.3.2.3. Other ‘Fibrous’ Biominerals in Invertebrates
Interestingly, fibrous arrays of biomineral “rods” are also found in the teeth of invertebrates, such as sea urchins,3,167,168 as well as chitons and limpets (of the molluscan class).4 The teeth of the sea urchin consist of “rods” of calcite embedded in an amorphous CaCO3 matrix (Figure 9c & d).3 More recent analysis indicates that the matrix is microcrystalline Mg-bearing calcite.168 It is noted once again that calcite does not normally form a rod-like morphology (at least not when grown inorganically), and recalling the fenestrated structure of the spine and test, it is amazing that all of the mineral elements in this organism display curved surfaces, which should be energetically unfavorable for calcite. Overall, one finds that the phase, composition, texture, morphology, and crystallographic orientation, are highly regulated in the urchin.
In the case of chitons and limpets, the teeth are even further hardened by adding in an iron oxide phase, which presumably provides additional abrasion resistance since these creatures use their teeth to scrape algae off of rocks (Figure 9e).169 These organisms were some of the first examples found of biomineralization occurring within a preformed organic matrix. In addition, the teeth are interesting composite structures, were the chiton uses a combination of magnetite and dahllite (carbonate apatite), while the limpets use goethite and an opaline silica and/or a hydrous ferrous phosphate counterpart.170 Recent cryo-TEM studies on limpet teeth find that organic fibrils do not seem to form a pre-formed compartment per se, and ultimately get pushed aside, but some seem to be overgrown by the crystals, which form hollow tube-like structures of the mineral.171 They suggest that the orientation of the crystals is dictated during the nucleation event associated with the fibrillar matrix.171 Hollow tube-like structures have also been identified in the “fibrous silica” portion of limpet teeth.7,9 A recent addition to the fibrous teeth category is the bloodworm, which reinforces its teeth with an unusual copper-containing mineral (Cu2(OH)3Cl) called atacamite, which provides the weakly mineralized protein matrix with high abrasion resistance (Figure 9f).17
2. Crystallization Pathways
Until recently, a review article on biomineralization nearly always started off with a discussion on classical crystal growth theory. This was because it was assumed that crystalline biominerals formed via the conventional mechanism of crystal nucleation and growth. In other words, ionic crystals that precipitate from an aqueous physiological solution were assumed to form a critical cluster size that initiated nucleation, which then might grow via ion-by-ion addition to the surface, such as at a screw dislocation defect (if at moderate supersaturation). In recent years, we have come to learn that this classical crystallization pathway is not followed by a variety of biomineralizing organisms; instead many biominerals seem to form by an amorphous precursor pathway (as will be summarized in Section 2.1).
Although the majority of older biomineralization literature described the crystallization reaction in terms of the classical nucleation and growth regime, a notable exception can be found in the works of Stephen Mann, such as his 1983 paper on Mineralization in Biological Systems. 78 This paper had a profound impact on my view of biomineralization. He stated that “Since amorphous phases are more soluble than crystalline phases at equilibrium, it seems possible that the formation of amorphous precursors could be widespread in biomineralization.” This statement was based on his discussion of crystallization via a step-wise precursor pathway, which was illustrated with a reaction coordinate diagram, as shown here in Figure 10, with some modification. Empirical observations of such reaction pathways have long been observed, and are often attributed to the Ostwald-Lussac rule of stages, which specifies that if a solution is supersaturated with respect to more than one phase, the more soluble (least stable) phase is often the first phase to form.78,172 Given sufficient time and energy, the products should ultimately transform into the lower energy, more thermodynamically stable state. Mann’s example was focused on calcium phosphate, which at that time was commonly known to form from an amorphous gel or an octacalcium phosphate (OCP) crystalline precursor. As illustrated in Figure 10 (for CaCO3), the pathway followed for the multi-step kinetically driven reaction will depend on the relative heights of the energy barriers between each of the metastable states. For example, while calcite is the most thermodynamically stable phase, it can be preceded by the less stable phase(s) due to their lower energy barriers,173 which could be metastable vaterite or aragonite, or even the unstable amorphous calcium carbonate (ACC) under conditions of high supersaturation.174–178 Note- in the diagram in Figure 10, I have added a phase prior to the ACC phase in the pathway, called a polymer-induced liquid-precursor (PILP) phase, which is a highly hydrated phase considered to be even more labile than the solid amorphous phase. The reason for considering this a distinct phase will be discussed further in the following sections.
Figure 10.
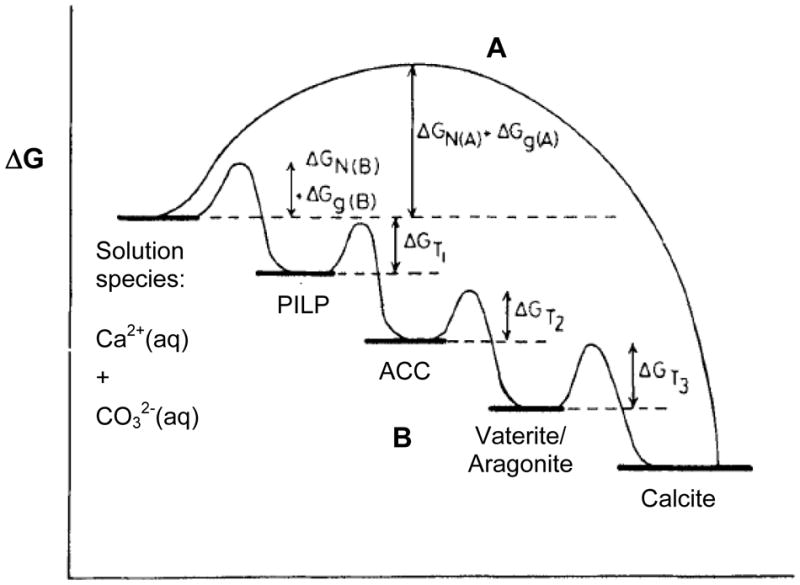
Reaction coordinate diagram representing different pathways to lowering the Gibbs free energy. Pathway A, which I will refer to as the conventional crystallization pathway throughout this manuscript, occurs for mineral precipitation de novo from solution, without passing through any intermediate phase(s) that require subsequent structural modifications. Pathway B demonstrates formation of a crystalline mineral from metastable phase(s) of different crystal structures, or even the amorphous phase. The energy barriers for the pathways differ, where the first step requires a ΔGNucleation + ΔGGrowth, while the subsequent steps only require a ΔGPhase Transformation. Calcium carbonate (CaCO3) is used as an example here, but it should be noted that the energy levels of the different CaCO3 phases and the heights of the energy barriers are not drawn to scale. A variety of pathways are possible, depending on the relative heights of the energy barriers, such as formation of ACC followed by transformation directly to calcite, and so on. Note- I have proposed a new step in the ACC pathway, which consists of a polymer-induced liquid-precursor (PILP) phase being a potential intermediate to the solid ACC phase.
One important contributor to such a multiple-step reaction is the surface energy, which is the hurdle that must be overcome whenever forming a new phase. In the case of an amorphous phase, it is reasonable to suggest that a less organized and therefore less dense phase will have a lower surface energy (due to fewer dangling bonds and uncompensated charges, and facilitation of surface relaxation), particularly if it is hydrated and more similar in chemistry to the surrounding aqueous environment.78
Another consideration is how the intermediate phase(s) transform to the final crystalline product. This will be discussed in detail in Section 3.4.2, but as presented by Mann,78 the growth of crystals from precursor phases of different crystallographic structure requires reorganization of the ions to new lattice coordinates. Phase transformations can occur via surface dissolution of the precursor followed by reprecipitation of a second phase, often upon particles of the initially formed solid (analogous to secondary nucleation). Alternatively, the second phase can be formed via an in situ solid state transformation, particularly when there is a close structural match and low interfacial energies between the two phases.
The presence of additives/impurities within the crystallizing medium can influence this crystallization pathway in several ways. If the additives are incorporated into the crystal, this can alter its solubility, thereby shifting the relative free energies of the phases. For example, Mg2+ion is known to raise the solubility of calcite, and because it doesn’t get incorporated into the aragonite lattice as readily, the aragonite phase will be favored when Mg2+ is present at sufficient levels (e.g., calcite is inhibited starting at around 2:1 of Mg2+:Ca2+).76,179,180 An additive can also influence the relative energy barriers between each reaction step, thus altering the kinetics of the transformation pathway. In this way, an additive can induce and/or stabilize the precipitation of a less stable phase. For example, Wan et al.181 examined the dynamic process of calcium carbonate crystallization by microcalorimetry, and found that L-Aspartic acid both accelerated the induction of vaterite, as well as stabilized the vaterite phase. Likewise, vaterite can be stabilized with polymers, presumably by capping the surface to avoid dissolution-recrystallization.182–185 The amorphous CaCO3 (ACC) phase, which is highly unstable, can also be stabilized, but usually for more limited lengths of time, using a variety of polymer additives.184,186–189 Huang et al.184 examined the influence of a series of molecular weights of polyacrylic acid (PAA- Na salt) on the formation of vaterite and ACC, and found that the higher molecular weights stabilized vaterite more effectively. On the other hand, the ACC product induced by the lowest m.w. (PAA1.2k-Na) was more stable than that induced by the higher m.w. (PAA25k-Na), in both the aqueous solution and dry state. They found that the strong inhibiting effects of PAA-Na could lead to CaCO3 particles through a nano-aggregation mechanism, and suggested that the spherulitic growth of vaterite seemed to be due to the insufficient inhibitory efficiency.
Navrotsky,190 who has rationalized the Ostwald step rule in terms of thermodynamic and kinetic principles, has shown that in addition to the kinetic stabilization of a metastable phase, the order of thermodynamic stability can also be inverted by a number of factors, including particle size and the presence of impurities (or structure-directing agents). For example, she argues that the three anhydrous polymorphs of CaCO3 are close enough in free energy that changes in surface chemistry and impurity effects can conceivably cause a cross-over in stability, which means that the phase that crystallizes may be controlled by thermodynamic as well as kinetic factors. This is primarily the case at the nanoscale, in which some precipitations and crystallizations occur by accumulation of clusters and nanoparticles. She describes a possible scenario for silica based biominerals (in diatoms and sponge spicules), but this may also be true of the calcific biominerals that appear to form from nanoclusters, as will be discussed in Section 5.2.1.
Before leaving this section, it should be noted that there are other non-classical crystallization pathways, including the interesting mesocrystal assembly system,191,192 which has also been postulated as playing a role in biomineralization. Cölfen and Antonietti have recently put forth a book entitled Mesocrystals and Nonclassical Crystallization,193 which provides an excellent overview of the subject. Such systems quite often make use of amorphous precursor nanoparticles, and as such, will be discussed further in Section 5.2.2.
2.1. Evidence of the Precursor Pathway in Biomineralization
I am presenting this review from a historical perspective, because it has been exciting to see how quickly this new paradigm has evolved. Virtually no one was talking about amorphous precursors when I entered the field as a graduate student in 1990. Now- the literature abounds with reports of amorphous phases, as I soon came to discover when I undertook the task of writing this review. I can hardly keep up with the incoming papers, and therefore offer my sincere apology to anyone I have left off.
2.1.1. From a Historical Perspective
A recent paper by Addadi51 notes that the first suggestion of a transient amorphous CaCO3 precursor in mollusk biominerals was by Towe and Hamilton, who back in 1968 observed hollow “crystals” in the developing nacre of the bivalve, and suggested that “a possibility exists that the incipient calcification is not in the form of aragonite but rather in some other phase”. Around that same time, several other groups had also been discussing an amorphous calcium phosphate precursor in bone formation.155,194,195 Nevertheless, these studies were evidently considered inconclusive, and the biomineralization community turned its attention to the more conventional crystallization pathway. To my knowledge, the first fully accepted demonstration of the amorphous precursor pathway being utilized in biomineralization was that of the chiton teeth, in which Lowenstam and Weiner196 who showed with IR that “the first-formed calcium phosphate mineral is amorphous. Over a period of weeks the mineral transforms to dahllite” (a poorly crystalline carbonated apatite, as found in bone). This biological organism was particularly valuable because the radula (a tongue-like organ) contains a continuously growing array of teeth (Figure 11a & b),170 which allowed them to locally examine the various stages of the biomineralization reaction. It was readily determined that the mineral is formed within a preformed organic matrix that is constructed in the shape of the tooth (Figure 11c), which then becomes surrounded by conduits that somehow supply the mineral precursor. Another interesting feature about this organism is that iron oxide mineral is formed in the posterior (scraping) edge of these teeth,197 which was later found to be formed from an amorphous precursor as well.170 In this case, the amorphous iron oxide seems to be stored as loose aggregates of ferritin-like assemblies called sidersomes (Figure 11c–e). Ferritin is a well-known globular protein used for intracellular iron storage in both prokaryotes and eukaryotes, which keeps the iron in a soluble and non-toxic form. The ferritin assemblies seem to attach to the fibrils (Figure 11d), which apparently builds up to generate the fibrillar texture of the magnetite. Thus it seems these organisms found a way to manipulate that operable source of iron for other functions, such as mechanical reinforcement. Redox potentials must also be taken into consideration in the materials chemistry of the iron oxides, such as during the storage and transport, and then subsequent crystallization of the mineral precursor.
Figure 11.

Biomineralization of chiton teeth, which are composed of sections containing dahllite and magnetite. (a) Optical micrograph of a chiton’s radula, which is a ribbon-like organ containing transverse rows of teeth. The teeth are progressively being formed along its length, which can be seen by the transition from orange (‘rusty’ ferrihydrate) to black (magnetite). (b) SEM of the rows of radular teeth. (a & b) Bar = 500 μm. (c) The preformed compartment of a chiton tooth, with a large collection of siderosomes (ferritin assemblies) gathering around the compartment, which are somehow transported through the surrounding tubules into the compartment to provide the amorphous iron oxide precursor. Bar = 1 μm (d) Mineralization within the cusp of chitons begins with the deposition of very small spherules which contain ferrihydrate, the amorphous iron oxide phase. The amorphous phase crystallizes into regions containing goethite and lepidocrocite or magnetite (which is black). The spherules appear to have an intimate relationship to the organic matrix, which consists of parallel arrays of fine fibrils that seem to organize the resulting acicular crystals. Bar = 1 μm (e) TEM of one siderosome is shown here, which is composed of an aggregate of ferritin cores. Bar = 0.2 μm. (a, b) (Reprinted with permission from ref 170. Copyright 1989 Wiley-VCH Verlag GmbH & Co. KGaA.) (c-e) (Reprinted with permission from ref 343. Copyright 1985 with kind permission of Springer Science and Business Media.)
With respect to CaCO3, the possibility of an amorphous precursor, however, was more elusive. While it had been known for some time that some organisms could produce and stabilize ACC phase,54,198 this was generally considered a separate biomineralization feature, and the relevance of the precursor pathway to the crystalline biominerals was not yet recognized. This time delay was due, in part, to the difficultly in examining biomineralization reactions in vivo, so most studies rely on ex vivo examination of extracted biomineral elements, and the presence of an unstable and transient phase can be difficult to detect. The amorphous phases that had been established were found because they are stabilized, either by a vesicular compartment or by macromolecules, and therefore were more readily detected. For example, some plant leaves contain cystoliths, which are grape-like bundles of spheroidal CaCO3 that provide storage of the calcium.199 Likewise, some crustaceans also store CaCO3 in the form of amorphous spherules,87 presumably because its higher solubility allows it to be more readily mobilized during the molting and rebuilding of the calcified exoskeleton.
Another reason the biomineralization community did not consider the precursor pathway is because much of the research had become focused on the role of ‘specific’ interactions between the organic matrix and the inorganic crystals, as mentioned in Section 2.1. This seemed to change in the late 90s, when Aizenberg et al.,200 who had been examining the occlusion of such proteins in the spicules of the sponge Clathrina, made an interesting observation. She found that the sponge spicules were composed of a crystalline calcitic core and an outer layer of amorphous CaCO3. This intriguing composite, composed of different phases of the same material, evidently prompted them to take a different look at the associated proteins, where instead of examining the morphological influence of the protein extracts, they now demonstrated that proteins extracted from this outer layer could stabilize the ACC phase in vitro.200 Right around this same time, experimentalists using in vitro model systems (such as myself) demonstrated that crystals grown in the presence of polypeptides mimicking the acidic proteins associated with biominerals, such as polyaspartic acid, followed a different mechanistic pathway- an amorphous precursor pathway.201–203 In this case, rather than stabilizing an amorphous phase indefinitely, it was found that by utilizing a transient amorphous precursor, a variety of the enigmatic features found in biominerals could, for the first time, be reproduced in the beaker! Similarly, Qi and Colfen found that complex morphologies of CaCO3 and BaSO4 were formed in the presence of double-hydrophilic block copolymers, and these were also found to be formed from aggregation of amorphous nanoparticles.204,205 Thus, it was becoming clear that the benefits of forming an amorphous phase could be extended well beyond ion storage, and that the role of soluble proteins in biominerals might differ from what had previously been considered. From my perspective (which may be biased), this series of events seemingly started the search for the elusive amorphous precursor in biomineralization.
As mentioned above, evidence of an amorphous phase can be difficult to detect, particularly when in the presence of crystalline phase, due to broadening of signals from the less homogeneous structure of the amorphous phase (such as in x-ray diffraction). In addition, the amorphous phase is usually unstable, particularly so for CaCO3, so it may convert to the crystalline phase during biomineral sample preparation for ex vivo examination. This has been directly observed in the spicules of sea urchin larvae, which were purified for examination by x-ray absorption spectroscopy (XAS).206 Because of the size and accessibility of the larval spicules, they could be examined by XAS in situ as well, which was found to more accurately reflect the amorphous content in the forming spicules. Likewise, Weiss et al.207 report that the entire larval shell of mollusks was susceptible to radiation damage when examined in SEM, presumed to be caused by dehydration or internal conformational changes. Given this assortment to hurdles in detecting the amorphous phase, it is perhaps not surprising that this discovery was so long in waiting, because it basically required someone to go looking for it. And indeed, now that people are looking for it, they keep finding it.
To my knowledge, the first report to describe a transitory ACC phase in biominerals was that by Beniash et al. in 1997,208 who examined spicule formation in sea urchin larvae (Figure 12). The spicules had been known to be comprised of single-crystalline calcite, but upon finding the presence of an additional ACC phase which transformed into calcite with time, this group indicated that “this observation significantly changes our concept of mineral formation in this well-studied organism.” Indeed, with this new insight, further work from this group has gone on to show ex vivo evidence of an amorphous precursor in the regeneration of the adult urchin spine,209 as well as the teeth of sea urchins,168 and they suggest that deposition of transient amorphous phases may be a useful strategy for producing single crystals with complex morphology. Furthermore, this group has identified proteins, as well as magnesium ions, as playing a role in the formation and stabilization of the ACC phase in the urchin larval spicules.210 For this, in vitro studies were used to determine that macromolecules extracted at the early stage of spicule growth could induce the formation of a transient ACC phase when in the presence of Mg-ion, while macromolecules extracted from the later stage, more crystalline biomineral, induced the formation of single crystals. Interestingly, their studies show that the biogenic transient amorphous phase is both structurally and compositionally different from the known stable ACC phase, and what is particularly surprising is that the transitory biogenic ACC reportedly does not contain bound water.210
Figure 12.
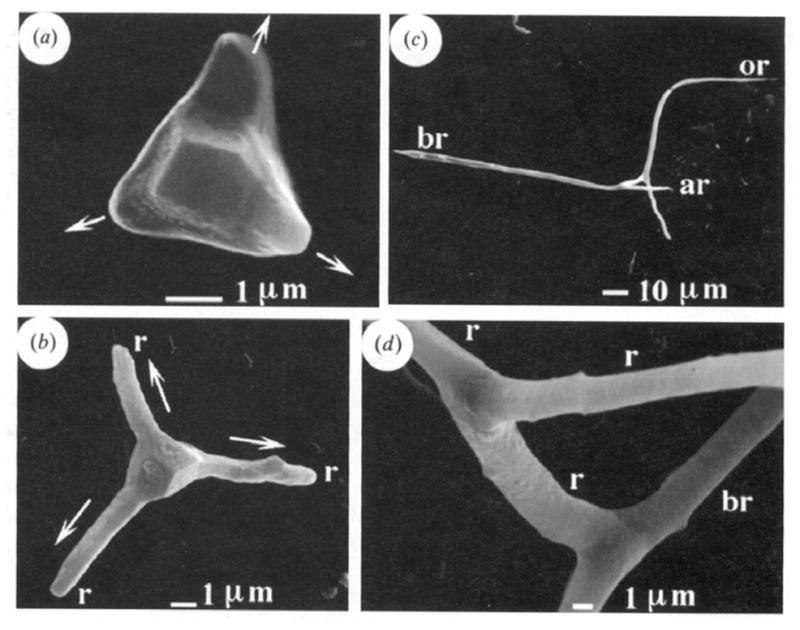
Developmental stages of endoskeletal spicule formation in sea urchin larvae. (a) At the earliest stage (20 hr), a rhombohedral-shaped calcite crystal is formed, and three branches are starting to emerge along the a-axes of the seed crystal (arrows). (b) At 25 hr, a well defined tri-radiate spicule is seen. (c) At 48 hr, the spicule is fully developed, where a change in growth direction created 3 rods growing roughly in the c-axis direction. (d) The central tri-radiate portion no longer shows the faceted calcite seed. (Reprinted with permission from ref 208. Copyright 1997 The Royal Society.)
Gayathri et al.211 have also examined the influence of the soluble, intracrystalline protein fraction on calcite growth, in this case, using proteins extracted from the ossicles of a sea star. These organisms are from the same phylum as the sea urchin (echinodermata), and similarly, the ossicles have a bicontinuous strutted morphology which closely resembles the inner-most region of the urchin spine. They find that the intracrystalline proteins contain unique glycine-rich polypeptides, and these seem to accelerate the conversion of the metastable ACC phase into its final crystalline phase of magnesium calcite. The in vitro system did not, however, lead to as high a level of magnesium incorporation as biogenic calcite, which can reach 20 mol% Mg (Mg incorporation is an important issued, and will be discussed further in Section 5.1).
The precursor story became even more exciting when an ACC precursor was found to also occur in the mollusks, which are from an entirely different branch of the animal phylogenetic tree than the echinoderms, such as the urchin (Deuterosomes vs. Protostomes, respectively).210 Weiss et al.207 found that the larval shells of two marine bivalves contain an ACC precursor for aragonite. Likewise, Marxen et al.212 found that shell formation starts with accumulation of calcium below the periostracum (the outer shell organic lining) at an age of about 60 hours, with the first precipitate being ACC. Interestingly, X-ray absorption spectroscopy (EXAFS) studies revealed that an aragonitic structural order was already present in this x-ray amorphous phase. In adult nacre, Nassif et al.213 have since shown, using HR-TEM, that the aragonite platelets in adult nacre are surrounded by a continuous layer of ACC. They point out that because there is a layer of ACC between the protein matrix and underlying crystal, the popular assumption of an epitaxial match between a structural organic matrix and nucleating crystal could not be responsible for the controlled [001] crystal orientation. They argue instead that the simple presence of charge interactions could extend across the 3 – 5 nm ACC layer, providing a less specific physicochemical mechanism for crystal alignment. In contrast, Addadi et al.,51 who also consider the tablets to be formed via some colloidal ACC phase from within a silk-like protein gel, maintain that specific aragonite nucleating proteins are used to control crystal orientation.
In support of the indirect evidence of ACC in adult bivalves mentioned above, the Weizmann group40,214 has also considered the prismatic layers as being formed from an amorphous precursor. Asprich proteins were extracted from the prismatic layer of the mollusk Atrina rigida, and using an in vitro crystallization assay, they find that the Asprich-c protein can induce and transiently stabilize the deposition of ACC. This is a relatively low M.W. protein of 21.8KDa, and contains ~ 60% acidic residues. They also examined mineralization of a chitin scaffold in the presence of the Asprich (as well as polyaspartate for comparison), and found that the chitin fibers were decorated with small spherical mineral particles, similar to their ex vivo observations of prism formation.
Complimentary to the work on bivalves, Hasse et al.215 have found ACC in the other class of molluscs, the gastropods. They examined the shells of a tropical freshwater snail, Biomphalaria glabrata, with high resolution synchrotron X-ray powder diffractometry and X-ray absorption spectroscopy (Ca K-edge EXAFS), and found the presence of aragonitic structure in the X-ray amorphous sample of 72 hour old eggs.
Bringing even more diversity into the picture by considering vertebrates, Lakshminarayanan et al.216 have examined quail eggshell formation (which rapidly forms polycrystalline calcite), and suggest the possibility of an amorphous precursor because ACC was precipitated in vitro using the soluble, intracrystalline organic matrix extracted from the eggshells. Eggshells are interesting because they are the most rapidly forming biomineral, and the calcitic mineral is formed in extracellular matrix. They found that the two major constituents of the organic matrix, acidic glycoprotein Ovomucoid and basic protein lysozyme, were not effective at inducing the amorphous phase, while the fraction containing low molecular weight peptides did induce ACC. This matrix was rich in Glu(n) and Asp(n) amino acids, as is true for the soluble matrix of many biominerals.
With respect to bone formation, there seems to be a new found interest in searching for an amorphous precursor here as well, where Weiner162 has recently speculated on this possibility in a literature review of Crane’s work,161 who recently found evidence of an OCP precursor. We have also proposed this possibility based on our in vitro model work,160 which not only suggests that ACP is likely involved in bone formation, but demonstrates an unsuspected mechanism that might help to explain how intrafibrillar mineralization is achieved (described in Section 4.4).
3. Biomimetic Model Systems Incorporating the Amorphous Precursor Pathway
The biomimetic approach provides a toolbox for materials engineers who wish to utilize biological processing methods for the development of advanced materials. However, before we can make the most out of this approach, we need to understand how to use these tools. Therefore, there is another category of biomimetic experiments, where the materials scientist is trying to model biological systems in order to elucidate the mechanisms involved in those biological processing methods in the first place. In this case, the value of biomimetic model systems is that it allows one to examine crystallization reactions in a well defined environment, where a particular response can be more readily correlated to the organic interactions that are responsible, and in turn be correlated to the features observed in the biological system that is being modeled. This is the general approach that (hopefully) will enable us to sort out the roles of the various additives or matrices in biomineralization. The following categories are fairly representative of the historical types of biomimetic studies:
the modulation of crystal morphology via selective binding to crystallographic faces
the inhibitory or promotory influence of an additive on crystal nucleation or growth
the templating of crystal nucleation to yield specific crystal phase or orientation
For such systems, the influence of the soluble additive has traditionally been considered to arise from molecular recognition between some specific structure of the additive with the organized lattice of the crystal. Now that we have convincing evidence that a multitude of biominerals utilize an amorphous precursor pathway, one must ask- how do these older experiments correspond to the mechanisms in biomineralization? For example, the following three cases illustrate that with this paradigm shift, we may need to rethink some of the conclusions derived from the older studies.
Case 1: For the first case, it was mentioned earlier that it is no longer clear how stereoselective protein binding to crystal faces would play a role in the morphogenesis of biominerals, since the crystal shape may already be dictated by the amorphous precursor (note- I am referring to morphological control only). On the other hand, the evidence for this hypothesis seemed quite strong. In the original reports, proteins extracted from the spines of sea urchins were found to specifically bind to different crystallographic planes,70,137 as evidenced by the expression of a new {10l} set of crystal faces. Proteins were occluded within the final crystals as determined by quantitative amino acid analysis.71 In support of this claim, ex-vivo synchrotron x-ray diffraction studies of biological specimens showed anisotropic defect texture of the urchin spine,61,67,71,130,141 suggesting an anisotropic distribution of proteins along select crystallographic directions within the biomineral. Our work, using an in vitro model that incorporates an amorphous precursor pathway, may help shed some light on these seemingly contradictory hypotheses (i.e., shape regulation through stereoselective adsorption versus molding an amorphous precursor). We argue that, based on the observations of transition bars during the amorphous-to-crystalline transformation, the anisotropic distribution of proteins occluded within biominerals may result from the anisotropy of polymer exclusion during the precursor transformation stage, rather than anisotropic inclusion of the proteins via stereospecific adsorption (see Section 5.2.3).142
Case 2: An analogous scenario can be described for the second case. Inhibitory/promotory experiments are particularly prevalent in studies on pathological biomineralization, such as kidney stone formation. It would be nice to be able to say that proteins that inhibit crystal nucleation of CaOx and CaP in vitro play a protective role against stone formation. This issue came up once before, when it had been shown some time ago that acidic macromolecules, which are inhibitory when in solubilized form, can promotory nucleation when adsorbed to a substrate.153,217,218 In fact, some additives exhibit different behavior depending on their concentration.219 But now one must consider another possibility, that those same ‘inhibitory’ molecules (such as acidic proteins) may in fact be responsible for generating an amorphous precursor, which throws an additional level of complexity into the picture because this pathway can lead to pronounced aggregation tendencies, as will be discussed in Section 4.5. Thus, one may need to take a different approach to examining the so called “inhibitory” proteins, and examine these proteins in vitro with an appropriate amorphous pathway model.
Case 3: Considering the third case, it is not clear how organic matrices will influence crystals that nucleate from within an amorphous precursor, rather than from the conventional pathways (i.e., ion-by-ion deposition from solution). Clearly there are diffusion and transport issues associated with the ability of ions to re-organize within a densified precursor phase. As an example, we find that the same organic templates (Langmuir monolayers) that lead to well oriented calcite crystals can lead to aragonite tablets when the crystallization proceeds via an amorphous precursor.119
Hopefully, I have convinced the reader that in vitro model systems that incorporate an amorphous precursor can provide new insights into potential biomineralization mechanisms, enlightening us to special issues that, in all probability, were not even considered in the model systems of the older literature. That is not to say that all the older experiments were not valid, and indeed much valuable information has been gained from model systems examining organic-inorganic interactions in general, but studies that utilized specific proteins extracted from biominerals may be less reliable in terms of relating the action seen in vitro to the protein’s real function in vivo. As described in several examples given in the Historical Perspective of Section 2.1, some of the more recent in vitro studies are now focused on trying to understand how an amorphous precursor phase is formed, or stabilized through interactions with extracted proteins (and/or Mg-ion). In this section, I will describe other in vitro systems which utilize synthetic analogues to the proteins found in the biominerals, which might be considered rather simplistic systems, but because of this simplicity, may help to further clarify the crystallo-chemical mechanisms involved in such precursor pathways, and without some of the conflicting issues described in Section 3.6, which discusses with issues associated with trying to model biomineralization reactions.
My group has been examining an amorphous precursor model system, but as was eluded to in the Crystallization Pathways Section 2, this system is somewhat different because it passes through a liquid-phase amorphous precursor. This does more than just add on an extra step along the kinetic pathway; it can have a profound effect on the crystal products. Therefore, I will start off by describing this crystallization process, which we call a polymer-induced liquid-precursor (PILP) process. There are some similarities to, as well as some differences from, the conventional view of an amorphous phase, as will be pointed out. It remains to be proven if this PILP process plays a role in biomineralization, and this will likely prove to be a difficult task given the metastable and dynamic nature of the phase (see Section 3.3). Nevertheless, the value of having such an in vitro model system is that it can provide mechanistic information to suggest how certain biomineral features might be created. The general concept of an amorphous precursor has finally taken hold and become well accepted; but the community may not be as convinced as I am that a liquid-like amorphous phase provides an even further revelation in understanding biomineral formation. Therefore, the following sections of this paper will emphasize how the liquid-like character of a “PILP” amorphous phase, as opposed to the more conventionally accepted (solid) ACC phase, could play an important role in the crystallization process. First, a brief description of the PILP process will be provided for those who are not familiar with our work. Those who are may wish to skip ahead to Section 3.2, for other related systems, or Section 4 for a discussion on its potential relevance to biomineralization.
3.1. The Polymer-Induced Liquid-Precursor (PILP) Process
The PILP process was first discovered for CaCO3, and therefore the observations for this system will be described first, before discussing other inorganic systems that we and others have examined. In the CaCO3 system, a popular way to slowly raise the supersaturation of the crystallizing solution is to use the vapor diffusion technique,218 which capitalizes on the decomposition of ammonium carbonate powder into ammonia and carbon dioxide, which gradually diffuse through the vapor phase of an enclosed chamber into a Petri dish containing a solution of calcium chloride (typically 10 to 20 mM Ca2+). In our experiments, prior to diffusing in the carbonate counterion species, a polymeric process-directing agent, such as polyaspartic acid-sodium salt, is added to the calcium solution so that it can interact with the calcium ions. The concentration of the polyaspartate additive we use depends on a variety of factors, such as its molecular weight, the use of other additives (e.g., Mg-ion), and the inorganic system and organic matrix, etc.; therefore, it may range anywhere from 1 μg/ml to 100 μg/ml (micromolar quantities), as described in our various literature reports.
A schematic illustrating the steps of the PILP process is provided in Figure 13, and representative images that correspond to those steps are shown in Figure 14. As the supersaturation is gradually increased with the addition of carbonate counterion (via infusion of NH3 and CO2), a critical concentration is reached at which point the solution undergoes liquid-liquid phase separation (Figure 13a and 14a). This can be observed macroscopically as streaks of cloudiness appear throughout the solution, which arises from the scattering of light off droplets of an amorphous precursor.220 Note- I refer to these as “droplets”, rather than particles, because this amorphous phase is so highly hydrated that it behaves as a liquid (to be described more fully below). The droplets settle and adsorb to the substrate (such as a glass microscope coverslip), and if the substrate is favorable (i.e., can be wetted), the droplets coalesce into a film or coating (Figure 13b and 14b).202,203,221 The amorphous film can then be observed to crystallize using polarized light microscopy (as well as diffraction techniques), eventually leading to a birefringent film of CaCO3 (Figure 13d, and 14d). The important point to recognize here is that the crystals retain the shape of the precursor phase (which is delineated by phase boundaries of the precursor). Whether it be round crystal ‘drops’,203 or thin films and tablets;119,180,202,203 or templated221 and molded222 crystals and fibers223,224, this mechanism, in principle, provides a means for forming an endless array of crystal morphologies!
Figure 13.
Illustration depicting the stages and features of the PILP process. (A) As a critical concentration is reached during the infusion of the carbonate species, isotropic droplets (~2 - 5 μm in diameter) phase-separate from the solution and accumulate on the substrate. (B) The droplets coalesce to form a continuous isotropic film. Some late-forming droplets may be partially solidified, or crystalline, and do not fully merge with the film. (C) Patches within the isotropic film become birefringent as crystal patches nucleate and spread across the precursor film. The transformation sometimes progresses in an incremental fashion, where prominent transition bars form from diffusion-limited exclusion of the polymeric impurity. The linear bars delineate sectors within single-crystalline calcite tablets, but are concentric within spherulitic films that transform in the radial direction. (D) The tablets continue to transform as the crystals grow laterally to form a continuous film. The transformed film is about half a micron thick, and composed of single-crystalline patches of calcite, or spherulitic patches of vaterite, which range from tens to hundreds of microns in diameter. (Reprinted with permission from ref 203. Copyright 2000 Elsevier Science B.V..)
Figure 14.
Optical micrographs of stages of the PILP process representing some of features illustrated in Figure 13. Micrograph (a) was taken in brightfield mode (to see the amorphous phase), while (b–d) were taken with crossed-polars to highlight the birefringence of the crystalline phase. (a) PILP droplets floating in the solution (~2 μm in size) are just barely visible in the early stage, when they are not dense. There seems to be some tendency to aggregate, such as the short chains seen here. (b) Coalescence of PILP droplets leads to smooth films, which become birefringent as they crystallize. In this case, a stream of PILP droplets flowed by the slide to form streaks of ACC film. Using the 1st-order red (gypsum) wave-plate, one can see both the amorphous regions (which are the same color as the magenta background), and the crystallizing patches, which display orange-n-blue birefringence. Large brown aggregates are also seen, which are common side products in this system. (c) During the amorphous-to-crystalline transformation, prominent transition bars (of micron periodicity) can be seen, and in this patch of film, they clearly divide the tablet into sectors as it transforms into single-crystalline calcite. (d) A mosaic film composed of single-crystalline patches of calcite. The dark regions are crystals oriented in the extinct position. This film was removed from the solution partway after it had started to crystallize, and exhibits a more pronounced defect texture in the outer regions where it crystallized after being air-dried. The regions that crystallized while still in solution are seen as the brighter rhombic patches in the center. (c & d) (Reprinted with permission from ref 203. Copyright 2000 Elsevier Science B.V..)
As the amorphous phase crystallizes, it tries to exclude the polymeric impurity. While much of this polymer goes back into solution,225 some polymer is not able to escape and becomes entrapped in the films as they solidify. We have considered this to be a diffusion limited process because of the in situ observations of transition bars during the amorphous to crystalline transformation.142,203 The transition bars appear as periodic striations in the birefringence because the polymer-enriched zones crystallize more slowly, and therefore remain isotopic (non-birefringent) longer (Figure 13c and 14c). The incremental exclusion of polymer has been confirmed using fluorescently labeled polyaspartate, where it was found that the transition bars are highly enriched with entrapped polymer (see Figure 58h in Section 4.5).142 A somewhat similar periodic texture, albeit polycrystalline, has been observed for BaCO3 grown in the presence of double-hydrophilic block copolymers, and an alternative explanation based on the Beloussov-Zhapotinsky reaction was suggested as a possible mechanism leading to the concentric banding pattern in this system.226 Although the mechanism underlying the transition bars may not be fully understood, the relevance of these features to biomineral textures will be discussed in Section 4.5.2.1 and Section 5.2.3.
Figure 58.

Features of PILP formed CaOx and CaP crystals of potential relevance to kidney stones. (a) Polarized light micrograph (with gypsum wave-plate) of CaOx particles formed with polyaspartate, with morphologies suggestive of a PILP precursor. Film patches are similar to those seen in the CaCO3 PILP system, and appear to have been formed by precursor droplets, many of which are not yet birefringent. A few large spherulitic dumbells are also present, which is common for CaOx, and often found in urine. (b) Numerous small spherical particles (~2 μm in diameter) appear to be single-crystalline ‘droplets’ (they do not contain Maltese crosses). A depression can be seen in many of particles, which is likely from dehydration of the original precursor droplet. They seem to be aggregating around and adsorbing to a large, spherulitic structure in the center. (c) SEM of a CaP spherulite which is relatively densely filled, showing a core-shell texture and some concentric laminations. These spherulites differ from solution grown spherulites, which are generally composed of more isolated crystals, such as the spiky needles seen in left spherulite in (f). (d) Multi-laminated spherulites formed from a gelatinous CaP precursor to hydroxyapatite. The weak birefringence and near transparent texture indicates these particles are not yet very crystalline. A lot of these globby spherulites seem to have a particle in the center, which has either densified first, or may have served as a nidus for accumulating droplets in a way that seemed to be occurring in figure (b). (e) The CaP globules are also fairly soft at this point, where they collapsed when dried on a substrate. (f) These highly birefringent CaP spherulites appear to be more crystalline, but are not yet dark brown, which would be expected for transmission optical microscopy on fully dense structures of this size (roughly 50 microns). The dark brown regions appear to be undergoing surface dissolution and recrystallization into needles. The concentric laminations are pronounced, and in the oblong agglomerate on the right, the laminations follow the contours of the coalesced globules. (g) Spherulitic films of CaCO3 also show concentric laminations midway in the A-to-C transformation, which we refer to as transition bars. (h) Fluorescently-labeled polymer (FITC-polyaspartate) demonstrates that the transition bars are created by diffusion-limited exclusion of the polymer during the A-to-C transformation. (Reprinted with permission from ref 386. Copyright 2006 John Wiley & Sons, Ltd..)
The liquid-like character of the early-stage amorphous precursor is evidenced by the coalescence of the droplets, which grow from tens of nanometers to a couple of microns. More direct evidence can be obtained by physically probing larger accumulations of the precursor, which flows like a slightly viscous liquid.203 Our light scattering studies suggest that aggregation of precursor droplets, rather than atom-by-atom growth, is the dominant mechanism of particle growth at this stage of the reaction.220 In other words, nanoparticles that come together do not agglomerate into a multi-particulate cluster, but instead coalesce to form a larger droplet, until they eventually become too solidified to fully coalesce. Once the droplets or films solidify and crystallize, they become highly birefringent. The particles shown in Figure 15a crystallized while in solution, and exhibit a uniform birefringence, suggesting that they are single crystalline, even though they have a spherical morphology (they are not spherulites, which would exhibit a Maltese -Cross pattern). Interestingly, these spherical crystal “droplets” have threaded disclination-like defects (the black lines move as the sample is rotated). They are, however, not liquid crystals. They are fully solidified and crystalline calcite at this point. The disclination-like defects probably arise from dehydration stresses which cause a steady shift in the crystal lattice, resembling the splay seen in liquid crystals.
Figure 15.

Crystallization of CaCO3 via colloids. (a) Single-crystalline ‘droplets’ formed from crystallization of PILP droplets floating in solution (~3 μm in size). As seen by the cross-polarized light, these crystals are not spherulites (no Maltese-Cross), and instead display thin curving lines that resemble disclination defects seen in liquid crystals. Bar = 50 μm. (b) Brightfield micrograph of a globular aggregate of PILP droplets collected at a late time-point, where the particles had grown quite large (5 to 10 μm), yet still exhibited substantial coalescence. (c) AFM of calcite films patterned on microcontact-printed SAMs. The colloidal texture is only apparent in the non-preferred region of the pattern, where the droplets did not ‘wet’ the substrate. A very smooth film was formed in the preferred region (COO−- functionality), where early stage precursor droplets coalesced into a smooth film, while the late stage droplets that deposited in the non-preferred regions solidified without ‘wetting’ the substrate or coalescing into a film. (e & f) AFM height mode images of a CaCO3 film deposited under a resorc[4]arene Langmuir monolayer (using 4 μg/mL PAA and 10 mM Mg2+), showing a colloidal texture, with nanograins ~30nm in diameter. The top surface of the film (e) was somewhat rougher than on the bottom surface of the film (f). (a) (Reprinted with permission from ref 203. Copyright 2000 Elsevier Science B.V..)
The crystal ‘drops’ are rare. More commonly, the droplets deposit on a substrate and coalesce into continuous films or coatings, as shown in Figure 14. The fluidity of the precursor is more obvious when the droplets only partially coalesce, such as the lobular aggregate shown in Figure 15b, where the large (micron sized) droplets were apparently partially solidified by the time they joined together. By extracting droplets from a reaction vial at various time points, one can see that as the droplets grow with time, they also solidify with time; therefore, the films that are formed by early stage droplets, which are nanoscopic in size, are very smooth at the micron scale, while late stage droplets only partially coalesce, and yield very bumpy films. This is also evident in the ‘wetting’ behavior of the droplets, which form very smooth films on substrates favorable for deposition, but form rounded particles on unfavorable substrates (Figure 15c & d). Notably, when the amorphous film crystallizes, the single-crystalline patches of calcite that form are orders of magnitude larger than the nanoparticulate constituents, remnants of which can only be detected by the colloidal texture when visualized at high magnification with AFM (Figure 15e & f).221 Section 5.2.1 will discuss the significance of the colloidal texture with respect to the nanogranular textures observed in biominerals.
If the reaction solution is not fully inhibited, large aggregates of crystals will also form, which are either composed of vaterite spherulites or calcite rhombs (as seen in Figure 14b, along with the films). The calcite rhombs often appear heavily distorted, and the aggregates seem to be ‘cemented’ together, which suggests the involvement of some contribution of precursor phase in leading to these hybrid ‘molten’ structures. As a side note- this aggregation tendency of crystals that grow from an amorphous phase prompted us to suggest that such a process may be responsible for some pathological biominerals, such as kidney stones (see Section 4.5).
3.1.1. Supporting Literature on a Liquid-Phase Amorphous Precursor
There are now other reports that also describe a liquid-like character for some amorphous phases, and this seems to have become a more widely accepted concept. In fact, it appears that the liquid phase may even occur without the use of polymeric process-directing agents; however, it is quite short lived, making it more difficult to detect, and more importantly, more difficult to utilize for crystal control. For example, Rieger et al.227,228 have examined the earliest formed precipitates (milliseconds to minutes) in the CaCO3 reaction using cryo-TEM and in situ x-ray microscopy, and describe the first-formed products as resembling an emulsion, even though no organics were present (Figure 16). Their system used a rapid mixing of CaCl2 and Na2CO3 solutions and, because it appeared to undergo spontaneous phase separation, they suggest that the reaction may follow the spinodal route because homogeneous supersaturation may be achieved fast enough in their system to pass through the binodal region of the phase diagram. The early stage liquid-like phase is short-lived in this system, and subsequently densifies and decomposes into nanoparticles (Figure 16b–c), which aggregate to form vaterite spheres several microns in diameter (Figure 16d). These spheres then undergo dissolution-recrystallization to form the more thermodynamically stable state of calcite rhombohedra (Figure 16d). These findings show that that even when the crystal products appear to have grown by the conventional process, as judging by the faceted, equilibrium type of morphology, the underlying path may be far more complex than previously realized.
Figure 16.

Stages of CaCO3 precipitation induced with rapid mixing of reactants, without additives. (a) Cryo-TEM shows an “emulsion-like” precipitate at 100 ms, and (b) after minutes, the densification and breakdown into nanoparticles, which as shown in (c) are non-diffracting ACC. (d) SEM of the products at 60 minutes shows vaterite spheres and calcite rhombs. (Reproduced by permission from ref 227. Copyright 2007 The Royal Society of Chemistry.)
They also examined the reaction in the presence of polycarboxylates. A 70:30 copolymer of acrylic:maleic acid (Mw = 70,000 g/mol) was included in the reaction, which was found to stabilize the nanoparticles against compact aggregation, forming flocs, the stability of which depended on whether there was sufficient polymer to cover the particles. Although the polymer stabilized the amorphous phase in the state of nanoparticles, it is not clear that it helped to stabilize the liquid-like state of the reaction. I typically judge this ability by the presence of coalesced mineral films; but this may not be possible where rapid mixing conditions are used, or an unfavorable substrate is not wetted by the precursor phase. In addition, we have observed a strong dependence on polymer molecular weight, where more granular precipitates are formed using a higher molecular weight polymer such as that used in Rieger’s studies. These observations may suggest that lower molecular weight polymer contributes more to the hydration character of the precursor; but further work is needed to verify this.
Wolf et al.229 have also found that a liquid-like amorphous CaCO3 phase can be formed, even without polymer, and without rapid mixing of reactants. Their system was raised in supersaturation using the Kitano method (CO2 escape) so that the reaction could be performed within an acoustically levitated droplet to avoid heterogeneous nucleation on surfaces. The reaction was monitored in situ via synchrotron WAXS, correlated with cryo-SEM examination of the intermediate reaction products. They conclude that “the formation of an amorphous liquid-phase mineral precursor seems to be a characteristic of the truly homogeneous formation of calcium carbonate itself,” and suggest that the non-crystalline phase is favored because of the variable coordination geometries and coordination numbers that are possible with the various carbonate species, creating a network with a distribution of local structures.
Zhang et al.182 have described an “amorphous liquid-like CaCO3–polypeptide precursor” using polyaspartate additive. A slight spreading was observed on the bottom of their particles, demonstrating the “soft” character of the microspheres. However, it seems that continuous films do not form in their full reaction (unless the solution is dried down); but instead, spherical assemblies of primary nanoparticles are formed, which then aggregate and reorganize into larger spheres of vaterite (seemingly similar to Rieger’s studies mentioned above). Their reaction conditions are very similar to our PILP system, with slow vapor diffusion and polyaspartate of moderate molecular weight (MW = 14,900), but they used a 100-fold higher polymer concentration. This apparently caps the primary nanoparticles and keeps them from fully fusing into films. In fact, the nanoparticles could be re-dispersed upon sonication. In comparison of their system to ours, which employ quite similar reaction conditions, one can conclude that the concentration of polymer can play a role in determining the fusion characteristics of the early-stage nanoparticles. While these authors were interested in the assembly and re-organization capabilities afforded by the higher polymer concentration, our focus has been on determining the optimal polymer concentration for allowing coalescence of the precursor droplets. For this, we find a more optimal concentration is in the range of 10 – 100 μg/ml, an order of magnitude lower than the 1 mg/ml used in their study. However, even at this lower polymer concentration, a colloidal texture can be observed by AFM in our products, although not nearly as pronounced as in the Zhang system. This may be related to a small amount of polymer capping, or it could arise from other factors, as described in Section 5.2.1.
Xu et al.230 have examined ACC hemispheres which deposit on mica or PDADMAC modified surfaces, and find that the ACC exhibits wetting behavior similar to that of liquid droplets. They suggest that “ACC in highly supersaturated solutions spontaneously forms liquidlike colloids with an open structure in solution, tends to coalesce, and deforms into hemispheres or forms continuous films when deposited on substrates.” They conclude (as do we) that “many different structures of CaCO3 biominerals in organisms may derive from the character of liquidlike ACC colloids.” Their work on ACC films is discussed further in the following section.
Faatz et al.231,232 have prepared amorphous spherules under basic conditions (0.5M NaOH) using the hydrolysis of dialkyl carbonates as a source for slow introduction of the carbonate counterion, which slows the precipitation down to a timescale of minutes to hours, allowing the experiments to be monitored by light scattering. They suggest that “liquid–liquid separation occurs followed by rapid gelation to give a glassy state.” The liquid-like character appears to be more limited in their system as compared to the PILP system, where the evidence of coalescence of particles is not obvious in the data provided (Figure 17a), and the purported liquid droplets apparently do not deposit and coalesce into films. Quantitative determination of CO2 and water loss by thermogravimetric analysis found the formula of the initial amorphous calcium carbonate to be CaCO3 • 0.75 H2O. This is clearly not enough water to provide liquid-like behavior, so I assume when they say “initial particles”, they are measuring the particles that have already solidified. Because their system did not use a polymeric additive, this seems to support the notion that the polyanionic polymer we and others use enhances the hydration characteristics, and thus fluidity of the amorphous phase.
Figure 17.
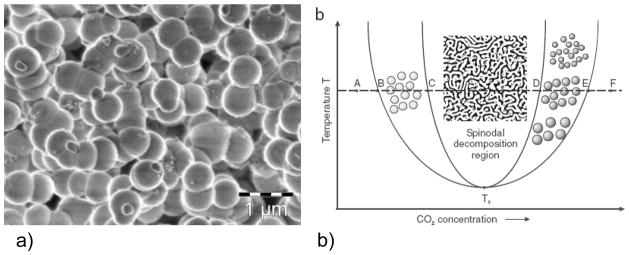
ACC particles prepared by Faatz et al.231 using the hydrolysis of dialky carbonates to release carbon dioxide at a timescale suitable for light scattering investigation. (a) Based on the partial coalescence of the particles, the authors postulated that the system was subject to liquid-liquid phase separation. Bar = 1 μm. (b) A “virtual” phase diagram was proposed for the ACC, considering the kinetically suppressed thermodynamically stable crystalline phase. A lower critical solution temperature was predicted for the ACC in their system to be around 10°C. (Reprinted with permission from ref 231. Copyright 2004 Wiley-VCH Verlag GmbH & Co.)
This team put forth a hypothetical “virtual” phase diagram for ACC formation (Figure 17b), and suggest that liquid-liquid phase separation occurs in their system via binodal decomposition into a calcium carbonate poor and rich phase. The phase diagram is “virtual” because a thermodynamic description is applied to a kinetic metastable phase, where the calcite is suppressed. Spinodal decomposition is also as a possibility, but was suggested to only occur under turbulent, high speed conditions, which is not the case in their quiescent system (in contrast to the Rieger system). In their system, this suppression is not considered to be due to an inhibitory polymer because none was added (however, an anionic polymer surfactant of (EO)68-(MAA)8-C12H25 was used to prevent sedimentation, although it was claimed to not influence the process). Instead, the crystalline phase is kinetically not allowed (if I understand their system correctly, the rapid decomposition of the reactants delivers the carbonate species in a time scale of seconds to minutes). The pH is not provided in this paper, so I wonder if a calcium hydroxide precursor might have formed since their reaction seems analogous to the reaction performed by Hardikar and Matijevic,233 who used 1 M NaOH. This might lead to a gel-like precursor that could condense into spherical colloids, as demonstrated in other hydroxide based systems. Based on particle size and number density analysis, they determined a lower critical solution temperature (LCST) of 10°C, below which ACC should not form. However, this is apparently system dependent, because we and others find that the ACC phase readily forms at 4°C, which is below their predicted LCST.
Comparison between these different systems is difficult in terms of deciphering the role of the polymer on ACC formation/stabilization. In our system, given the non-homogeneity of the crystallizing solution due to the sequestering ability of the polymer, I am not comfortable speculating about what type of phase transformation (i.e., binodal or spinodal) is occurring within the polymer-localized regions of high supersaturation. In other words, even though this is a quiescent and slowly evolving system, where spinodal decomposition might not be anticipated, there is presumably a high concentration of calcium that is sequestered/chelated by the polymer, which could then become locally supersaturated upon introduction of the carbonate counterion, but without forming a crystal nucleus because of the presence of the inhibitory polymer, until the system ‘crashes out’. This would be consistent with the seemingly spontaneous formation of the new phase upon addition of a critical concentration of counterion (as observed by light scattering), and the subsequent densification of the phase, as seen by Rieger227 and ourselves.225 Although the particles do grow in the PILP system, dynamic light scattering found that the PILP droplets grow by aggregation and coalescence, rather than atom-by-atom growth.220 On the other hand, we have not observed a worm-like morphology typical of spinodal decomposition; but in general, the classic spinodal morphology is difficult to observe because the bicontinuous structure often quickly breaks down into particulates. Further studies, at the nanoscale, are needed to resolve this issue. As indicated by Wang et al.234, the polymer-directed mineralization process is complicated because it appears to be a kinetically driven complex-formation process rather than a thermodynamically driven one. This is best illustrated by the compositional studies described in Section 3.3.
As a final note- the formation of liquid metastable phases was documented long ago in the Ostwald–Lussac rule of stages,235 but mention of a liquid phase seems to have been lost in translation in most recent references to this empirical rule. In the classic book On Growth and Form,236 D’Arcy Thompson discusses the formation of rounded concretions, and states the following:
In accordance with a rule first recognized by Ostwald, when a substance begins to separate from a solution, so making its first appearance as a new phase, it always makes its appearance first as a liquid.
3.1.1.1. Liquid-Phase Precursor Pathway to Organic Crystals
A liquid amorphous precursor has now been demonstrated for organic systems as well, where Cölfen and coworkers have shown an analogous process for various amino acids (Figure 18).237 For this system, they used oppositely charged polymeric process-directing agents, such as polyethyleneimine (Mw = 600 gmol−1) for the negatively charged amino acids d,l-glutamic acid and aspartic acid (IEP = pH 4.25), while polyacrylic acid (PAA2000 ; Mw = 2000 gmol−1) was employed to form PILPs of the positively charged amino acids l-lysine (IEP = pH 8.88) and l-histidine (IEP = pH 6.78). The system differs from the inorganic PILPs we produce because ethanol is added to induce the phase separation (as opposed to increasing the supersaturation via addition of reactions). The liquid character of the AA-PILP droplets (Figure 18b) was demonstrated by a preparative ultracentrifugation experiment on stained PILP droplets, which coalesced to form a two phase system. The AA-PILP phase was a viscous liquid with a honey-like consistency. Upon crystallization of the AA- PILP droplets, when they became birefringent (Figure 18c), porous microspheres were formed upon drying, which consisted of nanoplatelets (Figure 18d). Thus, it seems that the overall spherical morphology is retained, and the precursor crystallized in a spherulitic fashion (as opposed to the faceted single crystals in the control reaction), but the spheres become porous due to the higher level of polymer used in the process.
Figure 18.

Wohlrab et al.237 demonstrate an organic PILP process in the crystallization of amino acids. (a) Conventional crystallization of d,l-glutamic acid, after adding 10 mL ethanol to 1 mL saturated solution of d,l-glutamic acid (at 208°C). (b) Liquid-liquid phase separation of precursor droplets formed under the same conditions, but in the presence of 1 wt% PEI (Mw = 600 gmol−1) (c) Polarized light micrograph of the d,l-glutamic acid crystals grown from a concentrated PILP precursor phase. (d) SEM image showing the porous architecture of the spheres. Scale bars: a) 50 mm, b) 20 mm, (c) 10 mm, (d) 200 nm. (Reprinted with permission from ref 237. Copyright 2005. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.)
A multistep crystallization pathway involving liquid-liquid phase separation in some protein crystallizations (such as lysozyme and hemoglobin, as show in Figure 19) has been demonstrated by Vekilov’s group.238–243 In the lysozyme system, the generation of a dense liquid phase occurs in high-concentration solutions after temperature quenches into thermodynamically defined metastable and unstable regions, where they have fully mapped out the spinodal and binodal regions (e.g., Figure 19b). The “quasidroplets of a disordered intermediate” is followed by nucleation of ordered crystalline embryos within these dense liquid droplets. The crystals range from faceted tablets of lysozyme (Figure 19a), to loose spherulitic dendrites of ‘polymerized’ hemoglobin (Figure 19c), so it appears that neither of the protein liquid precursors follow a pseudomorphic transformation of the whole phase (i.e., spherical drop crystals are not formed), as seen in the CaCO3 PILP system. Even though the components of this system are not related to biominerals, or the temperature quench decomposition, the insights provided by this well-characterized system (such as influence of viscosity on structural fluctuations within the quasidroplet, chemical potentials involved in the driving force, interfacial contributions, etc.) may provide further mechanistic understanding of the thermodynamic and kinetic factors involved in multistep crystallization pathways in general.
Figure 19.

Vekilov and coworker’s 238 demonstration of a multi-step crystallization pathway involving liquid-liquid phase separation in protein solutions. (a) Faceted crystals of lysozyme forming within a dense liquid precursor droplet. (b) A section of the phase diagram of lysozyme solutions in the presence of 4% NaCl. For many proteins, the L-L coexistence line is below the liquidus line because of the very short range of attractive interactions. (c) A link between L-L separation and formation of ordered linear arrays in ‘polymerization’ of Hemoglobin S (deoxy-HbS). Concentration of HbS is 22 g dL−1. (top to bottom) When temperature is lowered from 42 to 35°C, the smaller of the dense liquid droplets disappear, while the larger ones serve as nucleation centers for HbS spherulites. Spherulites also appear at the locations where smaller droplets have been, apparently because of the undissipated locally higher concentration. (Reprinted with permission from ref 238. Copyright 2004 American Chemical Society.)
3.2. Other in vitro ACC Systems
The literature has been rapidly growing with a variety reports on systems using an amorphous precursor. In most cases, the early stage formation of the phase is not monitored, so one cannot say whether or not the precursor has liquid-like character, as in the PILP system. I suspect that the continuous film-like morphologies are related to this process, but cannot say with certainty. Therefore, I will present the review on these systems separately; but it should be kept in mind that there are likely many analogous issues (such as influence of substrate, additives, etc.).
3.2.1. ACC Films
Right around the same time as our discovery of CaCO3 thins films,202 two other groups (Guofeng Xu & Aksay244 and Takashi Kato245) also reported on the formation of thin CaCO3 films. These three studies were independent, and the mechanisms that were proposed differed. Notably, though, we all used reaction conditions that contained acidic polymer.
Aksay’s group found that continuous CaCO3 films could be deposited under Langmuir monolayers (Figure 20), and the templating effect of their novel porphyrin-based monolayer was considered an important contributor, coupled with growth inhibition by the use of poly(acrylic acid) as a soluble inhibitor.244 CaCO3 films have since been deposited on less specialized monolayers, such as stearic and arachidic acid,119,246–248 but the one thing in common is the use of the soluble acidic polymer. Their films were shown by FTIR, electron diffraction, and lack of birefringence, to be formed from a multistep, amorphous precursor pathway. The films exhibited different surface textures, where the side facing the monolayer was smooth and featureless, while the side facing the subphase “was covered with numerous particles (ca. 100 nm) that showed considerable intergrowth (Figure 20b).” We observe a similar colloidal texture on PILP films deposited under monolayers (Figure 15c–e), although the nanoparticle substituents are somewhat smaller (ca. 30 nm), yielding smoother films that appear to be more fully coalesced.249 Significantly, Xu’s porphyrin monolayer promoted preferential orientation of the crystals, where the calcite {00.1} plane was parallel to the film and hence, the porphyrin template. This same orientation was found for conventionally grown calcite crystals (of 3-D morphology) formed under the same porphyrin template in the absence of the inhibitor (Figure 20c).115 As noted, “these observations indicate that the control of crystal orientation, though occurring at a later time, is staged by the porphyrin template during the phase transformation.” They suggest that the porphyrin monolayer offers a pure negatively charged surface that can compensate the pure positively charged (00.1) face of calcite, as well as some flexibility and matching of the Ca-Ca distances on this plane. This (00.1) face, although not normally expressed because of the high interfacial energy associated with surface charge, is commonly expressed in biogenic crystals such as mollusk nacre.
Figure 20.

Demonstration by Xu et al. 244 of the formation of CaCO3 films under porphyrin-based Langmuir monolayers. (a) Schematic depicting the template-inhibitor approach to formation of oriented calcite films deposited under Langmuir monolayers. (b) SEM of the CaCO3 films, with one piece of film placed on top of another to provide a view of the two different sides of the film simultaneously. The smooth side was facing the template and the rough and particulate side was facing the subphase. The inset shows the rough side at higher magnification. (c) Calcite crystal nucleated on the same porphyrin monolayer, but without addition of polymeric inhibitor. The crystal orientation was the same as the CaCO3 films shown in (b), but the crystal grew into solution with a faceted 3D morphology since it was formed via the conventional nucleation and growth process. (a & b) (Reprinted with permission from ref 244. Copyright 1998 American Chemical Society.) (c) (Reprinted with permission from ref 115. Copyright 1997 American Chemical Society.)
Using this “template-inhibitor” approach, this group was also able to form continuous carbonated, calcium phosphate (CaP) films on an arachidic acid monolayer.250 In this system, the amorphous film had to be calcined to form a crystalline carbonated apatite film. As with the CaCO3 films, they suggest that the “observations indicate that the film formed through a sequence of particle deposition and fusion process. Primary particles (with diameters of 40–50 nm) formed first directly under the acidic template and condensed into a thin layer, which acted as the template for the deposition and condensation of the next layer of particles.” The proposed mechanism seems to be quite similar to what we had proposed for the PILP process, although a fluidic precursor was not described at this time.
Kato’s group have formed thin films of CaCO3 on biomimetic polysaccharide substrates, at first with chitosan,245 and more recently on cholesterol-modified polysaccharide of pullulan (Figure 21). In their first reports, both PAA and poly(Glu) were found to produce the mineral films, but under the conditions employed, films did not form on the glass substrate. In their 2000 paper,251 they state that “neither insoluble chitin nor a soluble acidic macromolecule can alone induce such thin-film development, and that the cooperation of the polysaccharides and the acid-rich macromolecules is essential.” In contrast, we did find that films could be formed on glass substrates (perhaps due to the higher molecular weight polymer); however, they tend to be less pure, with aggregates of 3-dimensional crystals present as well. Nevertheless, we have found this simpler system useful for detecting the amorphous precursor. In these earlier papers, Kato’s group did not originally consider the ACC mechanism to be operable in their system, and instead proposed that the adsorbed acidic macromolecules “can bind calcium ion on the surface of the chitin films, which results in a local high ion concentration of the calcium ion on the surface, inducing nucleation of calcium carbonate.” I agree that the adsorbed polyelectrolyte may promote heterogeneous nucleation, but would argue that the ‘nucleating’ phase is more likely to be that of ACC (as opposed to homogeneous nucleation of solution borne droplets that are seen in our system). Given that the polymer concentration they use is an order of magnitude larger than ours, which in turn leads to a localized high concentration of ions, both of these conditions should favor an amorphous phase. In their 2003 paper,252 they state that an amorphous to crystalline transformation is not observed in their process, but the ACC films can be hard to see, even on glass. Sometimes the surface needs to be scratched to detect it (e.g., see Figure 24 below),253 so on polymer-coated substrate, it might be particularly difficult to detect. The more recent papers by this group now include consideration of an amorphous precursor, where they discuss tensile stresses upon transformation from the amorphous-to-crystalline state.254 In their recent report using peptides isolated from the exoskeleton of a crayfish, the discussion is based upon an amorphous-to-crystalline transformation, which enabled them to prepare uniaxially-oriented CaCO3 thin-film crystals on chitin matrices.255 This group has had several other nice accomplishments in this area which can contribute useful mechanistic insights. In particular, they have found reaction conditions that enable polymorph control, and produced high purity aragonite thin films by adding Mg2+-ion (Mg:Ca of 6:1) along with the acidic polymer and chitosan.256 They then found that aragonite films could be templated on crystalline PVA substrates without Mg2+-ion, as well as vaterite films using a different polymer additive.252 Particularly impressive was the fabrication of a triple-layered organic-inorganic composite by sequential deposition of the chitosan-CaCO3 layers.257
Figure 21.

Kato and co-worker’s254 thin films of CaCO3 grown on polysaccharide substrates in the presence of soluble PAA (Mw=2,000). (a) CaCO3 films deposited on a hydrogel matrix of cholesterol-modified pullulan (a glucose-based polysaccharide) yielded a periodic relief structure. The underlying texture of the film is spherulitic, as seen in the partial spherulite in the inset. (b) At higher magnification, one can see the radial growths of the polycrystals. (c) Films that were formed rapidly produced a corrugated structure, such as the film here, where the reaction was carried out at 50°C (for 4 hours), as compared to the film shown in (b), which was formed for 2 days at 10°C. (a–c) (Reprinted with permission from ref 254. Copyright 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.)
Figure 24.

Sommerdijk et al.253 demonstrate that ACC films can also be induced with the polyanionic additive of DNA. (a) The control reaction, without DNA, produces discrete crystals after 3.5 h. (b) An ACC film produced with DNA at 3.5 h, with a scratch provided for visualization. The inset shows the film after 4 weeks. (c) Same as (b), but after 5 days some crystalline spherulites start to emerge. (d) Same as (c) but with the substrate modified with a DNA-surfactant double layer, which increased the crystallization rate. (Reproduced by permission from ref 253. Copyright 2007 The Royal Society of Chemistry.)
Recent work by Ulčinas et al.258 provides convincing evidence that this PAA-chitosan system does form from an amorphous precursor. They examined the formation of spherulitic thin films on chitosan substrates in the presence of PAA using AFM and SNOM (scanning near-field optical microscopy), and describe the formation of the spherulites within a “PAA-stabilized ACC gel”. This sounds similar to the PILP system, and in fact, the other features they observe also correlate with our observations, such as a colloidal texture, a concentric banding pattern (which appears similar to PILP transition bars), and the “smoothing” out effect of the small-scale features with time (Figure 22).
Figure 22.

Ulcinas et al.258 scanning probe microscopy study of the features observed during formation of CaCO3 thin film spherulites on chitosan substrates, using PAA (15 kDa) additive. (a) At high magnification, a nanoscale “polycrystalline texture” is described. (b) Concentric banding is observed at the micron scale, which appears similar to the transition bars observed in the PILP system. (c) The growth stages include a PAA-stabilized ACC gel, which nucleates spherulitic branches that build into lobes, followed by ‘ripening’ into a smooth film. (d) Quantitative analysis shows a decrease in surface roughness as well as film thickness during the “ripening” process. These observations are consistent with coalescence of precursor droplets (nanogranular texture), and a transformation that includes transition bars, with an eventual smoothing out effect and reduction in thickness due to dehydration of the precursor. (Reprinted with permission from ref 258. Copyright 2007 Elsevier B.V..)
In Sugawara and Kato’s paper in 2003,254 they also observe regular relief structures in CaCO3 films that were formed on the cholesterol-modified polysaccharide of pullulan (Figure 21a–c). These periodic, submicron relief structures appear similar to the ridged transition bars we have observed;142 however, their ridges seem to be permanent (or samples were removed prior to maturation), while our transition bars seem to smooth out with time, which was also the case in Ulčinas system.258
Cho’s group (Xurong Xu et al.)230,259–261 has also contributed to further understanding of ACC film formation. In their 2005 paper,230 hemispherical deposits of ACC (on inorganic substrates of mica and silicon) were treated according to their “wetting” behavior associated with interaction of the “liquidlike colloids” of amorphous phase with the surface chemistry of the substrate (Figure 23). Modification of the surface chemistry through adsorption of positively charged polyelectrolytes could alter the film formation properties (films form without soluble polyanion at their high supersaturation), such as density and coalescence of the ACC particles. They suggest that the nature of the ACC deposit on the substrate depends on the relative strengths of the cohesive force of ACC on each other and the adhesive force between ACC and the substrate. On the other hand, films were always formed when using the PAA additive, despite variations in surface charge of the substrates. In this case, they suggest that “the adhesive force between ACC and the substrates is then stronger than the cohesive force, so ACC tends to spread over the surface, which results in a continuous ACC film on the substrate.” In their system, the supersaturation is quite high (50 mM Ca2+), and they suggest that “ACC in highly supersaturated solutions spontaneously forms liquidlike colloids with an open structure in solution, tends to coalesce, and deforms into hemispheres or forms continuous films when deposited on substrates.” Further work by this group has focused on the transformation pathway of the ACC films, as described in Section 3.4.2.
Figure 23.

Xu et al.230 demonstrate the combined effect of substrate interaction with ACC colloids produced by soluble polymer, as illustrated in the schematic in (a). (b & c) The SEM images reproduced here were for the substrate of poly(diallyldimethylammonium chloride) (PDADMAC), which lead to significantly different wetting behavior of the ACC colloids produced without PAA (b) versus with PAA (c). (Reprinted with permission from ref 230. Copyright 2005 American Chemical Society.)
Ajikum et al.262 have tested three types of acidic polymers for the deposition of CaCO3 films onto synthetic fabrics of Nylon 66 knits, as well as demineralized eggshell membrane. In their system, the acidic polymers were pre-adsorbed to the substrates (after acid or base treatment), rather than added in soluble form. The possibility of an ACC precursor was not considered, but the film-like morphology suggests this might be the case. Their results demonstrate some of the complexities involved with cooperative effects between the adsorbed polymer and scaffold. For example, on the Nylon scaffolds, polyaspartate led to fairly continuous CaCO3 films, while polyacrylate deposited patches of calcite crystals, and polyglutamate produced a few small crystal aggregates (not film-like). In contrast, polyglutamate was able to deposit a film-like coating on the eggshell membrane, as did polyaspartate, while polyacrylate suppressed mineralization entirely. Interestingly, the polyglutamate-induced coating crystallized into aragonite, while the polyaspartate-induced coating crystallized into vaterite. In line with the cooperativity model first proposed by Addadi back in 1991 (with peptides adsorbed to sulfonated polystyrene matrices), the authors suggest that the polymorph selectivity might be due to the combined effect of the pre-adsorbed peptides (which may adopt a β-sheet conformation) with the surface functionalities of the treated scaffolds.
As a related side note, we found that polyglutamate was not as effective in depositing mineral films on glass substrates, yet a thick mineral coating did form on some crystal aggregates that were present. Again, the substrate-polymer combination seems to play a role in where the amorphous phase is formed, which may arise from stimulation of heterogeneous nucleation, or preferential adsorption homogeneously formed droplets.
Most recently, Sommerdijk et al.253 have even used DNA to deposit CaCO3 films via the amorphous phase (Figure 24). Although DNA is not directly associated with biominerals, it is a readily available and inexpensive polyelectrolyte, and is biocompatible for biomaterials applications. In their system, quite high concentrations of calcium were used (1M), along with correspondingly high polymer concentration (2.0 × 10−2 wt%), which formed a 2 μm thick ACC film within 3.5 hours. These films were slow to crystallize, but did seem to follow the pseudomorphic transformation, forming smooth and continuous calcite films. In comparison to their counterpart reaction using PAA (M.W. = 2000Da), which crystallized within an hour, the DNA induced film was only 5% crystalline after one week. Thus, the polymeric additive not only induces the ACC phase, but has a pronounced effect on the transformation kinetics. The substrate was also found to influence the transformation, where a more crystalline polymer (UHMWPE versus polycaprolactone) led to faster crystallization of the ACC film. In addition, the adsorption of DNA through a double-layer approach led to a 48% preferred (11.0) orientation. As was the case for Ajikum,262 they found that the thin film technique could be adapted to depositing thin CaCO3 coatings onto fibrous polymeric scaffolds, particularly after surface treatment of the fibers.
Aizenberg and coworkers have also been examining crystal formation via an amorphous precursor film, and were able to fabricate millimeter-sized single crystals of calcite around a micropatterned framework.263 Recently this group has developed an interesting approach to regulating crystallization by generating an ACC (particulate) film that can be stored in a dry atmosphere as a reservoir for ions, which can subsequently be induced to crystallize on command by the introduction of water and a secondary surface that is functionalized with a secondary surface that acts as a template for oriented and patterned nucleation. These studies are discussed in more detail in Section 3.4.2.1., as they also pertain to the regulating the transformation pathways of the amorphous phase.
3.2.1.1. CaCO3 Films from Biomineral Proteins
Biomineral protein extracts that have been examined in vitro not only lead to ACC, but some of the morphologies that are obtained clearly look like they were formed from an amorphous precursor (and quite possibly a liquid-phase amorphous precursor). I first noticed this in an earlier paper by Belcher et al.,264 where “flat polycrystalline plates” of aragonite were formed using the soluble anionic proteins extracted from the nacre of abalone shell (their Figure 1d is shown in Figure 25a). At this time (1996), the presence of an amorphous precursor was not even looked for since the suggestion of a precursor process in biomineralization had not yet been brought forth, and the focus of the paper was on the phase switching between calcite and aragonite. Therefore, I cannot say with certainty that these films were formed via the ACC pathway, but the film-like morphology is certainly typical of PILP formed ACC films. Even more-so, their figure shows a centralized 3D aggregate with an underlying film that has a radial extinction pattern (like a pin-wheel), and such course-grain spherulitic film structures are commonly seen in PILP formed films (Figure 25b).
Figure 25.

Comparison of PILP-formed film to the “tablets” produced by proteins extracted from the mollusk shell. (a) Flat “tablets” produced from proteins extracted from mollusk nacre. (b) Polarized light micrograph of PILP films which contain a large centralized aggregate. The aggregate often extends upward into tall towers, and is too thick to focus on in the light microscope (and therefore appears brown). The underlying film radiates from this central aggregate as a connected series of single-crystalline plates, arranged in a succession of crystallographic orientations such that when the microscope stage is rotated under crossed-polars, the tablets sequentially extinguish, generating a pin-wheel birefringence pattern (i.e., they are very course spherulites). The same pin-wheel pattern was described by Belcher. (a) (Reprinted with permission from ref 264. Copyright 1996 Nature Publishing Group.)
Samata et al.265 isolated a new matrix protein family (N16) which is specific to the nacreous layer of the Japanese pearl oyster, Pinctada fucata, and this protein induced sheets of platy aragonite when immobilized onto the insoluble membrane. Similarly to Kato’s work,251 the soluble protein only induced the mineral film when it was adsorbed to the insoluble membrane, and was mostly inhibitory with used with a glass substrate. A tablet morphology was described, but judging by the Maltese-cross patterns shown in their images, the individual plates appear to have a spherulitic texture, unlike the single-crystalline tablets in nacre.
More recently, Gayathri et al.211 used proteins extracted from seastar ossicles (Echinodermata, Asteroidea), and in this case, were looking for an amorphous precursor since this had already been discovered in the closely related taxa of the sea urchin. Using the soluble organic matrix, the ACC formed into a film-like morphology, as shown in Figure 26a (which resembles PILP films of coalesced droplets). Interestingly, the intracrystalline macromolecules consisted predominantly of glycine-rich polypeptides, which were shown in vitro to accelerate the conversion of ACC into its final crystalline product, and promoted the formation of magnesium calcite relative to aragonite (as compared to control sample with Mg-ion but without the soluble proteins). In this system, however, the initial ACC film transformed through elongated ellipsoids into dumbbell-shaped magnesium calcite crystals (Figure 26b), and apparently did not undergo a pseudomorphic transformation (like it presumably would in the biomineralizing environment, in order to generate the non-equilibrium morphology of the ossicles).
Figure 26.

ACC precursor induced in vitro with extracted proteins from seastar ossicles. (a) Gayathri et al.211 found that “Islands of thin films of ACC” were formed with the soluble proteins, in conjunction with Mg-ion. Note how this film appears to have formed from coalesced droplets, consistent with a PILP-like precursor phase. Without protein, only spherical particles were produced. (b) Spindle-shaped crystals emerged after one hour, which then transformed into the dumbbell-shaped spherulites of aragonite shown here. (a & b) (Reprinted with permission from ref 211. Copyright 2007 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim.)
As mentioned in the Introductory Section 3.2.1.1, Lakshminarayanan formed ACC by dissolving quail eggshell and then re-precipitating mineral in the presence of the remnant protein fraction (Figure 27). Neither ovomucoid (an acidic glycoprotein) or lysozyme (a basic protein), the major components of eggshell proteins, separately induced the precipitation of the ACC phase, while the fraction containing low molecular weight peptides induced the precipitation of ACC. This seems to correlate with our observations that fairly low molecular weight polymer is more potent in this regard (at least in forming a PILP ACC phase). The ACC that formed had a film-like morphology at first (Figure 27a), before it then transformed into spherulites (Figure 27b). In the case of eggshell, spherulitic textures are the natural form of the biomineral; therefore, a pseudomorphic transformation may not be necessary in this system.
Figure 27.

ACC precursor induced in vitro with the soluble organic matrix extracted from bleach-treated quail eggshell. Lakshrminarayanan et al.216 describe the precipitation reaction as follows: “A mesh of 400 ± 50 nm-sized spherical particles was formed on the microscopy slides within 1 h. These particles grew in size and formed larger particles of 700 ± 50 nm in diameter after 2 h. The submicrometer particles sintered together and formed a film-type structure that looked like a mosaic of polycrystalline patches in 4 h (shown in (a)). After a 6 h period, well-ordered, lozenge-shaped calcite crystals were emerged from these patches (shown in (b)). The surface of these crystals contained an array of microscopically aligned interdigitated calcite rods oriented along the c-axis that were reminiscent of the palisade layer.” This description and the images are consistent with a PILP-like process, but with insufficient stabilization or other factors to undergo a pseudomorphic transformation. (a & b) (Reprinted with permission from ref 216. Copyright 2006 American Chemical Society.)
Overall, I think these studies show that ACC will generally deposit as a film-like coating on relatively simple substrates or under the simplified reaction conditions employed in vitro, and that other factors, such as scaffolds and compartments, are needed to shape and mold it into the elaborate morphologies observed in biominerals. On the other hand, it may be that sequential depositions of this film-like coating are used to build up the larger scale structures, which often exhibit laminated microstructures (see Figure 49, to be discussed in Section 4.2.3).
Figure 49.

Examples of the concentrically-laminated textures seen in some large biominerals, such as the urchin spine and sponge spicules shown here, and their nanogranular subtextures. (a) SEM of an EDTA-etched fracture surface of an immature sea urchin spine showing the concentric mineral deposition lines. Bar = 1 μm. (b) AFM deflection image showing a nanogranular texture on a naturally grown trabecular surface of the urchin spine. (c) The nanogranular subtexture is also seen in this fracture surface, which may be relevant to the unusual conchoidal fracture patterns seen in urchin spines. (AFM height image, height range ~ 50 nm). (d) SEM of a siliceous sponge spicule. Etching with sodium hypochlorite reveals the concentric laminations of deposited silica, and also removes the organic axial filament that runs down the middle of the spicules. (scale bar 1 μm). (e) AFM surface plots of a spicule cross-section reveals the nanogranular subtexture within the concentric layers. (scan size 3.8 μm, height data scale 275 nm). (f) AFM surface plot of a longitudinal section revealing details of the annular nanoparticulate substructure (scan size 2 μm, height data scale 75 nm). (a) (Reprinted with permission from ref 54. Copyright 1997 The Royal Society of Chemistry.) (b–c) (Reprinted with permission from ref 345. Copyright 2008 Elsevier Ltd..). (d–f) (Reprinted with permission from ref 346. Copyright 2003 Elsevier Inc..)
3.2.2. ACC Particle Systems
ACC particles (ranging from 10s of nanometers to micron size) have been formed using a variety of additives, both biogenic (e.g., egg white lysozyme266) and synthetic. -Although there is a wealth of literature available in this area, of which studies on ACC particles began many years ago, the papers are too numerous to cite here. Some recent reviews discuss this work from the new found perspective of amorphous precursors in biomineralization and biomimetic systems.25,193,261 Admittedly, I am less familiar with this body of literature because my focus has been more on the liquid-phase amorphous precursor, and this was not usually considered in the older literature. While liquid droplets can lead to round particles (as shown in Figure 15), and thus may have occurred in many of those systems, the round morphology is not necessarily indicative of a liquid-like phase, since amorphous solids are isotropic and present a homogeneous surface energy as well.
3.3. Composition and Structure of ACC Phase(s)
One of the first questions that come to mind when examining a new phase is- what is its composition and structure? Thus- this is what we wanted to define for the PILP system since it seems to deviate substantially from the amorphous particle systems mentioned above. The molecular structure of an amorphous phase in general can be difficult to define, and in the PILP system, the problem is confounded by the variability in structure that seems to depend on the reaction conditions and solidification time. For example, even the consistency of the ACC phase generated with polymer is quite variable, with textures observed via optical microscopy that range from a slightly viscous fluid, gelatinous globule, elastomeric sheet, granular sand, spongy solid, to brittle glass (Figure 28). Therefore, it is suspected that there is not one particular molecular structure of the phase. Likewise, the composition of the PILP ACC phase seems to be just as difficult to define.
Figure 28.
Optical micrographs demonstrating the variety of physical textures observed for amorphous CaCO3 phases induced with polymeric process-directing agents under various conditions. (a) An accumulation of PILP droplets at the interface of an air bubble. The large drop behaved like a slightly viscous liquid, which readily deformed and flowed as the slide was shaken. (b) A thick spongy amorphous film deposited on a Langmuir monolayer using a micropump to raise the supersaturation. Partially coalesced droplets have also accumulated in the crack in the middle. Bar = 100 μm. (c) A rigid glassy ACC film formed on a Langmuir monolayer with the CO2 escape method. The cracks formed when the film was dipped off the monolayer onto a solid substrate. (b & c) Bars = 100 μm. (a) (Reprinted with permission from ref 203. Copyright 2000 Elsevier Science B.V..) (b,c) (Reprinted with permission from ref 386. Copyright 2006 John Wiley & Sons, Ltd..)
We set out to measure the composition of this novel amorphous phase by centrifuging out the precipitates at early time points in the reaction, and measuring the elemental composition throughout the process.225 We found that the composition was dynamic throughout the entire reaction. The polymer content starts off high, as does the water content, and both decline with time as the phase gradually solidifies into ACC. The amount of water in the phase was found to be correlated with the amount of entrapped polymer, supporting our hypothesis that the polymer enhances the degree of hydration of the amorphous phase. The calcium to carbonate ratio also changes with time because the carbonate is slowly added into the solution, and apparently joins in with the polymer/calcium-ion complex as the phase is formed. The carbonate ions effectively compete with the anionic polymer, which aids in its expulsion, along with the water, from the phase. A small amount of polymer may remain entrapped in the phase, and is sometimes seen within transition bars as the crystallization of phase further tries to expel the polymeric impurities (see Figure 58g & h).142
The concentration of the polymer was also found to affect the rate of precursor formation, where the higher polymer concentration promoted a faster reaction. In fact, the polymer even promoted the formation of the amorphous phase faster than mineral formation in the control reaction (without polymer). FTIR data indicated that an amorphous precursor was formed in both the control reaction and the polymer containing reaction, but the ACC phase was short-lived without the presence of polymer. The promotion of the phase should not be confused with crystallization, in which this same polymer (polyaspartate) is well known as being an inhibitor of crystal nucleation.
One of the most striking features of the PILP process is the evolution of the composition of the amorphous phase. In other words, the ACC composition is continuously changing and there is no single ACC phase for which the composition can be measured and compared to the singular ACC phases reported in the literature, which generally are found to have one mole of water per mole of CaCO3.209,267–270 For example, Weiner and coworkers271 distinguish between a transient anhydrous ACC form, with little to no water, and a metastable hydrated ACC with a composition of CaCO3·H2O. It is puzzling how an anhydrous ACC phase, which has little to no water, can reorganize into the crystalline structure at room temperature. Calorimetry studies on thermal transformation of synthetic ACC have shown crystallization exotherms at around 500 – 600K, with an activation energy in the ballpark of 150 to 300 kJ mol−1.272 Thus, it seems the only explanation is that short range order (as discussed below) in this unusual biogenic anhydrous ACC must be fairly close to the final lattice structure, and some entrapped protein (and/or inorganic impurities) in the biomineral provides this organization mobility.
An exciting development in this area has been provided by x-ray absorption spectroscopy studies, which have shown that in biogenic ACC, the local order can be different for different ACC samples, and this appears to influence the polymorph of the final crystalline biomineral.206,215,273–276 In earlier reports, prior to consideration of an amorphous precursor, two independent groups, Belcher et al.264 and Falini et al.,277 had shown that the soluble proteins extracted from the different layers of mollusk shells, corresponding to the calcite prisms versus aragonite platelets, could be used to switch between these corresponding crystal phases (calcite vs. aragonite, respectively) for crystals grown in vitro. As mentioned in Section 3.2.1.1, the thin tabular morphology that was created by these protein extracts look very much like our PILP formed tablets (Figure 25), and thus may indicate those proteins induced an amorphous precursor (which was not examined at that time), which would be consistent with the current view that the proteins influence the phase via regulation of short-range order of the ACC precursor. This is in stark contrast to the more traditional view that the crystal phase is modulated via templating interactions with an insoluble nucleating matrix.
More work in this area is needed to understand the molecular level interactions responsible for phase modulation. This will be challenging because of the difficulty in defining the structure of amorphous materials, along with an unstructured polymer that is presumably intertwined in the mix. Perhaps computational studies could be helpful for elucidating such organic-inorganic interactions; but this also poses challenges if one also includes water in the mix, which as mentioned above, may be significant with respect to the structure of the amorphous phase, and the kinetics of its transformation.
3.4. Role of Organic Constituents during Stages of Precursor Pathway
For the sake of discussion, it is convenient to consider the precursor pathway as a two-stage reaction, as shown below, where the first stage is the formation of the precursor, and the second stage is the amorphous-to-crystalline transformation.
The reason for breaking it down this way is because one can consider the roles of the organic matrix to be different for the two stages. For example, while the soluble polyanionic additive is responsible for inducing (or stabilizing) the precursor phase in Stage 1, the manner in which the phase subsequently transforms can be strongly influenced by other factors, such as the substrate upon which it is deposited (Section 3.4.2 below). One should recognize that this is an overly simplistic view, because some the soluble additive becomes incorporated within the ACC phase, which can then influence the kinetics of the transformation, or the resulting crystal phase. Likewise, the substrate may play a role in inducing the precursor phase, such as by attracting a high concentration of ions251 (described by some as an ionotropic effect278–280). Nevertheless, while keeping in mind that there can be overlap, this discussion will examine the molecular level interactions in a separate manner.
3.4.1. Stage I- Precursor Formation
3.4.1.1. Templating Location of Mineral
In biomineralization, Stage I of the precursor pathway is presumably when locational control is accomplished For example, biomineral location could be controlled by cellular processes that vectorially direct the secretion of mineral constituents or a precursor phase, or control the location of a mineral deposition vesicle; but it also may be influenced by insoluble matrices.1,4,13 Although we cannot emulate cellular deposition vesicles in the beaker, the latter case can be mimicked in vitro with templating studies, which demonstrate that the location of crystals formed from an amorphous phase can be patterned with micro-contact printed templates of patterned functionality (Figure 29).221 Unlike Aizenberg’s 1999 study, which beautifully demonstrated the patterning of oriented calcite crystals onto SAM templates,102 our goal here was not to use the template to stimulate nucleation of CaCO3 (crystals or ACC phase), but to see if we could control the location and shape of crystals through preferential adsorption of PILP droplets onto a template. In other words, we wanted to ‘mold’ calcite crystals through a colloidal crystallization process. For this, SAMs prepared from alkane thiols on gold were used to preferentially adsorb the precursor droplets to regions of carboxylate-terminated SAMs, which were favored over the surrounding regions of gold substrate (Figure 29a & b). Somewhat surprisingly, the droplets were found to bind to gold regions of a pattern (the regions previously avoided), if the remaining regions were patterned with methyl-terminated SAMs (Figure 29c & d). Thus, a specific functionality is not required to template the colloids, but the deposition does tend to be selective, depending on combined surface chemistry/energy of the substrate.
Figure 29.

Patterning mineral location using microcontact printed SAMs of variable functionality. Top Row- Polarized light micrographs with gypsum λ-plate; Bottom Row- AFM 3-D height images of corresponding patterned films. (a,b) Preferential film formation occurred on the carboxylate-functionalized SAM (birefringent grid pattern) over the gold surface. Some Mg-ion was used to stabilize the ACC phase, so the film has a polycrystalline texture. (c,d) Preferential film formation occurred on the gold surface relative to the methyl-functionalized SAM. The film is still amorphous here (non-birefringent), so the location of the mineral film is better seen in the AFM image and line scan below, which show the mineral deposited preferentially in the interior boxes of the grid pattern (the bare gold surfaces). Both micrographs were taken 42 days after being removed from the reaction solution. (Reprinted with permission from ref 221. Copyright 2007 American Chemical Society.)
The preference for a charged surface over a hydrophobic surface is not surprising considering that these droplets are highly concentrated clusters of ions, but the deposition onto the gold substrate in the second case was not expected. This seemed to suggest that the mineral formation was not a surface nucleation phenomenon (where ‘nucleation’ refers to a new phase, not a crystal); but instead, the droplets had already formed in solution, and needed some place to deposit, and thus chose the more favorable of the two regions available, which apparently was gold over methyl-terminated SAM. This statement needs further justification, because it is not inherently obvious why the PILP droplets would form homogeneously in solution, because one would expect a lower energy barrier for heterogeneous nucleation. Therefore, we wondered if the amorphous phase was nucleating on the surfaces. In other words, it might be the case that the smooth films we observe are from heterogeneous nucleation, and the bumpiness that forms later could be from the homogeneously solution born droplets that formed later. To study this issue further, we ran a similar micro-contact printing experiment, but placed the patterned substrate within the vial in a vertical stance (Figure 30). With this setup, we felt we could then distinguish between the two possible scenarios as follows: 1) if the amorphous films form from heterogeneous ‘nucleation’ on the surfaces, then the film would be expected to be thicker near the top of the reaction vial where the ion concentration would be the greatest (due to vapor diffusion of carbonate across the air-solution interface); 2) if the amorphous films form from adsorption of droplets that had first formed homogeneously in solution, then one would anticipate thicker films near the bottom of the reaction vial because the PILP droplets grow in size and number with time. The results showed thicker films near the bottom of the slide, which supports the second hypothesis. Although heterogeneous nucleation still cannot be ruled out, it appears that the majority of the film thickness is from solution born droplets. In other words, the polymeric process-directing agent appears to induce homogeneous liquid-liquid phase separation within the crystallizing solution. This is consistent with our light scattering studies as well;220 not to mention the microscale visual observation of droplets in the solutions.203 This is also consistent with Clarkson’s studies on ACC formation, which in his system were formed under conditions of high supersaturation, not with polymer.174 He found that the ACC formed homogeneously above a well defined ion activity product, while at concentrations insufficient to form the ACC phase, the crystalline phases nucleated heterogeneously. These observations may not necessarily be universal. One can imagine that some substrates may promote heterogeneous nucleation of ACC, which may be the case (although not determined) in Aizenberg’s system with SAMs and Mg-ion,281 or with the chitosan films used by Kato.251
Figure 30.
Schematic illustrating an experiment to determine if the ACC films are forming by heterogeneous nucleation of ACC on the SAM surface, or adsorption of PILP droplets formed homogeneously in solution. A wafer with microcontact printed SAMs (in a grid pattern) was placed in a vertical stance within the reaction vial. Scenario A- a thicker film is expected at the top because the supersaturation is highest near the air-water interface in the vapor diffusion technique. Scenario B- a thicker film is expected at the bottom because the droplets of PILP phase grow in concentration and size with time, the further they descend down the vial. On the right, AFM images and line profiles show that the films were over 3 times thicker on the bottom, supporting the proposed Scenario B. (Reprinted with permission from ref 221. Copyright 2007 American Chemical Society.)
To examine the effect of surface energy on ACC films, Xu and Cho259 functionalized their substrates (silicon wafers) with –OH, -Cl, and -CH3. They found that ACC films could be produced on the –OH and –Cl monolayers, even without PAA additive, while the –CH3 monolayer only yielded remnants of a film. They suggest that “the more hydrophilic surfaces may interact with the water molecules within the ACC phase, preventing them from escaping, which results in formation of good ACC films.” We found that the ACC films deposited more readily on the regions of –COOH terminated SAMs, than on –CH3 terminated SAMs, which is in accord with Xu’s observations of film formation. In contrast, though, they found that the more hydrophobic surfaces (-CH3 functionalized) crystallized more quickly, although film quality was somewhat poor. In our system, when ACC films deposited on both regions, the more hydrophilic regions crystallized more quickly than the films deposited on gold or –CH3 terminated SAMs (Figure 29a versus d). Since Cho did not examine charged functional groups, I am not sure if the carboxylate functionality in our system played a role in these differing observations.
3.4.1.2. Role of Soluble Polymer
Although I have been operating under the assumption that the soluble anionic proteins associated with biominerals are responsible for the ACC pathway, it should be kept in mind that an inhibitory polymer is not a requirement for formation of an ACC phase. Xu and Cho259 have also done some work in this area, comparing ACC film formation with and without polymer additive. In general, higher ion concentrations have to be used without the inhibitory polymer. For example, ACC films could not be formed from 5 mM and 10 mM CaCl2 solutions in the absence of PAA; yet in the presence of just 5 μg/mL PAA, these solutions yielded ACC films, highlighting the importance of the inhibitor’s role. The polymer also imparts strong kinetic effects to the process, in which films form, as well as transform, more quickly without polymer. Therefore, the films have to be intercepted more quickly for the non-polymer system if they are to be retrieved from solution to inhibit dissolution-recrystallization. They also found that magnesium ions behave as excellent inhibitors in the preparation of ACC films, exhibiting a stronger inhibiting effect on the transformation of the unstable ACC phase than PAA. Their findings correlate well with our findings on the PILP system as well.180 In fact, we find that when using a combination of these two types of inhibitors, one can drop the concentration of polymer in the presence of Mg-ion, and vice versa (although at the concentrations we used, at least some small amount of polymer was required to generate the films). The downside of using Mg-ion to form the amorphous phase is that it tends to generate a higher defect texture, and spherulitic films are formed with Mg-ion at moderately high levels.
Another way to generate pure ACC film (without crystal aggregate side products) is to carry out the reaction at lower temperatures (such as at 4°C, which can conveniently be performed in a refrigerator). Even though the solubility of the ACC phase increases at decreasing temperature,173,174,272,282 it can still be formed at 4°C (albeit more slowly than at 25°C), and preferentially over any crystalline products (which are more strongly averted at the lower temperature), thus providing a opportune way to form pure ACC films with fewer crystalline side-products. The ACC phase formed at the lower temperature is more stable (i.e., it transforms/crystallizes more slowly), so the film is typically removed from the fridge in order to promote its crystallization to the more thermodynamically stable phase. Interestingly, the ACC film formed at 4°C crystallizes far more quickly than an ACC film synthesized under the same conditions, except at the higher temperature of 25°C. Thus the amorphous phase formed at different temperatures differs. We are currently trying to determine the reason for this, but speculate that the lower temperature films may have more water, which could enhance the transformation rate; but other reasons could include a different content of entrapped polymer, or a different stoichiometry of ions. In the latter case, there is a temperature dependence to the ion pairing association between calcium and carbonate ions.174
One would like to understand what characteristics of the anionic polymer contribute to inducing and/or stabilizing the amorphous phase. In the PILP system, the molecular weight of the polymer, as well as its charge density, are primary factors in generating ACC PILP droplets that are able to coalesce into continuous films. For example, we have found an optimal molecular weight range of around 5000 to 15,000Da for generating smooth and continuous CaCO3 films. On the other hand, others have used lower molecular weights, such as 2000Da, but often employ higher polymer and/or ion concentrations. Higher molecular weights are less commonly used, perhaps due to their lower solubility, which in the presence of divalent ions, may precipitate out of solution. In our system, more granular precipitates are produced when using higher molecular weight polymer. Divalent ions are known to collapse polyelectrolyte chains, such polyacrylate in the presence of calcium salts.283,284 Therefore, with the lower molecular weight polyaspartate chains, which behave similarly to polyacrylate, we may be operating just at the brink of solubility, but at a size large enough to coordinate many cations. It deserves mention that many of the acidic proteins associated with biominerals also have relatively low molecular weights.40,87,214,216,285,286
In addition to the commercially available polymer, we have synthesized a series of peptides to study the characteristic features that can help to induce and optimize the PILP process, and found that peptides with higher charge density were most potent (as measured by the ability to form continuous film-like structures). Of course most proteins are not as highly charged as pure polyaspartate, but on the other hand, they quite commonly are highly phosphorylated. Therefore, we also prepared peptides containing phosphoserine, and the phosphorylated peptides enhanced the overall PILP formation process relative to peptides only containing carboxylate groups.142,287 We assume this is due to the higher acidity of the phosphate groups providing a greater affinity for calcium ions. Marsh’s studies on dentin phosphophoryn measured a maximum binding of 1.33 calcium ions per organic phosphate residue (one assumes that even with fully deprotonated carboxylate groups, the binding capacity of polyaspartate peptides would be less than 1).
In contrast to our findings of enhanced ACC formation with higher charge density peptides, Politi et al.214 found that the Asprich proteins extracted from the prismatic layer of the mollusk (described in Section 2.1.1) were more potent at inhibiting de novo calcite nucleation and produced ACC at lower concentrations than similarly sized polyaspartate, even though the Asprich protein contains around 30% hydrophobic residues.214 They discuss ordered water contributions, which may prevent expulsion of the hydration waters, as playing a possible role. The hydrophobic regions of the additive could also provide for more interactions of the sequestered ionic mineral precursor with the relatively hydrophobic chitin scaffold, thus promoting scaffold mineralization. Based on their in vitro observations, they propose that “the in vivo function of Asprich would be to induce and stabilize ACC particles and to inhibit uncontrolled calcite nucleation, until the mineral particles come into contact with the preexisting calcite prism…… followed by their fusion to growing calcite crystals.” Although the word ‘fusion’ is used, they do not suggest a fluidic amorphous precursor, and instead suggest that “when the colloids come in contact with the crystal, they diffuse along the surface to steps, where they undergo secondary nucleation to become part of the growing single crystal.”
When discussing polyanion-mediated mineralization, credit must be given to Mary Marsh, who had some time ago identified the significance of the biomineral associated phosphoproteins.84,288–290 Marsh has also shown that high-capacity calcium-binding proteins, such as those found in bivalve phosphoproteins and dentin phosphophoryn (with apparent M.W. of around 100,000Da), can self associate in the presence of calcium ions. In the case of the highly phosphorylated rat dentin phosphophoryn (HP), it forms aggregates at a calcium concentration of about 5 mM, and although this concentration is higher than physiological conditions, these aggregates are formed at much lower calcium ion concentrations (physiological values) in the presence of inorganic phosphate.289,291 In this case, for each P043− ion bound, the protein could then bind about 1.5 additional Ca2+ ions (this is consistent with our observations that the counterion can help bring in additional calcium loading in the PILP process). The protein-mineral ion complex was described as “a protein with two different ligands, Ca2+ ions and calcium phosphate clusters having a stoichiometry of about Cal,5P04.” This is similar to the stoichiometry of amorphous calcium phosphate, and no crystallinity was detected in these clusters. In fact, these sequestered mineral ions did not promote crystallization of hydroxyapatite (HA) in solution either, even though others217 had shown that phosphophoryn immobilized on substrates could promote the nucleation of HA. Marsh attributes this difference to the fact that self-association cannot occur for the immobilized protein, and thus different surfaces are exposed. Given that these ACP complexes did not convert to, or promote nucleation of, hydroxyapatite, Marsh suggested that the aggregates could provide a locally high concentration of bound mineral ions, so that degradation of the protein would lead to spontaneous precipitation of a mineral phase or to growth of preexisting mineral. An alternative suggestion was also provided, that the protein might become immobilized on the collagen fibrils in the dentin matrix before aggregation occurs, where the protein could conceivably catalyze the formation of hydroxyapatite crystals. The literature at this time did not consider vertebrate biominerals to be formed via an amorphous phase, but in retrospect, another explanation can be provided for her observations, where we propose that amorphous CaP complexes may play an active role in the intrafibrillar mineralization of collagen fibrils (as discussed in Section 4.4). On the other hand, I do suspect that this high molecular protein would need to be enzymatically cleaved to provide this activity.
Marsh visualized the protein aggregates via TEM, and Ca2+-protein complexes alone were found to be predominantly linear at low calcium content, but assume a dense compact globular structure (~25 nm discrete particles) when fully saturated with calcium. When the complexes were prepared with both charged calcium and phosphate, the protein remained in an aggregated state even in phosphate-free buffers having a calcium ion concentration too low to support free protein aggregation. We don’t observe aggregates (PILP droplets) with the simple polyaspartate additive until the carbonate counterion is added, which at our lower concentrations is consistent with the stabilizing influence of the counterion described by Marsh. Plus, the molecular weight of our polyaspartate is much smaller than the phosphophoryn.
It is clear that much more work is needed in this area before we can fully understand the structure-function relationship in these proteins, or even their synthetic analogues. Towards that goal, our group is trying to find ways to quantify the properties of the ACC phase (e.g., degree of hydration, kinetic stability), which is sorely needed to provide a basis for applying more systematic studies of the roles of the various reaction parameters and polymer properties on the precursor process.
3.4.2. Stage II- Precursor Transformation
This second stage of the precursor pathway is important for several reasons; the more obvious reason being that the transformation stage may dictate the resulting crystal orientation and phase. Relatively little is known about nucleation within an amorphous phase, and this will likely be an active area of research in the future; but it is certainly anticipated that epitaxial-like relationships can direct the transformation process, even though the results may differ from the same material crystallized via the conventional pathway. There is another, less obvious reason that the second stage of the precursor pathway is important, and that deals with the stability of the precursor phase, which can have profound impact on the resulting crystal products because it can influence the A-to-C transformation pathway. Therefore, this section will be broken down into these two key issues associated with Stage II of the process, (i) the stability of the amorphous precursor phase, and (ii) the orientational and phase-directing influence of the precursor phase.
3.4.2.1. Variable Transformation Pathways
Based on the literature and in situ observations in our own work, there seems to be (at least) three transformation pathways that an amorphous precursor can follow. The first possibility is a dissolution-recrystallization pathway. This seems to the route taken when the ACC phase is short-lived, particularly when it is not stabilized by a polymeric additive. This was mentioned in Section 3.1.1 when describing Rieger’s studies of transient ACC formation,227 where shortly after the emulsion-like structures are formed, they break down into nanoparticles that aggregate into micron sized vaterite spheres, and then dissolve and recrystallize into calcite rhombs (recall Figure 16). If polymer additive was included, flocs were formed if the polymer sufficiently coated the primary nanoparticles to stabilize them against compact aggregation. Although such a pathway is important to understand for a variety of commercial applications (such as filler particles and encrustation of pipelines), it is less valuable towards developing biomimetic strategies for molding an amorphous precursor to form non-equilibrium crystal morphologies, as occurs in biomineralization. In biomineralization, an insoluble matrix is likely to be involved, and when an amorphous phase is deposited on a substrate, the substrate can influence its transformation.
Han and Aizenberg292 have capitalized on this recrystallization pathway in a clever experiment which directs the recrystallization of the ACC phase on command. First, colloids of ACC were produced on OH-terminated SAMs (Figure 31a), and then a secondary nucleating substrate was placed in contact with the ACC phase, in the presence of a small amount of water, to lead to patterned locational and orientational control of the calcite crystals (Figure 31b). The crystals exhibit a faceted equilibrium habit since they amorphous phase underwent dissolution and recrystallization. Interestingly, Lee et al.,293 who examined similar crystal templating from an ACC phase deposited on mercaptophenol SAMs (using near-edge X-ray absorption spectroscopy (NEXAFS), photoemission spectroscopy (PES), XRD, and SEM), found that the precipitation of the ACC film brought about substantial structural disorder within the previously ordered monolayer, yet a preferred orientation of the calcite resulted from this ACC precursor despite the static disorder of the SAM template (Figure 31c & d).
Figure 31.

Examples of the dissolution-recrystallization pathway for transformation of ACC precursors. (a & b) Han & Aizenberg292 show that a transient ACC phase can be deposited onto hydroxyl-terminated SAMs, which can then be stimulated on command to recrystallize into calcite crystals of controlled orientation by contacting with a secondary nucleating surface in the presence of a small amount of H2O. (c & d) Lee et al.293 demonstrate that formation of ACC on mercaptoethanol SAMs leads to structural disorder in the monolayer which originally consisted of well-defined orientations; yet a preferential face of nucleation occurred upon recrystallization of the ACC into calcite on the SAM surface, despite the static disorder. A depletion zone in the original ACC granular film surrounds the newly crystallized (104) calcite rhomb. (a–b) (Reprinted with permission from ref 292. Copyright 2008 American Chemical Society.) (c–d) (Reprinted with permission from ref 293. Copyright 2007 American Chemical Society.)
A second transformation pathway is demonstrated by Sethman’s work (Figure 32),294 in which in situ AFM was used to examine new mineral formation on a calcite substrate in the presence of polyaspartate additive (5–15kDa). They found that that some type of gelatinous phase formed on the surface of the calcite substrate (Figure 32c & d), which seemed to densify and evolve into numerous nanocrystals (Figure 32e & f)). It does not appear to be a dissolution-reprecipitation reaction, but rather an overall physical change in shape of the precursor phase as these structures emerge (Figure 32d). Whether this gelatinous precursor is amorphous or not cannot could not be determined by AFM, but I think it is reasonable to assume that the originating ion-enriched non-descript gel phase is some type of amorphous phase (similar to that described by Ulčinas et al.,258 as shown in Figure 22). The iso-epitaxial relationship with the underlying calcite crystal presumably allowed for these multi-nucleating polycrystals of uniform orientation to combine into a “calcitic single-crystalline nano-cluster of semicoherent domains”. The mechanism was considered to be analogous to the mesocrystal assembly mechanism proposed by Colfen and coworkers,295 although separate nanocrystals do not need to rotate and align to form a semi-coherent crystal (as in assembly of solution borne nanocrystals), because they are already well oriented from secondary nucleation on the underlying calcite crystal.
Figure 32.

Sethman et al.294 demonstrate nano-cluster calcite growth from within an polyaspartate-mediated ACC gel phase that was deposited on a calcite substrate. (a,b) AFM of additive-free calcite growth shows step advancement typical of the conventional crystal growth pathway, with growth dominated by step advancement on the {10.4} faces. (c) With pAsp, the substrate crystal becomes covered with a gelatinous pAsp-ACC film. (d) The gelatinous precursor seems to densify and evolve into structures with multiple nucleation sites. (e) Substrate-conformable, homoepitactic nucleation of local growth domains occurs synchronously at numerous locations. (f) The growth domains mature and fuse into a mesocrystal, which is suggested to occur through a continuous nucleation of growth domains on top of each other to yield a semicoherent, dendritic cluster-growth pattern. (g–j) The crystallographic orientation of the substrate is preserved continuously throughout the whole precipitate, yielding a uniform lattice orientation conformable to the underlying (10.4) calcite substrate lattice. (i) Crystallographic orientation of the substrate calcite cleavage rhombohedron. (j) Cleavage of the substrate crystal oblique to the cluster layer reveals a rough fracture pattern of the precipitate (A = substrate, B = precipitate, C = surface; height range in area B = 40 nm). (Reprinted with permission from ref 294. Copyright 2005 Mineralogical Society of America.)
A third transformation pathway is the pseudomorphic transformation of the amorphous precursor. Pseudomorphic refers to the case where the crystalline product retains the shape of the precursor phase, and therefore is analogous to a solid-state transformation. However, without thermal mobilities providing for the reorganization of ions, it is assumed that some mobility is provided by the hydration waters of the ACC phase (therefore, I refer to it as a pseudo-solid-state transformation). This pathway was demonstrated by the PILP system in Figure 14d, and other thin films described in Section 3.2.1, where the crystals retain the shape of the originating precursor film. This is also nicely demonstrated by Aizenberg’s 2003 paper,263 where calcite crystals were molded around a micropatterned substrate by regulating the A-to-C transformation (Figure 33). They used SAMs composed of alkanethiols of different lengths terminated in phosphate, methyl, and hydroxyl groups to create a disordered organic surface that suppresses the nucleation of calcite and favors the formation of ACC. Before depositing the ACC film, nanoregions of a SAM that was known to induce oriented nucleation of calcite was deposited on the gold substrate by the tip of an atomic force microscope. By integrating these specialized nucleation sites, they were able to fabricate large (up to a mm) single crystals of calcite that were molded around the posts of the substrate. Had the ACC film undergone dissolution and recrystallization, such large molded single crystals could not have been formed under.
Figure 33.

Aizenberg et al.263 have fabricated micro-patterned single-crystalline calcite using specially designed structural templates with integrated nucleation sites to control the amorphous-to-crystalline transformation. (a) A micropatterned mold prepared by photolithography is treated with SAMs of functionality that suppresses calcite nucleation (such as a disordered phosphate-, methyl- and hydroxy-terminated layer), along with a high supersaturation (1 M Ca2+) to promote ACC. (b) A nanoregion was printed into each template using an AFM tip coated with a SAM of HS(CH2)nA (where A = OH, CO2H), to provide a singular site for calcite nucleation, which then propagated throughout the ACC film, leading to a large single crystal of calcite molded around the array of photolithographically produced posts. (c) SEM of a micromolded and [001] oriented single crystal of calcite. (d) Polarized light micrograph of a large porous single-crystalline patch of calcite. At this time-point, the surrounding ACC film “ruptured” into numerous birefringent spots, thus demonstrating the dimensional limitations that might be encountered in the precursor pathway. The dimensions and chemistry of the template were found to be important for regulating the initial ACC stabilization, as well as its transformation into crystalline calcite. (Reprinted with permission from ref 263. Copyright 2003 American Association for the Advancement of Science.)
The pseudomorphic transformation pathway has been of particular interest to our group because it enables the formation of non-equilibrium morphologies, which is the hallmark of biomineralization. However, we do see evidence for all three of these pathways in our PILP model system, and in fact, the significant challenge in this system is in trying to avoid the other pathways. For example, PILP formed films will often dissolve and form large aggregates in the vicinity (Figure 34a). The second pathway of restructuring of the amorphous phase may be responsible for the micro-faceting that is sometimes found in PILP formed products, such as the calcite fibers shown in Figure 34b. Alternatively, the microfacets could arise after the transformation, from surface reorganization as a means of lowering the surface tension of the underlying calcite (which is often seen for non-equilibrium morphologies that are left in solution for extended periods of time (Figure 34c & d). In the biological realm, it is assumed that vesicular membranes and biomolecular matrices help protect such non-equilibrium structures from dissolution or reorganization.
Figure 34.

Transformation of PILP formed ACC precursors. (a) An ACC film that was deposited on a Langmuir monolayer seems to be transforming by two pathways. The brownish aggregates are spherulites which likely formed from dissolution-recrystallization of the pre-existing ACC film, while the orange and blue patches of film are undergoing a pseudomorphic transformation. The amorphous film surrounding the spherulites can be seen by the rough texture in this particular example, but smooth ACC films are more difficult to image because they are not birefringent. (b) Calcite fibers emanating from a calcite seed crystal. When formed by the SPS mechanism, the fibers usually have fairly smooth curved surfaces, but sometimes microfacets are seen, as shown here. This may have arisen from mesocrystal assembly (from within the precursor phase), or from surface restructuring of the curved crystal surfaces. These fibers appear to be oriented along the [001] direction (a common growth direction in biominerals), as judging from the vertices of the rhomb-shaped microfacets on the tips of a few fiber (zoom in on top right fibers). Bar = 1 μm. (c) A patch of PILP deposited film that was left in solution for several weeks shows pronounced reorganization. The film was originally smooth and single crystalline. (d) At higher magnification, microfaceting is seen, or even full recrystallization has occurred (although it has remained localized to the film-like structure, unlike the typical recrystallization, which altogether obliterates the non-equilibrium morphology and generates aggregates of large rhombs or spherulites. I believe this was because the solution was slowly evaporating and maintaining a high supersaturation. The interesting channel patterns are probably from reorganization at sites of previous defects in the films.
The second and third pathways are perhaps not all that different, where either a gelatinous or fluidic precursor (in the case of PILP ACC) are formed in the presence of anionic polymer, and both seem to provide a means for forming smooth non-equilibrium morphologies. I think the different observations may arise from substrate effects, given that the reaction conditions in Sethman’s study are nearly identical to our PILP system, where we both used a polymeric directing agent of poly(L-aspartic acid)• sodium salt (Mw = 5–15 kDa). For example, both pathways lead to a final nanogranular texture, which will be discussed in Section 5.2.1; but the originating nanoclusters in Sethman’s model become triangular as nanofacets develop (as partial rhombs). They suggest this may be due to preferential adsorption of polymer onto those faces; but I offer an alternative suggestion. The nanogranular texture in our system does not display this triangular morphology, and instead resembles dense-packed spherical colloids. The primary difference between our systems is the substrate, where they use a calcite seed crystal, while we use glass or SAMs. The calcite seed could rapidly stimulate iso-epitaxial nucleation within the amorphous phase, such that it quickly crystallizes into the nanofaceted subunits, faster than the infilling with further amorphous phase. In our system, a smooth and continuous ACC film is formed through continuous deposition of precursor droplets, long before it crystallizes, since it is not stimulated by a calcite seed substrate. Their pyramidal subunits appear to have a rather low packing density once the gel phase has densified and evolved into what appears to be crystalline subunits. They suggest that “a continuous nucleation of growth domains on top of each other may easily yield a semicoherent, dendritic cluster-growth pattern.” This mechanism appears somewhat similar to conventional crystal growth under conditions of very high supersaturation, which leads to numerous overlapping nucleation islands. Indeed, this type of growth process can lead to rounded crystals with somewhat curved surfaces, but it seems like it would leave a lot of voids, and it is not clear to me if it is amenable to molding crystals with smooth surfaces (see for example their other model system presented in Figure 63, which shows a surface that is rough and heavily microfaceted). An alternative view is that the smoothing of the surface could occur via further deposition of amorphous phase, which could fill the void space left by the former contracted precursor phase. This seems more likely to form a smooth surface and single-crystalline structure than repeated crystal nucleation and overgrowths, which I would think would leave a lot of voids and microfacets. We have suggested that a similar ‘annealing’ effect might occur for the infilling of rough transition bars that form during the amorphous to crystalline transformation.142
Figure 63.

Sethman’s analysis of Octocoral sclerites and an in vitro model.381 (Top Row) SEM of fractured octocoral sclerites reveals the internal fibrous crystals of Mg-calcite, which as judged by the transition in texture, was suggested grow by aligned aggregation and fusion of nanoparticles. Concentric layers can also be seen here, and at higher magnification in AFM, the fine-grain nanogranular texture can be seen, which is thought to enable the smooth shaping of the sclerites. (Bottom Section) A model system of calcite crystals grown in polyacrylamide hydrogel. (a–c) SEM images; (d–f) AFM images. (a) Total view of a typical crystal with cluster-like surface morphology. (b) to (c) The fracture surfaces reveal a change in growth pattern, developing from a completely coherent crystal (core), towards a dendritic semicoherent cluster. (d), (e) Nano-granular surface structure and (f) sub-surface structure of the crystals. (g) Fracture surface in the transition area of dendritic splitting with predominantly smooth {101̄4} cleavage planes. (Reproduced by permission from ref 381. Copyright 2007 The Royal Society of London.)
I note that similar observations of a smoothing effect have been described by Ulčinas et al.258 in their scanning probe microscopy study on spherulite formation in a PAA-stabilized ACC gel that formed on chitosan substrates. In this case, the small scale features that were initially observed, such as fibrous growths, lobes and concentric bands (that appear similar to transition bars), became smooth with time, which could be quantified as a decrease in both surface roughness and height (recall Figure 22). They describe this as a ripening effect, and suggest dissolution of the ACC precursor occurs during crystallization, which then further necessitates the suggestion of solution PAA to adsorb to the growing crystals to maintain surface crystallization. I believe this latter aspect (again, the traditional view of surface-limiting inhibition) is not necessary because their system seems very much like our observations, which I regard as a pseudomorphic transformation (the thin film morphology of the precursor is retained). Even though intermediate features may be observed, the ‘wrinkles’ somehow become ‘ironed’ out during the transformation. Thus, highly smooth surfaces can be formed from such an annealing effect, even though we don’t fully understand how this occurs.
The observations by Tao et al.296 of hydroxyapatite nanocrystal re-orientation when encapsulated in an amorphous coating may also be relevant to this discussion on transformation pathways, but it isn’t known how this re-organization occurs. Can there be rotation of the nanocrystallites within the amorphous phase (this could be more readily envisioned if it has some fluidic character; but in their system, this presumably wasn’t the case since the particles were preformed). Or do the entrapped nanocrystals undergo dissolution-recrystallization (their system did sit for extended periods of time), leading to a ripening type of effect?
3.4.2.2. Transformation Kinetics
Overall, I believe the stability of the metastable amorphous phase is critical for capitalizing on the advantages of a precursor process, and therefore is an area we are actively pursuing. We have performed in situ x-ray reflectivity studies with collaborator Elaine DiMasi at the National Synchrotron Light Source (NSLS) to examine mineral film formation under Langmuir monolayers,246,297 such as stearic and arachidic acid. Studies thus far have shown that the kinetics of deposition of the precursor phase strongly impacts it stability, in which it was observed that PILP films that are formed quickly at the air-water interface will also dissolve quickly (Fig. 7).297 The rate of film formation was influenced simply by a change in geometry of the crystallizing trough, in which a larger surface to volume ratio of the container increased the CO2 escape rate, and therefore raised the supersaturation more quickly.
Xu and Cho’s studies are consistent with this finding, where the ACC films prepared without polymer additive (at high supersaturation) formed more quickly, but also had to be intercepted more quickly to avoid dissolution-recrystallization.259 They were able to form high quality, continuous films simply by retrieving the films from solution before they crystallized. They examined the transformation of ACC films in air, and found that it was still influenced by environmental factors, such as relative humidity and temperature.260 For example, the relative humidity was found to strongly increase the rate of transformation shown, yet those films became quite porous as the spherical subunits grew in size and partially coalesced. On the other hand, the ACC films could be kept under vacuum at room temperature for more than 3 months without showing evidence of crystallization. They suggest that increasing the humidity speeds up the transformation by supplying water as a matrix to promote the dissolution-recrystallization mechanism of transformation. I would argue that this is only partially the case, where full dissolution-recrystallization should lead to microfacets (as observed in other systems, and demonstrated in Figure 34c & d, for example). The spherical subunits of these films remained spherical, even though they grew, and (in my opinion), the moisture appeared to provide an “annealing” or Ostwald ripening effect. Perhaps a reasonable compromise is to suggest that these processes can occur to different degrees, where humidity may provide some intermediate level of each (i.e., the non-equilibrium film morphology is still retained, along with spherical constituents, while some reorganization leads to ripening effects).
In our system (which includes inhibitory polymer), some films do transform while still left in solution, and they stay smooth and continuous, without forming these pores. Thus, even when being surrounded by water, they still seem to be able to follow a pseudo-solid-state transformation pathway. Comparison of these systems suggests that the stability of the ACC phase is important for determining how it will transform, and this can be influenced by the presence of inhibitory polymer, substrate, and/or inhibitory Mg-ion, etc.. In the aqueous physiological solutions present in biomineralization, this stabilization is presumably accomplished through appropriate interactions with the organics. Thus, it is desirable to understand what features of these proteins might help to provide this type of stabilization. In our model system, features of the polypeptide additive that seem relevant include polymer molecular weight, concentration, charge density, etc..
Aside from moisture, temperature was also found to increase the transformation rate in Xu’s studies. The heated films exhibited a mosaic texture of single-crystalline patches, whereas the room temperature crystallized films had spherulitic textures. Given the relatively small ΔH values measured in the DSC (e.g., −0.86 and −2.49 kJ/mol), they argue that the thermodynamic energy barrier of the solid-solid transformation process can be easily crossed with the thermal energy provided. An interesting observation was that the films transformed at low temperatures (100°C) exhibited some preferred [110] orientation, which was lost in the films transformed at higher temperatures (300°C). Because the DSC/TGA data shows two water peaks, they suggest that the tightly bound waters, which are retained longer at the lower temperatures, may stabilize the (110) crystal faces. The kinetic differences also led to different textures, where films crystallized at the higher temperature, which presumably underwent a rapid dehydration and crystallization, caused the colloidal constituents to retain the nano-dimensions of the original ACC film.
Based on the observation that film formation rate affects it stability, one must now wonder what is different about the ACC phases that affect their stability? One possibility is that there could be compositional and/or structural differences in the precursor films. For example, the fast formed films may not collect the proper stoichiometry of ions for ease of crystallization, or they may lack some type of short or intermediate-range order that aids in crystallization. In the former case, our compositional studies show that the composition of the PILP phase changes with time,225 starting with a high calcium concentration that is sequestered by the polymer, which is gradually displaced by the carbonate ions that enter via the vapor diffusion method. It is not known what concentration of ions is required to induce the liquid-liquid phase transition, and therefore the stoichiometry of captured ions may differ depending on the how the supersaturation was raised, as well as characteristics of the polymer. And even when the PILP phase has formed, it may still be dynamic and continue to recruit the appropriate ions until a more stable phase can crystallize. Quite frankly, much more work is needed before we can begin to understand what is happening within the amorphous phase.
In addition to the stabilizing influence of the soluble polymer, the substrate might also be expected to play an important role during this stage as well, because if it promotes crystallization, it could help the unstable amorphous phase to more quickly reach the more stable crystalline phase, making it less prone to dissolution. For example, it is often observed that a fully continuous amorphous film may be deposited over the entire substrate in the early stages, but much of this film dissolves away except in the regions where crystallization has occurred. This then leaves behind thin platy crystals (i.e., tablets) on the substrate. This dissolution is exacerbated if a large crystal aggregate grows in the vicinity, which depletes the surrounding ion concentration making the amorphous phase even more prone to dissolution. The formation of aggregates has been a significant hurdle in controlling the process because it can be difficult to find the proper conditions that cause all of the ions to precipitate via the amorphous pathway, and not form solution crystallization by-products (which are not regulated for morphological control for example). As a side note- this aggregation tendency of crystals that grow from dissolution-recrystallization of an amorphous phase prompted us to suggest that such a process may be responsible for some pathological biominerals, such as kidney stones (see Section 4.5.2).
The patterning experiments described in the above section demonstrate that the substrate has a pronounced influence on the transformation rate of the precursor (Figure 29).221 The ACC films that formed on regions coated with carboxylate-terminated SAMs crystallized within days, while the ACC films that formed on the gold regions took months (these were both after removal from the solution). If we assume that the ACC phase that formed on the two regions was the same (because the formation of the droplets occurred during the Stage I, which used the same process-directing agent), then this variation in Stage II is apparently caused by the variation in substrate chemistry. The substrate chemistry effect is also clearly seen in Aizenberg’s study, where she was able to control the nucleation event from within the ACC film using specialized SAMs.263 In this case, not only was the transformation kinetics regulated, but crystal orientation was also regulated with the substrate chemistry.
Most of the studies seem to suggest that a polymeric process-directing agent is needed to provide sufficient stability to favor the pseudomorphic transformation. There is, however, a possible exception to this rule, where in the Aizenberg study mentioned above, the micropatterned single crystal of calcite was transformed from a phase supposedly produced by the SAM and high supersaturation.263 However, it was mentioned in passing that “In several experiments, we also added macromolecules extracted from biologically formed ACC-containing tissues, magnesium, or phosphates to stabilize the deposited ACC layer further.” So apparently there were some stabilizing additives present, and perhaps even polymer. In any case, it seems there is a counterplay between the stability of the amorphous phase against dissolution, versus its metastability that allows it to transform and crystallize into the more stable crystalline phase. By using a favorable substrate to stimulate transformation of the amorphous phase, one might be able to promote the pseudomorphic transformation before the metastable phase dissolves and recrystallizes.
3.4.2.3. Modulating Crystal Orientation and Phase
It seems reasonable to anticipate that a variety of phases could be produced from an amorphous precursor because the interactions responsible for inducing the precursor phase are non-specific (i.e., from ion binding of anionic polymer). Indeed, the ACC films we deposit on glass generally transform into a mixture of calcite and vaterite, where the calcite films have a mosaic texture from impinging domains of single-crystalline calcite, whereas the vaterite films always have a spherulitic texture.202,203 To enable control over the crystal phase, one presumably needs to find a way to promote and/or stabilize the more metastable phases. Returning to the reaction coordinate diagram in Figure 10, one might be able to accomplish this through modulation of the kinetic energy barriers (to pass through an intermediate phase of interest), and/or relative stabilities of the final crystal products. Such control could conceivably be derived from a templating substrate (during Stage II), or the influence of species intercalated within the amorphous phase, such as entrapped polymer or ionic impurities (created during Stage I).
We have explored conditions for modulating crystal phase from the PILP reaction, and find that nearly all of the phases of CaCO3 can be formed (e.g., films of calcite, aragonite, vaterite, monohydrate, possibly hexahydrate, and of course ACC). This was done simply by changing only the reaction kinetics and stoichiometry of ions (with the same substrate).287 Therefore, there is reason to suspect that there are compositional and/or intermediate-range structural differences in the precursor phase (created in Stage I) which could influence the subsequent steps of the precursor pathway. Because of this plausible effect, the obvious study to perform is to measure the composition and structure of the precursor phase. But this has proven difficult in our PILP system because the amorphous phase is dynamic (see Section 3.3). Both the polymer and water are seen to decrease with time, presumably as the phase tries to reach the more thermodynamically stable state by excluding these impurities. Likewise, the stoichiometry of ions is changing as the carbonate ions are slowly introduced into the system. Other model systems that don’t rely on the vapor diffusion method, or the changing composition of the PILP phase, might be more useful for this purpose.
Other groups have been more successful at producing high-purity thin films of either calcite or aragonite.256 Hosoda and Kato298 propose that the thin film polymorphs are dependent on the density of calcium ions on the surface of the nucleating polymer matrix, which is influenced by polymer concentration as well as polymer molecular weight, thus leading to metastable polymorphs with higher localized calcium. In the case of aragonite films on chitosan, Mg2+ ion was needed to form aragonite. In contrast, aragonite films could be templated on substrates of crystalline poly(vinyl alcohol) without addition of Mg2+ ion. Changing the soluble additive to polyGlu led to vaterite films on the same PVA substrates. For this system, they suggest a templating effect of the PVA matrix could align the COO- moieties to match the spacing of the ab plane of aragonite. However, there was no indication of preferred orientation of the aragonite, and it appeared to be of spherulitic texture. Aragonite readily undergoes polytwinning, and almost inevitably forms spherulites, which in the case of the films, generates a spherulitic texture from the needle-shaped polycrystals.
We also examined the influence of Mg-ion on our PILP reaction, thinking that it would promote the aragonite phase. Instead, we found that high magnesium calcite could be produced instead,180 which is an interesting feature in and of itself (see Section 5.1). However, we did eventually find a lower Mg-ion, in combination with a resorcarene monolayer, led to mix of calcite and aragonite tablets.119 The interesting feature here was that the aragonite tablets were single-crystalline, which is the first time that single-crystalline aragonite with a morphology similar to nacre has been produced synthetically (discussed in Section 4.1.3). On the other hand, the system was not well regulated, where some regions of the film transformed into calcite and aragonite patches, while some film dissolved and recrystallized into aggregates.
Perhaps with a more selective organic matrix, it might be possible to regulate the nucleation event (from within the amorphous phase), which could then regulate the crystal phase and orientation. Alternatively, more ‘specific’ interactions from the polymeric process-directing agent within the amorphous phase might influence which polymorph is formed. As mentioned in Section 3.3, the soluble proteins extracted from biominerals seem to provide control over crystal phase,264,277 and relatively simple peptide analogues have also been found to promote aragonite phase (although via conventional crystal growth).299 In addition, x-ray absorption spectroscopy has provided evidence of some type of pre-organizational short-range order in the x-ray amorphous material of biominerals, which may prescribe the phase of the final crystalline product.206,215,273,274 Similar observations have been made on synthetic ACC prepared with Mg-ion or poly(aspartic acid) (pAsp), where the Mg-induced ACC showed short-range structures most similar to aragonite (which developed into monohydrocalcite short-range structures after incubation in solution), while the pAsp-induced ACC possessed short-range structures resembling vaterite.300 These results suggest that the first stages of recrystallization involve the expulsion of water from the structure, rather than significant changes in the short-range structure around the calcium ions. Thus, this nice body of work shows that a variety of factors can influence the phase transformation, which go far beyond the traditional view of crystal phase being regulated by the insoluble organic matrix. Now one must consider the influence of entrapped polymer or ionic impurities, ion stoichiometry, or hydration waters, all of which can be present in the complex mixtures that comprise the amorphous phase(s).
In conclusion, the transformation route that is followed in Stage II can have important consequences because if one can mold and shape the precursor, and that shape is retained, then one has a means of forming crystals with an infinite array of morphologies. The hallmark of biomineralization is the hugely diverse and elaborate morphologies that are created! Therefore, I consider this to be an important area for pursuing further developments in biomimetics, where we need to examine at the molecular level how organic-inorganic interactions can dictate the transformation pathway of the amorphous precursor.
3.5. Related Ceramic Processing Techniques
There are some analogies between polymer-induced ACC processes and sol-gel processing of ceramics. Namely, the use of precursor species provides a means to mold and shape ceramics into fibers, patterned coatings, micromolded articles, etc., at low temperatures. However, the precursors used in the sol-gel process are chemical precursors which require polycondensation, such as organo-metallics, rather than physical precursors, such as PILP phase generated with polymer coordinated ions. A network can be formed from either colloidal sol particles, or hydrolysis and condensation of metal alkoxides to a ‘polymeric gel’. These materials, typically metal oxides, generally have to be heat treated to fully densify the gel-derived ceramic, which is not the case for the biomimetic PILP system. Nevertheless, there is a conceptual comparison that can be made between these low temperature ceramic processing techniques, and information from this more mature field could shed some light on the various processing issues associated with the molding and shaping of fluidic precursors.
Another ceramic processing technique that bears some similarity to the polymer-induced ACC process is the Polymer Complex Solution (PCS) method. The PCS method is a ‘gel’ processing method performed in aqueous solutions with water-soluble metal salts (e.g., ZrOCl2•8H2O and Ce(CH3COO)3•H2O), with addition of water soluble, cation-coordinating polymers, such as polyvinyl alcohol (PVA), polyacrylic acid (PAA), and polyethyleneimine. The metal-polymer complexes behave as crosslinking centers to from a 3-dimensional network entrapping water as a gel. On the surface, this sounds similar to the PILP process, but in the PCS system, the water is physically removed to concentrate the gel, and then the gel is heated to several hundred degrees to remove polymer and organics (such as citrate). These steps are not needed in the PILP process since it is thermodynamically driven to lower its energy by excluding polymer and water, and this advantage has apparently been capitalized on in the biological world, which does not have the luxury of applying heat or vacuum to make its ceramic materials. One promising aspect in the field of biomimetics is that further advances in understanding biomineralization may lead to new approaches toward mimicking Mother Nature’s ‘green chemistry’.
3.6. How Well Can We Mimic the Complex Biological Reaction Environment?
3.6.1. Mineralization Reactions
One of the first difficulties that arise when trying to develop an in vitro model system of biomineralization is the means by which the ions are introduced to generate the precipitation reaction. In biological systems, the biominerals are often formed within a compartment or macromolecular matrix, which allows for cellular control over entry of ions and additives, both in time and space. For example, ions can be pumped across the cellular membrane into a mineralization chamber, or as has been physically observed, small vesicles are delivered to a primary mineral deposition compartment1,4,72,301 (or to an extracellular matrix in the case of matrix vesicle in bone formation); or in some cases, the ions may be generated on location through enzymatic reaction (e.g. alkaline phosphatase to produce phosphate or carbonic anhydrase to produce carbonate). Therefore, when trying to develop an in vitro model system, it is sometimes desirable to slowly add one of the ions. This helps to avoid rapid precipitation that can result with direct addition of the two reactants, because at physiological concentrations, the supersaturation is often sufficiently high that rapid precipitation often results. It is also desirable to have sufficient time for the ions to interact with the polymer, which may either be a substrate that promotes heterogeneous nucleation or a soluble acidic macromolecule.
An in vitro system quite often used for CaCO3 precipitation (and that we use for most of our PILP experiments) involves the slow entry of the carbonate counterion into a Ca2+ containing solution, which is often where the polymeric additive is included to provide time for cation-polymer interactions. The slow entry is provided by the decomposition of ammonium carbonate powder, which generates a vapor phase of NH3 and CO2 that diffuses through a chamber and enters into the calcium containing solution to provide the carbonate counterion. I believe this nifty technique was first developed by Addadi and co-workers,85 and it has become popular because it leads to a very uniform product of calcite crystals with rhombohedral habit. Although vapor diffusion of counterion is obviously not the way supersaturation is achieved in biological systems, it could be considered analogous to addition of ions across a membrane, since the ions are added directly rather than through addition of a counterion solution, which dilutes the initial solution as it is added. There are alternative ways to slowly raise the supersaturation, such as through changes in pH (e.g,. the CO2 escape technique302 or decomposition of urea303), or by slow addition of a counterion solution (which can be accomplished by a burette or mini-pump), or double-diffusion of ions across a gel or porous membrane.304 We have found that the PILP process can be generated with several of these techniques; but most of our data for the CaCO3 system has been obtained with the vapor diffusion technique because we believe it most closely mimics biological systems (with slow but direct addition of a counterion, and not as a diluting solution).
An analogous vapor diffusion system is not available for the calcium phosphate system since ammonium phosphate does not decompose as readily as ammonium carbonate, so most people use direct addition or pH regulation. In the case of the CaP PILP reaction, we found that it could be performed without the gradual introduction of counterion, and direct addition can be used.160 The inhibitory effect of the polymer and a tris buffer solution keeps the ions from immediately crashing out into a precipitate. We perform this reaction at 37°C to more closely mimic the physiological conditions of vertebrates in bone formation.
Most of the reaction parameters, such as ion and polymer concentrations, temp., pH, etc., influence the reaction kinetics, and since the ACC pathway is kinetically driven, changes in such reaction parameters can lead to considerable variation in the products. Nevertheless, the generation of an amorphous phase (Stage I) is not that difficult to achieve, but the transformation stage (Stage II) is often times more profoundly influenced, as discussed in Section 3.4.2.
There is another reaction parameter that is generally not taken into consideration in crystallization reactions, but can have a profound impact for the PILP amorphous precursor pathway, and that is the reaction volume. And this becomes particularly pertinent when trying to correlate in vitro observations to biomineralization. One must keep in mind that the size scale of biological reactions is generally much smaller that the crystallizing dishes we utilize. For example, if biominerals do use a PILP type of precursor phase, it would only require short travel distances and time frames, so the ability to keep the amorphous material in a fluidic state would not be a problem. On the other hand, this means the presence of a PILP type phase in vivo may be very difficult to detect. The discovery of the liquid-phase precursor in vitro was fortuitous because the size scale of the reaction system we use just happened to provide dimensions sufficient for the droplets to grow to a micron size scale that could be more readily observed.
3.6.2. Protein Mimics
We are operating under the assumption that the acidic proteins associated with biominerals will behave similarly to the polyanions studied in vitro, if an appropriate model system can be designed. Although polyaspartate seems to be a rather crude mimic to the aspartic acid rich proteins found in biominerals, the amorphous precursor pathway does not require specific structural interactions with crystal faces since the organized lattice is not even present during the polymer-ion interactions which generate the amorphous phase. In other words, we are interested in the proteins that may serve as process-directing agents, and not considering proteins that may serve as structure-directing agents. These proteins would presumably have more specialized amino acid compositions and sequences, and protein conformations, than cannot be reproduced with a simple polyaspartate or polyacrylate model. Such structure-based interactions are likely involved in biomineralization, but arguably are less relevant to morphological control, and more important for other aspects of the process, such as control over crystal phase and orientation. As mentioned in Section 3.4.2, more work is needed in terms of understanding the influence of the organic matrix on transformation of an amorphous phase.
In the complex biological environment, there are numerous proteins which may act in concert to dictate the desired product. Even an individual protein could serve dual roles, including actions that go beyond the crystal modulation process. For example, there may be one domain of the protein that modulates crystal growth, such as through a collection of acidic residues to induce the amorphous phase, while another domain on the protein may bind to the organic matrix or crystals to provide cell recognition, or trigger some biochemical signaling, etc.. The multifunctionality of proteins could be a significant problem when one is examining extracted proteins in in vitro assays. In principle, it seems like the actual biological protein would provide more information than a simple polypeptide model, but if that protein is no longer in its natural surroundings, it may not even function the same. It is already well known that the polymers can behave differently when adsorbed to a substrate, where they may promote nucleation, versus free in solution, where they may inhibit crystallization altogether. By throwing this new function into the mix (the process-directing action), this situation is further confounded. Even something as simple as inclusion of Mg-ion, or providing the proper insoluble matrix, can have a pronounced effect on the activity of the soluble polymer. Thus, it is not surprising that those same proteins, which were once shown to modify crystal growth through stereoselective adsorption, are now shown to induce or stabilize an amorphous phase. I think one might expect a range of outcomes from the same proteins, where it all depends on how close the experiment is to mimicking the biological reaction environment overall (and what the experimenter is wanting to find).
Another issue that quite commonly ignored by biomimetics community is the role of the carbohydrate groups that are attached to proteins. While this is understandable considering the additional level of complexity involved with such hybrid polymers, it is not acceptable (in my opinion) for the cases where one is trying to determine if there is specific interactions, such as molecular recognition between a protein and crystal. In the case of generating an amorphous precursor, specific interactions are probably not required, and can perhaps be better modeled with minimal consequence from ignoring carbohydrate moieties (except the highly acidic ones, which could be responsible for inducing the phase). But as we try to go beyond simply forming an amorphous phase, but modulating its transformation, this may become an issue to deal with.
Enzymatic reactions might be used to regulate the inhibitory action of a large protein, which upon enzymatic degradation of the protein, allows crystallization to proceed. In the case of dental enamel, most of the organic matrix is removed through such degradative processes, leading to the most highly mineralized biomineral tissue.2,4,165 Such temporal control of crystallization additives is another area that is extremely difficult to model.
There is evidence that some biominerals might be formed in a gel-like matrix, such as the silk-like proteinaceous gel described for nacre,51,305 or the pre-formed matrix within the compartment of chiton and limpet teeth,171,196,197 etc.. This very likely has a pronounced impact on the amorphous precursor pathway, but yet adds an additional level of complexity that has not yet been addressed in model systems. Li and Estroff have initiated studies in this direction (on conventional crystal growth) by examining calcite crystals grown within agarose gels as a model of biomineralization.306,307 It will be interesting to see how such gel-matrix model systems influence an amorphous precursor. Do ACC nanoparticulates or PILP-like droplets adsorb to chains in the network, perhaps yielding anisotropic structures? Does the network influence their coalescence and transformation? Does the network become entrapped as a hybrid composite or excluded to form heterogeneous textured composites? Can such issues be controlled with specific structure-based interactions?
In conclusion, after having raised the host of issues that make it problematic when trying to model biomineralization mechanisms with biogenic protein additives, I conclude that using the more simple in vitro systems, such as crude polyaspartate additive, is not necessarily a bad thing, at least as a starting place. We first need to lay the foundation for understanding some of the principle crystallochemical mechanisms, and then we can start to build in higher levels of complexity by adding in components that work in combination. For example, adding in well defined templates is useful for studying how nucleation is effected in an amorphous phase, which itself is stimulated by other additives. With respect to the utilizing information from actual proteins, it would be valuable if functional domains, or specific chemical features, could be identified to study the more specific molecular-level interactions that may be responsible for the higher levels of control found in a biomineral. For example, the soluble proteins that help to form an amorphous phase may also provide the short-range ordering that regulates crystal phase, as suggested by studies of the ACC precursor in biominerals,206,212,215 and this type of crystallographic control may require more specific, molecular recognition types of interactions. Identifying the different functions of a protein, and how they correlate to the chemistry of the protein, may be tricky, for all the reasons mentioned above (and those not yet identified). Nevertheless, there may be cases where the correlations between protein chemistry/structure and function can be identified. For example, Aizenberg’s studies on proteins from antler sponge spicules and ascidians, both of which have regions of crystalline and amorphous CaCO3, suggest that the difference in amino acid composition between Glu versus Asp enriched acidic residues in these proteins can influence the stability of the ACC phase.268 An interesting feature has been identified, now we need to determine at the molecular level how this is accomplished, which may require a systematic set of polymers of well defined chemistries.
3.7. Concluding Remarks on the Role of Organics
Before I head into the next section on mimicking biominerals, let me first summarize how I view the roles of the different types of polymers that might be involved in the multistep crystallization pathway. Although further studies are needed, I currently consider the function of the soluble anionic polymer to be fourfold: 1) The polymer sequesters ions, while inhibiting crystal nucleation, which leads to a localized pool of ions that induce phase separation of the amorphous phase (which might be enhanced with Mg-ion, temp., etc.). 2) The polymer helps to stabilize the amorphous phase against dissolution, allowing the morphology of the precursor phase to be retained in the final crystalline product via the pseudo-solid-state transformation. 3) The polymer may help to bring in extra waters of hydration, generating liquid-liquid phase separation to induce the PILP process. The polymer may help to retard the solidification of the PILP amorphous phase (i.e., retain the hydration waters) long enough to allow manipulation of the phase (such as ‘coating’, ‘molding’ and ‘extrusion’). Overall, the role of the polymer can be described as a process-directing agent, which provides a powerful means of morphological control. This is in contrast to structure-directing agents, which achieve morphological control through specific ‘molecular-recognition’ types of interactions. I would argue that such structure-based interactions are less relevant to the morphogenesis of most biominerals, but more relevant to the regulation of other crystallographic features, such as phase, orientation, and texture.
The insoluble organic matrix could serve many functions, including the traditional view of templating nucleation of crystals from within the amorphous phase, which provides a means for regulating crystal phase and orientation. It may also be significant for inducing the amorphous phase, or attracting droplets of the amorphous phase, both of which could influence the location of the mineral deposit. The matrix can have a major influence on the A-to-C transformation, which can proceed through a variety of pathways, and thus require a high degree of control over the kinetics of the formation and/or transformation.
In many cases, the organic matrix in biominerals may consist of some type of gel-like network, which could vary from the dense-packed fibrillar arrays of collagen in bone formation, to the more minor constituents found in nacre and many other biominerals. The role of these matrices is less clear. Do they simply help to create a scaffold or compartment, or do they play a more active role in directing the mineralization process. In our biomimetic bone model system, collagen is seen to play an active role in directing the crystallographic orientation of the hydroxyapatite nanocrystals. The insoluble matrices found in other organisms are intriguing as well, such as the chitin fibrils and the silk-like proteins found in nacre. Rousseau et al.55,308 examined the intracrystalline matrix of individual nacreous tablets with AFM phase contrast and TEM darkfield mode, and find evidence of a fibrous interconnected network that “responds like a single crystal”, with some association to the orientation of the mineral phase, such that the nacre tablet is finely divided in the form a hybrid single-crystalline composite (Figure 35). This diffracting intracrystalline network would seemingly have to be formed by self-assembly of the macromolecules after secretion from the mantle cells. This sounds familiar to the fibrillogenesis of collagen which occurs after secretion of the procollagen peptides into the extracellular matrix, or the secretion of amelogenins in dental enamel (although there is far more organic matrix in bone, and far less in dental enamel; but this is due to enzymatic removal during maturation). The organization of these frameworks seems to be well correlated to the crystallographic orientation of the mineral, by some presently undefined mechanism(s). Rousseau proposed that each nacre tablet results from “the coherent aggregation of nanograins keeping strictly the same crystallographic orientation thanks to a hetero-epitaxy mechanism,” analogous to mesocrystal assembly.55 This is an interesting concept, although not necessarily one I agree with (I believe only one nucleation event per single-crystalline tablet is needed, as postulated in Section 4.1.).
It is clear that there is much more to learn when it comes to understanding the matrix-mineral interactions involved in the precursor pathway. In the next section, I will elaborate on how I think some of the issues tie together, as based on correlations between biomineral features with features observed in the in vitro model systems.
4. Mimicking Biomineral Features using the Amorphous Precursor Pathway
Biominerals often are organized into complex hierarchical structures, so in the biomimetic approach, rather than duplicate the entire biomineral, one is generally trying to see if a particular feature observed in the biomineral can be generated in the beaker. For example, no one is currently trying to reproduce all the layers in a mollusk shell, which presumably requires cellular control for spatial and temporal addition of additives and matrices. Instead, one wishes to understand how one primary component is formed, such as the laminated structure of nacre (or even its primary component, a single-crystalline tablet of aragonite); or how non-equilibrium crystal morphologies such as the urchin spine can be molded at room temperature, etc.. This section will demonstrate the wide variety of features that can be accomplished via the precursor pathway, as outlined by the following list:
Single-crystalline composites
Lattice strains
Nanogranular textures
Non-equilibrium compositions
-
Non-equilibrium morphologies
Tablets, films, coatings
Fibrous crystals
Molded crystals
Templated crystal location
Intrafibrillar mineralization of collagen
Cementatious minerals
Concentrically laminated spherulites
Crystal textures, including nanogranular, laminated, lattice distortion, anisotropic inclusions.
This section will also include discussions that propose new perspectives on the mechanisms by which these biomineral features are formed. Each of the new concepts will be presented as a hypothesis, which is then supported by evidence from the literature on the biomineral of interest, and how such features might be explained from an understanding of the crystallochemical mechanisms determined by the corresponding in vitro model system. As of yet, many of these ideas have not been proven, but the intent is to stimulate new ideas based on our current understanding of crystallization via the amorphous precursor pathway. Many of these new ideas are stimulated by my own group’s studies of the PILP process, and therefore the discussion includes the possibility of a fluidic amorphous precursor. It should be pointed out that there is currently no proof that a liquid-phase mineral precursor is involved in biomineralization, but I argue that it may be a necessary ingredient for generating some of the crystal features, and therefore deserves consideration. It took the biomineralization community many years to find the transient amorphous precursor in the first place, and determining whether an unstable ACC nanoparticle is solid or liquid in vivo is an even more challenging task. Nevertheless, because the in vitro models tell us that a fluidic amorphous phase can exist, and that the presence of anionic polypeptides that are similar to the proteins associated with biominerals can induce and stabilize such a phase, then it is logical to consider its possible relevance to biomineralization.
4.1. Non-Equilibrium Morphologies: Mineral Tablets, Films, and Coatings
4.1.1. Morphology of Nacreous Tablets
The first realization that the amorphous precursor pathway could yield features found in biominerals was upon examination of the CaCO3 crystals formed via the PILP process, which were the same thickness as the mineral tablets and sheets found in mollusk nacre (around 500 nm). They were also very similar to the mineral tablets/films deposited by Belcher et al.264 using protein extracts from nacre (as discussed in Section 3.2.1.1, with Figure 25). The terms tablets and films will be used interchangeably because, in some cases, isolated tablets are formed from PILP phase, but if a lot of precursor is deposited onto a continuous substrate, the ‘tablets’ will be connected into a continuous film-like morphology (I generally refer to these crystals as patches of film). This original discovery of CaCO3 thin films was for calcite, but similar tabular and film-like morphologies have now been formed from aragonite as well,119,256 which is the phase more pertinent to mollusk nacre. The discussion in this section is concerned with the thin tablet/film morphology, while the issue of crystal phase will be discussed in Section 4.1.3.
The surface of sheet nacre, which was shown in Figure 1b & c, has the appearance of a continuous thin film, where the tabular morphology of the individual crystals is not even apparent unless one separates the tablets by removal of surrounding organic matrix. As mentioned above, the PILP formed crystals are often connected in a film or sheet-like morphology, composed of connected ‘tabular’ crystals of dimensions similar to nacreous tablets. Given the exclusion of polymer that occurs during crystallization from within the precursor film (see Section 3.1), one would expect polymer enriched zones at the grain boundaries between the single-crystalline patches/tablets. In the mollusk, this would be more pronounced due to the organic rich environment (described as a hydrated gel-like medium of silk fibroin proteins, which become sandwiched between mineral and chitin).305 These proteins could also be excluded to the edges of crystals forming within a precursor film, thus yielding a mineral sheet that maintains a smooth surface topography, but is composed of patches/tablets that appear isolated due to the accumulation of surrounding organics.
The inherent thickness of the PILP formed CaCO3 films,203 as well as the CaCO3 thin films described by others,244,251,253,256,259,262,309–311 is not derived from a preformed compartment, but is a consequence of crystallization from within a the predefined boundaries of the precursor phase. Thus thickness of these tablets/films is determined by the quantity of mineral precursor produced per given volume, which in turn must be derived from the supersaturation of the CaCO3 solution and the sequestering capability of the inducing polymer. Most of these studies have been done using ion concentrations that are similar to sea water, which is around 12 mM Ca2+. Thus tablets and films are produced which are generally around 500 nm thick, similar to nacre and seminacre tablets found in mollusks and bryozoans (which are composed of aragonite and calcite, respectively).
In some reactions, separate tablets are deposited, rather than a continuous film, and in the PILP system, we find that calcite tablets oriented in the [001] direction express a roughly hexagonal morphology that is comparable to the seminacre tablets (Figure 36). The hexagonal shape results when a tablet nucleates off the (001) plane because the basal plane has hexagonal symmetry, and the two-dimensional film restricts the crystal from growing outward into the three-dimensional rhombohedral structure. The aragonite tablets in mollusk nacre express a pseudo-hexagonal morphology (pseudo because aragonite has an orthorhombic lattice, so the shape is not quite hexagonal), and apparently always nucleate on the basal plane, presumably due to some templating influence of the organic matrix,52 or dynamically grow into a preferred orientation.56 In our system- a variety of other crystal orientations can be present because the substrate is glass, which does not lead to preferred orientation. Therefore, the hexagonal shape is only occasionally seen; instead, the other randomly oriented tablets express rhomboidal type shapes dictated by the rhombohedral crystal symmetry projected onto the 2-dimensional plane of the film (again, the crystals are restricted to two-dimensional growth by the precursor film within which they grow).
Figure 36.

Comparison of PILP tablets to seminacre tablets. (a) These PILP formed calcite tablets nucleated off the (001) basal plane, and therefore exhibit a hexagonal morphology. Linear transition bars are seen in the wide sectors, while wavy transition bars are seen in the alternate narrower sectors. Some surrounding amorphous film can also be seen above the tablets, which appears to be dissolving in lieu of the crystal tablet. (b) Etching patterns are seen in cyclostome bryozoan seminacre (composed of calcite), where the narrower sectors are preferentially etched. Note the granularity in the etched regions, and finely laminated appearance in the wide sectors. Bar is 2 μm. (c) Schematic illustration by Weedon and Taylor,314 showing the disorganized layers of hexagonal tablets in seminacre, which grow by accretion on all sides, where ‘screw dislocations’ are the dominant mechanism for wall thickening in the older parts of the skeleton. (d) SEM example of the typical surface of seminacre, as described in c). (e) Schematic illustration of sectorization patterns: seminacre tablet divided into six roughly equal, alternately etched sectors (left); nacre tablet divided into a pair of sub-triangular, less-soluble sectors that meet at the center and two rhombic (or paired triangular), more soluble sectors (middle); and gastropod or cephalopod nacre tablet divided into polysynthetically twinned sectors (crystal individuals). The central hollow represents the site of the central organic accumulation (right). (a) (Reprinted with permission from ref 203. Copyright 2000 Elsevier Science B.V..) (b–e) Reprinted from ref 314 with permission from the Marine Biological Laboratory, Woods Hole, MA..)
As mentioned above, Kato’s group was one of the first to deposit thin films of CaCO3,245 in this case on a biomimetic substrate of chitosan film, which is the deacetylated form of chitin, the primary matrix found in the conchiolin membrane of nacre. The thin film morphology is similar to nacre in terms of thickness, however, the films appear to be composed of spherulites rather than hexagonal tablets. This group has been able to prepare layered composites by sequential deposition of PAA-induced mineral film and chitosan (spin coated).251,256 In principle, this sounds relatively straightforward, as one might anticipate being able to just sequentially deposit numerous layers to build up a laminated composite, as is done in nacre. They were able to sequentially deposit up to three layers. Three layers may not sound impressive relative to nacre, but in our experience, this is not as easy as it sounds, where any side products (such as crystals not grown via ACC phase, or adventitious droplets) can easily protrude from such thin films. They note that more precise control of the homogeneity of film thickness and flatness of the surface is needed to prepare multilayered composite films.
The PILP system was the first to duplicate the hexagonal tablet morphology of CaCO3 (Figure 36a).202 However, this was duplicated with calcite tablets, not aragonite tablets. Therefore, a more exacting comparison should be made with the seminacre of bryozoan organisms, which form thin tablets of calcite, rather than aragonite (Figure 36b). This biomineral is referred to as seminacre because, while the individual mineral tablets are quite similar in morphology to nacreous tablets, they are not as well organized, with tablets randomly stacking upon each other as non-uniform layers are deposited (Figure 36c & d). One feature of this hexagonal morphology that deserves further discussion is the fact that these tablets are expressing the unstable (001) crystallographic plane of calcite. Thus, it is pertinent to discuss the interfacial consequences of these thin tablet/film deposits.
4.1.1.1. Expression of Unstable Crystallographic Planes
One puzzling feature of biominerals is that the crystals often express unstable crystallographic planes. This is seen in the nacreous tablets, which expose the unstable (001) planes of aragonite. It is also seen in the vast array of calcitic biominerals that adopt an overall non-equilibrium morphology (discussed below in Section 4.2), where curved surfaces are expressed on single-crystalline calcite.54 A significant feature of the PILP process is that it can also generate crystals that expose unstable crystal planes (if the amorphous phase undergoes a pseudomorphic transformation, as described in Section 3.4.2.1). For example, calcite normally expresses its (104) planes when grown via the conventional crystallization process, but the surfaces of these thin films express a wide variety of planes. This is because the crystals are restricted to grow within the phase boundaries of the precursor, as mentioned above. Therefore, crystals that nucleate off the (001) plane will express this plane (if the phase boundaries are parallel to a planar substrate), as is seen in the hexagonal tablet in Figure 36a. This phenomenon is conveniently illustrated using the rapid overgrowth technique, where small crystals, grown via the conventional growth process, nucleate and grow on the films in an isoepitaxial fashion, yielding faceted overgrowths that represent the crystallographic orientation of the underlying film (Figure 37a & b). This technique was first developed by Okazaki,63 who beautifully demonstrated the crystallographic orientation and single-crystalline nature of the sea urchin spine. Figure 37a shows a PILP formed film with differently oriented domains that expose a variety of crystal surfaces, including the (001) planes in the magenta colored tablet (the magenta color also indicates that it is oriented along the [001] isotropic axis of calcite). Figure 37c shows that a conformal coating can be deposited on a curved surface as well, in this case a metal wire, generating single-crystalline domains that express curved surfaces. Kato312 and Sommerdijk253 have also deposited conformal calcite coatings onto curved surfaces, in this case using polymeric substrates, such as chitin fibers and a porous fibrous scaffold of poly(caprolactone), respectively.
Figure 37.

Polarized light micrographs with gypsum λ-plate of PILP formed calcite crystals. (a) Expression of unstable crystallographic planes in PILP formed crystals is demonstrated with the rapid epitaxial overgrowth technique applied to a calcite film. With zoomed view, one can see a variety of crystal orientations on the faceted overgrowths (which grow by isoepitaxy on the underlying calcite film). The magenta colored patch (top edge) is oriented at, or near, the [001] isotropic orientation, which is generally unfavorable for calcite to expose this high energy (001) crystallographic plane. In some cases, the crystal orientation is seen to gradually shift across a single-crystalline patch, thus indicating that the underlying film has a gradual shift in crystallographic orientation. This can be readily seen in the blue patch. (b) Overgrowths demonstrate biaxial strain in these PILP formed tablets. In addition to the coloration, one can see the directionality of the facets on the overgrowths. The magenta tablet to the right was mostly expressing the (001) face, except for two regions of alternative birefringence (the central spot, and the blue region on the left). Bar = 100 μm. (c) A conformal PILP coating deposited on a metal wire. The uniform birefringence of the patches shows large regions of single-crystalline calcite which are exposing curved surfaces (such as the yellow patch in the center). (d) A continuous CaCO3 film produced with addition of Mg-ion and polyaspartate shows a high degree of defect texture. Nevertheless, each patch can be considered a single crystal (derived from one nucleation event), even though the crystal orientation has shifted the further growth proceeded from the central point of nucleation. This is seen as a shift in birefringence towards the edge of the patches, which appear dark from extinction as the crystal orientation has fully shifted to about 45° at the periphery of the patches. This film is quite bumpy from precipitation of late stage droplets that did not coalesce into the film, where the colors of the droplets indicate a higher order interference color at the thicker bumps. (b) (Reprinted with permission from ref 203. Copyright 2000 Elsevier Science B.V..)
4.1.2. Defect Texture of Nacreous Tablets
4.1.2.1. Preferential Etching and Sectorization
The tablets in nacre and seminacre sometimes exhibit interesting sectorization patterns.41,313,314 In the sectors of seminacre calcite tablets, Weedon and Taylor describe a preferential etching pattern, where the narrower sectors of the hexagonal tablets seem to be more heavily etched (Figure 36b).314 The etching is presumably due to aging or sample preparation, but it is not clear why the sectors differ in their resistance to etching. The older literature has attributed this effect to selective binding of additives. We offer an alternative explanation based on the PILP model system. Sectorization patterns are commonly observed in this system midway through the amorphous-to-crystalline transformation, where the temporary transition bars delineate sectors within the tablets. In the hexagonal tablets (those that were oriented in the [001] direction), there are wide and narrow sectors, and these alternating sectors display different textures (Figure 36a). The wide sectors display transition bars that are roughly linear, while the transition bars in the narrower sectors are quite wavy. Because the transition bars can generally be correlated to the crystallographic lattice,203 the waviness seems to suggest that these narrower sectors contain more microdefects. Microdefective sectors would be expected to enhance the etching of the narrower sectors, thus providing an explanation for the preferential etching patterns observed in the nacreous tablets.
We postulate that these microdefects might result from more pronounced shrinkage stresses in the narrower sectors, as they are laterally squeezed in by the wider sectors during dehydration of the precursor phase. Evidence for shrinkage stress is provided in Figure 37. In general, there seems to be a relationship between these textures and the symmetry of the crystal, where biaxial strains are observed for crystals of rhomboidal morphology (as seen by the overgrowths in Figure 37b). Therefore, triaxial strains would be expected for the trigonal symmetry of the hexagonal tablets. Shrinkage stresses would not be expected for crystals grown via the conventional crystallization process; therefore, this could be a mineralogical signature of crystals grown via the amorphous precursor pathway (discussed further in Section 5.2.3).
There has been recent evidence to support the premise that the nacre in mollusks forms via an amorphous precursor (as mentioned in Section 2.1),51,207,212,213,315 and sectorized etching patterns are found in the aragonite tablets of nacre as well,41,314,316 but in different symmetry related patterns (Figure 36e). Such mineralogical ‘signatures’ may be providing supporting evidence of the amorphous precursor pathway, and the in vitro models can provide an explanation for the basis of these textures. There are other crystal textures that have been described for nacreous tablets, such as mesocrystalline,55,315 bridged nanocrystals,317 nanogranular and nanograin inclusions,318,319 etc.. These features will be addressed in Section 5.2.2 on Crystal Textures.
4.1.3. Crystal Polymorph
It should be pointed out that the bryozoan organisms are from an entirely different genera than mollusks (they are very small, communal organisms that build a layer of crust on algae), so it is interesting that they form tablets with such similar morphology, but out of a different crystal polymorph. The historical view on mollusk nacre has been that an additive could selectively bind to the (001) plane to generate this tabular morphology.86,138,218 If this were the case, a similar additive would need to have evolved for the bryozoan seminacre as well, which has the same morphology. Of course convergent evolution is possible; but on the other hand, if these biominerals both utilize a precursor pathway, then tablets and films are a relatively ‘natural’ morphology. The question then becomes, can different crystal polymorphs be deposited by the precursor pathway?
This question was already addressed in Section 3.4.2.3, where nearly all of the CaCO3 polymorphs have been produced from the amorphous precursor pathway. Of relevance to nacre, Sugawara and Kato have been successful at producing high-purity thin films of aragonite on chitosan substrates (in the presence of Mg-ion).256,298 While progress has been made in modulating crystal phase, it should be noted that the aragonite films in all of these studies form with a polycrystalline texture, and do not reproduce the single-crystalline texture of the tablets found in nacre.
We also added magnesium ion to our PILP reaction to try and promote the aragonite phase, in this case on glass substrates, and initially found that high magnesium calcite was produced instead (see Section 5.1).180 However, we did eventually find some conditions that led to aragonite tablets (low Mg-ion and a resorcarene monolayer), and to our delight, they were single-crystalline (Figure 38a–c).119 To my knowledge, this was the first time that single-crystalline aragonite with a morphology similar to nacre has been produced synthetically. Interestingly, the tablets sometimes exhibited a nanolaminated textures (Figure 38e), which resembled the nanolaminated texture that has been observed in nacre tablets as well (Figure 38f). On the down side, while the morphology was roughly nacre-like, the tablets were not of controlled orientation, as in nacre, and the overall film transformed into both calcite and aragonite tablets, as well as recrystallized into aggregates in various regions. Thus, this example serves to illustrate that while the amorphous precursor pathway can provide morphological features that match biominerals, we are still a ways off from regulating all the levels of control found in the biological system. Perhaps with a more selective organic matrix, it might be possible to also modulate the nucleation event (from within the amorphous phase), which could then regulate the crystal phase and orientation. Alternatively, more ‘specific’ interactions from the polymeric process-directing agent within the amorphous phase might influence which polymorph is formed. As mentioned in Section 3.3, the soluble proteins extracted from biominerals seem to provide control over crystal phase,264,277 so this seems a distinct possibility.
Figure 38.

Comparison of crystals formed on a Langmuir monolayer of specialty amphiphile tetracarboxyresorc[4]arene, prepared by Volkmer’s group108 (molecular structure shown in central box). (a) A pseudo-hexagonal patch of single-crystalline aragonite transforming within the surrounding ACC film, as observed between crossed-polars and gypsum λ-plate. The crystal exhibited a uniform extinction direction indicative of a single crystal. (b) Templated crystals could be dipped off the monolayer for examination with selected area electron diffraction, and this tablet, which shows Bragg contours in brightfield TEM, exhibited a single-crystalline spot pattern with aragonite d-spacings (c). (d) Using the same monolayer, calcite crystals grown by the conventional crystallization process (without Mg-ion) had previously been shown to nucleate with a preferred (012) orientation (and are large, faceted 3D crystals that grow into the solution). (e) Some of the monolayer templated CaCO3 films exhibited an interesting layered texture, as seen here in using AFM height mode. The thickness of this film was determined in deflection mode (not shown here) to be 890 nm at 48 hours of reaction, while a film measured at 18 hr only had two layers, and was 190 nm thick. (f) A nanolaminate texture has been observed in sheet nacre as well. (g) Schematic illustration of a functional model to explain the nanogranular and laminated textures observed in PILP formed CaCO3 films. (a–c) (Reprinted with permission from ref 119. Copyright 2006 American Chemical Society.) (d) (Reprinted from ref 108. Copyright 2002 Reproduced by permission of The Royal Society of Chemistry.) (f) (Reprinted with permission from ref 55. Copyright 2005 Elsevier Ltd..)
4.1.4. Crystal Orientation
In columnar nacre, it has been proposed that the uniform crystallographic orientation within an entire column of tablets is accomplished through mineral bridges between the layers.44,46,58,60 The interconnectivity of the biogenic organic scaffold is demonstrated in vitro by Cölfen’s group,320 who did a retrosynthesis of nacre, where the nacre mineral was dissolved and then the remnant organic scaffold was re-mineralized using an amorphous precursor induced with poly(aspartic acid). They were able to reproduce the brick-n-mortar morphology of the original nacre using this scaffolding to mold the amorphous phase (Figure 39). It was, however, filled in with calcite tablets rather than aragonite, which were small and not of uniform crystallographic orientation. Nevertheless, this experiment supports the hypothesis that this tabular biomineral morphology may be molded by a pre-formed scaffold, and it also supports the notion that the holes within the organic matrix allow for infiltration of the amorphous precursor. They argue that nanoparticles, which are seen throughout the scaffold, can pass through the 5–50 nm sized pores of the membranes, or can enter from the outer rim. It is also possible that droplets of a fluidic PILP precursor could be transported through these holes, and precursor droplets would also leave remnant nanoparticles upon subsequent examination. And additional consideration is that the matrix may have a gel-like consistency, as suggested by Nudelman’s environmental and cryo-SEM studies,305 so a liquid-phase precursor might be more amenable to transport through a gel than nanoparticles via capillary action, as proposed in Section 4.1.5.
Figure 39.

Retrosynthesis of nacre, performed in Cölfen’s lab320 by dissolving away the original mineral and remineralizing through the amorphous precursor pathway using polyaspartate additive. (a & b) TEM micrographs of highly mineralized regions of the synthetic nacre after 24 h reaction time. The platelet thickness is in part lower than in the natural archetype due to partial collapse of the demineralized matrix during its preparation and handling. (c) Electron diffraction pattern of the platelets in panel b, with d-spacings that match calcite. (d) Original nacre from Haliotis laeVigata. (Reprinted with permission from ref 320. Copyright 2005 American Chemical Society.)
Even with mineral bridging, this still does not provide an explanation for the originating crystal orientation. Therefore, it has long been assumed that the crystal orientation must be regulated by some specific interactions with the organic matrix, at least on the first nucleating layer. Gilbert and coworkers,56,321,322 however, have recently argued against this traditional view, and adopt a more passive mechanism. This group used synchrotron spectromicroscopy and x-ray absorption near-edge structure spectroscopy to probe the tablet orientation in nacre, and they find that orientational order of aragonite tablets in nacre is established gradually and dynamically (over a distance of about 50 microns). They developed a model of controlled assembly based on suppression of the crystal growth rate along a specific directions (such as by confinement within a layered structure), which yielded a tablet pattern consistent with their physical measurements. They propose that “the organism’s control of crystal orientation in nacre occurs via regulation of crystal nucleation and growth as opposed to direct templation or heteroepitaxial growth on organic molecules on the organic matrix sheets.” Congruent with this more passive mechanism, DiMasi and Sarikaya39 used synchrotron microbeam x-ray diffraction (XRD) as well as electron microscopy to examine the nacre-prismatic interface of red abalone shell, and found that the calcite and aragonite crystals lose their bulk orientational order in the region within 100 μm of the boundary. With respect to crystallization via the amorphous phase, neither hypothesis (epitaxial templating versus dynamically preferred directionality), seems unreasonable. However, there is an important point to be made about these studies which show a change in crystal orientation of the nacre tablets, and that is that the surface that is expressed on these tablets is then not always the (001) plane. In fact, this surface may not be a true crystal facet at all, since the tablets that are at other crystal orientations still have the same tabular shape. In other words, these observations seem to suggest that the crystal surface may simply be whichever surface was delineated by the phase boundary of the precursor (as seen in the PILP system, as mentioned in Section 4.1.1.1). On the other hand, facets do sometimes develop in the lateral direction as the amorphous phase crystallizes across the precursor film, while sometimes more non-descript shapes are formed. Interestingly, if one looks in the older literature, a variety of shapes were pictured for the forming nacre tablets, ranging from perfectly round tablets, to euhedral rectangular and hexagonal platelets.36,43,46,47,323–325
Blank et al.326 have shown that a preferred orientation of aragonite crystals will form on the fracture surface of nacre, but the overgrowths were polycrystalline needles (which is common for aragonite), and did not duplicate the tabular structure of the original nacre. It would be interesting to see this same experiment done under conditions to deposit an amorphous film, which might provide both this preferred orientation and thin tabular morphology.
It has become increasingly apparent that relatively little is known about nucleation from an amorphous phase. Some groups have found a partial preferred orientation evolve from ACC films, such as in Sommerdijk’s calcite films generated with the DNA polyelectrolyte, 48% of the crystal domains were (11.0) oriented.253 This is not a common orientation found in biominerals, but this particular orientation has been documented in a few other synthetic studies as well (calcite not from ACC phase).279,311,327 A variety of reasons for the preference were suggested, such as spacing of carboxylate groups, ionotropic effect, and non-specific chemical interactions; but this still remains a largely unresolved issue.
Aizenberg has been at the forefront of determining routes towards regulation of crystal orientation, with many of these studies using well-defined SAM templates. Her templating work originally examined crystals formed via the conventional route,102,103,281,328 and more recently has turned to the amorphous precursor route.263,292,329,330 She has found that the SAM templates that promoted specific crystal orientations via the conventional crystallization route can promote the same orientation in ACC films that transform to calcite.263 In this case, however, the specific SAMs were applied in only small nanoregions using an AFM tip, in order to avoid multiple nucleation sites within the ACC film. As described more fully in Section 3.4.2.1 (see Figure 33), this enabled the formation of large (up to a mm in diameter) single-crystalline calcite films of controlled orientation.
In summary, thus far the following features of nacre have been mimicked, but not all together in a combined system that provides all the levels of crystallographic control found in the biomineral (i.e., morphology, texture, orientation, polymorph, location):
Thin CaCO3 sheets of calcite or aragonite, with some control over polymorph, but aragonite is polycrystalline
Hexagonal tablet morphology of calcite, but not all of controlled [001] orientation
Single-crystalline tablets of aragonite, but not of controlled orientation or purity
Oriented aragonite overgrowths, but not of tabular morphology
Laminated composites via sequential deposition, but not with controlled orientation, polymorph, or tablet location
Laminated composites via infiltration of biogenic scaffold, but without controlled orientation or polymorph
Molded single crystals of calcite of preferred orientation.
4.1.5. An Alternative Perspective on Nacre Formation
Hypothesis: The mantle cells that are secreting the necessary ingredients for biomineralization could be secreting droplets of PILP phase, or a protein that induces PILP droplets in the extrapallial fluid, along with the various proteins that make up a viscous hydrogel matrix. The natural tendency of the PILP droplets to coalesce and form thin tablets and films could then lead to the similar tabular morphology observed in the differing nacreous type structures, as follows:
Sheet Nacre
In the case of sheet nacre, the mantel cell secretions could then coalesce to form a thin and continuous precursor film, much like the films formed in the PILP process, which would then be followed by subsequent deposition of a new layer of conchiolin membrane, and so on. The excluded organic impurities, including the acidic proteins inducing the process, as well as the gel-like silk-fibroin matrix,305 would accumulate between the interlamellar chitin layers, as well as laterally between the tablets, so that the resulting sheet would be composed of tablets with very uniform thickness (since this is delineated by the originating precursor film), separated by a thin organic layer from excluded proteins. This could provide an explanation for extraordinarily flat and coherent surface in sheet nacre (Figure 1c). The crystal phase may be influenced by either a templating matrix or various soluble ingredients, while the preferred crystal orientation is more likely influenced by the chemistry of the matrix, or the dynamic growth processes mentioned in the prior section.
This hypothesis is supported by the biomineral literature as follows. Some of the early work done by Watabe indicated that the deposition of the organic matrix layer occurs after, or about the same time as, the tablets come in contact with one another in sheet nacre.46 This indicates that the crystals have already formed in a thin tabular morphology before any membrane is deposited to limit their vertical growth.
This is also supported by the more recent work of Rousseau et al.,59,308 who examined sheet nacre by embedding and hardening the soft matter at the forming surface. Although they argue in favor of the “compartment” theory, the compartment that they refer to is not membrane bound, but consists of some type of viscous liquid gel whose overall volume defines the limiting thickness of the tablets that form within this so called “biofilm” (Figure 40). This film seems to be fairly rigid as subsequent layers can be deposited on top in a stair-case arrangement (i.e., a new layer is deposited before the prior layer has formed tablets). They suggest that the film consists of a macromolecular gel with calcium carbonate ionic species, because the Raman spectrum of the film surrounding the tablets (Figure 40d) exhibits the same aragonite peaks as the well-defined tablets (but with more background fluorescence, suggested to be from organics).59 They consider this to be from nanocrystals resulting from a preparation artifact.308 While I agree with this proposition, the large size of the CaCO3 peak suggest that the original film is comprised of a large amount of some type of mineral precursor phase (in combination with some fibrous structures and/or the silk-like gel described by Addadi et al.51). Thus, the data shown is consistent with the deposition of an ACC-organic hybrid film which transforms via restructuring and growth of the tablets within the confines of this precursor film. I consider this to be different than growth of crystals within a preformed compartment, in which that terminology generally implies crystals growing from solution in the compartment via the conventional crystallization pathway, where the preformed compartment delimits the shape of the crystals. In the mechanism I propose, the shape is delineated by the phase boundary of the viscous macromolecular-ACC precursor phase, and this type of mechanism has been demonstrated in several in vitro model systems of CaCO3 thin film formation via an amorphous precursor (discussed in Sections 3.1 and 3.2). For this hypothesis, there is then connectivity between mineral in the overlapping layers as they are secreted directly on top of the prior layer (before the mineral becomes sandwiched between excluded matrix), thus an iso-epitaxial relationship could account for the strikingly consistent orientation of the crystals in all three crystallographic directions in some bivalve nacre.49
Figure 40.

Rousseau et al.308 study on the formation of sheet nacre, with the incipient layer of sheet nacre visualized from mantle and shell side. This type of deposition has been referred to as “staircase” pattern, which leads to the imbricate backsteinbau structure. (a) From the mantle side, a fracture surface was created by forming a hardened replica of the viscous films from which the aragonite tablets evolve (lighter round patches). (b) From the shell side, the forming tablets can be seen within the film, which has separated due to drying artifacts. (c) SEM of shell side showing location of Raman analysis, and (d) corresponding Raman spectra from the region of the tablet (star) and the surrounding film (circle). Both regions show aragonitic peaks. The slope on the top spectrum is from autofluorescence of some unknown species in the film. (Reprinted with permission from ref 308. Copyright 2005 Elsevier Inc..)
Columnar Nacre
In the case of columnar nacre, the location of the columns would likely be dictated by the locale of the secreting epithelial cells in the mantel, where each cell secretes a load of droplets, and because of the fluidity of the phase, the droplets would accumulate as a mound on the matrix beneath the cells. The images presented for the newly forming ‘tablets’ in columnar nacre often appear as little round mounds of mineral,44,46,324 and do not resemble crystals in the early stages. If the precursor phase is a liquid (like the PILP phase), it could also get pulled into deeper sub-surface layers by capillary action. This is a new concept, which is supported by some of the older literature with respect to how these supposed ‘preformed compartments’ in columnar nacre are made. In the pioneering work by Nakahara325 and Watabe,46 it was found that a thick surface sheet forms as the first deposit of organic structure, and then thin, ordinary sheets separate one by one from the surface sheet to form the regularly spaced compartments that sandwich the mineral layers.46,325 In the TEM images provided in the Nakahara paper (reproduced in Figure 41),46,325 one can see that there is not really a preformed compartment until some mineral starts to push it up and separate it out from the underlying sheets. In fact, I don’t recall seeing any images in the various papers on columnar nacre that show fully empty compartments until after the fact, when the mineral has been dissolved out (Note- some regions could look like empty compartments if one does not image in the region of the tallest stacks. For example, in Nakahara’s micrograph shown in Figure 41a, if someone were to take an image in the region of the shorter stacks, the tablets would appear to be surrounded by empty compartments). In the images in Figure 41b–d, it can be clearly seen that the newly forming mineral is pushing up the membrane and thus creating the compartment.
Figure 41.

Nakahara’s325 TEM study of the growing surface of columnar nacre from two gastropods. The samples were carefully fixed with a buffered 5% glutaraldehyde solution, and then embedded in araldite to preserve the delicate structure of the growing surface. In images 4–7, the mineral was removed with EDTA, which then appears as white voids in the electron micrographs. (a) Low power TEM (decalcified and stained with uranyl acetate), showing three nacreous stacks that had a cut through the top crystals (T) in Turbo. (b) High power TEM of the top of the crystal stacks. A delicate network of organics can be seen in the space between the pre-existing stacks, as well as multiple layers of packed sheets at the top membrane. The newly formed crystals at the top of the stack (T) appear to be pushing up the membrane as the sheets become separated into a new compartment. (c) Unstained section cut vertically to the growth surface of nacre of Tegula shows stacks of crystals (C), with interlamellar organic sheets (S), and a dense surface sheet (D). (d) Enlarged view of left image in b) shows the emergence of some form of mineral in the newest forming compartments. Nakahara indicates that “the growing crystals (C) and top crystals are always surrounded with envelope (E).” Rather than assuming this envelope is arising from specific protein adsorption, I suggest that this could simply be condensed organic matter as the mineral phase penetrates into the dense organic matrix, and as it crystallizes, it will further exclude the polymeric impurities such that they become concentrated at the surfaces and edges of the forming tablets. (Reprinted with permission from ref 325. Copyright 1979 Malacological Society of Japan.)
Note- this argument does not oppose the mineral bridging hypothesis, but rather, I think the pores in these membranes provide a route for capillary infiltration of a liquid-phase mineral precursor phase, and thus mineral bridges and connectivity between tablets would be formed as a consequence. Therefore, I propose that the formation of these purported ‘compartments’ might arise from capillary infiltration of a PILP type precursor phase, which first accumulates in fairly localized regions just below the mantle epithelial cells that secrete the precursor, thus dictating the location of the columns. This may be in combination with other gel forming ingredients, such as the silk-like proteins described by Addadi et al.,51 but for this proposed mechanism, the capillary infiltration would require a fluidic precursor, and thus need to be accomplished prior to gelation, or nanoparticle formation.
Seminacre
In the case of seminacre, it also seems unlikely that a membrane-bound compartment exists because it forms with a random layering of tablets (Figure 36d & e). Seminacre is also interesting because the growing surface has a lot of spiral steps and terraces (Figure 36d),314 resembling AFM images of crystal growth at screw dislocations; yet these steps are several hundred nanometers thick, and resemble macrosteps, which tend to form in crystals grown far from equilibrium.331 Importantly, spiral growth steps are not limited to the calcite in seminacre. Some of the older literature shows similar “screw dislocations” in the aragonitic nacreous tablets (e.g., mussel shells of Geukensia and Mercenaria;332 pelecypod shells of genus Pinctada;47 and the Atlantic pearl oyster, Pinctada radiate Leach49), and these seem to be associated with micron-scale ‘growth pyramids’,49 where nucleating ‘islands’ and ‘terraces’ are euhedral tablets of aragonite. It is not clear how a limiting membrane or inhibitory additive could create these thin tabular structures since the layers are not truly separate layers in these regions.
Collective View
A thin tabular morphology is found in these three different systems, yet the observations of the forming biominerals do not support the theories of a preformed compartment, or a subsequently deposited membrane or inhibitory protein. Therefore, I propose that the thin tabular morphology in all of these systems is dictated by the inherent thickness of a precursor phase. Perhaps the less regulated design of the seminacre in the simpler bryozoan organism evolved into the more uniform structures found in mollusks through the use of more organized scaffolds, which provided enhanced mechanical properties or some other evolutionary benefit. From this perspective, one can propose a common mineralization mechanism that leads to this convergent tabular morphology found in seminacre (calcite), and both types of nacre (aragonite). This does not preclude the other types of more specific organic-inorganic interactions that may be responsible for higher levels of crystallographic control, such as crystal phase, texture, and orientation.
4.2. Non-Equilibrium Morphologies: ‘Molded’ Single Crystals
Many biominerals do not adopt a faceted crystal habit, even though the same mineral formed at the same supersaturation in the beaker would express well-defined crystallographic planes to minimize the surface energy. Such biominerals appear to have been molded or shaped in some fashion, such as by growth within a membrane-delineating compartment. This is particularly the case in the calcitic biominerals, which will be discussed further below. There are some notable exceptions to this rule, such as the euhedral crystals found in the well-faceted rhomb crystals covering some types of coccoliths (Figure 42),333 and the otoconia found in the gravity receptor organs of vertebrates (see Figure 66 in Section 5.3), and perhaps the dense-packed prismatic crystals that often form the basis of a hierarchical structure of the hard tissue.
Figure 42.
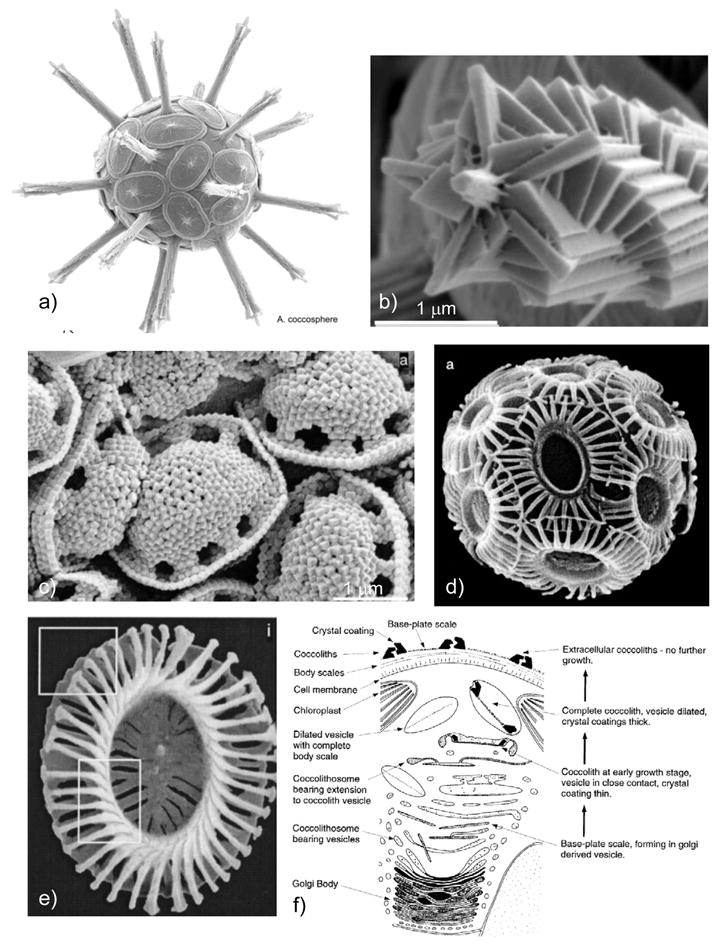
Coccolithophores are a group of algal plant plankton that produce exoskeletons (coccospheres) consisting of a collection of minute calcite plates, called coccoliths. (a) A coccosphere from Rhabdosphaera clavigera beautifully demonstrates the high degree of organizational control exerted during coccolith formation, which occurs within membrane-bound vesicles that are then transported to surface of the cell. The unicellular organism is encased in calcitic base plates, which are narrow and enclose a large mass of lamellar crystal units, called heterococcoliths. b) A central spine emerges from some base plates, which consist of intergrown calcite crystals that exhibit rhombic faces with perfectly regular alignment spiraling up the structure. The penta-radiate apical structure is consistently developed. Bar = 1 μm. (c) Calyptrosphaera pirus contains holococcoliths, which are plates composed of a large number of minute, closely-packed, morphologically simple crystallites. This is a common arrangement, with the rhombohedral crystallites to be organized in hexagonal arrays with their c-axes directed perpendicularly to the coccolith surface. Bar = 1 μm (d) The complete coccosphere of Emiliania huxleyi, a well studied example. Bar = 1 μm (e) An individual coccolith shows the chiral symmetry and anticlockwise imbrication of the inner tube elements. (f) Schematic section through part of a cell of Pleurochrysis carterae, illustrating the sequential development of the coccoliths. (a & b) (Reprinted with permission from ref 72. Copyright 2003 Mineralogical Society of America.) (c–f) (Reprinted with permission from ref 333. Copyright 1999 Academic Press)
Figure 66.

Lin et al.420 study of the composite calcite crystals of otoconia. A guinea pig utricle (the vertebrate gravity receptor organ) was treated with a freeze-fracture and deep-etch procedure to examine the fibrous network and interconnections with the otoconia crystals. (a) Numerous, barrel-shaped otoconia are embedded in a loose filament matrix. Bar = 1 μm. In other images (not shown here), the otoconia appear to be linked together by filamentous cross-links. (b) Freeze fracture of an individual otoconium shows an organic central core (CO) formed by a tight meshwork of filaments, and a well-faceted, dense crystalline outer shell (OS) of calcite. Bar = 0.5 μm. (c) On the surface of the otoconia, pore-like openings can be seen in the mineral (arrows ), which appear near or under the surface filaments. Bar = 0.1 μm. (d) The fracture plane through the mineralized outer shell of this otoconium shows several deep radial channels (arrows) that seem to connect the central core with the otoconium’s outer surface. Bar = 0.2 μm. (Reprinted with permission from ref 420. Copyright 2000 Academic Press.).
4.2.1. Coccoliths: A Combination of Equilibrium and Non-Equilibrium Forms
In the case of the coccoliths, which are exoskeletal plates formed by coccolithophorids (unicellular algal organisms), the amazing feat of this biomineral example is the high degree of uniformity in size and shape, as well as organization of crystals (Figure 42a–c). In addition to the organized faceted crystals seen in the holococcoliths, the heterococcoliths also contain calcite crystals with non-equilibrium morphologies (Figure 42d–e). Even when regular crystallographic faces are not expressed in these non-faceted crystals, there is still a consistent relationship between crystallography and element morphology.72 For example, in Emiliania huxleyi, which is the organism that produces the heterococcolith shown on Figure 42e,333 the distal shield hammer handles are elongated parallel to the crystallographic c axis, while the proximal shield surfaces correspond to the prismatic [12̄01] faces. Coccolithophorids were one of the first organisms to have the sequence of biomineralization events carefully examined at the microbiological level, which nicely illustrates the use of compartmentalization within membrane-bound mineral deposition vesicles (Figure 42f).72,333 Van der Wal et al.334 demonstrated that the coccolith base plates are formed first in Golgi-derived vesicles, which remain in close contact with the mineral as the shape forms in parallel with the forming mineral development, suggesting it may have a shape-forming function imparted by cytoskeletal pull.72 Subsequent crystallographic extensions are formed by further addition of mineral deposition from small vesicles called coccolithosomes. These plates and crystal extensions are then transported and exocytosed to the cell surface, where they apparently provide protective armor to the organism. The coccolithosomes appear to play a key role in calcification. They have been shown by Marsh to be formed of a complex of acidic polysaccharides and calcium ions,335 and it was suggested that that they function as calcium vectors during biomineralization while the polysaccharide phase forms the crystal coatings.
Smith et al.96 examined the influence of polysaccharides extracted from coccoliths on calcite growth via AFM, and found that these extracts had a tendency to aggregate into long strings, and when a CaCO3 buffer solution was added, rounded particles formed as well as “crystals budding off the strings”. In the images provided, however, the blebs on the strings do not appear to be faceted, like crystals, but instead, the morphology of these ‘buds’ appears (to me) to be of an amorphous form, with rounded blebs that have coalesced into stringy agglomerates. Therefore, it is possible that the discrete clusters formed in the presence of these biopolymer extracts might be mineral-associated PILP-like clusters.
4.2.2. Sea Urchin Spicules and Spines
In addition to the coccoliths, the spicules of urchin larvae, and the spines of the adult urchin, are fabricated into convoluted non-equilibrium morphologies that present smoothly curved surfaces (recall Figure 3). No facets can be seen, yet they behave optically and diffract as though they are single-crystalline calcite. As mentioned in the Historical Perspective Section 2.1, the larval spicules were one of the first calcitic biominerals found to utilize an amorphous precursor pathway because the small organism could be examined in situ during spiculogenesis (Figure 12). An important aspect of this biomineral system is that the spicules are formed within a membrane-bound compartment, which evidently shapes the mineral as it is being formed. In contrast to the chiton tooth, where a preformed compartment consisting or organic matrix is shaped like the tooth, the vesicular compartments in many biominerals are not preformed into the final biomineral shape, as one might envision for the molding of an object. Instead, the membrane is closely apposed to the forming biomineral element, and expands dynamically as the mineral is shaped into its elaborate morphology. Even in the large biomineral elements, such as the urchin spine, the synctium expands during mineral formation.4 In fact, the limited space surrounding these forming biominerals was perhaps a clue that there was not enough solution to provide the ions necessary to form the biomineral. Thus, it was generally assumed that a continuous supply of ions would need to be pumped across the membrane-bound compartment.
4.2.3. An Alternative Perspective on the Molding of Biominerals
Hypothesis: The issue of limited compartment space could be reconciled by an accretion of a highly concentrated pool of ions which is sequestered as an amorphous phase (such as nanoparticles or PILP droplets). This concentrated pool of ions could be added via vesicular transport to the growing element in a spatially and temporally controlled fashion. Such directed transport could be envisioned as providing a high degree of spatial control over mineral deposition, as opposed to ions diffusing freely in solution. If the precursor has fluidic character, it could be slathered onto a forming crystal and rapidly coalesce, eventually transforming into a single-crystalline element (if a pseudomorphic transformation occurs), even if deposition occurs over separated time intervals.
The urchin example seems to be the norm for biominerals formed within compartments, which generally take on the shape of the compartment as both are being dynamically shaped by cellular processes. Although it is certainly clear that the compartment is important for dictating the shape of the biomineral, to my knowledge the ability to mold solution borne crystals within a vesicle has not been demonstrated in vitro. For example, Mann and Hannington 336 grew iron oxides in unilamellar vesicles, but the crystals adopted the faceted habit typical of magnetite. In contrast, the magnetite nanocrystals formed by magnetotactic bacteria, which form in a membrane-bound vesicular compartment (which is unusual for prokaryotes), can be produced with acicular morphologies, including bullet and tear drop shapes (Figure 43). Although one cannot expect to dynamically mold a vesicle in vitro to generate shapes other than spheres, there still should be the possibility to form a round crystal in a vesicle. This may highlight the significance of the fact that the magnetotactic bacteria first transport an amorphous ferrihydrite precursor into the vesicle.337 Usually, the discussions regarding this ferrous hydrous Fe(III) oxide precursor are centered around the redox reaction, where one-third of the irons then need to be reduced to Fe(II) in order to nucleate the magnetite crystal; and it is just assumed the compartment shapes the crystal. But it may be that the amorphous precursor is also significant with respect to providing the ability to mold such a non-equilibrium shape within the vesicle (as well as in the acicular ‘fibrous’ magnetite seen in chiton teeth).
Figure 43.
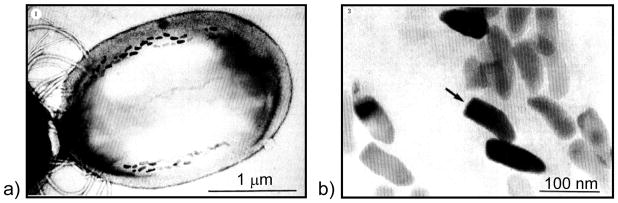
Iron oxide biominerals in magnetotactic bacteria. (a) This ovoid bacterium contains 3 multi-stranded chains of magnetosomes (the 3rd strand in the middle is faint as it is in the back of the cell). There are multiple flagella at the left end of the cell, which propel the cell as it is guided by magnetotaxis from the earth’s geomagnetic field. The TEM sample was stained with uranyl-acetate. Bar = 1 μm. (b) Bullet-shaped anisotopic inclusions of magnetite isolated from digested cells. Bar = 100 nm (Reprinted with permission from ref 439. Copyright 1987 The Royal Society.)
In 2001, Loste and Meldrum304 demonstrated in a synthetic system that CaCO3 crystals could be ‘molded’ by forming an ACC precursor in a constrained volume (pores of a track-etch membrane), which transformed into calcite with a morphology governed by compartment. The amorphous phase was generated using a double-diffusion technique of counterions across a U-tube, and at low temperature (4–6°C) to stabilize the ACC. They found that if the ACC precursor particles entirely filled the pore channels prior to crystallization, the pore channel could be replicated to yield rod-shaped single crystals of calcite.338 In their first papers of crystal growth in confinement, they surmised that the amorphous precursor was a necessary ingredient; however, Meldrum’s group later found this was not the case. In this case, they performed an elegant experiment in which the shape of the urchin spine was replicated by growing calcite within an inverse replica of the spine.339 This inverse replica was prepared by forming polymer within the interconnected pore space of the spine, then de-mineralizing to leave behind a purely organic scaffold, which could then be re-mineralized back to the original shape of the biomineral (Figure 44). They stated that “All evidence suggests that the templated calcite single crystals grew in the absence of an amorphous calcium carbonate (ACC) precursor phase, demonstrating that the presence of ACC is not a necessary prerequisite to the formation of single crystals of calcite with complex morphologies.” This group has since shown that SrSO4, PbSO4, PbCO3, NaCl and CuSO4.5H2O, can all be grown into sponge-like complex morphologies, where it seems that “the principal requirement for templating single crystals is simply that the ‘natural’ size of the crystal under the given growth conditions must exceed the length scale of the template.” For example, the PbCO3 replica shown in Figure 44b was polycrystalline due to the lower solubility of this mineral.340
Figure 44.

Meldrum and coworkers339 demonstrate the ‘molding’ of crystals grown in confinement using the double-diffusion technique of ions across a porous membrane. (b) A single crystal of calcite was grown in a polymer mold (PMMA/EA) created by inverse replication of the sea urchin skeletal plate. No evidence of an ACC precursor was found (although a vaterite precursor was prevalent in this system). (b) This PbCO3 replica was polycrystalline due to the lower solubility of the mineral and its naturally smaller crystal size. (a) (Reprinted with permission from ref 339. Copyright 2004 The Royal Society of Chemistry.) (b) (Reprinted with permission from ref 340. Copyright 2006 The Royal Society of Chemistry.)
In this counter-diffusion system, the replicas are generally less than a mm in size, and the ions can be slowly and continuously added to avoid random nucleation events such that a single crystal can be formed. This system seems like a nice analogue to the biological system because one can imagine that the vesicular membrane could contain active pumps for controlled addition of ions across the membrane. However, since this 2004 experiment, it was discovered by Politi et al.209 that the urchin spine in fact forms via the amorphous precursor route. This biomineral was more difficult to examine in situ than the spicule, but they were able to examine the regeneration of a fractured spine, and found that it was being formed from the accumulation of a hydrated amorphous phase. Thus, even though Meldrum has shown that one can mold the sponge-like morphology of the spine via the conventional crystallization pathway, it apparently is not the preferred pathway used by the organism. Why?
To investigate this issue, we chose to follow Meldrum’s strategy for replicating the urchin spine, but instead infiltrated the inverse replica with an amorphous precursor. During my doctoral work, I had inadvertently discovered that the PILP precursor had space-filling properties when I tried to form a mineral coating on some glass beads (Figure 45), and instead found that the precursor seeped in between the beads to form space-filling calcite crystals (Figure 45b), unlike the control reaction that did not contain polymer (compare Figure 45a).201 This seemed to indicate that the precursor could be molded within a compartment, but to examine this in a more regulated fashion, my student adopted Meldrum’s more elegant technique for replicating the urchin spine (Figure 46).222 He formed the inverse replica with poly(hydroxyethylmethacrylate), pHEMA, which was readily available in the lab (Figure 46a–c). Note, the control reaction without polymer, which proceeded by the conventional crystallization process, did not lead to spatially delineated crystals (Figure 46d–e), unlike Meldrum’s system. This is because he used the direct addition of reactants rather than the double diffusion technique. In contrast, the same reaction conditions with polymer did lead to space-filling crystals, and enabled replication of the overall morphology of the spine (Figure 46f); however, the element was not single-crystalline or uniform in texture. This is probably because nothing was done to control the nucleation event (from the amorphous phase); the pHEMA scaffold was a hydrophilic porous hydrogel, and probably too favorable for calcite nucleation for this large replica (~5 mm in length). In contrast, the space filling crystals that formed in between the glass beads were single-crystalline calcite (Figure 45b). One lesson that can be learned here is that, even though space-filling crystals can readily be molded with the amorphous precursor, an important aspect of gaining higher levels of control (such as texture) is to control the transformation stage of the reaction.
Figure 45.
Capillarity, filling space, and mineral cements created with PILP-formed crystals, as demonstrated by calcite crystals grown in the presence of glass beads (75 μm in diameter). (a) Crystals grown by the conventional solution crystallization process nucleated calcite rhombs randomly on the surface of the glass beads. (b) With the addition of polyaspartate, PILP phase formed and seeped down between the beads and filled the space, leading to spatially-delineated calcite crystals. The single-crystalline texture is evidenced by a singular extinction direction; and the bulls-eye birefringence pattern is due to the thickness gradient across the crystals, demonstrating that the crystal is filling the space between the interstices of the beads. For example, the arrow points to one spatially-delineated single crystal of calcite. The beads became ‘cemented’ to each other and the glass substrate in this system as well, unlike those in the control reaction of (a).
Figure 46.

Replication of the sea urchin spine by micromolding PILP phase in an inverse replica of the urchin spine. (a) Monomer of 2-hydroxyethyl methacrylate (HEMA) was polymerized within a bleached spine of the urchin Arbacia punctulata, to create the poly(HEMA) inverse replica, which looks very similar to the original spine microstructure (shown in Figure 3b & c). (b) Higher magnification shows the bicontinuous microstructure, which is what allows for infiltration of polymer to make the replica, followed by removal of the original biomineral, and subsequent infiltration of the mineral precursor, to mold a replica. (c) Energy dispersive spectroscopy (EDS) shows the replica is all organic, once the original mineral of the spine has been removed, leaving behind a pure poly(HEMA) scaffold. (d) Photographic comparison between the Poly(HEMA) scaffold, replicas, and the original spine, to demonstrate dimensional changes that occur when the hydrogel scaffold is dried. The original polymer scaffold is shown in (A); while (B) shows a scaffold containing crystals grown via the conventional process; while (C) shows the scaffold mineralized with the PILP process; and (D) shows an original urchin spine, which is purple from impurities typical of the purpuratos species. Note, the dimensions of specimen (B) are much smaller because the crystals did not infiltrate and fill space in the scaffold, so the poly(HEMA) hydrogel collapsed upon drying. Only a few crystals nucleated on the surface of the scaffold, which can be seen in the SEM in (e). On the other hand, the dimensions of the original spine were retained for specimen (C) because the precursor phase fully infiltrated the porous scaffold, and replicated the morphology of the spine. (f) The molded replica of filled space and produced a similar morphology, but had rougher surfaces, presumably because of the hydrogel nature of the template, and was not single crystalline, due to multiple nucleation events. (Reprinted with permission from ref 222. Copyright 2006 American Chemical Society and American Institute of Chemical Engineers.)
In biominerals, it seems that one way of accomplishing this is to use a seed crystal. For example, in the first stage of spicule formation in the urchin larvae, a small crystal is formed that initially has the shape of rhombohedral calcite (see Figure 12). This rhomb apparently serves as a seed upon which the amorphous phase is deposited and subsequently transforms by isoepitaxy with the calcite seed. At this point, the shape of the amorphous phase has already been defined by the compartment such that a non-equilibrium morphology is attained. Some type of ‘seed crystal’ is also seen in the formation of coccoliths and sponge spicules,341 which in addition to regulating crystal orientation and phase, may provide this regulatory influence over the single-crystalline texture of the biomineral.
Li and Qi developed a synthetic system that inadvertently mimicked this approach, where a calcite seed crystal emerged from an initially amorphous “tuber”, which subsequently stimulated the surrounding amorphous phase to crystallize, such that single-crystalline calcite was formed (Figure 47).342 In their system, the amorphous phase was infiltrated into the pore space of a 3D colloidal assembly of carboxylate-functionalized polystyrene nanospheres (styrene–methyl methacrylate–acrylic acid colloidal spheres, ~ 450 nm in diameter) using vacuum assisted filtration of a dispersion of ACC. Both the carboxylate functionality and vacuum assisted filtration were found to be important for achieving effective infiltration into the template, enabling them to fabricate a “3D ordered macroporous material (3DOM)” of single crystalline calcite.
Figure 47.

Li and Qi’s342 vacuum assisted infiltration of an ACC dispersion into a colloidal film of PS-MMA-AA colloids. (A) The amorphous to crystalline transformation of the inadvertent seed crystal. (B) The 3D ordered macroporous calcite crystal formed by this process. In (A), the images in (a) and (b) show the initial formation of a “tuber” of ACC using different amounts of ACC, which are surrounded by ACC that had infiltrated into the surrounding pore space of the colloidal template. Images (c) and (d) show a centralized rhombohedral calcite that had transformed from the amorphous precursor. Inset in (d) is a enlargement of the central rhomb (inset bar is 1 μm). (Reprinted with permission from ref 342. Copyright 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.)
An alternative way to gain control over the nucleation event in a synthetic ACC system was demonstrated by Aizenberg et al. (as previously shown in Figure 33).263 Rather than using a crystal seed, they were able to control the nucleation event by AFM printing nanoregions of a functionalized SAM with integrated nucleation sites, enabling fabrication of a molded calcite crystal nearly a millimeter in diameter.
In a variation on Aizenberg’s two-dimensional calcite crystals (i.e., films), we have shown that there does not necessarily have to be a rigid compartment to mold crystals when using an amorphous precursor, because the phase boundaries of the precursor itself can delineate the shape of the crystal if it crystallizes via a pseudomorphic transformation. In this case, one can ‘mold’ 2-D calcite crystals of defined morphologies by using a SAM template to control where the precursor phase is deposited (Figure 48). In the PILP system, the droplets preferentially adsorb to the charged carboxylate regions of a microcontact printed SAM, forming patterned films of calcite with non-equilibrium morphologies (Figure 48b–d).221 This strategy of regulating colloid adsorption could be used in biomineralization, even for simple tasks such as templating crystal location, as in the nacreous tablets in columnar nacre, or patterned silica in diatoms, etc.. Even in chiton teeth, the amorphous iron oxide deposits seem to adsorb to the fibrous organic matrix (Figure 11d),343 allowing the mineral to be templated into its fibrous morphology.
Figure 48.

Polarized light micrographs (using first-order red gypsum λ-plate) of calcite templated on patterned SAMs, first printed with COO−-terminated SAMs in a grid pattern, and then CH3-terminated SAMs were coated on the remaining square areas. (a) Patterned rhombohedral polycrystals of calcite formed by the conventional solution crystallization. (b) Patterned films of calcite generated with polymeric process-directing agent. The films are composed of large single-crystalline domains of calcite (e.g., one crystal is aqua blue and the other green), but also contain many late-stage PILP droplets and crystalline aggregates due to the incomplete inhibition of solution crystallization byproducts. Note the preference for adsorption of the late-formed PILP droplets at the edges of the films (arrows). (c) Well-defined patterned film of calcite composed of single-crystalline domains on the order of 10s of microns (regions of uniform retardation color and extinction direction). A lower reaction temperature reduced the number of crystal side-products, but a few small aggregates are still present. (d) Well-defined patterned films of calcite using a combination of polymer and Mg-ion inhibitors. The Mg-ion inhibitor, while eliminating crystal byproducts, also acts as an impurity that causes a more polycrystalline texture in the film. (Reprinted with permission from ref 221. Copyright 2007 American Chemical Society.)
Another feature that is distinctly different in the biological systems versus all of our in vitro models is that the organic compartment is often times not preformed. For example, when the urchin spicules and spine are formed, cellular processes dynamically shape the compartment as the biomineral is formed. The reason for doing it this way is not known, and it may just be related to the how vesicular compartments are made. On the other hand, one issue to consider is that a bulky amorphous phase may be more difficult to regulate. For example, we have seen that bulk accumulations of PILP phase do not crystallize well (or at all), and generally lead to the formation of spherulitic clusters. We believe this is because it is more difficult for the exclusion of polymer and water during the transformation. Likewise, even in the relatively thin crystal formed in Aizenberg’s study,263 they found that after 3 to 4 hours, the remaining metastable ACC film ruptured spontaneously into a polycrystalline layer. They determined that the micropattern played an important role by providing a “microsump action” for the release of water and other impurities (a dye added to the reaction) during the amorphous-to-crystalline transition, as well as relaxation of stress in the forming crystal. They draw an analogy to the biomineral systems, and suggest that the self-assembling biological macromolecules can provide sites for impurity discharge and stress relaxation. I suggest another feature of relevance to this dimensional limitation is the laminated textures in some biominerals, where sequential depositions may help to avoid these problems. As mentioned previously, we find that dehydration stresses can be significant for crystals formed from a hydrated amorphous precursor, and even in a thin film format, microdefects can be pronounced; thus, the size and thickness of the precursor phase can be a significant consideration in the A-to-C pathway. In the case of the regenerating urchin spine, Politi et al.209 found that the tip of the spine underwent a reduction in volume as it crystallized under the electron beam, and they considered this ACC likely to be a hydrated phase, as suggested by the broad IR peak at 3500cm−1. In fact, Addadi (or the journalist for Discover magazine) described the ACC phase as a “gel-like material that is densely packed with calcium carbonate molecules”.344 Thus, the issue of volume reduction may explain why nearly all biominerals are constructed out of small components, and when large structures are built, they appear to be laid down sequentially as thin layers (Figure 49). Ex vivo evidence of mineral layering is seen in many biominerals, such as the spicules of sponges (including both CaCO3345 and biosilica sponge spicules346–349), sea urchin spines,54 calcareous tubes of serpulid worms,46 kidney stones,350 and even the concentric lamellae comprising osteonal bone. In the last case (and possibly others), the concentric laminations are likely due to the limited size of cells that are constructing these crystals, and their secretory capacity.
Before concluding this section, I should probably address the question I posed above- if single crystals can be molded by the conventional crystallization pathway, why are so many molded biominerals formed via the amorphous precursor pathway? Of course I don’t know the answer to this, but one suggestion is that the unique textures that are formed via the precursor pathway (which will be discussed more in Sections 5.2 and 5.3), such as occluded polymers or high-magnesium calcite, or the hierarchical structures from nanograins to nanolaminates, may provide enhanced mechanical properties. Alternatively, it may be that it is just plain difficult to grow conventional crystals in the presence of all that organic matter found in organisms.
Keeping in mind that one of the first biomineral functions to evolve in organisms was the ability to regulate ion homeostasis, this likely initiated the utilization of an amorphous phase because it can be more readily mobilized by dissolution for transport. This is certainly the case for Ferritin in iron oxides,351,352 and ACC in cystoliths in plants,199,274,353,354 and storage granules in crustaceans,355–357 etc..
Hypothesis: The use of biopolymers to stabilize the amorphous phase likely evolved early on, and if such biopolymers were secreted from the cell, the result may have been to generate an amorphous phase, which with sufficient fluidic character could deposit on the cell membrane in the form of a simple mineral coating. This, then, could have provided the desirable evolutionary characteristics of endoskeletal protection. Further evolutionary advances of this pathway, such as vesicular compartments and matrix templating with more specific interactions, would then lead to the higher levels of structure and complexity that we now admire for their beautiful structures and enhanced mechanical properties.
From this perspective, the complexity of biomineral features may be viewed differently, which could provide a new understanding of the evolutionary tree. For example, instead of considering a highly complex array of structure-directing proteins as being responsible for the shaping of an urchin spine (which would require precise control over protein sequence and conformation, which would have to be varied as the mineral shape changes in different regions and across different skeletal elements of the organism), the molding an amorphous precursor with non-specific charged proteins is considerably less complex. The real complexity lies the in the shaping of the vesicular compartment or assembly of organic matrix (more traditional bio features), and the higher level features of the biomineral phase that subsequently evolved would include control over crystal phase, orientation and texture. In addition, since many proteins have evolved to serve multiple functions, the biomineralizing proteins could contain domains for binding to specific components of the organic matrix, or even to serve basic cellular functions (such as peptide signaling domains).38
4.3. Non-equilibrium Morphologies: Rod and Fibers
As mentioned in the introductory Section 1.3.2.3, there are several examples of biominerals that have ‘fibrous’ morphologies. This is particularly prevalent in the teeth of a variety of organisms, including the hydroxyapatite “prisms” in vertebrate dental enamel (Figure 9a & b),4 the calcitic “rods” in urchin teeth (Figure 9c & d),3 the magnetite needles in chiton teeth (Figure 9e),169 and the atacamite fibers in the blood worm jaw (Figure 9f).17 Apparently this fibrous architecture provides mechanical properties well suited for the rigors of chewing, 167,358,359 which in the case of the invertebrates, can entail scraping algae off of rocks. The chiton and limpet teeth are continuously formed, thus providing a convenient system for analyzing the stages of mineralization, as was shown in Figure 11.
4.3.1. An Alternative Perspective on’ Fibrous’ Biominerals
In the case of the urchin tooth, Ma et al.,168 suggest that “the central ‘stone part’ of the tooth is built and performs as a fiber-reinforced composite material, where the soft matrix is the polycrystalline filler phase, and the needles are the fibers.” The literature more commonly refers to these elongated biominerals as “rods” or “prisms”, but I consider them to be fibers for several reasons. In images of rat enamel, such as the ones reproduced in Figure 9b, the immediate impression that one gets is that the material looks like a woven fabric, which of course implies that the weave is constructed with fibers. In fact, when I show this picture to people without telling them what it is, they assume it is a woven fabric of polymeric fibers. They are quite amazed to learn that the ‘woven’ structure is made out of bioceramic crystals. Given the extremely long length (~ 800 μm) and high aspect ratio of these crystals, which form in polycrystalline bundles that span across the entire thickness of the enamel layer, one can see that the crystals bend and curve and follow contours, unlike what one would describe as a needle or rod. Perhaps there is some opposition to this terminology because fibers are generally made by extrusion or spinning, and of course such a process is not relevant to solution borne crystals. Or is it?
Hypothesis: The fibrous morphologies found in biominerals may occur through a biological type of extrusion process. Clearly biominerals are not melted and extruded at physiological conditions; but fibers can also be formed in liquid and gel systems, and given that we now know that a liquid-phase mineral precursor can be formed, the fluidic character could enable some type of extrusion processing, as discussed below.
The Wikipedia definition of extrusion is as follows: Extrusion is a process used to create objects of a fixed cross-section profile. Note- I am not suggesting that a mineral precursor is extruded thru a die or spun as a fiber, but rather droplets may be secreted from the cell, which continuously accrete in a one-dimensional fashion to lead to a fibrous morphology. Thus, this would be a slow and templated form of extrusion (from the biological realm). Although evidence to support this hypothesis from the biomineral literature is limited; the in vitro model systems demonstrate that mineral fiber formation is possible through the precursor route.
4.3.2. Calcite Fibers and the Urchin Tooth
Our group was the first to grow calcite fibers in an aqueous-based system (an accidental discovery, to be honest).224 In fact, there have since been at least three different sets of reaction conditions that have led to inadvertent formation of calcite fibers, and their mechanism of formation seems to differ (Figure 50).223,224,360 The first time we observed fibers was when we used a PILP system with a polymer of higher molecular weight than usual (35,600 g/mol polyaspartic acid), and fibers seemed to emanate from some large gelatinous globules (Figure 50a).224 We assumed these large globules were an accumulation of the precursor phase since they were initially non-birefringent and somewhat transparent, before they changed in shape and became birefringent upon crystallization (as observed with time-lapse video micrography). SEM analysis after drying showed fiber diameters of around 600 – 1000 nm. The non-rigid nature of the fiber precursor is apparent in the image shown in Figure 50b, where in the middle of the micrograph a small fiber is ‘draped’ over a larger fiber. In other words, these fibers are clearly not crystalline needles or whiskers. We knew from earlier work that some of the PILP products have a core-shell structure, such as the helical vaterites, which are spherulites of vaterite surrounded by a shell with a more dense texture.202 Therefore, it seemed reasonable to expect that the gelatinous globules might also have some type of membranous “skin” or encrusting shell, from which the interior contents of the globule might be extruded upon rupture. This would be analogous to the silicate gardens described in the literature, where a seed crystal of a soluble metal ion salt is introduced into a sodium silicate solution. In that system, a gelatinous coating of hydrous metal silicate surrounds the seed to form a semi-permeable membrane which ruptures due to osmotic pressure and “extrudes” the high concentration of metal salts within, which re-precipitate at the wall of the jets.361,362
Figure 50.

The prevalence of calcite fibers formed inadvertently in the PILP system. (a & b) Calcite fibers that grew off gelatinous globules of precursor phase. (a) Once the fibers crystallize, they appear single-crystalline in cross-polarized light (the image shown here is with the gypsum wave-plate). (b) In this SEM image, it can be seen that the small fiber in the center is draped across the bigger fiber, and apparently was “soft” when it formed. It clearly is not a needle. (c & d) Calcite fibers that grew off preformed seed crystals of calcite when a PILP coating was applied. In (d), bulbous tips on the fibers are suggestive of a remnant flux droplet, which in this system, would consist of a droplet of PILP phase. (e) Dense-packed fibrous overgrowths that formed at the edge of calcite films deposited via the PILP process on SAMs. (f) Schematic depiction of the solution-precursor-solid (SPS) mechanism proposed for the formation of these seeded fibers. We propose that PILP droplets deposit on the calcite seed substrate and possibly accumulate at high energy sites, such as surface defects and edges. A sufficient collection of PILP phase may form a “flux” droplet, which provides the one-dimensional growth through either a continued collection of PILP droplets, or ion replenishment of the flux droplet, such that fibers are ‘extruded’ from the surface of the seed substrate. (g) Polarized light microscopy of a single crystalline fiber broken off one of the SPS formed aggregates, for comparison to (h), a fiber extracted from the tooth of a sea urchin. Both fibers, of similar dimensions, appear single-crystalline across a bend in the overall fiber direction, which suggests that the fibers are still amorphous when they are constructed, and subsequent crystallization then proceeds across non-equilibrium morphology. (a,b) (Reprinted with permission from ref 224. Copyright 2003 Taylor & Francis.) (c,d,f–h) (Reprinted with permission from ref 223. Copyright 2004 American Chemical Society.) (e) (Reprinted with permission from ref 366. Copyright 2003 Materials Research Society.)
At this point, we considered the fibers as likely being formed by extrusion, but then we came across another two sets of reaction conditions that led to calcite fibers, but these systems did not form any gelatinous globules. In one set of experiments, our original intent was to deposit a PILP mineral coating on calcite seed crystals in order to regulate the crystallographic orientation of the mineral film by stimulating iso-epitaxial nucleation on pre-existing calcite seeds.223 This idea was based upon biomineral systems that use a rhomb-shaped calcite seed, such as during the early stages of spiculogenesis in sponges and urchin larvae,.208,363 It is thought that this rhomb seed crystal dictates the crystallographic orientation of the spicules, which is retained even after the spicules branch off into different directions as they grow into their non-equilibrium morphology. Thus, our goal was to regulate crystal orientation in calcite films deposited on a seed crystal. The calcite ‘seed’ crystals we used were rather large (a fortunate misunderstanding between advisor and student), approximately 20 – 40 μm in dimension. They were grown on glass coverslips which could then be placed in a fresh crystallizing solution for PILP deposition. As expected, when no polymer was used, iso-epitaxial overgrowth of polycrystalline layers of calcite formed on the rhomb seed crystals. But to our surprise, when PILP phase was deposited onto the calcite rhombs, high aspect-ratio fibers (100 – 800 nm in diameter, 5 – 80 μm in length) were found emanating from all directions on the seed rhombs (Figure 50c & d).223 We also found that a seed substrate with higher defect texture (a gel grown calcite rhomb) led to a very high fiber density, which suggests that the fibers might somehow be stimulated to nucleate at defect sites. In addition, it had been previously observed in another system, where we were patterning calcite films on self-assembled monolayers, that short fibrous arrays would grow off the edges of the preformed calcite films (Figure 50e). Interestingly, these fibrous arrays look remarkably similar to the morphology of the atacamite fibers seen in the bloodworm jaw (recall Figure 9f).
Based on the observation of remnant “bobbles” at the tips of some of these fibers (Figure 50d), as well as the occasional serpentine growth, these features seemed similar to those found in semiconductor fibers produced via the vapor-liquid-solid (VLS)364 or solution-liquid-solid (SLS)365 mechanisms; therefore, we proposed an analogous mechanism called the solution-precursor-solid (SPS) mechanism (Figure 50f).223 Unlike the VLS and SLS mechanisms, however, the SPS mechanism occurs in an aqueous-based solution under ambient conditions, where a molten metal flux drop is not required. Instead, the accumulation of PILP droplets, possibly at surface defect sites, is hypothesized to create a “molten flux droplet”, which contains a high concentration of ionic reactants. The significant mechanistic aspect of the VLS and SLS processes is that the size of the flux droplet restricts the crystal to one-dimensional growth. The fiber then grows continuously as long as the flux droplet is replenished with reactants. When the reactant supply becomes limited, growth stops, and the flux droplet solidifies, leaving a remnant bobble on the tip of the fiber. Thus, by analogy, the bobble tips on some of the CaCO3 fibers seemed to suggest they were formed by an analogous mechanism, but with a ‘molten’ flux droplet of PILP phase (Figure 50d).
When comparing ‘fibers’ isolated from a sea urchin tooth to our synthetic calcite fibers, there is a striking resemblance, and we suggested that an SPS type of mechanism might play a role in their formation.223 They both appear to be single-crystalline calcite (optically and diffracting), and retain their crystallographic orientation even when there is a bend or change in fiber direction (Figure 50g and h). While our synthetic fibers are randomly organized, the biogenic “needles” are aligned in parallel arrays (embedded in a microcrystalline matrix of Mg-calcite). Interestingly, the “needles” seem to grow off the edge of the preformed plate, which is consistent with our observations of SPS fibers growing off the edges of seed crystals and calcite films (Figure 50e).366 In the case of the urchin tooth, recent studies by Ma et al.168 show that not only are the individual needles single crystalline, but they say that “when the needles grow off the edge of a primary plate, this complex from the very first stages of formation behaves as a single crystal of calcite, despite the fact that the two structural elements have completely different shapes and form at different times.”
The urchin teeth have now been shown to form from an amorphous phase as well;168 but whether any of these mechanisms observed in vitro are relevant to their formation is an unresolved question. On the one hand, the biomineral fibers do not have bobbles at the tips, which we consider to be evidence of a flux droplet; but neither do ours in many of the experiments (e.g., Figure 50c), so this may only mean the flux droplet has thickness similar to the fiber, or it may even dissolve at the end of the reaction due to excessive impurity accumulation. Another issue that argues against the SPS mechanism is that these biomineral elements reportedly form within a pre-formed cytoplasmic sheath,367 which implies the non-equilibrium morphology is molded, and not constrained to one-dimensional growth by a flux droplet. However, if I understand this literature correctly, these compartments contain the entire needle-plate complex,168 and I have not seen evidence of compartments for each of the individual needles. Thus, if the needles grow directly on the primary plate (such as via an SPS type of growth), this could yield a uniform crystallographic orientation across the entire plate-needle complex element. This type of natural assembly process, perhaps organized with an organic matrix, would seemingly be far more energy efficient than having to mold each of the fibrous elements within a large array of vesicular compartments. On the other hand, some regions of the tooth show fibrous elements show well-defined branch points, or have small barbs on their surfaces (personal observations), so these types of features would be more consistent with a molding process.
We have recently shown that fibers can also be formed with BaCO3368 and SrCO3,369 but a remnant flux droplet is not seen, and they have a nanodomain texture, so at this point, we are not sure if they form by the same mechanism (although the reaction conditions are quite similar). Note- other groups have formed similar BaCO3370,371 and SrCO3372 fibers, but propose a mechanism based on mesocrystal assembly.
In conclusion, while I think it is clear that these calcitic biominerals have fibrous morphologies, it is not clear if an SPS or other mechanism is operative. But at the very least, it is now well documented that mineral fibers can be readily formed in aqueous-based reactions. In fact, it seems so easy to form calcite fibers, where they inadvertently form under a variety of conditions, that I favor the hypothesis that this tendency was capitalized on through evolution to ultimately provide enhanced mechanical properties. More work is needed to determine the fiber formation mechanism with certainty, both in vitro and in vivo.
4.3.3. Rods of Hydroxyapatite in Dental Enamel
I have been discussing the SPS mechanism for CaCO3, which may seem somewhat removed from an extrusion process. Let me address this by returning to the biomineralization of calcium phosphate biominerals in dental enamel, which is called amelogenesis. The enamel forming cells, called ameloblasts, change their shape to tall cells and become polarized as they form a cone-shaped Tomes process at the end of the cell.373 During the secretory stage of amelogenesis, mature secretory granules rapidly move to the Tomes process, where there appears to be fusion of large numbers of these secretory granules as they are released from the Tomes process. The enamel crystals form in direct apposition to the cell, and always form with their long axes perpendicular to the membrane of the Tomes process, thereby forming aligned polycrystalline bundles of hydroxyapatite referred to as “rods” or “prisms”. This occurs primarily at two sites, leading to the rods and interods (i.e., mineral formed in between Tomes processes of neighboring cells). The Tomes process thus seems to provide control over the shape and orientation of the rods and interods. The ameloblasts, which are lined up in a row (as is typical of epithelial cells), slowly move away from the dentino-enamel junction, such that their path controls the interweaving structure of the bundles of rods/interods (Figure 9a & b).
Hypothesis: From a materials science perspective, one could envision this site-directed secretion as extrusion, where the ‘die’ that controls the uniform cross-section of the rods would be the Tomes process. Actually, the linear array of ameloblast cells resembles a set of dies in a spinneret, which then ‘spin’ the fibrous bundles into the complex woven fabrics found in dental enamel. Of course the granules that are secreted are generally thought to contain the protein components, such as amelogenins and enamelins; but it is possible that the cells are also secreting a mineral precursor. This would offer a viable explanation for how the crystals form in direct apposition to the cells, in such a site-directed one-dimensional fashion, to enable weaving of the crystal bundles.
Although rarely discussed in recent literature, there has been some evidence of an amorphous mineral precursor in dental enamel. Nakahara and Kakei’s TEM studies of developing enamel find that the ribbon-shaped structures that appear in the earliest mineralized enamel are composed of an inner mineral portion devoid of a crystalline lattice.374 There also appears to be a surrounding organic envelope. They state that “Unstained sections of the same materials show moderately electron dense rod-shaped structures that apparently represent the inner mineral portion appearing as a somewhat fuzzy outline. These observations indicate that the earliest mineral laid down in developing enamel is not crystalline; further, the non-crystalline mineral and organic envelope are probably forming a molecular complex.”
The enamel system seems to be substantially different than the chiton teeth, which are formed in a pre-assembled tooth-shaped matrix. In amelogenesis, it seems the matrix is formed synchronously with the mineral phase. This matrix includes a well-studied component of amelogenin proteins, which in vitro form spherical subunits that self assemble into arrays that are thought to modulate the shape of hydroxyapatite into these highly elongated crystals through selective interactions with specific crystal faces.164,165 For example, in the in vitro model system of Iijima and Moradian-Oldak, elongated and c-oriented crystals of octacalcium phosphate (OCP, often considered a precursor to HA biominerals) have been formed by the double diffusion technique across a dual membrane device, where the ions diffuse from mutually opposite directions through a cation selective membrane and dialysis membrane.375 On the other hand, Moreno and Aoba376 measured the calcium content of the fluid from porcine enamel during the secretory stage, and found a value of the degree of saturation close to unity. Given this low value, they suggested that the driving force for crystallization might occur in intermittent fluxes. They also noted that about 85% of the calcium present in the fluid was in non-ionic form, which they presumed to be bound up by the peptides of the matrix. Their data seems consistent with a localized secretion of ion-enriched droplets, because an overall higher supersaturation throughout the matrix would not be required to initiate crystallization, and instead localized secretions could promote mineralization in the desired regions while the remaining regions contain only metastable solution.
Iijima and Moradian-Oldak were also able to produce elongated apatite crystals with the addition of fluoride and amelogenins,377 and indeed they do resemble the individual crystallites in enamel, although they are not formed in densely-packed bundles as in enamel prisms. An intriguing feature of the dense-packed prisms in enamel is the deviation in crystallographic orientation across each prism. The texture resembles ‘splay’ seen in liquid crystals,378 or the splay seen in densely infilled spherulites.258 Similar splay textures are found in many biominerals, including both CaCO3 and CaP, as judging by the schematics provided in Carter’s comprehensive book on Skeletal Biomineralization: Patterns, Processes and Evolutionary Trends.3 This texture is particularly prevalent in prismatic structures, which Carter defines as “mutually parallel, elongate, adjacent structural units that do not interdigitate strongly along their mutual boundaries.” We have observed a similar splay in crystal orientation of CaCO3 crystals growing within the confines of narrow streaks of amorphous precursor film (Figure 51). Although these are only two-dimensional film structures, they demonstrate the principle of splay and constrained growth textures from an amorphous precursor. Such textures seemingly arise from a high density of nucleation sites, while solution grown crystals are generally not as densely packed (with the exception of dense-packed surface coatings, where the crystals do not grow long enough to develop such splay).
Figure 51.

Crystallographic texture of splay, which is found in a variety of biominerals with prismatic structures, including dental enamel. (a) A two-dimensional CaCO3 film analogue, showing the radiating growth pattern of polycrystalline CaCO3 emanating from 2 central ridges in the streaks of PILP film. For some reason, the crystal transformation within inhomogeneous PILP films usually starts in the thicker regions, as seen here (the brighter birefringent yellow is thicker). On the top streak, however, there appears to have been a scratch or crack, which will also stimulate the nucleation within these metastable ACC films. (b) TEM of one of these films, which is composed of aragonite, as indicated by the spacings in the SAED pattern (inset). The splay texture leads to arcs in the diffraction pattern of localized regions.
Regarding the amelogenins, which have been shown in vitro to self-assemble into ribbons of spheres,164 such an assembly could be locationally-directed through such a cellular secretory/extrusion process. The amelogenin matrix would then help constrain the location of the secreted mineral precursor and its subsequent crystallization. Given that the organic matrix is not a preformed compartment, and would therefore be secreted with the proposed precursor droplets, the function of the amelogenins might be described as a co-extrusion processing aid (when viewed according to this hypothesis).
The studies of Tao et al.296 seem to be in line with this concept of assembling nanoparticle CaP precursors. They have demonstrated the formation of elongated hydroxyapatite crystallites through an assembly of preformed nanospheres of calcium phosphate (Figure 52). The core of the mineral nanospheres contained a tiny crystallite of hydroxyapatite (~ 5 nm), which was surrounded by an ACP shell, and this ACP shell was shown to be essential for the coalescence and moldability of the particles into chains. Interestingly, elongated needle-like shapes formed even without organic additive (although PAA was present initially to form the nanospheres), as shown in the insert in Figure 52, which is consistent with the one-dimensional growth we observe in the SPS calcite fibers. Aggregate chains are even apparent in the micron-sized PILP droplets shown in Figure 14a. Polarizability and various things have been proposed to explain anisotropic assemblies of nanoparticles in the formation of mesocrystals, but in the case of isotropic amorphous droplets, the assembly mechanism is not clear. In Tao’s system, the kinetics of this anisotropic nanoassembly was promoted with glycine, and was about 20 times faster with amelogenins. Interestingly, even though the internal crystallites of the initially coalesced aggregates were of random crystallographic orientation, as the amorphous phase crystallized, the whole complex aggregate managed to eventually evolve into single-crystalline needles (or plate-like structures were assembled with glutamic acid) by some undetermined mechanism. In the case of the biominerals, it may not be necessary, or even likely, that the secreted precursor phase will have initially formed nanocrystals; but one significant point that can be made from these studies is that the amorphous precursor provides both a mechanism for self-assembly into elongated structures, which can be enhanced in combination with organic aids (processing aid), and it provides the moldability and cementatious properties for assembling into single-crystalline structures.
Figure 52.
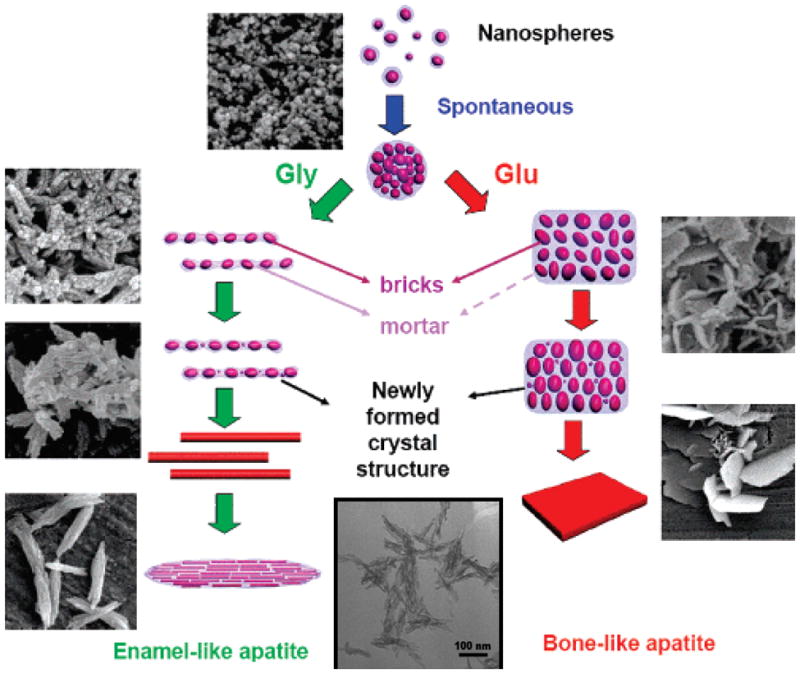
Tao et al.296 model of apatite evolution via the assembly of nanocrystallites (5 nm) of hydroxyapatite (purple) encapsulated with an ACP phase (light blue). The corresponding experimental states are shown with SEM (and TEM in the manuscript). The nanospheres that were nucleated in a metastable supersaturated solution could spontaneously aggregate into clumps, or needles given sufficient time (TEM insert at bottom center), but the assembly process was modified in the presence of additives, such as Gly, Glu, and amelogenin. The modifiers could determine the different forms that evolved, e.g., one-dimensional linear assemblies with glycine (and amelogenin) versus two-dimensional plates with glutamic acid, which were moldable and coalesced due to the ACP coating. The ACP transformed into the thermodynamically stable hydroxyapatite phase with time, and the individual hydroxyapatite domains somehow re-oriented and fused to form single crystals (red). These single crystals could assemble into the next level of architecture of parallel arrays, forming enamel-like prisms. (Reprinted with permission from ref 296. Copyright 2007 American Chemical Society.)
Other evidence to take into consideration is the texture of the biomineral, which can often give clues about the formation mechanism. For example, Kirkham et al.379 examined the early development of enamel with AFM and found that the crystal surfaces display morphological features resembling regular bead-like swellings, while at later stages a regular patterning was seen in terms of charge density and polarity on the crystal surfaces, features which are not seen in solution grown crystals. This group (Robinson et al.)380 has also shown with Chemical Force Microscopy on polished sections that the individual enamel crystallites have a chemically heterogeneous subtexture composed of transverse bands across the crystals (Figure 53a,b), as well as spherical subunits that stack in hexagonal arrays surrounding a dark central pit (Figure 53c,d). The dissolution of the crystal centers may be related to the central dark line that has been an enigmatic feature observed in high resolution TEM studies, so they suggest that the spherical subunits “may represent a junction region between such initiating entities as they fuse to form the developing crystal.”
Figure 53.
Nanoscale textures in dental enamel crystallites, as observed by Robinson et al.380. (a & b) Frictional force imaging of maturation stage rat enamel crystals. (a) Friction obtained using hydroxylated cantilever tips, pH 7. Lighter areas, which correspond to regions of high friction, show transverse high friction bands ~40 nm in width. (b) At at pH 5.5, the banding is again visible, but some bands exhibited patches of much higher friction. (c & d) High resolution AFM height images of polished human enamel sections. Images were obtained with unmodified tips, tapping mode in air. (c) Longitudinal/oblique sections of enamel crystal. Some transverse segmentation of crystals is visible indicative of the banding structure seen on maturation stage crystals. Central dark lines represent holes or grooves along the crystal long axes. (d) Transverse sections of human enamel crystals. The crystals appear to be made up of a series of roughly spherical subunits surrounding a central hole or pit. (Reprinted with permission from ref 380. Copyright 2004 The Royal Society of Chemistry.)
These observations are consistent with the amorphous precursor pathway. For example, PILP formed crystals have been found to contain a colloidal texture (see Figures 15 and 61),221 and a similar texture is prevalent in calcitic biominerals (described in Section 5.2.1.1),381,382 many of which have now been found to be formed by an amorphous precursor. Therefore, it is plausible that the subtexture in dental enamel could arise from crystal growth via assembly of amorphous colloids of the mineral precursor. In the case of fibrous biominerals, however, an additional proposition is needed, where the one-dimensional growth requires an anisotropic assembly of the colloids. This could arise from the locality of the extruded precursor droplets at the Tomes process, or confinement of precursor within the synchronously forming amelogenin compartment (e.g., the “envelope-mineral complex” described by Nakahara374), or from a sticking coefficient, where droplets (or nanoparticles) favor deposition at the ends of the growing fibers due to a ‘sticky’ flux droplet,223 analogous to mesocrystal assembly in the case of solid nanoparticles,193 or deposition may occur laterally along a ‘sticky’ fibrous matrix.
Figure 61.

Comparison of calcite film topology when PILP phase is deposited on different surfaces. (a) Polarized light micrographs show that in this particular experiment, PILP phase deposited on both the COO−-terminated SAMs (grid regions) and on the bare gold surface (interior square regions), but was much thicker on the former, as indicated by the different degrees of birefringence. (b) Three-dimensional AFM height image scanned across an 80 μm × 80 μm surface area showing different film thicknesses for PILP phase deposited on COO−-terminated SAMs versus on bare gold surface. (c) and (d) AFM images scanned across the surface of a 3 μm × 3 μm area within each patterned region (left- 3-D “height mode” image to show quantitative height deviation; right “deflection mode” image to show lateral dimensions of colloidal surface). As can be seen in (c), PILP film deposited on COO−-terminated SAMs was formed from significantly smaller particles than that formed on the bare gold surface (d), even though the final film thickness was much greater. (Reprinted with permission from ref 221. Copyright 2007 American Chemical Society.)
The flux droplet suggestion provides the not-so-obvious link between extrusion and SPS, where the SPS mechanism provides a means for controlling where the numerous precursor droplets adsorb, which in turn defines the cross-sectional width that is typical of an extruded element. However, there is no indication of a flux droplet in enamel rods. Therefore, I suspect that organic matrices have evolved that provide the ‘sticking coefficient’ needed for anisotropic adsorption of the mineral colloids (as also suggested by Tao’s work296). This may be what is occurring in chiton teeth as well, where clumps of amorphous ferrihydrate precursor seem to attach to the fibrils in the preformed matrix (Figure 11d),343 such that a lateral accumulation of precursor leads to extended fibrous structures.
4.3.3.1. Enameloid
Regarding the issue of enamel crystal morphology, it is relevant to consider enameloid, which is an enamel-like tissue in the outer layer of teeth from boney fish and sharks (Chondrichthyes and Osteichthyes), which forms by both mesodermal and ectodermal cells of the tooth bud.3 Significantly, crystals with similar morphologies are formed in enameloid, even though amelogenins are not present.4 Instead, collagen is laid out in a preformed matrix by odontoblasts, and then some proteins, such as enamelins, are secreted by the inner epithelial cells.3 The initial crystals seem to line up and form in close association with the collagen, and as with the amelogenins, the collagen matrix is enzymatically removed at maturation to yield large crystals of hydroxyapatite (roughly 200 nm in width).4 Given that enamelins (highly acidic glycoproteins) are found in both tissues, while the amelogenins are not, and that both tissues form parallel arrays of highly elongated crystallites, this seems to suggest that the morphogenesis is not directly regulated by the amelogenins, but by the enamelins. The newly forming crystals do appear to be associated with the collagen fibrils, so one might assume they serve a scaffolding function analogous to the amelogenins. However, in vitro studies of hydroxyapatite growth with collagen do not lead to hydroxyapatite crystals with morphologies that resemble enameloid crystals in the least; nor do the crystals line up parallel to the collagen fibril (unless formed via a precursor process).
Hypothesis: The enamelins may be responsible for the elongated fibrous morphology, but not in the generally assumed role of acidic proteins as specific structure-directing agents. The enamelins are intimately associated with the crystals (extractable only by dissolution), and thus may have become entrapped in an amorphous precursor. Such acidic proteins seem like a possible candidate for inducing an amorphous PILP-like precursor, which could provide for a biological extrusion process for one-dimensional growth.
In enameloid, the inner epithelial cells double in height to become “tall cells”,383 similar to the occurrence of tall cells of ameloblasts, suggesting that they help to orchestrate the mineralization process. However, in enameloid, the matrix has already been formed, and the crystals are not laid out it direct apposition to the cells (in fact, mineralization is sometimes initiated on the odontoblast side of the preformed matrix). It has therefore been suggested that the epithelial cells function mainly to degenerate the organic (collagen) matrix during maturation, since this is a feature common to both enamel and enameloid. I suggest that, given this similarity in tall cell behavior to ameloblasts, it is also plausible that the cells in both these systems are secreting precursor droplets, which provide a mineralization mechanism that leads to extremely long crystals. However, the crystals in enameloid are aprismatic and less organized, which is why I believe the directed secretion of amelogenins from the Tomes process may have evolved to provide this higher level of organizational control.
In conclusion, it seems clear that some form of directed secretion is occurring in enamel formation. This could only entail secretion of the organics, which then assemble into a directional scaffold; but the continued accretion and densification of the mineral phase is also consistent with a secretion/extrusion process applied to a mineral precursor phase.
4.4. Vertebrate Bone
As described in the Introductory Section 1.3.2.1, there has been recent evidence to support our original hypothesis (which was proposed in a MSE-Report)160 that bone formation involves an amorphous precursor. An ACP phase has now been found by Mahamid et al.163 in newly forming zebrafish bone, which later transforms into the mature crystalline mineral. This model species was particularly valuable because the long-fin bones are continuously formed, allowing for examination of the different stages of the mineralization process. But this still leaves an important question unanswered-how does the amorphous phase get inside the collagen fibrils?
4.4.1. An Alternative Perspective on Bone Formation
As described in the Introductory Section 1.3.2.1, the biominerals of bone do not exhibit elaborate molded morphologies. Nevertheless, the nanocrystals of hydroxyapatite (HA) in bone do seem to be molded by growth within confined nanoscopic compartments delineated by the interstitial space of self-assembled collagen fibrils. But, as mentioned above, it is still not known how the amorphous phase, and thus the final crystallites, become infiltrated within the fibrils.
Hypothesis: The interpenetrating nanostructured architecture of bone, which is a result of intrafibrillar mineralization of collagen fibrils, may be accomplished by a fluidic mineral precursor, in which capillary forces provide the long-range forces needed to draw the amorphous precursor into the interstices of the collagen fibrils.
Most in vitro crystallization assays that grow crystals via the conventional pathway (without polymeric process-directing agent) find that crystals only nucleate on the surface of the collagen scaffold. In contrast, we have shown that simply by adding anionic polymer, intrafibrillar mineralization can be achieved because the collagen becomes saturated with an amorphous precursor phase (Figure 54). The experimental evidence for this is described in detail in our MSER review paper,160 so it won’t all be duplicated here. In that report, we proposed that the mechanism for accomplishing intrafibrillar mineralization is through capillary action, in which the liquid-like amorphous precursor can be drawn into the gaps and grooves of collagen fibrils by capillary forces (schematic of proposed mechanism presented in Figure 55).
Figure 54.

Intrafibrillar mineralization of type-I collagen scaffolds using the PILP process. (a) The control reaction, without polymer, leads to random spherulitic clusters of hydroxyapatite on the surface of the collagen fibrils, typical of the many reports in the literature. At the relatively high ion concentrations we use, an amorphous CaP gel does form in the solution (which become cloudy), but this type of ACP does not lead to infiltration of the fibrils. (b) With the addition of polyaspartate, the collagen becomes well infiltrated with mineral, but no apparent clusters are seen on the surface. To demonstrate that mineral is actually present, EDS, XRD, and TGA all show very high degree of mineralization (up to 70wt% hydroxyapatite, as in bone). (c) TEM of collagen fibrils prior to mineralization, stained with PTA to show the periodic banding pattern typical of type I collagen. (d) After mineralization, the fibrils take on a striated appearance due to the presence of platy intrafibrillar crystallites. There appears to be some PILP droplets adsorbed to the fibrils (arrows). (e) Thick dark crystallites can be seen in the TEM of this fibril, which at such high degrees of mineralization, obscure the periodic banding pattern. (f) Selected area electron diffraction of this fibril (and all fibrils examined) show that the crystallites are uniaxially oriented in the [001] direction, roughly parallel to the collagen fibril axis, with similar degrees of tilting and rotation disorder, as in bone. (g) Dark-field TEM of the same fibril, using the (002) reflection, shows the prevalence of oriented crystallites within the fibril. (Reprinted with permission from ref 160. Copyright 2007 Elsevier B.V..)
Figure 55.
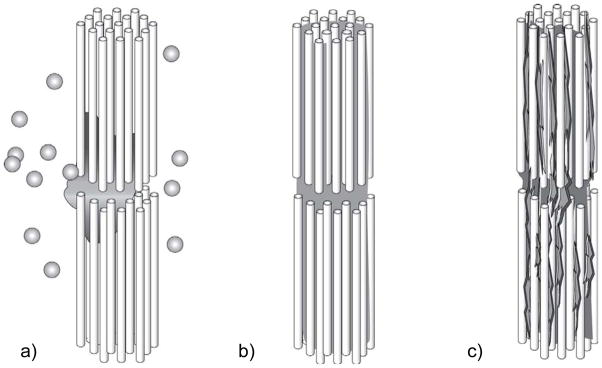
Schematic illustration of the proposed mechanism of collagen mineralization via the PILP process. As a scaffold, type I collagen triple-helical “molecules” self assemble into fibrils that exhibit a periodic banding pattern due to hole and overlap zones. The rods shown here represent the tropocollagen triple helices. (a) Nanoscopic PILP droplets adsorb to the collagen fibril and are drawn into the gap zones of the fibril by capillary forces. (b) The amorphous phase solidifies once it has fully infiltrated the interstices of the fibril, including the gap zones as well as the regions between the collagen molecules. (c) Crystallization of the ACP phase into hydroxyapatite leaves the fibrils embedded with nanocrystals of hydroxyapatite that are aligned roughly parallel to the fibril. (Reprinted with permission from ref 160. Copyright 2007 Elsevier B.V..)
Additional support for the general concept of an amorphous precursor playing a key role in intrafibrillar mineralization is also provided by Deshpande and Beniash,154 who examined mineralization of isolated collagen fibrils on TEM grids. They used reaction conditions very similar to ours, with the addition of polyaspartate (but with a different buffer); but they propose a different mechanism, which is based on the more traditional view that the polymer interacts with the collagen to promote heterogeneous nucleation. No experimental evidence was provided to support this hypothesis. The data we provided to support the hypothesis of capillary infiltration was based on tracking the infiltration of fluorescently-labeled polyaspartate into dense-packed collagen scaffolds (turkey tendon) using confocal microscopy, where simple diffusive transport of polymer was limited (polymer + calcium), while polymer that induced the PILP phase (polymer + calcium & phosphate) was transported into the collagen to a far greater extent (Figure 56). Thus, while it is conceivable that a polymer could diffuse into an isolated collagen fibril, as proposed by Deshpande and Beniash, its transport was far more limited in the case of a dense-packed collagen scaffold (except when the precursor phase was generated). This is the more biologically relevant environment since collagen is densely packed into lamellae in secondary bone formation. Moreso, while the binding of proteins to the surfaces of collagen fibrils has been demonstrated in vitro (such as with phosphophoryn),152 to my knowledge, diffusive transport of such proteins to the interior of fibrils has not been shown. This would seemingly be necessary for the proposed hypothesis that the protein promotes nucleation of amorphous phase within the fibril.
Figure 56.

Confocal fluorescence microscopy showing depth of penetration of FITC-labeled polyAsp into dense collagen scaffolds (sliced turkey tendon). Micrographs on the left were imaged with 400 PMT, while micrographs on the right were imaged at 500 PMT and offset to a greater depth to capture the fluorescence below the line of excessive intensity near the surface. (A) Control samples (without phosphate counterion) consistently yield penetration depths of around 100 μm. (B) Tendon mineralized using the PILP process show very high intensities near the surface, and then continued penetration at larger depths, but with significant loss of intensity. A z-offset of 120 μm was used for the control sample (overall depth of 590 μm) and 180 μm was used for the mineralized sample (overall depth of 650 μm). Note: all the control samples had much lower intensity profiles and hence the offset was lower to include the faint edge of the highest intensity. All depth scale bars = 500 μm. (Reprinted with permission from ref 160. Copyright 2007 Elsevier B.V..)
Interestingly, under the most optimal conditions for mineralization, where 70wt% mineral can be incorporated into a collagen scaffold, matching the degree of mineralization found in bone, the crystallizing solution stays clear throughout the mineralization process, while the collagen scaffold becomes white as it is selectively infiltrated with mineral (manuscript in preparation).384 The observation that the solution remained clear did lead us to consider the alternative hypothesis mentioned above, that the polymer may generate an inhibitory environment surrounding the collagen, and thereby promote nucleation within the fibrils. But this does not seem to be supported by the confocal depth-of-penetration study, because it is not clear why there would be such a pronounced difference in penetration of polymer into the collagen, particularly when the size of the polymer + calcium ions is smaller than polymer + both counterions. Instead, we find that the solution remains clear because the nanoscopic droplets of PILP phase are stable, and do not grow in size (as detected by light scattering), 384 unlike the CaCO3 PILP system.
Importantly, this system is one example where we argue that the fluidity of the precursor phase is essential for generating this biomineral feature (i.e., intrafibrillar crystallites). This is highlighted by the fact that the conventional gel-like ACP phase, which forms at the supersaturation we work with, does not infiltrate the collagen fibrils, and only leads to random crystal clusters on the surface of the fibrils (Figure 54a). Thus, the polymer seems to impart some special characteristics to the amorphous precursor, such as fluidity and/or surface charge.
An additional finding with this model system is that the crystals that grew (i.e., transformed from the amorphous phase) within the collagen fibrils inherently adopted the [001] crystallographic orientation found in bone, yielding diffraction patterns that consistently match those of bone (Figure 54f).160 This was a surprise because we had not added any of the specific proteins that had long been thought to induce epitaxial nucleation of the HA in bone formation. Our goal was simply to get mineral inside fibrils. Apparently, the collagen itself can direct the crystal orientation, but only when infiltrated with amorphous mineral. At this point, we cannot say if there are specific domains within the collagen fibrils (or adsorbed polyaspartate) that control crystal orientation, or if it is simply a constrained growth effect, where the rapid growth direction of HA squeezes out crystals at other orientations when growing within the narrow confines of the collagen fibril. This concept of competitive growth has been suggested for oriented crystal textures in other biominerals, such as avian egg shell385 and mollusk nacre,321 and computational simulations demonstrate the development of preferred orientations in polycrystalline materials growing from randomly oriented nuclei.56,57,322 This concept seems amenable to the bone system because of the anisotropy of hydroxyapatite crystals, which combined with the anisotropy of the organic compartment, could be envisioned as rapidly leading to a dominant growth direction.
In conclusion, the important point to be made here is that the amorphous precursor pathway provides, for the first time, a means for mimicking the nanostructure of bone (i.e., intrafibrillar nanocrystals of hydroxyapatite [001] aligned within the collagen fibrils). The ability to achieve a high degree of intrafibrillar mineralization did not require specific non-collagenous proteins, which have long been considered to function as epitaxial nucleating proteins. It required a means for infiltrating the collagen with amorphous mineral, and we propose that a fluidic precursor may be the modus operandi for accomplishing this important step in bone formation.
4.5. Pathological Biominerals
Pathological biomineralization also occurs in the presence of organic matrices and additives, so the relevance of the amorphous precursor pathway to these undesirable biomineral deposits is an area of emerging consideration. This could include stone formation, which occurs in many locations in the body (kidney stones, gout, pulpal stones, etc.), as well as atherosclerotic plaque, and biomaterial encrustation. The focus in our group has been on issues associated with urolithiasis (kidney stone formation); therefore, this discussion will be centered on kidney stones. But it should be kept in mind that amorphous phases could conceivably lie at the foundation of any of these pathological mineral deposits.
4.5.1. Kidney Stones
Kidney stones are polycrystalline aggregates, most often composed of some combination of calcium oxalate and/or some calcium phosphate, along with considerable organic matter from the complex urinary environment (Figure 57). Given that crystal growth is not a highly regulated process in such pathological deposits, there is not likely just one fundamental mechanism for all the different stone types that are observed. The additives (i.e., organic impurities in the urinary environment) could impact the crystallization process during the nucleation stage, or during crystal growth and/or aggregation. Thus, it is a highly complex problem. Nevertheless, there are several features of kidney stones that can be reproduced with the amorphous precursor model system; therefore, these studies provide the basis for proposing new ideas on urolithiasis (kidney stone formation).
Figure 57.

Scanning electron micrographs of a human urinary stone with a composite structure consisting of CaOx surrounding a CaP core. (a) The calcium oxalate crystals (O) exhibit a radial arrangement as they emanate from a core structure (P). This type of “polycrystalline aggregate” appears to be a spherulite. Energy dispersive x-ray analysis of the core region reveals calcium and phosphate peaks. Bar is 5 μm. (b) Higher magnification of the core structure shows rounded concretions with a fair amount of coalescence, which may hint that the CaP structures were formed from a fluidic amorphous precursor. Bar is 1 μm. (Reprinted with permission from ref 440. Copyright 1997 American Urological Association Inc..)
4.5.2. An Alternative Perspective on Stone Formation
Hypothesis: An amorphous mineral precursor may be induced by the macromolecules in the urinary environment, and play a role in kidney stone formation through several possible routes, as discussed in detail below:
solidification of an amorphous precursor can lead to dense-packed spherulites, and diffusion-limited exclusion of entrapped macromolecules leads to concentric laminations.
A fluidic mineral precursor can seep into cracks and crevices, forming a mineral ‘cement’, or can adsorb onto surfaces and coalesce into a continuous mineral coating, leading to ‘growth ring’ types of laminations
The first thing that attracted my attention to this field was the prevalence of crystal aggregates in our CaCO3 PILP model system (e.g., recall Figure 14b). This seems to result from the liquid-like character of the PILP precursor because the precursor droplets can adsorb to surfaces, and seep into cracks and crevices (capillarity revisited). This is demonstrated by the glass bead experiment shown in Figure 45b, where the precursor appeared to have seeped in between the beads, which then became glued together and to the glass substrate by CaCO3 PILP phase. Aside from glass beads, this is likely responsible for the pronounced aggregation tendency seen in many of our ‘hybrid’ crystal products, where it seems that part of the reaction may proceed via the conventional ion-by-ion crystal growth, while part of the crystal products are distorted and have a ‘molten’ appearance, presumably influenced by the fluidic properties of the precursor phase.202 We have observed that the precursor droplets preferentially like to adsorb to preformed crystals (relative to the surrounding glass or plastic substrate), and this can lead to coatings that ‘cement the crystals together into aggregates.
Kidney stones are not composed of CaCO3, so we first set out to determine if calcium oxalate (CaOx) and/or calcium phosphate (CaP) crystals could be formed by the PILP process. There was some evidence that CaOx PILP droplets were formed (although reproducibility has been problematic with this system), and these droplets had a tendency to aggregate and/or form spherulites (Figure 58a & b). In the case of CaP, a PILP precursor was already being developed for a biomimetic bone system, and it was observed that spherulitic aggregates were a common byproduct in this system as well (Figure 58c–f). Note- the spherulites in these systems consist of densely-packed polycrystals (Figure 58c), and we believe this space filling property arises from crystal growth from within an amorphous globule. For example, the early stage spherulites of CaP shown in Figure 58d are rather faint in birefringence, and they flatten when deposited on a substrate (Figure 58e). But as they crystallize, the birefringence becomes brighter until the spherulites fully densify, and can no longer transmit light (if they are 10s of microns in diameter), where they finally appear dark brown (Figure 58f). Needle-like crystals can also form from the globules (Figure 58f), and in this case, seem to form from a surface dissolution-recrystallization reaction (since they are not densely packed).
It should be noted that these two paths that lead to polycrystalline aggregates are derived from distinctly different mechanisms. The polycrystalline aggregate of a spherulite is formed from radial growth of polycrystals from a centralized nucleation point. The crystals can form branches which help to fill space, particularly when there is a preference for twinning or splay. If the crystals are transforming from an amorphous globule, the space surrounding the forming crystallites is already filled with mineral precursor, and will eventually crystallize as well, leading to a dense-packed spherulite. On the other hand, if the crystals are growing from solution, there is likely to be more space between the crystallites as they adopt their needle or platy crystal habits. In contrast to this well-defined spherulitic growth process, true ‘aggregation’ occurs when pre-formed crystals come together, and they can then become cemented together, either by an organic ‘glue’ or a mineral ‘cement’.
We hypothesize that the CaP minerals that form at the core of some stones might be generated by the first mechanism, in which they appear to form small spherulites, and these spherulites could then provide a nidus for subsequent deposition of the CaOx crystals.386 The CaOx crystals seem to form by both processes, where the randomly arranged and more faceted crystals appear to be true aggregates, while the concentrically-laminated spherical deposits appear to be textured spherulites (discussed below in Section 4.5.2.1).
The urinary tract contains many anionic proteins (as well as lipids) which could conceivably stimulate the amorphous precursor pathway. A more in depth review on this subject is provided in our chapter of the text on Biomineralization- Medical Aspects of Solubility.386 Many in vitro models of stone formation consider the acidic proteins as having an inhibitory action, which is therefore assumed to provide a protective action against stone formation. However, it should be remembered that it is the “inhibitory” polymers that tend to generate the amorphous precursor pathway. Thus, even though the function of these proteins may be to protect against urolithiasis, it is quite possible that this inhibitory action could be overwhelmed by the supersaturation or other factors in the reaction environment, and lead to this other mechanism of mineral deposition. In other words, a reaction medium that has inhibitory additives can eventually ‘crash out’ with spherulites, and/or droplets of a precursor phase. Spherulitic growth is rapid and can lead to relatively large structures in a short time; likewise, the aggregation tendency of crystals in a solution containing precursor droplets can also generate large structures quickly; so both of these aggregation phenomena could contribute to crystal retention in the renal tubules and augment stone formation. The crystal textures observed in the spherulites formed via the amorphous pathway in the in vitro model may also be relevant to some Randall’s plaque, which has taken the spotlight in recent years.
4.5.2.1. Randall’s Plaque and Multi-laminated Spherules
Recent evidence by Evan and coworkers387–391 implicates Randall’s plaque as playing an important role in idiopathic calcium oxalate (CaOx) stone formation. As originally described by Randall,392 and further examined in greater detail by the Evan team, this pathological mineralization starts with the deposition of calcium phosphate (CaP). The initial deposits appear as small electron-dense multilaminated spherules (roughly 50 nm in diameter), that form first at the basement membrane of the thin loops of Henle (Figure 59a),391 and then spread into the nearby interstitial space (Figure 59b), ultimately extending beyond the urothelial lining into urinary space, where the spherules somehow stimulate attachment and/or nucleation of the CaOx crystals that grow into a large stone.
Figure 59.
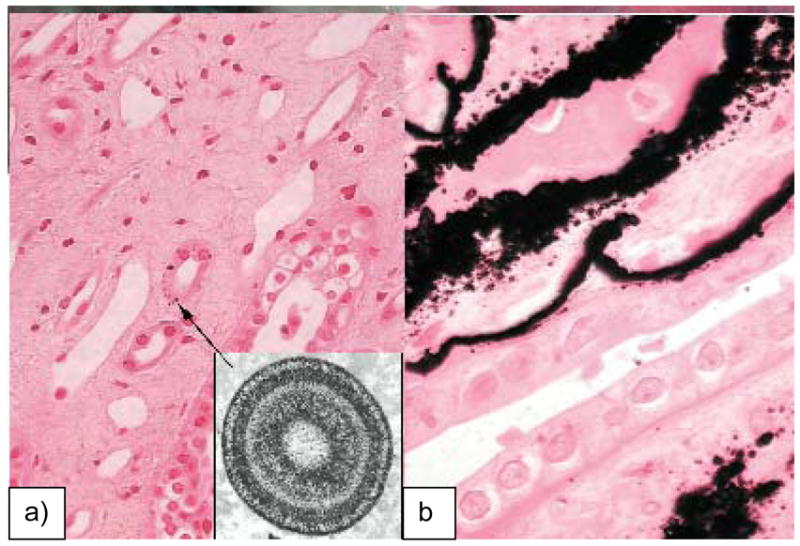
Evan’s391 work on idiopathic CaOx stone forming patients shows the location and morphology of newly forming precipitates found in Randall’s plaque. (a) Histologic optical image (Yasue stained) of a papillary biopsy section with minimal plaque shows spherically shaped, brown-black deposits only in the basement membranes of the thin loops of Henle (arrows). The insert shows a TEM micrograph of one of the deposits, which are typically multilaminated spherules containing alternating light and dark rings. (b) A denser region of plaque shows an accumulation of such deposits forming a continuous layer of calcifed matrix. (Reprinted with permission from ref 391. Copyright 2006 American Institute of Physics.)
The spherules in Randall’s plaque are quite small, but even full-sized stones often exhibit concentric laminations, and in this case appear to be spherulites (with a radial subtexture) composed of CaOx. For the larger multi-laminates, the layers are often attributed to daily (or dietary) growth rings, which are thought to form when the urinary environment, which is normally metastable, becomes excessively supersaturated (such as by dehydration, or hyperoxaluria). The in vitro models can provide an alternative hypothesis for how such multi-laminated spherical deposits may be formed. For example, multi-laminated spherulites can be generated with an amorphous precursor (Figure 58c–h), but notably, this does not occur by sequential deposition of mineral layers. These synthetic spherulites are much larger than the plaque spherules, but commensurate with the laminated spherulitic texture seen in many stones, such as the one shown in Figure 57a. Our 2-D model system of spherulitic CaCO3 films shows that the concentric laminations result from diffusion-limited exclusion of the polymeric impurity, which leads to polymer-enriched layers (Figure 58g & h).142 Ulčinas et al.258 also observed concentric bands in CaCO3 spherulites formed from a PAA-stabilized ACC gel (recall Figure 22 in Section 3.2.1), but since they were monitored by scanning probe microscopy, the role of polymer exclusion was not resolved. In the urinary environment, there are many proteins, lipids, and muco-polysaccharides, all of which could become entrapped in the amorphous precursor, and therefore would be expected to generate pronounced laminations.
An important consequence of this plaque is that it provides an anchored mineralization site, in which the CaP deposits apparently stimulate CaOx crystal formation into the urinary space.388,393 It is thought that the differences in pH between the urine and tissue could be responsible for the change in mineral composition because HA forms in a more basic pH, while the lower pH of urine favors CaOx.
We have done some in vitro studies on the overgrowth of calcium oxalate on calcium phosphate spherulites, where both minerals were formed in the presence of polyaspartate. The CaP precipitates grew from amorphous globules to form dense-packed spherulites. CaOx was then precipitated in the presence of these pre-formed spherulites, and with the polyaspartate additive, it was found that the amorphous CaOx PILP droplets liked to adsorb to the preformed CaP spherulites, and is some cases, they coalesced into a partial mineral coating (Figure 60a).386 This type of deposition, if performed daily, might be expected to lead to a ‘growth ring’ type of structure, analogous to the kidney stones that appear to have continuous, film-like multilayered coatings (Figure 60b). Smooth and continuous mineral films and coatings are typical of mineral formed by the PILP process, as seen in Figure 60a (and throughout this report). Thus, an amorphous precursor can lead to layered textures by two different pathways, diffusion-limited exclusion of organics, or sequential deposition of mineral coatings.
Figure 60.

Deposition of CaOx on pre-formed CaP spherulites in the presence of polyaspartate, to emulate the ‘growth rings’ that are found in some kidney stones. (a) The SEM micrograph shows a film-like coating of CaOx that deposited on the surface of a preformed CaP spherulite. We believe the coating was produced from a CaOx PILP phase induced by polyaspartate. There are also aggregates of small ill-defined crystals, which appear to also have been influenced by the polymer. The rough texture of the underlying CaP polycrystals of the spherulite can be seen, and clearly an epitaxial match was not required to stimulate the CaOx overgrowth layer. (b) This natural stone, which exhibits a pronounced laminated texture, is composed of whitlockite. Interestingly, the formation of metastable whitlockite is usually associated with the presence of Mg-ion, and in our experiments, Mg-ion dramatically enhances the ability to generate PILP films of CaCO3. Notably, the mineral layers in this stone are about the same thickness as the majority of films we have deposited with the PILP process (about half a micron). It is not clear whether or not this stone has a spherulitic texture, but the relatively separated structure of the laminations suggests they are caused by a sequential deposition of mineral coatings. (Reprinted with permission from ref 386. Copyright 2006 John Wiley & Sons, Ltd..)
Ryall350 has put together an interesting collage of multi-laminated spherules found in a wide variety of organisms, ranging from nanobacteria, to the electron-dense concretions in the midgut of various insects; and calcifying granules in the hepatopancreas of snails; and so on; all of which bear a strikingly resemblance to the multilaminated spherules found in Randall’s plaque. She draws the conclusion that “it is likely that the granules in human kidneys fulfill analogous functions to those in other organisms—particularly in calcium homeostasis.”350 While the function of the spherules (often times amorphous) in many of these organisms may be for ion storage, this seems (to me) to be a rather unregulated, and thus an undesirable process to occur in renal epithelial tissue. I would argue, instead, that this similarity in morphology and texture is simply a result of how an amorphous mineral precipitates, and particularly with entrapped organics that succumb to diffusion-limited exclusion, leading to concentric laminations. In support of this, the calcium phosphate in plaque has been described as containing amorphous calcium phosphate (ACP), as well as ‘biological’ apatite similar to bone.393 In the case of CaOx, Ryall found that crystals extracted from urine and treated with protease contained discrete, well-packed spherical subunits (similar features were observed in the CaOx crystals of plant raphides).350 As will be discussed in Section 5.2.1, this nanogranular texture may be a ‘mineralogical signature’ of an amorphous precursor in these CaOx biominerals as well.
Osteopontin, which is a prominent protein associated with mineral in the plaque394 (as well as in bone), is a highly acidic protein which could conceivably induce the amorphous phase in the biological environment. Such acidic proteins are generally considered to be inhibitors of crystal nucleation,85,86,218 and indeed, this is true of polyaspartate, our simple model protein, as well. It is therefore reasonable to postulate that osteopontin, which is prevalent in plaque, may be there for the purpose of inhibiting mineralization within the tissue if the cells sense an excessive ion concentration in the papillary tissue. However, if the inhibitory potential of such proteins is overwhelmed (such as by the hypercalciluria found in idiopathic stone formers), a metastable amorphous phase could be formed, which could instigate this unfortunate set of consequences.
4.6. Concluding Remarks on Mimicking Biomineralization
Hopefully, I have demonstrated that many of the unique morphological features found in biominerals that confounded the materials engineer can now be mimicked using the amorphous precursor pathway. In our system, these were mostly serendipitous discoveries that resulted from the natural consequences of depositing an amorphous phase on various substrates. Presumably, Mother Nature already made these ‘discoveries’ long ago, perhaps with just excretion of relatively simple acidic proteins, which originally served the function to regulate ion balance, but then evolved into more useful formats as the properties become more optimal for various mechanical or optical functions. For example, I would argue that the similar morphological attributes of nacre and seminacre, both consisting of layers of thin tablets, even though neither calcite nor aragonite form this morphology in a conventional crystallization, arises from the natural deposition of thin film-like morphologies of ACC (and/or PILP). Likewise, the fibrous morphologies of teeth, found in very different biomineral systems (CaCO3, CaP, Fe3O4), may be another natural consequence of the one-dimensional assembly of amorphous precursor particles (and/or PILP droplets), which again was capitalized on because of the enhanced mechanical properties of fiber-reinforced composites.
Although some may find the arguments presented in this section to be too speculative, it should be recognized that these are in fact no more speculative than the former hypothesis, that stereospecific interactions were responsible for modulating biomineral morphologies. It now seems likely that this is not the case for the biominerals that are formed by molding an amorphous phase within a compartment. While the science behind this hypothesis was sound, and there was clear evidence for such a mechanism from in vitro model systems, there was never direct proof in biomineralization, because of course such proof is difficult, if not impossible, to obtain in vivo. That is the case here for the new hypotheses presented here, where the small transport distances and limited time scales involved in biomineral formation make in vivo analysis difficult, and even ex vivo analysis complicated when trying to determine the fluidic character of an amorphous phase, particularly one of nanoscopic dimensions and short lifetime (which would be the case for a protein-induced PILP phase in biominerals). It is fortunate that the liquid-phase precursor induced with polyaspartate was stable enough to accumulate over the large distances of a crystallizing dish to be able to discover its existence.
There is another issue that is often raised in discussions involving in vitro model systems- just because a similar morphology is reproduced in the beaker does not necessarily mean that is how the biomineral is formed. This is true, and exaggerated by the fact that the morphology of calcite can be easily modified with just about any additive. The argument I present here is based on strength in numbers, where it is the sheer number of features that can be reproduced in the beaker, and under relatively simple reaction conditions, that command attention. And furthermore, it is the fact that the mechanisms proposed offer a viable explanation for the physicochemical features observed in all of these biominerals. The explanations are simple in concept, but in practice, are far more complicated, and will require much further study before we can fully capitalize on the toolkit that Mother Nature has devised.
The following section will discuss non-equilibrium compositions and crystal textures, both of which could have been included in this section since these are features observed in both biominerals and the model systems, yet are not typical of solution grown crystals. I separated them into a separate section because I think they are more representative of a particular crystal growth mechanism, whereas morphological features can sometimes be mimicked through different pathways.
5. Potential Mineralogical Signatures of an Amorphous Precursor Pathway
It would be nice to be able to identify mineralogical ‘signatures’ of the crystal growth mechanism, for several reasons. One reason is for geological and paleontological analysis of minerals. An interesting example to consider is the Martian meteorite ALH84001, which drew a lot of attention in the late 1990s because of the debate over whether some of the mineral constituents were of geologic versus biogenic origin, because they had features consistent with both high and low temperature deposition,395,396 such as the iron oxides with morphologies similar to magnetotactic bacteria, or the core-shell globules of carbonate minerals. Section 5.1 further discusses the geological significance of non-equilibrium mineral compositions and the “vital effect”.
For the biomimetics field, it would be helpful to be able to predict the crystal growth mechanism involved in a particular biomineral, which could then be mimicked more closely with the appropriate choice of in vitro model system. For example, it doesn’t make much sense to try and determine if a protein adsorbs to a specific crystallographic face when the morphology of that biomineral might simply be generated by molding the precursor in a vesicle, and thus the real function of that protein may be altogether different than the previously considered structure-directing shape modulation.54 From a materials chemistry perspective, in order to identify specific organic-inorganic interactions, we first need to identify the role that organic plays in modulating the crystallization process, and this may be best done by examining ‘features’ produced by the organic. In the case of pathological biomineralization, which could include disease conditions associated with bone or teeth formation, as well as kidney stones and atherosclerotic plaque, we need to have a better understanding of the underlying mineralization mechanism before we can even hope to find a way to solve the problems. Thus, such mineralogical signatures can help steer the biophysicists to search for further examples of the amorphous precursor. A case in point is our recommendation to re-open the search for an amorphous precursor pathway in bone formation,160 which I predict will eventually be demonstrated.
So what are the ‘signatures’ that may provide clues about the mineralization mechanism? As a first guess, the biominerals under most suspicion as being likely to have formed from an amorphous precursor pathway are those that have non-equilibrium compositions and morphologies. However, as Meldrum has shown,397 such non-equilibrium morphologies can be mimicked via conventional crystal growth in confinement (see Section 4.2.3), so this feature cannot be considered definitive evidence of an amorphous precursor. Likewise, while I would argue that the thin tablet and film morphologies are strong evidence to suggest an amorphous precursor, Kato’s view may differ (see Section 3.2.1).252 Other features that may hint at the precursor mechanism are the types of crystal defect textures, such as those produced from lattice strain caused by dehydration of the hydrated precursor, as well as the nanogranular colloidal textures that have been observed for the cluster growth mechanism. Thus, a better understanding of the mechanistic relationship between such textures to the crystal growth pathways may provide a stronger basis for identifying mineralogical signatures.
5.1. Non-equilibrium Compositions
With respect to non-equilibrium compositions, the CaCO3 system provides the best example. Inorganically grown calcite only allows for around 8% Mg2+ incorporation before it becomes so well inhibited that the aragonite polymorph is produced in its place.80,179 In contrast, some calcitic biominerals have far greater levels of Mg incorporation. For example, echinoderms form high magnesium calcite that contains around 10–15% magnesium in the primary elements,3 and red coralline algae secretes a high magnesium calcite of up to 30%.398–400 Robach et al.359 performed chemical mapping on urchin teeth of L. variegatus with secondary ion mass spectroscopy (SIMS), which enables both the organic and inorganic content to be examined. They find there is remarkable control of the local concentration of chemical composition, and show that the macromolecules associated with very high Mg calcite differ in amino acid composition from those found in the regions of high Mg calcite. Specifically, aspartic acid was found to be co-localized with the very high Mg columns, while the Ser distribution, an indicator of total protein, was uniform. They suggest that modulation of the Asp content of macromolecules secreted into the different growth spaces in the tooth may provide the mechanism controlling whether high Mg calcite (primary structural elements, 10–15mol% Mg) or very high Mg calcite (columns, 30–35mol% Mg) are formed, which they suggest occurs through Asp stabilization of an amorphous calcium carbonate precursor phase.
The in vitro models can support this suggestion. The older literature had shown that organic additives could increase the Mg incorporation.401 The possibility of an amorphous precursor being formed by those organics was probably not considered in the older works. More recently, we have come to learn that such high magnesium calcite is often (if not always) produced through the amorphous precursor pathway.180,268,402 In the case of using a polymeric process-directing agent to induce the amorphous precursor, one can imagine that Mg-ions could be chelated to the anionic polymer along with the calcium ions (at levels depending on their relative ion affinities), and thus both could get entrapped within the amorphous phase when it precipitates out. Some of it may become excluded during the amorphous-to-crystalline transformation, but in the PILP system (using polyaspartate additive), up to 37% magnesium can be found in CaCO3 precipitated from solutions based on sea water composition, containing a ratio of Mg:Ca = 5:1.180 However, not all of this Mg is incorporated directly into the lattice, so if the Mg content is determine by the reduction in x-ray lattice spacings, one finds that 26% magnesium is actually incorporated into the calcite lattice as a solid solution, where EDS shows 37% overall (the remaining Mg is assumed to be excluded to grain boundaries of remnant amorphous regions). Over-estimation of Mg content is likely the case for SIMS and ICP types of bulk analytical methods as well. Similar variance was found in octocoral sclerites examined by Sethmann et al.,381 who measured 15 mol% with ICP versus 9 - 11mol% via XRD. A range of composition in Mg content can be obtained via the PILP process (10–37%), which spans the range found in biologically formed magnesium-bearing calcite (12–40%); therefore, it seems reasonable to suggest that high magnesium calcite may be considered a mineralogical signature of probable crystallization via an amorphous precursor.
Kwak et al.329 have found values of Mg incorporation at 18% using a Mg2+/Ca2+ ratio = 4, which is comparable to the 20% found in system at the same ratio. In their SAM system, however, the amorphous CaCO3 was produced without using a polymeric process-directing agent, and a dual phase system resulted, where crystals templated by the SAM only incorporated only up to 8 mol% Mg, while spherical deposits from ACC had the high Mg content. Either the templated crystals did not form from an amorphous phase, or they could have undergone dissolution-recrystallization into the faceted crystals, resulting in exclusion of the Mg impurity. These results seem to suggest that impurity entrapment is more of a function of the crystallization pathway (i.e., proceeding through a stabilized amorphous phase), and is less dependent on the polymer-Mg ion affinity. In support of this, we found that increasing polymer concentration (from 2 to 20 μg/ml) had no significant effect on the Mg content when precipitated at an intermediate level of Mg2+/Ca2+ = 3.5.403 On the other hand, others have found that the Mg content in biominerals can be correlated with organic-rich regions,404,405 which means that there is either a pronounced affinity of the Mg ions to those particular organics, or in accord with these in vitro studies, that the organics stabilize the amorphous phase and/or its transformation, which consequently leads to the Mg enrichment.
The influence of the transformation pathway is highlighted by the Gayathri et al.211 study using the proteins extracted from seastar ossicles. As mentioned in Section 3.2.1.1, the soluble organic matrix promoted the formation of ACC with a film-like morphology, as shown in Figure 26a (which resembled PILP films of coalesced droplets). While the addition of the soluble proteins promoted the formation of magnesium calcite relative to aragonite, the high level of magnesium incorporation was only found in the early stages, when the material was still amorphous. For example, the magnesium levels were initially as high as those measured for the seastar ossicles (about 20 mol% Mg incorporated in the calcite lattice and about 0.01 weight% of organics), but they then dropped with time to around 7% as the ACC phase crystallized, with only a minor enhancement provided by the soluble protein addition. An important thing to note in this system is that the initial ACC film transformed through elongated ellipsoids into dumbbell-shaped magnesium calcite crystals, and apparently did not undergo a pseudomorphic transformation (i.e., the non-equilibrium film-like morphology was lost). This suggests that a solid-state type of transformation is required to entrap the magnesium ions (which were clearly already present in the initial ACC phase), while dissolution-recrystallization seems to effectively allow the impurity ions to be released before it recrystallizes. They note that “the relatively low levels of incorporation of magnesium ions into the calcite crystals obtained from in vitro crystallization experiments highlight the difficulty involved in simulating the microenvironment and control systems identical to those in the biological system.” I agree, and suggest that the important factor that is missing in this case is the stabilizing influence of some other ingredient, such as another soluble protein or insoluble matrix.
5.1.1. Significance of Impurity Incorporation and the “Vital” Effect
The incorporation of impurities is significant for several reasons. From a materials engineering perspective, the impurity could influence the properties of the mineral, such as mechanical or optical. Mg-ion certainly influences the texture of the mineral with respect to both lattice strain and defect density. In our PILP system, while Mg-ion enhances film formation by reducing or eliminating crystal side products, it also causes the film to break down into a more polycrystalline texture when at moderate levels (e.g. Figure 29a and 37d).180 The influence of Mg incorporation on the urchin spine has been described by Blake et al.406 as having a “mosaic texture due to small changes in the orientation of submicrometer-sized crystallites, which despite their differences in orientation, are largely coherent.” Tsipursky and Buseck407 find that the imperfections in biogenic calcites show numerous coherent and complex incoherent boundaries between the slightly misoriented mosaic blocks, and most of these defect zones contain dislocations that relieve stresses in the structure.
In the calcium phosphate biominerals, carbonate, fluoride, magnesium, and other impurities are also present in significant levels in the hydroxyapatite of bones and teeth, and the significance in this system derives from the influence of such impurities on solubility. Bone mineral needs to be bioresorbable for remodeling and repair, as well as general metabolic homeostasis. In contrast, fluoride is popular in toothpaste because it lowers the solubility of the mineral in teeth. Thinking beyond calcific biominerals, the issue of impurity incorporation may be relevant to potential commercial applications using a biomimetic approach to deliberately entrap inorganic ‘dopants’ in a crystal, which is desirable for semiconductor and other advanced ceramic materials.
Another reason impurity incorporation is significant is based on what geochemists refer to as the “Vital Effect”. This is related to the influence of vital (living) contributions on the trace elements and isotopes found in skeletal biominerals, which are studied because they provide paleoenvironmental measures of the climate that the organism was living in. As described by Rosenberg408 in Carter’s classic treatise on Skeletal Biomineralization:
The concentration of trace and minor elements within skeletons frequently diverges from predictions based on physical/chemical principles. Why should the increase in concentration of Mg within a skeleton increase with temperature in some marine invertebrates, but not in others? Why should the concentration of trace elements in skeletons change with ontogeny, and why should ontogenetic compositional patterns differ among taxa? Does the distribution of trace elements within a skeleton shed any light on the origination and diversification of biominerals?
Clearly the amorphous precursor pathway could play a vital role in this “vital effect”. Unfortunately, because the conditions are far from equilibrium (even though the sea water conditions are not), the influence of organic matter, along with Mg-ion (and other yet unresolved species), towards inducing this pathway, could make such desirable correlations very difficult to predict. I think it will require determining the contribution of this pathway (and/or organic matter) to each organism that is being used in these types of studies. The mechanisms may not even be consistent within one organism. As an example, Meibom et al.409 examined individual skeletal components in zooxanthellate coral, and found that the centers of calcification had higher trace element concentrations and distinctly lighter isotopic compositions than the fibrous components of the skeleton, and suggested that they must be formed by different mechanisms. I think even if they all form via a similar mechanism, the amorphous precursor pathway, Robach’s study mentioned above suggests that there can be substantial variation in composition depending on the interaction with organics.359 For example, the local Mg content changes abruptly at the interface between columns and plates in the urchin tooth, yet the whole structure diffracts as a single crystal. On the positive side, there is consistency within a species, and once the contributions of different mechanisms are better understood, perhaps the influence of temperature and other factors of interest can be derived from correlative in vitro models.
5.2. Crystal Textures- Biomimetic and Biominerals
5.2.1. Colloidal and Nanogranular Textures
In our PILP model system of an amorphous precursor pathway, the mineral films appear very smooth at low magnification (Figure 62a & b), yet a colloidal texture can be seen at high magnification with atomic force microscopy (Figure 62c & d). The dimensions of the colloidal substituents are dependent on numerous factors, and can range anywhere from 5 to 100 nanometers in early stage precipitates, but grow as large as several microns for late stage precipitates (recall Figure 15). The reason for this the nanocolloidal texture is not fully understood, because the single-crystalline patches of calcite can extend over tens to hundreds of microns, 3 orders of magnitude larger than the nanogranular constituents. They appear well coalesced to form a relatively homogeneous phase, but perhaps are outlined by minute amounts of the polymeric additive. It is also possible that the droplets are not each exactly alike in composition and texture when they deposit, which might lead to this non-homogeneous texture. For example, there may be differing polymer content, or the polydispersity of chain length in the commercial polyaspartate may influence the degree of ion binding or stoichiometry, as well as hydration level. Whatever the reason, it is clearly a different texture than crystals grown via the conventional crystallization process, and could conceivably be used as a ‘signature’ of crystals formed from accumulation of colloidal precursors.
Figure 62.

Nanogranular texture of calcareous sponge spicules, as examined by Sethman et al. 382. (a) Optical micrograph of the center of a large triactine spicule imaged in reflected light with the image focus beneath the spicule surface to show the finely segmented features that lie perpendicular to the long axes, which are the a*-axes in the sponge spicules (as illustrated in the schematic to the right). (b) AFM height image shows the nanogranular structure of a spicule surface in the (0001) plane of the actines. (c) and (d) Regular triangular etch figures in the nano-cluster material developed in etch pits on the (0001) surface. (c) AFM height image with triangle drawn as a guide to the eye, and (d) is the same area with phase image for enhancement of small-scale morphology contrast. (e) Relationship between orientations of etch figures seen in (c) and (d), to the overall spicule morphology and its associated crystallographic axes, showing the calcite {101̄4} rhombohedron, and the prismatic {112̄0} set of calcite crystal faces. (f) Schematic cross section through a spicule actine (grey) in relation to the crystallographic axes and crystal faces that approximate the cylindrical surface. (Reprinted with permission from ref 382. Copyright 2006 Elsevier Inc..)
Such a nanogranular texture could arguably arise after the fact, since our samples were not examined until after the reaction was complete. Ulčinas et al.,258 however, have observed similar features during in situ AFM and SNOM analysis of the growth of spherulitic films from an PAA-stabilized ACC gelatinous precipitate that formed on chitosan (see Figure 22, discussed previously in Section 3.2.1). Concentric bands were also observed, which seem analogous to the transition bars observed in the PILP system. All the features in these films, such as the nanoscale particulates, radial fibrous growth texture, and micron-scale concentric bands, became smooth with time (Figure 22c & d), which was described as a ‘ripening’ type of process.
Another question that is prompted by these nanoparticulate textures is whether they arise from heterogeneous nucleation of a colloidal ACC phase, or if the nanoparticles are solution borne, and then adsorb to the surface. Kato argues (for the analogous PAA- chitosan system in his experiments) that the soluble polymer adsorbs to the chitosan and stimulates nucleation of crystallites.251,298 The Ulčinas study suggests that the polymer may be stimulating nucleation of ACC colloids on the chitosan instead. There is also the possibility that the soluble polymer is inducing homogeneous nucleation of the colloids in solution, which then adsorb to the substrate, which is what we observe in our PILP system (recall Figure 30). Both could conceivably occur, which was a topic of discussion in Section 3.4.1.1.
5.2.1.1. Comparison to Biomineral Textures
In line with these in vitro observations, recent studies find a nanogranular texture in a variety of biominerals, which may hint that they form via a similar colloidal precipitation mechanism. See for example the AFM images in Figure 49 of the urchin spine and siliceous sponge spicules. In 2001, Dauphin318 reported on a nano-grain texture observed by AFM in mollusk nacre, which has also been seen via SEM in etched samples.51 Rousseau et al.55 also observe a nanogranular texture in the tablets of sheet nacre, and using phase contrast AFM, could image the intricate network of low modulus material (presumably intracrystalline proteins) surrounding each of the grains (recall Figure 35b). Clode and Marshall410,411 described “fusiform crystals”, and low temperature FE-SEM images show calcium-enriched nodular structures (23–48 nm in diameter) within a mesh-like network of organic matrix in the calcification zone of scleractian corals. Oaki et al.317 examined a variety of biominerals with FE-SEM and FE-TEM, and found a nanogranular texture in the nacreous layer of shells, skeletons of echinoderms, coral, foraminifera, and eggshells of hens and emus. The sizes of the aragonite- and calcite-type nanocrystals were within a range of approximately 20–180 nm and 10–80 nm, respectively. They discuss the relationship of the oriented nanocrystals from a perspective of mineral bridges across polymer-coated nanoparticles. Staolarski and Mazur412 demonstrate that a nanogranular texture exists in both calcitic and aragonitic fibrous skeletal elements found in a variety of sponges and corals. (Note- ‘fibrous’ refers to the texture from a spherulitic growth pattern412). They point out that “nanogranular organization of CaCO3 elements is not evidence, per se, of their biogenic versus abiogenic origin, or their aragonitic versus calcitic composition, but rather is a feature of CaCO3 formed in the presence of organic molecules.” Even in the calcium oxalate and calcium phosphate biominerals, similar textures have been demonstrated. Ryall shows examples of calcium oxalate monohydrate crystals, obtained from pathological precipitates extracted from urine, and raphide crystals extracted from plants, and both expose spherical subunits when treated with protease.350 Robinson and coworkers have observed “spherical subunits” in the hydroxyapatite crystals of dental enamel using chemical force microscopy (see Figure 53c,d, as discussed in Section 4.3.4).380 The subunits seem to somehow be responsible for the frictional bands that lie across the long axis of the crystallites (Figure 53a,b). Interestingly, the calcitic sponge spicules described below exhibit a similar segmental layering perpendicular to the long axes of the spicule crystal (Figure 62a),382 and the spherical subunits in the CaOx biominerals seem to link together into fibrous nanostructures.350
Sponges can be composed of silica or CaCO3, and both seem to exhibit a nanogranular texture (Figure 49 and Figure 62, respectively). Sethmann et al.,382 who examined the calcareous sponge spicules, describe the spicules as a nano-cluster composite structure (Figure 62b). Upon etching these calcitic spicules, they found triangulated stacks of granules that were correlated with the crystallographic symmetry of the single-crystalline element (Figure 62c–f). Through combined high-resolution and energy-filtering TEM, they were able to determine that there was carbon enrichment between the domain boundaries of the nanoclusters, which they conclude is an intercalated proteinaceous organic matrix. The etched triangular units appear similar to their in vitro model of polyanion-mediated mineralization, which was discussed in detail in the Transformation Pathways of Section 3.4.2.1.294 By monitoring the crystallization in situ via AFM, they found that polyaspartate (5–15kDa) additive created a gelatinous polyAsp-ACC film that covered the underlying calcite crystal, which then broke down into homo-epitactic nucleation of numerous local growth domains (recall the triangular subunits that evolved from a PAA-ACC gel, shown in Figure 32). Based on these observations, they proposed a nano-clustered crystal growth model, whereby “this controlled growth mechanism could facilitate the shaping of elaborate, rounded structures, as in sea urchin skeletons, without the need for a whole set of different plane-specific crystal growth inhibitors.”
More recently this group has examined the ultrastructure of octocoral sclerites (Figure 63: Top Row),381 and describe the formation of the internal fibrous crystals as “aligned aggregation and fusion of nanoparticles that are initially precipitated as nano-granular layers on the sclerite surface, enabling the smooth shaping of the sclerites.” Using the experimental approach, they demonstrated that analogous precursor nanoparticles can be generated within a polyacrylamide hydrogel via counter diffusion of ions (Figure 63: Bottom Section). In vitro, the nano-granular layers fused into continuous, but micro-faceted, calcite rhombs. Although the morphology differed from the fibrous sclerites, they used this model to explain how the crystallographic orientation can be preserved across a large element when sequential layers of nanoparticles are deposited, and make a comparison to the non-classical crystallization process described by Cölfen’s group as mesocrystal growth.295 Significantly, the nanogranular texture in this report was not induced by a soluble ionic polymer, but simply resulted from disrupted growth caused by the polyacrylamide hydrogel, which they explain as being caused by expulsion and condensation of the gel in the form of a porous membrane on the growing surface of the crystal. They compare the nanogranular texture observed here to that produced with the soluble polyanionic additives described above, in which the polyaspartate formed a gelatinous calcifying membrane, and conclude that nano-granular precursor structures can be induced by the physical properties of gelatinous networks.294 It is not clear in this polyacrylamide system if the nanograins are amorphous or crystalline, but they argue that in either case (in line with Navrotsky’s discussion190), the fusion of softly bound particles to form coherent crystals can be from thermodynamic equilibration and the high surface charge of the nanoparticles.
As a side comment, there may be somewhat of a disconnect between the terminology used in various reports, such as ‘ripening’, nanoparticle ‘fusion’, etc.. I tend to use the term ‘coalescence’, because in the PILP system, we see that even large-scale droplets, several microns in size, can fuse together to become one single-crystalline entity (Figure 64a & b), which would presumably not undergo these ripening processes commonly ascribed to energetically unstable nanoparticles. It is definitively the fluidic character of the precursor phase that leads to coalescence and formation of such non-equilibrium morphologies in our system; therefore, it is not unreasonable to suspect that this might occur for the nanoparticle systems described by many others as well. While there may be nanograins formed by the PAM hydrogel, the other aspects of smoothing and shaping seem to be absent, where a rhomb formed with a very rough and microfaceted texture (Figure 63a).
Figure 64.

Mid-stage transformation of PILP formed assemblies, demonstrating crystal growth across a pre-defined shape of ACC phase. Images are light micrographs viewed between crossed-polars with gypsum λ-plate, to determine between amorphous and crystalline phases. (a) ‘Pearl necklace’ strands of PILP droplets have accumulated at the air-water interface, and partially coalesced. A few regions have begun to crystallize (birefringent orange and blue regions- go Gators!). Beaded strands of single-crystalline calcite are formed because the transformation traverses across the pre-assembled precursor droplets. (b) Collections of unusually large PILP droplets exhibit quite high coalescence considering the substantial size of the spherical subunits. Particles 8 microns in diameter surely did not fuse together due to surface energetics and ripening associated with nanoparticulate systems, and thus must have had fluidic character. Each of the differently colored assemblies are single-crystalline calcite. A few smaller ‘pearl necklace’ strands can also be seen in the top left corner, but they are still amorphous (non-birefringent). (c) Mid-stage transformation shows a birefringent patch of calcite with a dendritic morphology, but it is not growing by a dendritic process. It is single-crystalline and simply transforming across the precursor, which adopted a dendritic shape due the underlying resorcarane monolayer, which did have dendritic texture, and apparently stimulated deposition of PILP droplets as a textured matrix. (d) Mid-stage transformation of a large single-crystalline patch of calcite that grew around pores. The pores, which likely formed due to outgassing of CO2, can be seen in the amorphous region to the left as well. Note the breakdown in crystal orientation near the periphery of these large single crystals.
In addition to suggesting that this mechanism could facilitate the shaping of elaborate biominerals, Sethmann has used this in vitro growth model to offer an explanation for the mechanical isotropy observed in the fracture behavior of the urchin spine,345 which has been a long-standing enigmatic feature of these biominerals.68,413 Similar nanocluster domains were found to exist in the skeletal elements of the sea urchin (30–50 nm), both on the natural surface and on the fracture surfaces (see Figure 49a–c).345 They suggest that the typical {10.4} cleavage planes may be inhibited by the physical blocking of polymer inclusions or lattice defects, or due to lattice strain-induced crack deviation, or a combination thereof.
In conclusion, it is not clear if an amorphous phase of nanoscale dimensions necessarily requires the liquid-like character found in the PILP phase in order to form such fusiform crystals (although it certainly would help). In our in vitro system, we even see partial coalescence of micron-sized ACC droplets, which at this size scale would not be expected to occur without some fluidic character (Figure 64a & b). Sethman’s models indicate that a nanogranular texture is not limited to polyanion-mediated mineralization, but still seems to result from some type of organic interaction, where in both cases, the organics are probably being excluded from the crystallization of an amorphous precursor. It should be kept in mind, however, that an ACC phase can form with a colloidal texture even without organic constituents, but this typically transforms into vaterite spherulites and/or faceted rhombs (see Section 3.1.1). Even in Sethman’s example, while the polyacrylamide gel did lead to nanograins, they ultimately crystallized into faceted structures. Thus, I still maintain that the polyanionic additives play a significant role in directing the transformation such that non-equilibrium morphologies can be generated.
5.2.2. Comparison to Mesocrystals
Since the realization that many biominerals may not form via the classical crystallization process, a variety of non-classical crystallization routes have been suggested in the recent literature. One prevalent example is the suggestion that biominerals are formed via mesocrystal assembly. The evidence for this is based on analysis of the textures of such biominerals, which frequently exhibit a nanogranular texture, as described above. Depending on how one perceives this mechanism, it could be considered similar to what I have proposed, that amorphous colloids assemble and coalesce, which then crystallize into relatively coherent, single-crystalline structures. On the other hand, mesocrystals can also form through the assembly of nanoparticles that are already crystalline, and this requires an oriented attachment mechanism.414 For example, Gehrke et al.415,416 consider oriented attachment of nanoparticles as being responsible for the formation of rosetta- and lens-shaped vaterite superstructures (formed without organic additives), although formation and fusion of the plates was suggested to occur with the primary amorphous nanoparticles.
Tao’s recent work on calcium phosphate nanoparticle assemblies suggests that the coalescence and moldability of aggregated nanoparticles could arise from an amorphous coating surrounding the nanocrystals.296 I discussed this work previously in Section 4.3.4 with respect to hydroxyapatite fiber formation (Figure 52), but it seems relevant to the issue of mesocrystal assembly. In this system, the encapsulated nanocrystals somehow managed to rotate and reorganize into single crystals after they had aggregated and become entrapped within the superstructure aggregate. The superstructure could be modulated through addition of organics (such as platelets with glutamic acid, versus needle-like crystals with glycine or amelogenin), but some anisotropic assembles resembling needles were formed even without additive.
Generally there is an overall crystallographic relationship between the nanocrystallites that constitute the subunits of a mesocrystal and its overall symmetry and macroscale morphology (analogous to colloidal assembly). Therefore, I personally have had a hard time reconciling how nanograins with spherical or non-descript shapes could assemble into anisotropic macrostructures, particularly over such large length scales, without some type of structure-directing additive; but the Gehrke and Tao papers seem to show that it is possible since they form platy and needle-like structures out of roundish nanoparticles, and even without organic additives. In any case, given the observance of the amorphous phase in a variety of forming biominerals, and nanoparticles in the case of the regenerating urchin spine,209 it seems more likely that the nanoparticles that initially assemble are amorphous. This is what we observe in the PILP system, which is more readily determined due to the large dimensions of the amorphous precursor. Therefore, I have generally assumed that the nanograins must coalesce before crystallization commences in order to lead to single crystals that are orders of magnitude larger than their nanogranular constituents. But the issues of how the nanoparticles coalesce, and at what point they crystallize into a coherent structure, is not fully resolved for biominerals.
While on the subject of mesocrystals, I feel a word of caution is needed. Just because one observes nanoparticulate subunits, or organic material surrounding subunits, does not necessarily mean there was oriented attachment, or that the matrix arranged crystalline particles. It could be that crystallization simply proceeded across a collection of amorphous precursor particles, leaving behind remnants of the polymer-encapsulated nanoparticles. This is considered in the Oaki et al. model of bridged nanocrystals, as mentioned previously in the section describing nanogranular textures found in biominerals.317 Figure 64a demonstrates this at the micron scale, where we know that these beaded strands of conjoined particulates (analogous to “pearl necklace” structures described in the literature) of uniform crystallographic orientation are created by crystallization across a collection of partially-coalesced droplets of precursor phase. This is ability to crystallize across a collection of precursor phase is of course the key point in generating an endless variety of crystal shapes, as further illustrated in Figure 64c & d. While such morphologies may have rounded surfaces, facets can also emerge from transformation of the amorphous precursor, or there can be reorganization to present a lower the surface energy, particularly on curved surfaces (e.g., microfaceted calcite fiber in Figure 34b), or surfaces that expose the less stable crystallographic planes (Figure 34c & d). Thus, the presence of microfacets is not necessarily due to oriented attachment of nanocrystals. The mechanisms proposed in numerous papers, such as coherent orientation via heteroepitaxy, oriented attachment, bridging, etc., may in some cases be simply due to crystallization across a collection of precursor colloids, and/or a breakdown into semi-coherent nanocrystalline domains (as discussed below). Overall, one can see that a variety of crystal growth mechanisms can lead mesocrystal structures (single crystals composed of subunits), so it should be kept in mind that the observation of such structures is not necessarily indicative of oriented attachment or a templated assembly type of process.
5.2.3. Other Defect Textures from Amorphous Precursors
There is another potential contributor to biomineral defect textures that has not really been discussed in the literature, and this is the possibility of shrinkage stresses created during transformation of the precursor phase. Based on our observations of the PILP model system, although we do not have any quantitative structural data of lattice strain at this time, there is substantial macroscopic evidence demonstrating shifts in crystallographic orientation, which presumably arise to release the stress and encroaching lattice strain during the A-to-C transformation. As mentioned in Section 4.1.2.1, biaxial strains are observed for thin-film crystals of rhomboidal morphology which are partially constrained by the substrate during the A-to-C transformation (see Figure 37b). Other examples include crystals that undergo a directional transformation over long distances (Figure 37d), where a gradual shift in birefringence can be observed as the crystallization front moves further and further away from the initial point of nucleation. In other words, as the microscope stage is rotated, instead of a singular extinction direction expected for a single crystal, the crystal gradually becomes extinct over several degrees of rotation, even though it is clear that the overall crystal is from one nucleation point. This gradual shift in crystal orientation can be demonstrated using the rapid epitaxial overgrowth technique, where the overlaying microcrystal facets show a continuous shift in orientation across the underlying PILP formed films (Figure 37a).
Another pertinent observation is that these shifts in crystallographic orientation are often accompanied by a breakdown in the crystal structure as stresses build up during the directional crystallization of the amorphous phase (Figure 65). This is readily seen at the micron scale, where a single-crystalline patch (i.e., growing from a singular nucleation site) within a mosaic film will exhibit a relatively uniform extinction direction except at the periphery, where a pronounced defect texture is observed (Figure 65a & b). It is debatable as to whether such a patch should be considered a single crystal; but I consider it as such when it is derived from a singular nucleation event, and thus leads to a correlated defect texture of semi-coherent domains (Figure 65c). In addition, the nano- to micro-domains are more likely to have shapes that are correlated with the crystallographic lattice, rather than appearing as round grains, such as those that that arise from colloid accumulation.
Figure 65.

Examples of semi-coherent domain texture in crystals derived from an amorphous precursor (from the PILP process). (a & b) Polarized light micrographs of mosaic films often show a high degree of defect texture within the individual single-crystalline patches of calcite, particularly towards the periphery of the patches. (b) The texture on the bottom patch is suggestive of microtwinning. On the top patch, the bright white patches are from a thicker overlayer that has formed on the underlying film, and optical fringes surrounding these regions seem to suggest that this overlayer has caused stress in the thin underlying film. This patch has a single-crystalline extinction direction, with the white regions simply representing a higher order retardation color from thickness increase. (c & d) TEM micrographs of PILP formed single crystals with microdefect texture. (c) A film patch of calcite (CaCO3) shows a variety of textures in the TEM, including stretched pores (that resemble crazing), and microfacets at the periphery which show the change in crystal orientation, suggestive of microtwinning. (d) A BaCO3 nanofiber which shows a gradual shift in crystallographic orientation along its length. SAED patterns, taken from the middle of the fiber (inset), and to the left and right sides, were used to track the [002] direction along the length of the fiber (orange arrows). A white line parallel to the [002] in the middle of the fiber was added for visual reference. The size of the SAED aperature size was ~550 nm diameter.
For this type of defect texture, the breakdown leads to semi-coherent domains that might appear as nanocrystals or microcrystals, but they are still relatively close in orientation (Figure 65c & d), with an overall structure that appears roughly single-crystalline via optical extinction or electron diffraction (with some arcing of the spots from tilt disorder). In the recent literature, one might find such defect textures described as mesocrystals, but the underlying basis of the correlated textures seen in this system are quite different, and are created by the A-to-C phase transition, not an oriented assembly process.
Correlated textures are also observed in some of our mineral fibers as well, which exhibit a shift in optical extinction along their length, even though they locally display a single-crystalline SAED spot pattern. This is particularly pronounced in BaCO3 fibers, where the SAED pattern may show a combination of spots and arcs (Figure 65d). The witherite phase of BaCO3 (structurally analogous to the aragonite phase of CaCO3) is prone to twinning, which could contribute to this more pronounced breakdown of crystal structure.
This correlative defect texture is commonly observed in the PILP model system, but since this occurs in Stage II when the solidified amorphous phase crystallizes, it could be pertinent to crystallization via an amorphous phase in general, because in either case, stresses can arise from dehydration under volumetric constraints as a hydrated precursor transforms into an anhydrous crystal.
All of these observations are consistent with the textures observed in some biominerals. For example, Blake et al.406 examined the urchin spine, and describe a “mosaic texture due to small changes in the orientation of submicrometer-sized crystallites, which despite their differences in orientation, are largely coherent.” Tsipursky and Buseck407 find that the “imperfections in biogenic calcites show numerous coherent and complex incoherent boundaries between the slightly misoriented mosaic blocks. Most of these defect zones contain dislocations that relieve stresses in the structure.” Sethmann et al. examined the nanostructure of sponge spicules, and by selecting the streaked spots in the electron diffraction pattern, dark-field TEM highlighted spherical pockets in the sample with “distinctly accumulated textural misalignments”.382 Their HRTEM image showed crystal domains that appeared to be tilted randomly with respect to each other, but FFT revealed certain preferential crystallographic orientations.
In addition to dehydration of the precursor, entrapped polymer might also generate stresses in crystals. Zolotoyabko and Pokroy417 have shown (with high-resolution X-ray powder diffraction at the European Synchrotron Radiation Facility) that the biogenic aragonite of mollusk nacre has significantly altered lattice parameters relative to synthetic and geologic aragonite. Specifically, the highest and positive (tensile-like) lattice distortions were found along the c-axis (Δc/c up to 0.2% ), while lattice distortions along the a-axis were also positive but about two times lower than along the c-axis, whereas along the b-axis the distortions were negative (compression-like). They argue that this anisotropic strain is caused by the intra-crystalline organic molecules trapped within individual crystallites. They demonstrate that under mild ‘annealing’ (150–200°C), the lattice distortions are relieved, but this strain relief is accompanied by a reduction in coherence length (subdivision of crystal volume into much smaller blocks which coherently scatter X-rays), the opposite of what one would expect for annealing in a traditional ceramic, where thermally activated re-crystallization (at much higher temperatures) leads to grain growth. By analogy to strained heterostructures seen in band gap materials with structurally isomorphous materials of slightly different lattice parameters, they conclude that the lattice strain is relieved upon annealing the biogenic crystal by thermally-induced conformational changes or partial defragmentation of the occluded macromolecules. It is possible that this thermal annealing could also relieve stresses caused by the processes mentioned above.
While there have been several nice TEM and diffraction studies on the defect texture of biomineral crystals, there have been relatively fewer from the in vitro models for comparison. Nevertheless, these findings are in good agreement with other observations from the in vitro models. For example, the anisotropic exclusion of the polymeric impurity has been observed in transition bars that form during the amorphous-to-crystalline transformation in the PILP process.142 This also leads to a non-homogeneous texture that appears to be well correlated with the crystallographic lattice. In fact, we have proposed that this model system may help to explain the anisotropic domain texture observed in biominerals, which generally has been attributed to selective protein adsorption in the older literature (prior to consideration of the amorphous precursor pathway). Therefore, these findings are consistent with the argument made by Zolotoyabko and Pokroy,417 that entrapped proteins are responsible for such anisotropic lattice distortions. The in vitro model system may help us to understand why the polymeric impurities become preferentially excluded along specific crystallographic directions, which in turn leads to selective occlusion of polymer in association with specific crystallographic planes.
Shrinkage stresses would not be expected for crystals grown via the conventional crystallization process; therefore, this could provide a mineralogical signature of crystals grown via the amorphous precursor pathway. I cannot say whether this necessitates a fluidic PILP-type precursor, or if the conventional ACC precursor would also lead to this texture; but one might expect a higher degree of hydration to lead to more pronounced shrinkage effects. On the other hand, I assume some densification will also be encountered in the traditional amorphous phase because assemblies of the more solid-like particles should have interparticle pore space to reconcile with as they crystallize into a compact structure, in addition to exclusion of the one unit of water associated with conventional ACC (e.g., ACC density is 1.62 g/ml,418 whereas calcite is ca 2.7; therefore, about 40 % volume shrinkage would be expected). Regarding the degree of hydration of the precursor, it has been noted that when the transformation is performed in a dry atmosphere (the amorphous films are removed from the solution), the defect texture is more prevalent (see Figure 14d), suggesting that a more rapid dehydration of the precursor may exaggerate the stresses, whereas the incorporated water may allow more ion mobility and ‘annealing’ during the transformation. The situation becomes even more complicated when one considers that the solidification rate of the PILP precursor is influenced by the polymeric-process-directing agent, as seen in our compositional studies.225 In addition, the entrapped polymer also influences the rate of the A-to-C transformation, as do Mg ions (perhaps similar to the inhibitory effect seen for both of these additives in conventionally grown calcite). In the latter case, Mg-ions substantially reduce the rate of transformation, yet they promote a higher defect texture. This is presumably due to the presence of an impurity, which may cause lattice strain when incorporated into calcite, or accumulate at defect sites if excluded, since not all of the Mg is effectively incorporated into the lattice (see Section 5.1).
In conclusion, there are a variety of textures seen in biominerals and in crystals grown via the amorphous pathway, which are not found in the classical crystallization pathway. It is not clear if these result from different formation mechanisms or just different interpretations. The community perhaps needs to come up with more precise ways to define the processes (or determine the processes) since a nanogranular or mesocrystal structure can seemingly result from a variety of crystallization pathways. For example, the following reported processes seem to lead to crystals with such textures:
rotation and oriented attachment of polarized nanocrystals
oriented attachment via organic-templated nanocrystals
coherent aggregation of nanograins with hetero-epitaxy from organic matrix
assembly of amorphous solid colloids, with subsequent fusion, ‘ripening’, or recrystallization
assembly of amorphous-coated nanocrystals, with coalescence and reorganization into single crystals
assembly and coalescence of polymer-induced fluidic mineral precursor droplets
crystal bridging across polymer-stabilized particulate boundaries
semicoherent domains from breakdown of lattice strains during densification and/or crystallization
anisotropic lattice distortions from occlusion of polymer that selectively adsorbed to specific crystallographic planes
anisotropic lattice distortions from diffusion-limited anisotropic exclusion of polymer along transition bar zones
Thus, while some these defect textures seem to result from incorporation of polymer and/or densification, both of which can result from the amorphous precursor pathway, more work is needed to determine the various contributions and decipher the true mechanistic attributes.
5.3. Single-Crystalline Composites and Hybrid Crystals
As has been mentioned previously, many biominerals seem capable of forming single-crystalline structures that encapsulate an organic matrix. These often contain rather small concentrations of intracrystalline proteins, such as in the urchin spine, and it is perhaps not hard to imagine that crystal growth could proceed around these macromolecules and entrap them within the lattice, particularly if they are already entrapped in a solidified amorphous precursor where they cannot be readily excluded to the solution.
In addition to the urchin calcite, the aragonite in mollusk shells also contains intracrystalline proteins. As described in the previous section, Zolotoyabko and Pokroy suggest that such proteins are responsible for the anisotropic lattice distortions in biogenic aragonite.417 They note that the observed maximum strain values of about 0.2% are rather high for brittle ceramics and, in some materials of non-biogenic origin, are close to the strain magnitudes which cause fracture in a brittle ceramic material.
Rousseau et al.55 have examined the intracrystalline proteins in mollusk sheet nacre using TEM, and describe this biomineral as a hybrid composite (see Figure 35). The mineral phase was finely divided (nanogranular), interspersed with a “foam-like” intracrystalline matrix that behaved as a single crystal in TEM darkfield mode. There was a correlation between the bright matrix and mineral within select tablets, therefore, they propose that the single-crystalline nature of the mineral results from “coherent aggregation of nanograins keeping strictly the same crystallographic orientation thanks to a hetero-epitaxy mechanism.”
In the realm of biominerals, one cannot say with certainty whether such composite type crystals are deliberately designed for a purpose, or if they are simply artifacts of a crystallization carried out in the presence of macromolecules. Being that there are various degrees of organic occlusion, depending on the organisms needs, I tend to believe that when such hybrid structures were naturally formed, they were evolutionarily selected for because they provided some inherent advantages, such as reducing brittleness of the bioceramic in echinoderms, or providing connectivity in the otoconia. As is well stated by Zolotoyabko and Pokroy,417 “this important question remains unanswered: whether the discovered mechanical deformation is the fee that is paid for the effective route of polymorph selection by using an amorphous precursor, or that these deformations have an additional important function which is presently unknown to us?”
At this point, one might be tempted to conclude that the amorphous precursor pathway allows for the formation of these hybrid composites, since these two classes of biominerals are now considered to be formed from some type of amorphous precursor. There is, however, another biomineral example of a single-crystalline composite that may argue differently, and that is the otoconia of the inner ear. As mentioned in Section 4.2.1, these are well faceted CaCO3 crystals (calcite in mammals), and not particularly interesting in terms of morphology (Figure 66). Their organization is not that interesting either, where they are randomly arranged as they are embedded within a filamentous gelatinous matrix. They seem to be rather ‘dirty’ crystals with a fibrous organic matrix sticking all over their surfaces. The crystals themselves, however, are very intriguing, because while the crystals are well faceted, thus appearing to be single-crystalline, internally, each otoconium contains a compact central core meshwork of organic filaments (Figure 66b).419,420 Organic filaments within the crystals seem to radiate outward and emerge through channels and pores to the surface of the crystal (Figure 66c & d), where they then provide a network of filamentous crosslinks to other crystals and/or the surrounding matrix and gelatinous membrane. The purpose of making such a single-crystalline composite is not likely to be a need for enhancing the mechanical properties since they do not provide a protective or skeletal function (as in the urchin spine for example), but rather to provide interconnectivity between the crystal masses that are connected via filaments within the gelatinous matrix. In this biomineral, there is far more organic matrix, and particularly in the interior, at the central core, where the nucleation event must start. The faceted morphology seems to suggest that these crystals are formed via the conventional crystallization process; but it is surprising to me that this large quantity of organic matter doesn’t seem to disturb the overall growth habit.
Synthetically, single-crystalline composites have been achieved as well, demonstrating that crystal growth can proceed around obstacles that do not fit in the crystal lattice site. For example, Li and Estroff have produced nanoporous calcite crystals through crystal growth in an agarose hydrogel medium, which is demonstrated using SAED on thin sections, and etching and electron back-scattered diffraction (EBSD) studies on the bulk specimens.306,307 Although significant roughening of the facets occurred, there was no indication that these crystal formed from an amorphous phase. Even larger micron-sized intercalation can be achieved, as demonstrated by Meldrum and coworkers,421 who have grown both calcite and zinc oxide crystals around porous templates, such as polymer membranes and latex particles. As described previously in Section 4.2.3, Li and Qi have made 3D ordered macroporous calcite using a templating approach, where they first vacuum infiltrated the interstices of a colloidal assembly of functionalized polystyrene nanospheres with an ACC phase, and single-crystalline calcite was achieved due to the initial formation of a calcite seed crystal (Figure 47).
Such model systems demonstrate that single crystals with large internal surface areas between organic and inorganic phase can be produced without destroying the long-range order of the crystalline component, and particularly when the surface chemistry limits the nucleation event.397 Significantly, not all of these systems relied upon an amorphous precursor to generate the hybrid composite crystals. With respect to the biogenic counterparts, it seems that both crystallization pathways can lead to intracrystalline macromolecules. The echinoderms and mollusks, which contain occluded proteins, are now considered to be formed from an amorphous precursor. In contrast, the otoconia appear to be formed via the conventional crystallization pathway, as judging by the faceted morphology (although this remains to be determined, since the amorphous precursor route can also lead to faceted crystals).
In conclusion, while hybrid crystals can result from the amorphous precursor pathway, as seen in some of the biomineral examples, it does not appear to be a pre-requisite, as suggested by the otoconia, and demonstrated for several synthetic models. Therefore, I conclude that this is not a reliable mineralogical signature of the amorphous precursor route.
5.4. Concluding Remarks on Mineralogical Signatures
At this time, I think the most reliable signatures of the amorphous precursor pathway are the non-equilibrium compositions, and nanogranular defect textures. However, in the latter case, it still needs to be resolved if nanogranular textures are also formed via mesocrystal assembly, which in some cases apparently do not utilize an amorphous precursor. While I believe this is not the case in biominerals, some may disagree. The lattice distortions are another potential signature, since they have been identified with amorphous-formed biominerals, and the PILP model system. However, I don’t think it has been demonstrated whether or not similar anisotropic lattice distortions occur in the hybrid crystals formed via the conventional growth process. So this feature is still questionable. I think that some specific lattice distortions, such as biaxial strain, or a continuously increasing gradient in distortion, do argue for a solidification mechanism, and thus the amorphous precursor. As mentioned above, the hybrid crystals themselves do not seem to provide a reliable indicator of crystallization pathway, although the particular organization of the organics may provide further clues, such as if they are surrounding nanograins, or entrapped in diffusion-limited transition bars. While many questions remain, with few definitive answers, there has been some nice progress made considering that this is still a comparatively new area.
6. Outlook and Prospects
Throughout this paper I have shown examples of biomineral features that have been mimicked in the lab utilizing the precursor pathway. The question is, how far have we come, and how far do we have to go? One consideration with in vitro models is figuring out how to dissociate the processing capabilities afforded by cellular control versus those derived from crystallochemical processes, because we shouldn’t necessarily expect to duplicate a biomineral in its entirety. Although self assembly is a primary component of biomineral systems, and of keen interest to the biomimetics community, one surely cannot expect to throw all the ingredients found in a biomineral into a beaker and have them assemble into a sea shell or a bone. Therefore, my approach is to break down the biomineral structures into specific and readily defined features, to see if we can understand how the organic-inorganic interactions lead to each of those specific features. For example, the features I have pinpointed and discussed in this paper are the following:
Thin mineral films, coatings, and tablets
Mineral fibers
Patterned crystal location
Molded crystals
High-magnesium calcite
Single-crystalline composites
Crystal lattice distortions
Nanogranular and nanolaminated textures
Concentric lamination
Dense spherulites
Capillarity and interpenetrant composites (i.e., intrafibrillar mineralization of collagen)
It is through a combination of these features that the biological system achieves its hierarchical structure, and this often relies upon cellular processing, which provides spatial and temporal control. For example, the coccolith is molded and shaped within a vesicular compartment (Figure 42f), and this could conceivably be accomplished in vitro with the right mold and control over nucleation events. But the organism then transports this biomineral component to build itself a surrounding shell. This type of control would be pretty difficult to duplicate in the beaker, for example in trying to engineer an elaborate core-shell particulate system. On the other hand, we have been able to make ‘soft’-‘hard’ core-shell particles by depositing a mineral coating on emulsion particles (Figure 67a,b), mimicking the less elaborate biomineral of dinoflagellate cysts.422,423 This can still be a useful construct for controlled release applications, in which active agents can be encapsulated within the fluidic core, and released via the biodegradable CaCO3 shell.424 Likewise, we have been able to achieve intrafibrillar mineralization of collagen, reproducing the nanostructured architecture of bone (Figure 54).425 But to synthesize the higher levels of structure found in bone will require some way to organize the collagen matrix into osteons, which is clearly something that cells are better able to accomplish as they locally and sequentially deposit concentric layers around the blood vessels. But the materials engineer can sometimes adapt, and perhaps find that sequential deposition of laminates will suffice for generating the desired mechanical properties or biocompatibility for a biomimetic bone graft substitute.
Figure 67.

Biomimetic core-shell particles mimicking dinoflagellate cysts, for potential application as biodegradable microcapsules. Oil-in-water emulsion droplets served as the substrate for deposition of a CaCO3 precursor coating, which crystallized to form a smooth mineral shell that encapsulated a fluid, oil-based interior of the emulsion colloids. (a) The core-shell texture is evidenced by the birefringent rings. (b) The core-shell texture is also evident in SEM of particles that were lightly crushed, revealing the hollow interior which had been filled with oil.
6.1. Benefiting from Non-Specificity of the Precursor Pathway
The beauty of the precursor pathway is that the highly specific interactions that were once considered the basis of biomineralization are not necessary, at least for morphological control. This is advantageous for a variety of reasons. For one, it allows the Materials Chemist to design in vitro experiments without the need for particular proteins, which are not readily available to most of us biomimetic engineers. It also suggests that it will be easier for Materials Engineers to be able to mimic biomineralization processes for commercial applications, if one does not need to synthesize specific proteins or peptides in order to achieve crystal modulation via molecular recognition. Although we have more to learn before we will be able to mimic all the levels of control found in biomineralization, I feel that this new paradigm brings the field of biomimetics a sizeable step forward.
6.1.1. Non-Specificity of Polymeric Process-Directing Agents
The prior view of the role of the soluble acidic proteins of biominerals required very specific structural interactions of protein sequences with specific crystallographic planes.54,122,123 There was, therefore, much interest in the sequence and conformations of such proteins, where for example β-sheet or α-helix structures might display a regular array of carboxylate groups for interaction with specific planes of the crystal. In light of the fact that many biominerals have now been found to use an amorphous precursor pathway, the specificity of the soluble proteins needs to be looked at from this new perspective. That is not to say that specific structure-directing interactions are not involved in biomineralization, but they seem less necessary for generating the morphological features found in many biominerals, and instead may be more relevant to regulation of crystal phase, orientation, and texture (see Section 3.4.2.3). So perhaps it is not surprising that analytical studies have found that many of these soluble acidic proteins do not seem to adopt well-defined conformations in solution.285,426,427 For example, Kim et al.427 have examined the mollusk shell protein AP7, which is considered responsible for aragonite polymorph formation and stabilization within the nacre layer of red abalone. This group demonstrated that the 30 amino acid N-terminal domain, denoted as AP7-1, interacts with Ca(II) ions at -DD-sequence clusters, yet retains its unfolded, conformationally labile structure in the presence of Ca(II) ions. This protein, although not examined for ACC forming ability, possesses the capability of inhibiting calcium carbonate crystal growth in vitro via growth step frustration or interruption. Flexible conformations have also been found for some of the proteins associated with calcium phosphate biominerals as well, such as bone sialoprotein (BSP) and osteopontin (OPN).285 BSP and OPN are members of the SIBLING (Small Integrin-Binding LIgand, N-linked Glycoprotein) family, and the solution structures were solved by one dimensional proton NMR and transverse relaxation times. Fisher et al. found that the “polypeptide backbones rapidly sample an ensemble of conformations consistent with them both being completely unstructured in solution. This flexibility appears to enable these relatively small glycoproteins to rapidly associate with a number of different binding partners including other proteins as well as the mineral phase of bones and teeth.”
Given the high degree of charge character, it seems reasonable to not expect them to have regular chain folding as is found in the globular proteins for example, which bury the more hydrophobic residues at the interior (although there may be neighboring hydrophobic domains on the acidic proteins that also do this). Instead, the functional form of these proteins may be related to its ability to sequester ions, while inhibiting crystallization. Both the ion-binding affinity and strength are probably important, but at this point, it is not clear whether it is better to have strong or weak ion-binding interactions. But with this concentrated pool of localized ions, the polymer also needs to be able to inhibit or delay the nucleation of the crystalline phase, which would otherwise occur rapidly at such high local supersaturation. Another aspect that may be important for the polymer is its ability to retain hydration waters; otherwise, the ion-protein interactions might lead to ‘salting out’. At least in the PILP system, the high degree of hydration seems to be influenced by the polymer,225 and this allows it to form ion-enriched droplets, rather than solid precipitates.
Given this list of polymeric requirements, of which there certainly may be more, one still finds that it is possible to produce ACC films with a variety of polymeric processing-directing agents. In the literature described in the prior section, the ACC phase seems to be most often produced using polyaspartic acid, polyacrylic acid, and sometimes polyglutamic acid, along with DNA and other specialty polymers such as double-hydrophilic block copolymers and anionic dendrimers. One can also include in this list the variety of proteins that also induce the amorphous phase, which are described in Section 3.2.1.1. As can be seen, these polymers are usually negatively charged polyelectrolytes, and are generally considered inhibitory with respect to crystallization of calcite. I don’t believe the mechanism for inhibition is well understood, but the outcome is that it then allows the kinetic pathway to proceed through intermediate formation of metastable phases, and of interest here, the amorphous phase. What may be lacking at this point in these in vitro models is the higher levels of control, where specificity in protein sequence or conformation may influence the short-range order of the ACC phase that apparently controls crystal phase and texture (see Section 3.4.2.3).
6.1.2. Non-Specificity of Minerals Formed via the Precursor Pathway
Given that highly specific interactions are not required between polymer and mineral to induce the amorphous phase, we have considered that other mineral systems might be amenable to precursor processing as well. Therefore, we have been trying to determine if the PILP process can be induced in a variety of inorganic minerals. Thus far, we have found that a PILP type phase can be formed in the following inorganic systems: CaCO3,203 CaP,160 CaOx,386 BaCO3,368 SrCO3,369 CdS (unpublished results). However, the liquid-phase precursor is not always easy to detect or collect, so we presently are judging the PILP-forming capability by whether the mineral forms into morphologies typical of a liquid-like precursor, such as ‘molten’ crystals, single-crystal drops, continuous films, or fibers. We see some of these features in the list of minerals above, yet there are differences in each system. For example, amorphous films of CaP tend to transform by the recrystallization route, leading to the typical dense-packed array of nanoplatelets seen for hydroxyapatite films grown on a variety of substrates. The difference here could be related to the much lower solubility of CaP versus CaCO3, along with stronger surface energy contributions in hydroxyapatite crystals that lead to faceting. Perhaps a more potent polymer could be designed to help stabilize the amorphous-to-crystalline transformation more effectively; but given that the hydroxyapatite crystals in biominerals are also not large, this might just be an inherent difference in the transformation pathways followed by the different systems. Our reason for suggesting that the CaP phase does form via a PILP pathway, and that this pathway is distinctly different than the common ACP gel, is because of the ability to achieve intrafibrillar mineralization of collagen by capillary action, which we believe to be a direct consequence of the fluidity of the amorphous phase (discussed in Section 4.4). The gel-like precursor of CaP does not infiltrate the collagen fibrils, and only leads to spherulitic clusters on the surface.160
6.1.2.1. Non-Calcium Based Biominerals?
Returning to the biominerals, for which there are nearly 70 mineral phases reported,428 could some of these other biominerals be candidates for formation via the precursor pathway? I could envision a PILP type of process occurring for the other ionic salt types of biominerals, such as in the sulfates, where barium, strontium and calcium sulfates are found in a few biomineral examples (see Table 1). There have been papers showing a polymer modulated crystallization of BaSO4 that leads to fibers and cone-shaped morphologies that strongly resemble the BaCO3 structures we form with an amorphous precursor, thus these seem likely candidates.
6.1.2.2. Iron Oxide Biominerals?
How about the oxide-based biominerals, such as the iron oxides found in magnetotactic bacteria and chiton/limpet teeth; or the silicon dioxide biominerals found in sponges and diatoms? Can my argument be extended this far, where the chemistries are entirely different? Of course the iron oxides have already been found to form via an amorphous phase, at least in some cases, such as chiton teeth (the goethite in limpet teeth seems to grow from the direct crystallization route;171 perhaps because it is already an acicular form, and doesn’t need to molded into this fibrous form). The magnetite seen in the chiton teeth appears to be highly anisotropic, resembling fibers that are molded within the organic matrix (Figure 9e);169 yet acicular particles are not readily synthesized for crystals with cubic crystal systems such as magnetite. Likewise, the magnetite produced by magnetotactic bacteria can also be formed into acicular structures, such tear-drop and bullet-shaped morphologies (Figure 43).429 In this case, single domain magnetite nanocrystals are formed within a membrane-bound compartment called a magnetosome. Even though it was suggested long ago that an amorphous ferrihydrate precursor is present within this compartment, the molding of the amorphous phase is generally not considered responsible for these novel morphologies. Instead, the concept of structure-directing additives via preferential adsorption is generally assumed responsible for the non-equilibrium morphologies. In the iron oxide biominerals, however, it seems that the polymer responsible for sequestering the amorphous iron oxide phase is ferritin, which is a supramolecular assembly, quite different than the polyanionic proteins. On the other hand, acidic proteins have also been found associated with magnetite biominerals, but their function is usually considered to be a nucleating protein to control the crystal orientation, or structure-directing additives for controlling crystal shape, or alignment of the magnetosome particles into a chain to magnetically guide the microorganism.430 I don’t believe anyone has looked at the iron oxide systems from this new perspective, where an amorphous precursor might simply be molded prior to transformation into the crystalline magnetite. The more important issue for the magnetotactic bacteria is the crystal alignment and assembly into chains.
6.1.2.3. Silica Based Biominerals?
In the case of siliceous biominerals, I didn’t even consider them at first because the chemistry of a covalently polymerized network seemed too far removed from ionic salts. But I have become intrigued with the silaffin proteins found in diatoms, and their polyamine counterparts.431–436 These polymers promote precipitation by flocculation, a process that leads to non-porous close-packed silica spheres by “fusion” of oligomeric silicic acids.436 The silica precipitates in diatoms (Figure 68a) as well as those produced with extracted proteins (Figure 68b) and synthetic polyamine analogues are reminiscent of the ‘droplets’ of CaCO3 that can be formed with the polycarboxylates. Of course the silica system is already amorphous, and does not further transform into a crystal. But the feature that I am interested in here is the possibility of a fluidic precursor, which densifies into silica without high temperature processing (such as is typically required in conventional sol-gel processing). In contrast, the silicatein proteins extracted from sponge spicules nicely catalyze the condensation reaction, but they seem to lead to a porous friable networks of silica when used with synthetic chemical precursors (such as TEOS and phenyltriethoxysilane).437 The images of partially and fully coalesced colloids formed with the silaffins seem to suggest there may be some fluidic character in the initial precipitates (Figure 68), which could generate the more compact densified structures. As described by Sumper and Kroger,436 “Microscopic phase separation turned out to be essential for the polyamine-induced silica precipitation. Mono-, oligo- or poly-silicic acid molecules may be adsorbed on and/or dissolved in the polyamine microdroplets, thereby forming a coacervate (formation of a ‘liquid precipitate’) which finally hardens by silica formation.” As I have argued for the CaCO3 system, the fluidic character could provide a powerful means for achieving morphological control in siliceous biominerals as well, which being non-crystalline, is the primary feature of interest for these biominerals (in addition to catalysis of the condensation reaction). Sumper and Kroger also suggest that “the apparently elastic silaffin–silica phase observed at the early stages may represent the mouldable biosilica material used to form the silica elements of C. fusiformis (the diatom from which the silaffin was extracted).” Thus, there seems to be an interesting analogy between these seemingly different biomineral systems, and that relates to the sequestering capability of the polyelectrolyte (cationic proteins in the case of the silicification versus anionic proteins in the case of calcification), to concentrate a local pool of precursor species while inhibiting/regulating the polymerization (rather than crystallization). This is an intriguing prospect to consider- could there be one unifying mechanism amongst all these biominerals- the use of polyelectrolytes to sequester a highly concentrated pool of precursor species into a fluidic and moldable form?
Figure 68.

Silica precipitation in vivo, for comparison to Kroger’s in vitro studies using polyamine proteins extracted from diatoms.441 The silica particles in all of these systems appear to be quite dense, and they seem to exhibit varying degrees of coalescence suggestive of some fluidic character in the initial precipitates. (a) Early silicification process in the axial filament in a desme sponge spicule. Note the fluidic character of the “fused” precipitates. (b) SEM images of the morphogenesis of silica precipitated in vitro with native silaffin-1A protein, using 100 mM monosilicic acid solution buffered at pH 5.5 (50 mM sodium acetate). The first silica structures appear as extended flat networks of irregularly-shaped branched bands (the dark flat streaks in the image a). As described by the authors, the early phase (a silicified silaffin phase) appears to be elastic since within a few minutes, it transforms into spherical particles (400 – 700 nm in diameter). Scale bars = 2 μm. (a) (Reprinted with permission from ref 442. Copyright 2006 NRC Canada.) (b) (Reprinted with permission from ref 441. Copyright 2002 American Association for the Advancement of Science.)
6.2. A Unifying Hypothesis
There are some 70 different biologically formed minerals,4,13,428 and although many of the general concepts are similar between the different biominerals, such as the ability to control various crystallographic features, many of us working in the field tend to focus on a system of given chemistry. In my case, this is the calcium-based biominerals, which are thus emphasized more in this review. But even within this limited regime, there are certainly differing biomineral features that lead one to believe that the mechanisms involved in each are dissimilar. For example, as first described in Section 1.3.1, many CaCO3 biominerals are molded and shaped into elaborate single-crystalline morphologies, while the CaP biominerals consist of small platy nanocrystals, of which the more interesting feature is their organization into hierarchical structures. Therefore, it is perhaps not surprising that these two communities have not been all that closely linked. There is, however, one distinguishing feature that is found in both of these systems, and that is the presence of the acidic proteins. In fact, they seem to be ubiquitous in nearly all biominerals. But given the differing morphological features found in invertebrate biominerals versus vertebrate biominerals, their mechanistic influence has been perceived differently. In the CaCO3 biominerals, the acidic proteins had generally been considered to be responsible for selective adsorption to growing crystals, enabling morphological control; while in the CaP biominerals, the acidic proteins have been assumed to be responsible for selective nucleation of the hydroxyapatite crystals in order to promote crystals to form within collagen fibrils in a specific [001] orientation. But now, with this major paradigm shift in the field that highlights the fundamental role of an amorphous precursor pathway, is there a new way to view the role of the acidic polymers in these differing systems?
Let us return to making a comparison between the invertebrate CaCO3 biominerals, and the vertebrate CaP biominerals, where now, one might see that there are perhaps more similarities between these systems than first meets the eye. Yes, the crystal sizes and morphologies are very different; but this is due to an inherent difference between the surface energies of the two minerals. But in both cases, I would argue that the crystals are ‘molded’ within compartments, where the compartment that forms the bone crystallites is extremely small, being created by self-assembled collagen fibrils (or self-assembled amelogenins in enamel). And this ‘molding’ capability quite possibly derives from the amorphous precursor pathway (and arguably from a liquid-like precursor phase). And the amorphous precursor pathway quite possibly is induced by the anionic proteins (as is the PILP process). And the inducing capability quite possibly results from the combination of ion-binding capacity in conjunction with inhibitory kinetics provided by the macromolecular constituents (and arguably the hydration characteristics associated with the polymer-ion combination). In other words, I am suggesting that the functional role of certain anionic proteins in these different biominerals is the same, to induce the amorphous precursor pathway (and arguably, a liquid-like precursor).
In the case of pathological biomineralization, such as stone formation, many of the proteins extracted from the stones are known to be modulators of crystallization in vitro, which can occur during crystal nucleation, growth, or aggregation. Stones form in a complex urinary environment, and are composed of a heterogeneous population of mineral products, so all of these stages may be important in the formation or retention of the stone. One might assume that since there are many crystal types and crystal faces present, that highly specific interactions are not involved in the inhibitory and blocking potential of these proteins; instead, more generic crystal binding domains, such as domains of acidic functionality, could be useful for inhibiting both crystal nucleation and growth, as well as aggregation (such as via steric repulsion of a protein of appropriate size). My present thoughts on this topic are that these non-specific inhibitory proteins may be secreted by the renal epithelial cells, most often resulting in successful inhibition of the stone; but under certain conditions, the desired “inhibitory” action of the protein is overcome by the supersaturation or other environmental factors, leading to amorphous precursors, which seems to be evidenced by some of the features observed in stones, such as compact spherulites, core-overgrowth structures, concentric laminations, nanosphere subunits, and generalized aggregation tendencies and cementatious properties. Thus, while an amorphous precursor might be very useful in biologically-controlled mineralization, it could have detrimental effects in contributing to pathological biomineralization.
In conclusion, I propose the following unifying hypothesis- that the unique compositional and textural features found in many different biomineral systems, although not the same, may be derived from an amorphous precursor pathway. For example, a nanogranular texture is found in CaCO3 and CaOx biominerals, as well as high levels of impurity incorporation, versus the poorly-crystalline and non-stoichiometric apatite in found in bone. These features differ, yet the unpinning mechanism may be related. Actually, in the larger hydroxyapatite crystals of enamel, there also seems to be evidence of spherical subunits that may be related to the colloidal precipitation mechanism. We can achieve intrafibrillar mineralization of collagen with both CaP and CaCO3, which highlights the non-specificity of the processes involved in forming such an interpenetrating composite (i.e., doesn’t require a specific mineral-protein interaction). Such comparisons could be made with many of the other biomineral systems, such as a precursor sequestering capability of the polyamine based proteins in siliceous biominerals, with its associated densification and fusion of colloids, or the molding of anisotropic iron oxide biominerals, etc.. Thus, the point to be made is that one could view the role of the soluble-type biopolymers from a new perspective, in which they may serve as process-directing agents, whose function is to induce the amorphous precursor pathway (or in some cases, with a PILP phase as a precursor to the amorphous solid phase), thus providing a potent means for regulating biomineral structure and properties.
The biophysicists have now done the convincing work to show that indeed many biominerals are formed via an amorphous precursor pathway. I predicted this would be the case when I first discovered the PILP process, because it seemed to provide the missing piece of the puzzle. I felt that I could finally understand Mother Nature’s secret of how biominerals are formed! However, not all are in agreement on this concept, and it still remains to be determined if any of the biominerals are formed by a liquid-phase amorphous precursor. I believe it is the liquid-phase aspect that was, and still continues to be, a difficult concept for the community to grasp (yet one that will prove to be of vital significance). Now that others have confirmed its existence in vitro as well, I hope that the biophysicists will be intrigued enough to take on the challenge of searching for it in biological systems, because the fluidity of the precursor can have profound consequences on the morphology and texture of the resulting crystals. This is the message that I hope to have conveyed in this paper.
Acknowledgments
The work presented from my group’s studies was supported through the following grants, and I am ever so grateful for the hard work and insightful discussions contributed by my students, as well as the many images used throughout this manuscript. This material is based upon work supported over the last 10 years by the National Science Foundation, NIDDK, and NASA, as follows: The PILP templating studies were funded by NSF Grant No. BES-9980795; the collagen mineralization studies from a Biosystems at the Nanoscale grant ECS-9986333(CaCO3 PILP) and a NIRT grant BES-0404000 (CaP PILP); the PILP mechanistic studies through a CAREER grant DMR-0094209, and currently through a Materials World Network grant DMR-0710605; the mineralogical signatures work through the Exobiology program in the ROSS of NASA from grant NRA00-OSS-01-043; the biomimetic kidney stone work by NIH grant R01 DK59765-01; and the core-shell particles from NSF funded UF-Particle Engineering Research Center (PERC) grant EEC-9402989.
Biography

Laurie B. Gower is currently an Associate Professor of Materials Science & Engineering, and has developed an interdisciplinary research program in the area of biomimetic synthesis of materials. Growing up in Florida, collecting seashells was her favorite pastime. Now, as a Materials Scientist, she has come to learn that the beauty of these shells derives from their intricate microstructures, which in turn provides them with fascinating mechanical and optical properties. With this new-found admiration of shells, she wants to understand, as do many Materials Chemists, how they are made. Therefore, her work is two-pronged, with fundamental studies aimed at developing an understanding of the mechanisms involved in biomineralization, and applications-based work targeted at using the tools Mother Nature has devised to construct novel biomimetic composite materials. The research program started with her doctoral work, where she received her PhD in 1997 in Polymer Science & Engineering from the University of Massachusetts at Amherst, under the direction of Professor David Tirrell, who thankfully provided the freedom and support for her to explore new directions. She became fascinated with the role that biopolymers play in regulating crystal growth in biologically formed minerals, such as bones, teeth, invertebrate shells and exoskeletons, as well as pathological mineral formation in kidney stones and biomaterial encrustation. Prior to her doctoral work, she obtained a Master’s Degree in Bioengineering at the University of Utah, working under Professor Donald Lyman, where she examined segmented-polyether-urethane-urea (SPEUU) biomaterials for use as biocompatible elastomers for vascular graft applications. Her academic career brings her back to her alumnus school, the University of Florida, where she first began her bachelor’s studies in Engineering Sciences with a biomedical emphasis. Once a gator fan, always a gator fan! Acknowledgment should be made to the little urchin pet in the photo, who seemed totally unaware of the inspiration that he and his ancestors have provided by their intricately designed exoskeletons. He was just squirming and wanting to get back into the water. Gower still enjoys the hobby of collecting shells, as they continue to provide a source of amazement, from the inside out.
References
- 1.Mann S, Webb J, Williams RJP, editors. Biomineralization - Chemical and Biochemical Perspectives. VCH Publishers, Inc; N. Y: 1989. [Google Scholar]
- 2.Bonucci E, editor. Calcification in Biological Systems. CRC Press; Boca Raton: 1992. [Google Scholar]
- 3.Carter JG, editor. Skeletal Biomineralization: Patterns, Processes and Evolutionary Trends. Vol. 1 Van Nostrand Reinhold; N.Y: 1990. [Google Scholar]
- 4.Lowenstam HA, Weiner S. On Biomineralization. Oxford University Press; N.Y: 1989. [Google Scholar]
- 5.Crick RE, editor. Origin, Evolution, and Modern Aspects of Biomineralization in Plants and Animals. Plenum Press; New York & London: 1989. [Google Scholar]
- 6.Frankel RB, Blakemore RP, editors. Iron Biominerals. Plenum press; New York: 1991. [Google Scholar]
- 7.Skinner HCW, Fitzpatrick RW, editors. Biomineralization: Processes of Iron and Manganese- Modern and Ancient Environments. Supplement 21. Catena Verlag; Germany: 1992. [Google Scholar]
- 8.Khan SR, editor. Calcium Oxalate in Biological Systems. CRC Press, Inc; N.Y: 1995. [Google Scholar]
- 9.Simpson TL, Volcani BE. Silicon and Siliceous Structures in Biological Systems. Springer-Verlag; N.Y: 1981. [Google Scholar]
- 10.Kirschvink JL, Jones DS, MacFadden BJ, editors. Magnetite Biomineralization and Magnetoreception in Organisms : a New Biomagnetism. Plenum Press; New York: 1985. [Google Scholar]
- 11.Simkiss K, Wilbur KM. Biomineralization- Cell Biology and Mineral Deposition. Academic Press, Inc; N. Y: 1989. [Google Scholar]
- 12.Müller WEG, editor. Silicon biomineralization : biology, biochemistry, molecular biology, biotechnology. Springer; Berlin ; New York: 2003. [Google Scholar]
- 13.Mann S. Biomineralization: Principles and Concepts in Bioinorganic Materials Chemistry. Oxford University Press; New York: 2001. [Google Scholar]
- 14.Königsberger E, Königsberger L, editors. Medical Aspects of Solubility. John Wiley & Sons, Ltd; West Sussex, England: 2006. [Google Scholar]
- 15.Bäuerlein E, Behrens P, Epple M, editors. Handbook of Biomineralization. Wiley-VCH; Weiheim: 2007. [Google Scholar]
- 16.Dove PM, Yoreo JJD, Weiner S, editors. Biomineralization. Vol. 54 Mineralogical Society of America; Washington, D.C: 2003. [Google Scholar]
- 17.Lichtenegger HC, Schoberl T, Bartl MH, Waite H, Stucky GD. Science. 2002;298:389. doi: 10.1126/science.1075433. [DOI] [PubMed] [Google Scholar]
- 18.Feng QL, Cui FZ, Pu G, Wang RZ, Li HD. Mater Sci Eng C-Biomimetic Supramol Syst. 2000;11:19. [Google Scholar]
- 19.Sellinger A, Weiss PM, Nguyen A, Lu YF, Assink RA, Gong WL, Brinker CJ. Nature. 1998;394:256. [Google Scholar]
- 20.Zhang SM, Gao YJ, Zhang ZJ, Dang HX, Liu WM, Xue QJ. Acta Chimica Sinica. 2002;60:1497. [Google Scholar]
- 21.Zhang X, Liu CL, Wu WJ, Wang JF. Materials Letters. 2006;60:2086. [Google Scholar]
- 22.Tang ZY, Kotov NA, Magonov S, Ozturk B. Nature Materials. 2003;2:413. doi: 10.1038/nmat906. [DOI] [PubMed] [Google Scholar]
- 23.Estroff LA, Hamilton AD. Chem Mat. 2001;13:3227. [Google Scholar]
- 24.Cölfen H. Topics in Current Chemistry. 2007;270:1. [Google Scholar]
- 25.Xu AW, Ma YR, Cölfen H. J Mater Chem. 2007;17:415. [Google Scholar]
- 26.Aizenberg J, Livage J, Mann S. J Mater Chem. 2004;14:E5. [Google Scholar]
- 27.Dujardin E, Mann S. Advanced Materials. 2002;14:775. [Google Scholar]
- 28.Whaley SR, English DS, Hu EL, Barbara PF, Belcher A. Nature. 2000;405:665. doi: 10.1038/35015043. [DOI] [PubMed] [Google Scholar]
- 29.Sarikaya M, Tamerler C, Jen AK-Y, Schulten K, Baneyx F. Nature Materials. 2003;2:577. doi: 10.1038/nmat964. [DOI] [PubMed] [Google Scholar]
- 30.Dickerson MB, Naik RR, Stone MO, Cai Y, Sandhage KH. Chemical Communications. 2004:1776. doi: 10.1039/b402480j. [DOI] [PubMed] [Google Scholar]
- 31.Naik RR, Brott LL, Clarson SJ, Stone MO. Journal of Nanoscience and Nanotechnology. 2002;2:95. doi: 10.1166/jnn.2002.074. [DOI] [PubMed] [Google Scholar]
- 32.Huang Y, Chiang CY, Lee SK, Gao Y, Hu EL, De Yoreo J, Belcher AM. Nano Letters. 2005;5:1429. doi: 10.1021/nl050795d. [DOI] [PubMed] [Google Scholar]
- 33.Evans JS, Samudrala R, Walsh TR, Oren EE, Tamerler C. MRS Bulletin. 2008;33:514. [Google Scholar]
- 34.Tamerler C, Sarikaya M. MRS Bulletin. 2008;33:504. [Google Scholar]
- 35.Zaremba CM, Belcher AM, Fritz M, Li Y, Mann S, Hansma PK, Morse DE, Speck JS, Stucky GD. Chem Mat. 1996;8:679. [Google Scholar]
- 36.Mutvei H. Biomineralisation- Research Reports. 1972;6:96. [Google Scholar]
- 37.Gregoire C. Journal of Biophysical and Biochemical Cytology. 1961;9:395. doi: 10.1083/jcb.9.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gotliv BA, Kessler N, Sumerel JL, Morse DE, Tuross N, Addadi L, Weiner S. Chembiochem. 2005;6:304. doi: 10.1002/cbic.200400221. [DOI] [PubMed] [Google Scholar]
- 39.DiMasi E, Sarikaya M. Journal of Materials Research. 2004;19:1471. [Google Scholar]
- 40.Nudelman F, Chen HH, Goldberg HA, Weiner S, Addadi L. Faraday Discussions. 2007;136:9. doi: 10.1039/b704418f. [DOI] [PubMed] [Google Scholar]
- 41.Mutvei H, Omori M, Watabe N. The mechanisms of biomineralization in animals and plants. Proceedings 3rd International Biomineralization Symposium. 1980:49. [Google Scholar]
- 42.Wise SW. Science. 1970;167:1486. doi: 10.1126/science.167.3924.1486. [DOI] [PubMed] [Google Scholar]
- 43.Nakahara H, editor. Nacre formation in bivalve and gastropod molluscs. Springer-Verlag; New York: 1991. Vol. Biomineralization ‘90. [Google Scholar]
- 44.Lin AY-M, Chen P-Y, Meyers MA. Acta Biomaterialia. 2007 doi: 10.1016/j.actbio.2007.05.005. [DOI] [Google Scholar]
- 45.Meyers MA, Chen P-Y, Lin AY-M, Seki Y. Progress in Materials Science. 2008;53:1. [Google Scholar]
- 46.Watabe N. Prog Crystal Growth Charact. 1981;4:99. [Google Scholar]
- 47.Wada K. Nature. 1966;211:1427. [Google Scholar]
- 48.Gregoire C. Nature. 1959;184:1157. [Google Scholar]
- 49.Wise SW, de Villiers J. Trans Am Microsc Soc. 1971;90:376. [Google Scholar]
- 50.Watabe N, Wilbur KM. International Symposium on the Mechanisms of Mineralization in the Invertebrates and Plants. University of South Carolina; Georgetown S.C: 1974. p. 461. [Google Scholar]
- 51.Addadi L, Joester D, Nudelman F, Weiner S. Chem Eur J. 2006;12:980. doi: 10.1002/chem.200500980. [DOI] [PubMed] [Google Scholar]
- 52.Weiner S, Talmon Y, Traub W. Int J Biol Macromol. 1983;5:325. [Google Scholar]
- 53.Weiner S, Traub W. Journal of Rheumatology. 1981;8:1011. [Google Scholar]
- 54.Weiner S, Addadi L. J Mater Chem. 1997;7(5):689. [Google Scholar]
- 55.Rousseau M, Lopez E, Stempfle P, Brendle M, Franke L, Guette A, Naslain R, Bourrat X. Biomaterials. 2005;26:6254. doi: 10.1016/j.biomaterials.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 56.Metzler RA, Zhou D, Abrecht M, Coppersmith SN, Gilbert PUPA. ArXiv online article. 2008 [Google Scholar]
- 57.Rodriguez-Navarro A, Garcia-Ruiz JM. European Journal of Mineralogy. 2000;12:609. [Google Scholar]
- 58.Schaffer TE, Ionescu-Zanetti C, Proksch R, Fritz M, Walters DA, Almqvist N, Zaremba CM, Belcher AM, Smith BL, Stucky GD, Morse DE, Hansma PK. Chem Materials. 1997;9(8):1731. [Google Scholar]
- 59.Rousseau M, Lopez E, Coute A, Mascarel G, Smith DC, Naslain R, Bourrat X. Bioceramics-16. 2004;254–256:1009. [Google Scholar]
- 60.Song F, Soh AK, Bai YL. Biomaterials. 2003;24:3623. doi: 10.1016/s0142-9612(03)00215-1. [DOI] [PubMed] [Google Scholar]
- 61.Aizenberg J, Hanson J, Koetzle TF, Leiserowitz L, Weiner S, Addadi L. Chem-Eur J. 1995;1:414. doi: 10.1096/fasebj.9.2.7781928. [DOI] [PubMed] [Google Scholar]
- 62.Clarkson ENK. Invertebrate Palaeontology and Evolution. Chapman & Hall; New York: 1994. [Google Scholar]
- 63.Okazaki K, Dillaman RM, Wilbur KM. Biol Bull. 1981;161:402. [Google Scholar]
- 64.Donnay G, Pawson DL. Science. 1969;166:1147. doi: 10.1126/science.166.3909.1147. [DOI] [PubMed] [Google Scholar]
- 65.Nissen HU. Science. 1969;166:1150. doi: 10.1126/science.166.3909.1150. [DOI] [PubMed] [Google Scholar]
- 66.Blake DF, Peacor DR. Scanning Electron Microscopy. 1981:321. [PubMed] [Google Scholar]
- 67.Berman A, Hanson J, Leiserowitz L, Koetzle TF, Weiner S, Addadi L. Science. 1993;259:776. doi: 10.1126/science.259.5096.776. [DOI] [PubMed] [Google Scholar]
- 68.Berman A, Addadi L, Weiner S. Nature. 1988;331:546. [Google Scholar]
- 69.Berman A, Addadi L, Kvick A, Leiserowitz L, Nelson M, Weiner S. Science. 1990;250:664. doi: 10.1126/science.250.4981.664. [DOI] [PubMed] [Google Scholar]
- 70.Albeck S, Aizenberg J, Addadi L, Weiner S. J Am Chem Soc. 1993;115:11691. [Google Scholar]
- 71.Aizenberg J, Hanson J, Koetzle TF, Weiner S, Addadi L. J Am Chem Soc. 1997;119:881. [Google Scholar]
- 72.Young JR, Henriksen K. In: Biomineralization. Dove PM, Yoreo JJD, Weiner S, editors. Vol. 54 Mineralogical Society of America; Washington, DC: 2003. [Google Scholar]
- 73.Weiner S, Sagi I, Addadi L. Science. 2005;309:1027. doi: 10.1126/science.1114920. [DOI] [PubMed] [Google Scholar]
- 74.Aizenberg J, Hendler G. J Mater Chem. 2004;14:2066. [Google Scholar]
- 75.Williams RJP. In: Biomineralization- Chemical and Biochemical Perspectives. Mann S, Webb J, Williams RJP, editors. VCH Publ; Weinheim: 1989. [Google Scholar]
- 76.Lippmann F. Sedimentary Carbonate Minerals. Springer-Verlag; N. Y: 1973. [Google Scholar]
- 77.Titiloye JO, Parker SC, Osguthorpe DJ, Mann S. J Chem Soc, Chem Comm. 1991:1494. [Google Scholar]
- 78.Mann S. Structure and Bonding. 1983;54:125. [Google Scholar]
- 79.Falini G, Fermani S, Gazzano M, Ripamonti A. J Chem Soc-Dalton Trans. 2000:3983. [Google Scholar]
- 80.Berner RA. Geochim Cosmochim Acta. 1975;39:489. [Google Scholar]
- 81.Weiner S, Wagner HD. Annu Rev Mater Sci. 1998;28:271. [Google Scholar]
- 82.Veis A. In: Extracellular Matrix Biochemistry. Piez KA, Reddi AH, editors. Elsevier; Amsterdam: 1984. [Google Scholar]
- 83.Termine JD. Rheumatology. 1986;10:184. [Google Scholar]
- 84.Marsh ME. In: Biomineralization 93- 7th Int Symp on Biomineralization. 1 Allemand D, Cuif J-P, editors. Vol. 14. Bulletin de l’Institut oceanographique; Monoco: 1994. [Google Scholar]
- 85.Addadi L, Weiner S. Proc Natl Acad Sci USA. 1985;82:4110. doi: 10.1073/pnas.82.12.4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Weiner S, Addadi L. Trends Biochem Science. 1991;16(7):252. doi: 10.1016/0968-0004(91)90098-g. [DOI] [PubMed] [Google Scholar]
- 87.Hecker A, Testeniere O, Marin F, Luquet G. FEBS Letters. 2003;535:49. doi: 10.1016/s0014-5793(02)03856-5. [DOI] [PubMed] [Google Scholar]
- 88.Kamat S, Su X, Ballarini R, Heuer AH. Nature. 2000;405:1036. doi: 10.1038/35016535. [DOI] [PubMed] [Google Scholar]
- 89.Currey JD. Science. 2005;309(5732):253. doi: 10.1126/science.1113954. [DOI] [PubMed] [Google Scholar]
- 90.Akkus O, Jepsen KJ, Rimnac CM. Journal of Materials Science. 2000;35:6065. [Google Scholar]
- 91.Akkus O. Journal of Biomechanical Engineering-Transactions of the Asme. 2005;127:383. doi: 10.1115/1.1894204. [DOI] [PubMed] [Google Scholar]
- 92.Buehler MJ. Nanotechnology. 2007;18:295102. (9pp) [Google Scholar]
- 93.Ge J, Wang XM, Cui FZ. Mater Sci Eng C-Biomimetic Supramol Syst. 2006;26:710. doi: 10.1016/j.msec.2005.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Heim AJ, Matthews WG, Koob TJ. APPLIED PHYSICS LETTERS. 2006;89:181902. [Google Scholar]
- 95.Rijt JAJvd, Werf KOvd, Bennink ML, Dijkstra PJ, Feijen J. Macromolecular Bioscience. 2006;6:697. doi: 10.1002/mabi.200600063. [DOI] [PubMed] [Google Scholar]
- 96.Smith BL, Paloczi GT, Hansma PK, Levine RP. Journal of Crystal Growth. 2000;211:116. [Google Scholar]
- 97.Thompson JB, Kindt JH, Drake B, Hansma HG, Morse DE, Hansma PK. Nature. 2001;414:773. doi: 10.1038/414773a. [DOI] [PubMed] [Google Scholar]
- 98.Gutsmann T, Fantner GE, Kindt JH, Venturoni M, Danielsen S, Hansma PK. Biophysical Journal. 2004;86:3186. doi: 10.1016/S0006-3495(04)74366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fantner GE, Oroudjev E, Schitter G, Golde LS, Thurner P, Finch MM, Turner P, Gutsmann T, Morse DE, Hansma H, Hansma PK. Biophysical Journal. 2006;90:1411. doi: 10.1529/biophysj.105.069344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fantner GE, Rabinovych O, Schitter G, Thurner P, Kindt JH, Finch MM, Weaver JC, Golde LS, Morse DE, Lipman EA, Rangelow IW, Hansma PK. Composites Science and Technology. 2006;66:1205. [Google Scholar]
- 101.Love JC, Estroff LA, Kriebel JK, Nuzzo RG, Whitesides GM. Chemical Reviews. 2005;105:1103. doi: 10.1021/cr0300789. [DOI] [PubMed] [Google Scholar]
- 102.Aizenberg J, Black AJ, Whitesides GM. Nature. 1999;398:495. [Google Scholar]
- 103.Han YJ, Aizenberg J. Angew Chem-Int Edit. 2003;42:3668. doi: 10.1002/anie.200351655. [DOI] [PubMed] [Google Scholar]
- 104.Muller H, Zentel R, Janshoff A, Janke M. Langmuir. 2006;22:11034. doi: 10.1021/la061637k. [DOI] [PubMed] [Google Scholar]
- 105.Heywood BR. In: Biomimetic Materials Chemistry. Mann S, editor. VCH Publ., Inc; N.Y: 1996. [Google Scholar]
- 106.Mann S, Heywood BR, Rajam S, Walker JBA. J Phys D: Appl Phys. 1991;24:154. [Google Scholar]
- 107.Sato K, Kogure T, Kumagi Y, Tanaka J. J Colloid Interface Sci. 2001;240:133. doi: 10.1006/jcis.2001.7617. [DOI] [PubMed] [Google Scholar]
- 108.Volkmer D, Fricke M, Agena C, Mattay J. Cryst Eng Comm. 2002;4(52):288. [Google Scholar]
- 109.Volkmer D, Fricke M, Vollhardt D, Siegel S. J CHEM SOC DALTON Trans. 2002:4547. ? [Google Scholar]
- 110.Zhang L-J, Liu H-G, Feng X-S, Zhang R-J, Zhang L, Mu Y-D, Hao J-C, Qian D-J, Lou Y-F. Langmuir. 2004;20:2243. doi: 10.1021/la035381j. [DOI] [PubMed] [Google Scholar]
- 111.Buijnsters PJJA, Donners JJJM, Hill SJ, Heywood BR, Nolte RHM, Zwanenburg B, Sommerdijk NAJM. Langmuir. 2001;17:3623. [Google Scholar]
- 112.Fendler JH, Meldrum FC. Advanced Materials. 1995;7:607. [Google Scholar]
- 113.Berman A, Charych D. Journal of Crystal Growth. 1999;198/199:796. [Google Scholar]
- 114.Bekele H, Fendler JH, Kelly JW. Journal of American Chemical Society. 1999;121:7266. [Google Scholar]
- 115.Lahiri J, Xu GF, Dabbs DM, Yao N, Aksay IA, Groves JT. Journal of the American Chemical Society. 1997;119:5449. [Google Scholar]
- 116.Heywood BR, Mann S. Chem Mat. 1994;6:311. [Google Scholar]
- 117.Fricke M, Volkmer Dirk. Topics in Current Chemistry. 2007;270:1. [Google Scholar]
- 118.Litvin AL, Valiyaveettil S, Kaplan D, Mann S. Adv Mater. 1997;9(2):124. [Google Scholar]
- 119.Amos FF, Sharbaugh DM, Talham DR, Gower LB, Fricke M, Volkmer D. Langmuir. 2006;23:1988. doi: 10.1021/la061960n. [DOI] [PubMed] [Google Scholar]
- 120.DiMasi E, Kwak S-Y, Pichon BtP, Sommerdijk NAJM. CrystEngComm. 2007;9(12):1192. [Google Scholar]
- 121.Pokroy B, Aizenberg J. CrystEngComm. 2007;9(12):1219. [Google Scholar]
- 122.Addadi L, Berman A, Moradian-Oldak J, Weiner S. Croatica Chemica Acta. 1990;63(3):539. [Google Scholar]
- 123.Albeck S, Addadi L, Weiner S. Connective Tissue Research. 1996;35:419. doi: 10.3109/03008209609029213. [DOI] [PubMed] [Google Scholar]
- 124.Buckley HE. Crystal Growth. Chapman Hall; London: 1951. [Google Scholar]
- 125.Mann S, Didymus JM, Sanderson NP, Heywood BR, Samper EJA. J Chem Soc Faraday Trans. 1990;86(10):1873. [Google Scholar]
- 126.Feng QL, Pu G, Pei Y, Cui FZ, Li HD, Kim TN. Journal of Crystal Growth. 2000;216:459. [Google Scholar]
- 127.JIMENEZ-LOPEZ C, RODRIGUEZ-NAVARRO A, DOMINGUEZ-VERA JM, GARCIA-RUIZ JM. Geochimica et Cosmochimica Acta. 2003;67(9):1667. [Google Scholar]
- 128.Wierzbicki A, Sikes CS, Madura JD, Drake B. Calcif Tissue Int. 1994;54:133. doi: 10.1007/BF00296064. [DOI] [PubMed] [Google Scholar]
- 129.Orme CA, Noy A, Wierzbicki A, McBride MT, Grantham M, Teng HH, Dove PM, DeYoreo JJ. Nature. 2001;411:775. doi: 10.1038/35081034. [DOI] [PubMed] [Google Scholar]
- 130.Aizenberg J, Hanson J, Ilan M, Leiserowitz L, Koetzle TF, Addadi L, Weiner S. Faseb Journal. 1995;9:262. doi: 10.1096/fasebj.9.2.7781928. [DOI] [PubMed] [Google Scholar]
- 131.Bouropoulos N, Weiner S, Addadi L. Chem Eur J. 2001;7:1881. doi: 10.1002/1521-3765(20010504)7:9<1881::aid-chem1881>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 132.Levy-Lior A, Weiner S, Addadi L. Helvetica Chimica Acta. 2003;86:4007. [Google Scholar]
- 133.Meldrum FC, Hyde ST. Journal of Crystal Growth. 2001;231:544. [Google Scholar]
- 134.Teng HH, Dove PM, DeYoreo JJ. Geochimica et Cosmochimica Acta. 1999;63(17):2507. [Google Scholar]
- 135.DeYoreo JJ, Wierzbickib A, Dove PM. CrystEngComm. 2007;9(12):1144. [Google Scholar]
- 136.Albeck S, Aizenberg J, Addadi L, Weiner S. Journal of the American Chemical Society. 1993;115:11691. [Google Scholar]
- 137.Albeck S, Weiner S, Addadi L. Chemistry: The European Journal. 1996;2(3):278. [Google Scholar]
- 138.Addadi L, Moradian-Oldak J, Weiner S. Surface Reactive Peptides and Polymers- Discovery and Commercialization. In: Sikes CS, Wheeler AP, editors. ACS Symp Ser. Vol. 444. Washington, DC: 1991. [Google Scholar]
- 139.Addadi L, Weiner S. In: Biomineralization - Chemical and Biochemical Perspectives. Mann S, Webb J, Williams RJP, editors. VCH Pub; N. Y: 1989. [Google Scholar]
- 140.Berman A, Hanson J, Leiserowitz L, Koetzle TF, Weiner S, Addadi L. Journal of Physical Chemistry. 1993;97:5162. [Google Scholar]
- 141.Weiner S, Addadi L, Wagner D. Materials Science and Engineering C. 2000;11:1. [Google Scholar]
- 142.Dai L, Cheng X, Gower LB. Chem Mat. 2008 in press. [Google Scholar]
- 143.Glimcher MJ. Rev Mod Phys. 1959;31(2):359. [Google Scholar]
- 144.Murugan R, Ramakrishna S. Composites Science and Technology. 2005;65(15–16):2385. [Google Scholar]
- 145.Fisher LW, Termine JD. Clinical Orthopaedics. 1985;200:362. [PubMed] [Google Scholar]
- 146.Linde A. In: Dentin and Dentinogenesis. Linde A, editor. CRC Press; Boca Raton, FL: 1984. [Google Scholar]
- 147.Bianco P. In: Calcification in Biological Systems. Bonucci E, editor. CRC Press; Boca Raton: 1992. [Google Scholar]
- 148.Gericke A, Qin C, Spevak L, Fujimoto Y, Butler WT, Sorensen ES, Boskey AL. Calcif Tissue Int. 2005;77:45. doi: 10.1007/s00223-004-1288-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hoang QQ, Sicheri F, Howard AJ, Yang DSC. Nature. 2003;425:977. doi: 10.1038/nature02079. [DOI] [PubMed] [Google Scholar]
- 150.Landis WJ, Song MJ, Leith A, McEwen L, McEwen BF. Journal of Structrual Biology. 1993;110:39. doi: 10.1006/jsbi.1993.1003. [DOI] [PubMed] [Google Scholar]
- 151.Glimcher MJ. Philos Trans R Soc London Ser B. 1984;304:479. doi: 10.1098/rstb.1984.0041. [DOI] [PubMed] [Google Scholar]
- 152.Traub W, Jodaikin A, Arad T, Veis A, Sabsay B. Matrix. 1992;12:197. doi: 10.1016/s0934-8832(11)80062-4. [DOI] [PubMed] [Google Scholar]
- 153.Saito T, Arsenault AL, Yamauchi M, Kuboki Y, Crenshaw MA. Bone. 1997;21:305. doi: 10.1016/s8756-3282(97)00149-x. [DOI] [PubMed] [Google Scholar]
- 154.Deshpande AS, Beniash E. Cryst Growth Des. 2008;8:3084. doi: 10.1021/cg800252f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Harper RA, Posner AS. Proceedings of the Society for Experimental Biology and Medicine. 1966;122:137. [Google Scholar]
- 156.Termine JD, Posner AS. Nature. 1966;211:268. doi: 10.1038/211268a0. [DOI] [PubMed] [Google Scholar]
- 157.Termine JD, Wuthier RE, Posner AS. Proceedings of the Society for Experimental Biology and Medicine. 1967;125:4. doi: 10.3181/00379727-125-31999. [DOI] [PubMed] [Google Scholar]
- 158.Brown WE, Chow LC. Annu Rev Mater Sci. 1976;6:213. [Google Scholar]
- 159.Glimcher MJ, Bonar LC, Grynpas MD, Landis WJ, Roufosse AH. Journal of Crystal Growth. 1981;53:100. [Google Scholar]
- 160.Olszta MJ, Cheng X, Jee SS, Kumar R, Kim Y-Y, Kaufman MJ, Douglas EP, Gower LB. Materials Science & Engineering R-Reports. 2007;58:77. [Google Scholar]
- 161.Crane NJ, Popescu V, Morris MD, Steenhuis P, Ignelzi JMA. Bone. 2006;39:434. doi: 10.1016/j.bone.2006.02.059. [DOI] [PubMed] [Google Scholar]
- 162.Weiner S. Bone. 2006;39:431. doi: 10.1016/j.bone.2006.02.058. [DOI] [PubMed] [Google Scholar]
- 163.Mahamid J, Sharir A, Addadi L, Weiner S. Proceedings of the National Academy of Sciences USA. 2008;105:12748. doi: 10.1073/pnas.0803354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Fincham AG, Moradian-Oldak J, Slavkin HC. J Struct Biol. 1995;115:50. doi: 10.1006/jsbi.1995.1029. [DOI] [PubMed] [Google Scholar]
- 165.Fincham AG, Moradian-Oldak J, Simpson JP. J Structural Biology. 1999;126:270. doi: 10.1006/jsbi.1999.4130. [DOI] [PubMed] [Google Scholar]
- 166.Cui F-Z, Ge J. Journal of Tissue Engineering and Regenerative Medicine. 2007;1:185. doi: 10.1002/term.21. [DOI] [PubMed] [Google Scholar]
- 167.Wang RZ, Addadi L, Weiner S. Phil Trans R Soc Lond B. 1997;352:469. doi: 10.1098/rstb.1997.0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ma Y, Weiner S, Addadi L. Advanced Functional Materials. 2007;17:2693. [Google Scholar]
- 169.van der Wal P, Videler JJ, Havinga P, Pel R. Architecture and chemical composition of the magnetite-bearing layer in the radula teeth of Chiton olivaceus (Polyplacophora) Plenum Press; New York: 1990. [Google Scholar]
- 170.Webb J, Macey DJ, Mann S. In: Biomineralization- Chemical and Biochemical Perspectives. Mann S, Webb J, WIlliams RJP, editors. VCH Verlagsgesellschaft mbH; Weinheim: 1989. [Google Scholar]
- 171.Sone ED, Weiner S, Addadi L. J Struct Biol. 2007;158:428. doi: 10.1016/j.jsb.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 172.Leadbeater B, Riding R, editors. Biomineralization in Lower Plants and Animals. Clarendon Press; New York: 1986. [Google Scholar]
- 173.Ogino T, Suzuki T, Sawada K. Geochim Cosmochim Acta. 1987;51:2757. [Google Scholar]
- 174.Clarkson JR, Price TJ, Adams CJ. Journal of the Chemical Society, Faraday Transactions. 1992;88:243. [Google Scholar]
- 175.Sondi I, Matijevic E. J Colloid Interface Sci. 2001;238:208. doi: 10.1006/jcis.2001.7516. [DOI] [PubMed] [Google Scholar]
- 176.Wolf G, Günther C. Journal of Thermal Analysis. 2001;65:687. [Google Scholar]
- 177.Bolze J, Pontoni D, Ballauff M, Narayanan T, Cölfen H. J Colloid Interface Sci. 2004;277:84. doi: 10.1016/j.jcis.2004.04.029. [DOI] [PubMed] [Google Scholar]
- 178.Han YS, Hadiko G, Fuji M, Takahashi M. Journal of the European Ceramic Society. 2006;26:843. [Google Scholar]
- 179.Glover ED, Sippel RF. Geochim Cosmochim Acta. 1967;31:603. [Google Scholar]
- 180.Cheng X, Varona PL, Olszta MJ, Robbins LL, Gower LB. Journal of Crystal Growth. 2007;307:395. [Google Scholar]
- 181.Wan P, Tong H, Zhu ZH, Shen XY, Yan J, Hu JM. Materials Science and Engineering a-Structural Materials Properties Microstructure and Processing. 2007;458:244. doi: 10.1016/j.msec.2006.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhang ZP, Gao DM, Zhao H, Xie CG, Guan GJ, Wang DP, Yu SH. Journal of Physical Chemistry B. 2006;110:8613. doi: 10.1021/jp060838u. [DOI] [PubMed] [Google Scholar]
- 183.Naka K, Huang SC, Chujo Y. Langmuir. 2006;22:7760. doi: 10.1021/la060874k. [DOI] [PubMed] [Google Scholar]
- 184.Huang SC, Naka K, Chujo Y. Polymer Journal. 2008;40:154. [Google Scholar]
- 185.Cölfen H, Antonietti M. Lanqmuir. 1998;14:582. [Google Scholar]
- 186.Huang SC, Naka K, Chujo Y. Langmuir. 2007;23:12086. doi: 10.1021/la701972n. [DOI] [PubMed] [Google Scholar]
- 187.Gorna K, Hund M, Vucak M, Grohn F, Wegner G. Materials Science and Engineering: A. 2008;477:217. [Google Scholar]
- 188.Donners JJM, Heywood BR, Meijer EW, Nolte RJM, Roman C, Schenning APHJ, Sommerdijk NAJM. Chem Commun. 2000:1937. [Google Scholar]
- 189.Xu AW, Yu Q, Dong WF, Antonietti M, Cölfen H. Advanced Materials. 2005;17:2217. [Google Scholar]
- 190.Navrotsky A. PNAS. 2004;101:12096. doi: 10.1073/pnas.0404778101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Cölfen H, Antonietti M. Angew Chem-Int Edit. 2005;44:5576. doi: 10.1002/anie.200500496. [DOI] [PubMed] [Google Scholar]
- 192.Cölfen H, Mann S. Anew Chem Int Ed. 2003;42:2350. doi: 10.1002/anie.200200562. [DOI] [PubMed] [Google Scholar]
- 193.Cölfen H, Antonietti M. Mesocrystals and Nonclassical Crystallization. John Wiley & Sons, Ltd; The Atrium, Southern gate, Chichester, West Sussex, England: 2008. [Google Scholar]
- 194.Termine JD, Posner AS. Science. 1966;153:1523. doi: 10.1126/science.153.3743.1523. [DOI] [PubMed] [Google Scholar]
- 195.Posner AS. Physiological Reviews. 1969;49:760. doi: 10.1152/physrev.1969.49.4.760. [DOI] [PubMed] [Google Scholar]
- 196.Lowenstam HA, Weiner S. Science. 1985;227:51. doi: 10.1126/science.227.4682.51. [DOI] [PubMed] [Google Scholar]
- 197.Lowenstam HA. Geological Society of America Bulletin. 1962;73:435. [Google Scholar]
- 198.Simkiss K. In: Mechanisms and Phylogeny of Mineralization in Biological Systems. Suga S, Nakahara H, editors. Springer-Verlag; N.Y: 1991. Vol. Biomineralization ‘90. [Google Scholar]
- 199.Okazaki M, Setoguchi H, Aoki H, Suga S. Botanical Magazine- Tokyo. 1986;99(1055):281. [Google Scholar]
- 200.Aizenberg J, Lambert G, Addadi L, Weiner S. Adv Mater. 1996;8(3):222. [Google Scholar]
- 201.Gower LA. PhD Dissertation. University of Massachusetts; Amherst: 1997. [Google Scholar]
- 202.Gower LA, Tirrell DA. J Crystal Growth. 1998;191(1–2):153. [Google Scholar]
- 203.Gower LB, Odom DJ. J Crystal Growth. 2000;210(4):719. [Google Scholar]
- 204.Cölfen H, Qi LM. Chem-Eur J. 2001;7:106. doi: 10.1002/1521-3765(20010105)7:1<106::aid-chem106>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 205.Qi LM, Cölfen H, Antonietti M, Li M, Hopwood JD, Ashley AJ, Mann S. Chem-Eur J. 2001;7:3526. doi: 10.1002/1521-3765(20010817)7:16<3526::aid-chem3526>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 206.Politi Y, Levi-Kalisman Y, Raz S, Wilt F, Addadi L, Weiner S, Sagi I. Advanced Functional Materials. 2006;16:1289. [Google Scholar]
- 207.Weiss IM, Tuross N, Addadi L, Weiner S. Journal of Experimental Zoology. 2002;293:478. doi: 10.1002/jez.90004. [DOI] [PubMed] [Google Scholar]
- 208.Beniash E, Aizenberg J, Addadi L, Weiner S. Proc R Soc Lond B. 1997;264:461. [Google Scholar]
- 209.Politi Y, Arad T, Klein E, Weiner S, Addadi L. Science. 2004;306:1161. doi: 10.1126/science.1102289. [DOI] [PubMed] [Google Scholar]
- 210.Raz S, Hamilton PC, Wilt FH, Weiner S, Addadi L. Advanced Functional Materials. 2003;13:480. [Google Scholar]
- 211.Gayathri S, Lakshminarayanan R, Weaver JC, Morse DE, Kini RM, Valiyaveettil S. Chem-Eur J. 2007;13:3262. doi: 10.1002/chem.200600825. [DOI] [PubMed] [Google Scholar]
- 212.Marxen JC, Becker W, Finke D, Hasse B, Eppie M. J Moll Stud. 2003;69:113. [Google Scholar]
- 213.Nassif N, Pinna N, Gehrke N, Antonietti M, Jager C, Cölfen H. Proc Natl Acad Sci U S A. 2005;102:12653. doi: 10.1073/pnas.0502577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Politi Y, Mahamid J, Goldberg H, Weiner S, Addadi L. CrystEngComm. 2007;9(12):1171. [Google Scholar]
- 215.Hasse B, Ehrenberg H, Marxen JC, Becker W, Epple M. Chem-Eur J. 2000;6:3679. doi: 10.1002/1521-3765(20001016)6:20<3679::aid-chem3679>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 216.Lakshminarayanan R, Loh XJ, Gayathri S, Sindhu S, Banerjee Y, Kini RM, Valiyaveettil S. Biomacromolecules. 2006;7:3202. doi: 10.1021/bm0605412. [DOI] [PubMed] [Google Scholar]
- 217.Lussi A, Crenshaw MA, Linde A. Arch Oral Biol. 1988;33:685. doi: 10.1016/0003-9969(88)90124-0. [DOI] [PubMed] [Google Scholar]
- 218.Addadi L, Moradiam J, Shay E, Maroudas NG, Weiner S. Proc Natl Acad Sci USA. 1987;84:2732. doi: 10.1073/pnas.84.9.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Furedi-Milhofer H, Sarig S. Progress in Crystal Growth and Characterization. 1996;32:45. [Google Scholar]
- 220.DiMasi E, Liu T, Olszta MJ, Gower LB. Biological and Bio-Inspired Materials and Devices. In: Sandhage KH, Yang S, Douglas T, Parker AR, DiMasi E, editors. Mat Res. 873E Society Proceedings; Warrendale, PA: 2005. [Google Scholar]
- 221.Kim Y-Y, Gower LB. Langmuir. 2007;23(9):4862. doi: 10.1021/la061975l. [DOI] [PubMed] [Google Scholar]
- 222.Cheng X, Gower LB. Biotechnology Progress. 2006;22(1):141. doi: 10.1021/bp050166+. [DOI] [PubMed] [Google Scholar]
- 223.Olszta M, Gajjeraman S, Kaufman M, Gower L. Chem Mat. 2004;16(12):2355. [Google Scholar]
- 224.Olszta MJ, Odom DJ, Douglas EP, Gower LB. Connective Tissue Research. 2003;44:326. [PubMed] [Google Scholar]
- 225.Dai L, Douglas EP, Gower LB. Journal of Non-Crystalline Solids. 2008;354:1845. [Google Scholar]
- 226.Wang TX, Xu AW, Cölfen H. Angew Chem-Int Edit. 2006;45:4451. doi: 10.1002/anie.200601038. [DOI] [PubMed] [Google Scholar]
- 227.Rieger J, Frechen T, Cox G, Heckmann W, Schmidt C, Thieme J. Faraday Discussions. 2007;136:265. doi: 10.1039/b701450c. [DOI] [PubMed] [Google Scholar]
- 228.Rieger J, Thieme J, Schmidt C. Langmuir. 2000;16:8300. [Google Scholar]
- 229.Wolf SE, Leiterer J, Kappl M, Emmerling F, Tremel W. Journal of American Chemical Society. 2008;130:12342. doi: 10.1021/ja800984y. [DOI] [PubMed] [Google Scholar]
- 230.Xu XR, Han JT, Cho KL. Langmuir. 2005;21:4801. doi: 10.1021/la047069v. [DOI] [PubMed] [Google Scholar]
- 231.Faatz M, Grohn F, Wegner G. Advanced Materials. 2004;16:996. [Google Scholar]
- 232.Faatz M, Grohn F, Wegner G. Mater Sci Eng C-Biomimetic Supramol Syst. 2005;25:153. [Google Scholar]
- 233.Hardikar VV, Matijevic E. Colloids and Surfaces A: Physichemical and Engineering Aspects. 2001;186:23. [Google Scholar]
- 234.Wang T, Colfen H. Langmuir. 2006;22:8975. doi: 10.1021/la0609827. [DOI] [PubMed] [Google Scholar]
- 235.Ostwald W. Z Phys Chem. 1897;22:289. [Google Scholar]
- 236.Thompson DAW. On Growth and Form. Dover Publ; N.Y: 1942. [Google Scholar]
- 237.Wohlrab S, Cölfen H, Antonietti M. Angew Chem-Int Edit. 2005;44:4087. doi: 10.1002/anie.200462467. [DOI] [PubMed] [Google Scholar]
- 238.Vekilov PG. Crystal Growth & Design. 2004;4:671. doi: 10.1021/cg1011633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Vekilov PG. Journal of Crystal Growth. 2005;275:65. [Google Scholar]
- 240.Vekilov PG, Galkin O, Filobelo LF. Abstracts of Papers of the American Chemical Society. 2004;227:U302. [Google Scholar]
- 241.Kashchiev D, Vekilov PG, Kolomeisky AB. Journal of Chemical Physics. 2005:122. doi: 10.1063/1.1943389. [DOI] [PubMed] [Google Scholar]
- 242.Filobelo LF, Galkin O, Vekilov PG. Journal of Chemical Physics. 2005:123. doi: 10.1063/1.1943413. [DOI] [PubMed] [Google Scholar]
- 243.Pan WC, Kolomeisky AB, Vekilov PG. Journal of Chemical Physics. 2005:122. doi: 10.1063/1.1887168. [DOI] [PubMed] [Google Scholar]
- 244.Xu G, Yao N, Aksay I, Groves JT. J Am Chem Soc. 1998;120:11977. [Google Scholar]
- 245.Kato T, Suzuki T, Amamiya T, Irie T, Komiyama M, Yui H. Supramolecular Science. 1998;5(3–4):411. [Google Scholar]
- 246.DiMasi E, Gower LB. Mat Res Soc Symp Proc- Advanced Biomaterials -- Characterization, Tissue Engineering. 2001. p. HH3.38.1. [Google Scholar]
- 247.DiMasi E, Olszta MJ, Patel VM, Gower LB. Cryst Eng Comm. 2003;5(61):346. [Google Scholar]
- 248.DiMasi E, Patel VM, Sivakumar M, Olszta MJ, Yang YP, Gower LB. Langmuir. 2002;18:8902. [Google Scholar]
- 249.Amos F. Dissertation. University of Florida; 2005. [Google Scholar]
- 250.Xu G, Aksay L, Groves JT. Journal of American Chemical Society. 2001;123(10):2196. doi: 10.1021/ja002537i. [DOI] [PubMed] [Google Scholar]
- 251.Kato T. Advanced Materials. 2000;12:1543. [Google Scholar]
- 252.Hosoda N, Sugawara A, Kato T. Macromolecules. 2003;36(17):6449. [Google Scholar]
- 253.Sommerdijk NAJM, Leeuwen ENMv, Vos MRJ, Jansen JA. CrystEngComm. 2007;9(12):1209. [Google Scholar]
- 254.Sugawara A, Ishii T, Kato T. Angew Chem-Int Edit. 2003:5299. doi: 10.1002/anie.200351541. [DOI] [PubMed] [Google Scholar]
- 255.Sugawara A, Nishimura T, Yamamoto Y, Inoue H, Nagasawa H, Kato T. Angew Chem-Int Edit. 2006;45:2876. doi: 10.1002/anie.200503800. [DOI] [PubMed] [Google Scholar]
- 256.Sugawara A, Kato T. Chem Commun. 2000:487. [Google Scholar]
- 257.Kato T, Sugawara A, Hosoda N. Advanced Materials. 2002;14(12):869. [Google Scholar]
- 258.Ulcinas A, Butler MF, Heppenstall-Butler M, Singleton S, Miles MJ. Journal of Crystal Growth. 2007;307:378. [Google Scholar]
- 259.Xu X, Han JT, Cho K. Chem Mat. 2004 doi: 10.1021/cm035183d. WEB. [DOI] [Google Scholar]
- 260.Xu X, Han JT, Kim DH, Cho K. J Phys Chem B. 2006;110:2764. doi: 10.1021/jp055712w. [DOI] [PubMed] [Google Scholar]
- 261.Xu XR, Cai AH, Liu R, Pan HH, Tang RK. Progress in Chemistry. 2008;20:54. [Google Scholar]
- 262.Ajikumar PK, Lakshminarayanan R, Valiyaveettil S. Crystal Growth & Design. 2004;4(2):331. [Google Scholar]
- 263.Aizenberg J, Muller DA, Grazul JL, Hamann DR. Science. 2003;299:1205. doi: 10.1126/science.1079204. [DOI] [PubMed] [Google Scholar]
- 264.Belcher AM, Wu XH, Christensen RJ, Hansma PK, Stucky GD, Morse DE. Nature. 1996;381:56. [Google Scholar]
- 265.Samata T, Hayashi N, Kono M, Hasegawa K, Horita C, Akera S. Federation of European Biochemical Societies Letters. 1999;462:225. doi: 10.1016/s0014-5793(99)01387-3. [DOI] [PubMed] [Google Scholar]
- 266.Voinescu AE, Touraud D, Lecker A, Pfitzner A, Kunz W, Ninham BW. Langmuir. 2007;23:12269. doi: 10.1021/la701892v. [DOI] [PubMed] [Google Scholar]
- 267.Addadi L, Raz S, Weiner S. Advanced Materials. 2003;15:959. [Google Scholar]
- 268.Aizenberg J, Lambert G, Weiner S, Addadi L. Journal of the American Chemical Society. 2002;124:32. doi: 10.1021/ja016990l. [DOI] [PubMed] [Google Scholar]
- 269.Weiner S, Levi-Kalisman Y, Raz S, Addadi L. Connective Tissue Research. 2003;44:214. [PubMed] [Google Scholar]
- 270.Faatz M, Grohn F, Wegner G. Advanced Materials. 2004;16:996. [Google Scholar]
- 271.Weiner S, Sagi I, Addadi L. Science. 2005;309:1027. doi: 10.1126/science.1114920. [DOI] [PubMed] [Google Scholar]
- 272.Koga N, Nakagoe Y, Tanaka H. Thermochimica Acta. 1998;318:239. [Google Scholar]
- 273.Becker A, Bismayer U, Epple M, Fabritius H, Hasse B, Shi JM, Ziegler A. Dalton Transactions. 2003:551. [Google Scholar]
- 274.Levi-Kalisman Y, Raz S, Weiner S, Addadi L, Sagi I. J Chem Soc-Dalton Trans. 2000:3977. [Google Scholar]
- 275.Levi-Kalisman Y, Raz S, Weiner S, Addadi L, Sagi I. Advanced Functional Materials. 2002;12:43. [Google Scholar]
- 276.Neumann M, Epple M. European Journal of Inorganic Chemistry. 2007:1953. [Google Scholar]
- 277.Falini G, Albeck S, Weiner S, Addadi L. Science. 1996;271:67. [Google Scholar]
- 278.Greenfield EM, Wilson DC, Crenshaw MA. Amer Zool. 1984;24:925. [Google Scholar]
- 279.Archibald DD, Qadri SB, Gaber BP. Langmuir. 1996;12:538. [Google Scholar]
- 280.Thiele H, Awad A. J Biomed Mater Res. 1969;3:431. doi: 10.1002/jbm.820030305. [DOI] [PubMed] [Google Scholar]
- 281.Han YJ, Aizenberg J. Journal of the American Chemical Society. 2003;125:4032. doi: 10.1021/ja034094z. [DOI] [PubMed] [Google Scholar]
- 282.Brecevic L, Nielson AE. Journal of Crystal Growth. 1989;98:504. [Google Scholar]
- 283.Schweins R, Huber K. European Physical Journal E. 2001;5:117. doi: 10.1140/epje/i2006-10047-7. [DOI] [PubMed] [Google Scholar]
- 284.Schweins R, Lindner P, Huber K. Macromolecules. 2003;36:9564. [Google Scholar]
- 285.Fisher LW, Torchia DA, Fohr B, Young MF, Fedarko NS. Biochem Biophys Res Commun. 2001;280:460. doi: 10.1006/bbrc.2000.4146. [DOI] [PubMed] [Google Scholar]
- 286.Urry LA, Hamilton PC, Killian CE, Wilt FH. Developmental Biology. 2000;225:201. doi: 10.1006/dbio.2000.9828. [DOI] [PubMed] [Google Scholar]
- 287.Dai L. Dissertation. University of Florida; 2005. [Google Scholar]
- 288.Marsh ME. Journal of Experimental Biology. 1986;239:207. doi: 10.1002/jez.1402390208. [DOI] [PubMed] [Google Scholar]
- 289.Marsh ME. Biochemistry. 1989;28:346. doi: 10.1021/bi00427a047. [DOI] [PubMed] [Google Scholar]
- 290.Marsh ME, Sass RL. Biochemistry. 1984;23:1448. doi: 10.1021/bi00302a017. [DOI] [PubMed] [Google Scholar]
- 291.Marsh ME. Biochemistry. 1989;28:339. doi: 10.1021/bi00427a046. [DOI] [PubMed] [Google Scholar]
- 292.Han TYJ, Aizenberg J. Chem Mat. 2008;20:1064. [Google Scholar]
- 293.Lee JRI, Han TYJ, Willey TM, Wang D, Meulenberg RW, Nilsson J, Dove PM, Terminello LJ, van Buuren T, DeYoreo JJ. J Am Chem Soc. 2007;129:10370. doi: 10.1021/ja071535w. [DOI] [PubMed] [Google Scholar]
- 294.Sethmann I, Putnis A, Grassmann O, Lobmann P. American Mineralogist. 2005;90:1213. [Google Scholar]
- 295.Niederberger M, Cölfen H. Physical Chemistry Chemical Physics. 2006;8:3271. doi: 10.1039/b604589h. [DOI] [PubMed] [Google Scholar]
- 296.Tao JH, Pan HH, Zeng YW, Xu XR, Tang RK. Journal of Physical Chemistry B. 2007;111:13410. doi: 10.1021/jp0732918. [DOI] [PubMed] [Google Scholar]
- 297.DiMasi E, Kwak SY, Amos FF, Olszta MJ, Lush D, Gower LB. Physical Review Letters. 2006:97. doi: 10.1103/PhysRevLett.97.045503. [DOI] [PubMed] [Google Scholar]
- 298.Hosoda N, Kato T. Chem Mater. 2001;13(2):688. [Google Scholar]
- 299.Levi Y, Albeck S, Black A, Weiner S, Addadi L. Chem Eur J. 1998;4(3):389. [Google Scholar]
- 300.Lam RSK, Charnock JM, Lennie A, Meldrum FC. Crystengcomm. 2007;9:1226. [Google Scholar]
- 301.Beniash E, Addadi L, Weiner S. J Struct Biol. 1999;125:50. doi: 10.1006/jsbi.1998.4081. [DOI] [PubMed] [Google Scholar]
- 302.Kitano Y. Bulletin of the Chemical Society of Japan. 1962;35(12):1973. [Google Scholar]
- 303.Sondi I, Skapin SD, Salopek-Sondi B. Crystal Growth & Design. 2008;8:435. [Google Scholar]
- 304.Loste E, Meldrum FC. Chemical Communications. 2001:901. [Google Scholar]
- 305.Nudelman F, Shimoni E, Klein E, Rousseau M, Bourrat X, Lopez E, Addadi L, Weiner S. J Struct Biol. 2008;162:290. doi: 10.1016/j.jsb.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 306.Li H, Estroff LA. CrystEngComm. 2007;9(12):1153. [Google Scholar]
- 307.Li HY, Estroff LA. Journal of the American Chemical Society. 2007;129:5480. doi: 10.1021/ja067901d. [DOI] [PubMed] [Google Scholar]
- 308.Rousseau M, Lopez E, Couté A, Mascarel G, Smith DC, Naslain R, Bourrat X. J Struct Biol. 2005;149:149. doi: 10.1016/j.jsb.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 309.Kotachi A, Miura T, Imai H. Crystal Growth & Design. 2006;6(7):1636. [Google Scholar]
- 310.Tanaka Y, Nemoto T, Naka K, Chujo Y. Polymer Bulletin. 2000;45:447. [Google Scholar]
- 311.Zhang S, Gonsalves KE. Materials Science & Engineering: C. 1995;3:117. [Google Scholar]
- 312.Kato T, Amamiya T. Chemistry Letters. 1999:199–200. [Google Scholar]
- 313.Mutvei H, editor. Using plasma-etching and proteolytic enzymes in studies of molluscan shell ultrastructure. Springer-Verlag; New York: 1991. Vol. Biomineralization ‘90. [Google Scholar]
- 314.Weedon MJ, Taylor PD. Biol Bull. 1995;188:281. doi: 10.2307/1542305. [DOI] [PubMed] [Google Scholar]
- 315.Jäger C, Cölfen H. CrystEngComm. 2007;9(12):1237. [Google Scholar]
- 316.Mutvei H. Zoologica Scripta. 1978;7(4):287. [Google Scholar]
- 317.Oaki Y, Kotachi A, Miura T, Imai H. Advanced Functional Materials. 2006;16:1633. [Google Scholar]
- 318.Dauphin Y. Palaeontologische Zeitschrift. 2001;75:113. [Google Scholar]
- 319.Barthelat F, Li C-M, Comi C, Espinosa HD. Journal of Materials Research. 2006;21(8):1977. [Google Scholar]
- 320.Gehrke N, Nassif N, Pinna N, Antonietti M, Gupta HS, Cölfen H. Chem Mat. 2005;17:6514. [Google Scholar]
- 321.Metzler RA, Abrecht M, Olabisi RM, Ariosa D, Johnson CJ, Frazer BH, Coppersmith SN, Gilbert P. Physical Review Letters. 2007:98. doi: 10.1103/PhysRevLett.98.268102. [DOI] [PubMed] [Google Scholar]
- 322.Gilbert PUPA, Metzler RA, Zhou D, Scholl A, Doran A, Young A, Kunz M, Tamura N, Coppersmith SN. Journal of the American Chemical Society. 2008 doi: 10.1021/ja8065495. submitted. [DOI] [PubMed] [Google Scholar]
- 323.Erben HK. Biomineralisation - Research Reports. 1972;4:15. [Google Scholar]
- 324.Wada K. Biomineralisation - Research Reports. 1972;4:141. [Google Scholar]
- 325.Nakahara H. Venus. 1979;38:205. [Google Scholar]
- 326.Blank S, Arnoldi M, Khoshnavaz S, Treccani L, Kuntz M, Mann K, Grathwohl G, Fritz M. Journal of Microscopy. 2003;212(Pt3):280. doi: 10.1111/j.1365-2818.2003.01263.x. [DOI] [PubMed] [Google Scholar]
- 327.Volkmer D, Fricke M, Huber T, Sewald N. Chemical Communications. 2004;(16):1872. doi: 10.1039/b405613b. [DOI] [PubMed] [Google Scholar]
- 328.Aizenberg J. Journal of Crystal Growth. 2000;211:143. [Google Scholar]
- 329.Kwak SY, DiMasi E, Han YJ, Aizenberg J, Kuzmenko I. Crystal Growth & Design. 2005;5:2139. [Google Scholar]
- 330.Aizenberg TY-JaJ. Mater Res Soc Symp Proc. 2005;873E:K4.10.1. [Google Scholar]
- 331.Potapenko SY. Journal of Crystal Growth. 1995;147:223. [Google Scholar]
- 332.Wilkes DA, Crenshaw MA. Scanning Electron Microscopy. 1979:469. [Google Scholar]
- 333.Young JR, Davis SA, Bown PR, Mann S. J Struct Biol. 1999;126:195. doi: 10.1006/jsbi.1999.4132. [DOI] [PubMed] [Google Scholar]
- 334.Wal Pvd, Jong EWd, Westbroek P, Bruijn WCd, Mulder-Stapel AA. Journal of Ultrastructure Research. 1983;85:139. doi: 10.1016/s0022-5320(83)90103-x. [DOI] [PubMed] [Google Scholar]
- 335.Marsh ME. Protoplasma. 1994;177:108. [Google Scholar]
- 336.Mann S, Hannington JP. J Colloid & Interface Science. 1988;122(2):326. [Google Scholar]
- 337.Bazylinski DA, Frankel RB. Nature Reviews- Microbiology. 2004;2:217. doi: 10.1038/nrmicro842. [DOI] [PubMed] [Google Scholar]
- 338.Loste E, Park RJ, Warren J, Meldrum FC. Advanced Functional Materials. 2004;14(12):1211. [Google Scholar]
- 339.Park RJ, Meldrum FC. J Mater Chem. 2004;14:2291. [Google Scholar]
- 340.Yue WB, Kulak AN, Meldrum FC. J Mater Chem. 2006;16:408. [Google Scholar]
- 341.Mann S. In: Biomineralization- Chemical and Biochemical Perspectives. Mann S, Webb J, Williams RJP, editors. VCH Publishers; N. Y: 1989. [Google Scholar]
- 342.Li C, Qi LM. Angew Chem-Int Edit. 2008;47:2388. doi: 10.1002/anie.200705403. [DOI] [PubMed] [Google Scholar]
- 343.Nesson MH, Lowenstam HA. In: Magnetite Biomineralization and Magnetoreception in Organisms. Kirschvink JL, Jones DS, MacFadden BJ, editors. Vol. 5 Plenum Press; New York: 1985. [Google Scholar]
- 344.Rabinovich A. Discover. 2005;26(05) [Google Scholar]
- 345.Sethmann I, Wörheide G. Micron. 2008;39:209. doi: 10.1016/j.micron.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 346.Weaver JC, Pietrasanta LI, Hedin N, Chmelka BF, Hansma PK, Morse DE. J Struct Biol. 2003;144:271. doi: 10.1016/j.jsb.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 347.Weaver JC, Aizenberg J, Fantner GE, Kisailus D, Woesz A, Allen P, Fields K, Porter MJ, Zok FW, Hansma PK, Fratzl P, Morse DE. J Struct Biol. 2007;158:93. doi: 10.1016/j.jsb.2006.10.027. [DOI] [PubMed] [Google Scholar]
- 348.Aizenberg J, Weaver JC, Thanawala MS, Sundar VC, Morse DE, Fratzl P. Science. 2005;309:275. doi: 10.1126/science.1112255. [DOI] [PubMed] [Google Scholar]
- 349.Muller WEG, Eckert C, Kropf K, Wang XH, Schlossmacher U, Seckert C, Wolf SE, Tremel W, Schroder HC. Cell and Tissue Research. 2007;329:363. doi: 10.1007/s00441-007-0402-x. [DOI] [PubMed] [Google Scholar]
- 350.Ryall RL. Urol Res. 2008 doi: 10.1007/s00240-007-0131-3. [DOI] [PubMed] [Google Scholar]
- 351.Chasteen N, Harrison P. Journal of Structual Biology. 1999;126:182. doi: 10.1006/jsbi.1999.4118. [DOI] [PubMed] [Google Scholar]
- 352.Kim KS, Macey DJ, Webb J, Mann S. Proceedings of the Royal Society of London Series B, Biological Sciences. 1989;237:335. [Google Scholar]
- 353.Arnott HJ. In: The Mechanism of Mineralization in the Invertebrates and Plants. Watabe N, Wilbur KM, editors. Univ. South Caroline Press; Columbia, SC: 1976. [Google Scholar]
- 354.Setoguchi H, Okazaki M, Suga S. In: Origin, Evolution, and Modern Aspects of Biomineralization in Plants and Animals. Crick RE, editor. Plenum Press; New York: 1989. [Google Scholar]
- 355.Becker A, Ziegler A, Epple M. Dalton Trans. 2005:1814. doi: 10.1039/b412062k. [DOI] [PubMed] [Google Scholar]
- 356.Raz S, Testeniere O, Hecker A, Weiner S, Luquet G. Biological Bulletin. 2002;203:269. doi: 10.2307/1543569. [DOI] [PubMed] [Google Scholar]
- 357.Shechter A, Glazer L, Cheled S, Mor E, Weil S, Berman A, Bentov S, Aflalo ED, Khalaila I, Sagi A. Proc Natl Acad Sci U S A. 2008;105:7129. doi: 10.1073/pnas.0800193105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Currey JD. In: Skeletal Biomineralization: Patterns, Processes and Evolutionary Trends. Carter JG, editor. I Van Nostrand Reinhold; New York: 1990. [Google Scholar]
- 359.Robach JS, Stock SR, Veis A. J Struct Biol. 2006;155:87. doi: 10.1016/j.jsb.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 360.Kim Y-y, Gower L. 10th International Ceramics Congress- Part B- Non Conventional Routes to Ceramics; Florence, Italy. 2002. [Google Scholar]
- 361.Coatman RD, Thomas NL, Double DD. Journal of Materials Science. 1980;15:2017. [Google Scholar]
- 362.Jones DEH, Walter U. J Colloid Interface Sci. 1998;203:286. doi: 10.1006/jcis.1998.5447. [DOI] [PubMed] [Google Scholar]
- 363.Wilt F. Journal of Structual Biology. 1999;126:216. doi: 10.1006/jsbi.1999.4105. [DOI] [PubMed] [Google Scholar]
- 364.Wagner RS, Ellis WC. Jom-Journal of Metals. 1964;16:761. [Google Scholar]
- 365.Trentler TJ, Hickman KM, Goel SC, Viano AM, Gibbons PC, Buhro WE. Science. 1995;270:1791. [Google Scholar]
- 366.Kim Y-Y, Gower LB. Materials Research Society; San Francisco: 2003. [Google Scholar]
- 367.Kniprath E. Calcified Tissue Research. 1974;14:211. doi: 10.1007/BF02060296. [DOI] [PubMed] [Google Scholar]
- 368.Homeijer SJ, Olszta MJ, Barrett RA, Gower LB. Journal of Crystal Growth. 2008;310:2938. [Google Scholar]
- 369.Homeijer SJ, Barrett RA, Gower LB. 2008 in preparation. [Google Scholar]
- 370.Wang TX, Reinecke A, Cölfen H. Langmuir. 2006;22:8986. doi: 10.1021/la060985j. [DOI] [PubMed] [Google Scholar]
- 371.Yu SH, Antonietti M, Cölfen H, Hartmann J. Nano Letters. 2003;3:379. [Google Scholar]
- 372.Kuther J, Bartz M, Seshadri R, Vaughan GBM, Tremel W. J Mater Chem. 2001;11:503. [Google Scholar]
- 373.Nanci A, Smith CE. In: Calcification in Biological Systems. Bonucci E, editor. Chapter 13 CRS press; Boca Raton: 1992. [Google Scholar]
- 374.Nakahara H, Kakei M. In: Origin, Evolution, and Modern Aspects of Biomineralization in Plants and Animals. Crick RE, editor. Plenum Press; N.Y: 1989. [Google Scholar]
- 375.Iijima M, Moriwaki Y, Takagi T, Moradian-Oldak J. Journal of Crystal Growth. 2001;222:615. [Google Scholar]
- 376.Moreno EC, Aoba T. Adv Dent Res. 1987;1:245. doi: 10.1177/08959374870010021301. [DOI] [PubMed] [Google Scholar]
- 377.Iijima M, Moradian-Oldak J. J Mater Chem. 2004;14:2189. [Google Scholar]
- 378.Demus D, Richter L. Textures of Liquid Crystals. Verlag Chemie; N. Y: 1978. [Google Scholar]
- 379.Kirkham J, Brookes SJ, Shore RC, Bonass WA, Smith DA, Wallwork ML, Robinson C. Connective Tissue Research. 1998;38:91. doi: 10.3109/03008209809017025. [DOI] [PubMed] [Google Scholar]
- 380.Robinson C, Connell S, Kirkham J, Shore R, Smith A. J Mater Chem. 2004;14:2242. [Google Scholar]
- 381.Sethmann I, Helbigb U, Wörheide G. CrystEngComm. 2007;9(12):1262. [Google Scholar]
- 382.Sethmann I, Hinrichs R, Wörheide G, Putnis A. Journal of Inorganic Biochemistry. 2006;100:88. doi: 10.1016/j.jinorgbio.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 383.Sasagawa I, Ishiyama M, Akai J. Materials Science and Engineering C. 2006;26:630. [Google Scholar]
- 384.Jee SS. Dissertation. University of Florida; 2008. [Google Scholar]
- 385.Garcia-Ruiz JM, Navarro AR, Kalin O. Materials Science & Engineering C-Biomimetic Materials Sensors and Systems. 1995;3:95. [Google Scholar]
- 386.Amos FF, Olszta MJ, Khan SR, Gower LB. In: Biomineralization- Medical Aspects of Solubility. Königsberger E, Königsberger L, editors. Chapter 4 John Wiley & Sons, Ltd; West Sussex, England: 2006. [Google Scholar]
- 387.Evan AP, Lingeman JE, Coe FL, Parks JH, Bledsoe SB, Shao YZ, Sommer AJ, Paterson RF, Kuo RL, Grynpas M. Journal of Clinical Investigation. 2003;111:607. doi: 10.1172/JCI17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Evan A, Lingeman J, Coe FL, Worcester E. Kidney Int. 2006;69:1313. doi: 10.1038/sj.ki.5000238. [DOI] [PubMed] [Google Scholar]
- 389.Evan AP, Coe F, Lingeman JE. Kidney Int. 2007;71:83. doi: 10.1038/sj.ki.5002113. [DOI] [PubMed] [Google Scholar]
- 390.Matlaga BR, Coe FL, Evan AP, Lingeman JE. J Urol. 2007;177:31. doi: 10.1016/j.juro.2006.08.088. [DOI] [PubMed] [Google Scholar]
- 391.Evan AP. Renal Stone Disease, 1” Annual International Urolithiasis Research Symposium; Indianapolis, Indiana. 2006. p. 15. [Google Scholar]
- 392.Randall A. Intl Abst Surg. 1940;71:209. [Google Scholar]
- 393.Evan AP, Coe FL, Lingeman JE, Shao Y, Sommer AJ, Bledsoe SB, Anderson JC, Worcester EM. Anatomical Record-Advances in Integrative Anatomy and Evolutionary Biology. 2007;290:1315. doi: 10.1002/ar.20580. [DOI] [PubMed] [Google Scholar]
- 394.Evan AP, Coe FL, Rittling SR, Bledsoe SM, Shao YZ, Lingeman JE, Worcester EM. Kidney Int. 2005;68:145. doi: 10.1111/j.1523-1755.2005.00388.x. [DOI] [PubMed] [Google Scholar]
- 395.Treiman A. Recent scientific papers on ALH 84001 explained, with insightful and totally objective commentaries. NASA- Lunar Planetary Institute; 1999. http://cass.jsc.nasa.gov/lip/meteorites/alhnpap.html. [Google Scholar]
- 396.Treiman AH. Meteorit Planet Sci. 1998;33:753. doi: 10.1111/j.1945-5100.1998.tb01681.x. [DOI] [PubMed] [Google Scholar]
- 397.Wucher B, Yue WB, Kulak AN, Meldrum FC. Chem Mat. 2007;19:1111. [Google Scholar]
- 398.Garrels R, Wollast R. Am J Sci. 1978;278:1469. [Google Scholar]
- 399.Morse JW, Bender ML. Chem Geol. 1990;82:265. [Google Scholar]
- 400.Weber JN. Am J Sci. 1969;267:537. [Google Scholar]
- 401.Kitano Y, Kanamori N. Geochem J. 1966;1:1. [Google Scholar]
- 402.Raz S, Weiner S, Addadi L. Advanced Materials. 2000;12:38. [Google Scholar]
- 403.Cheng X. Dissertation. University of Florida; 2005. [Google Scholar]
- 404.Rosenberg GD, Hughes WW. Lethaia. 1991;24:83. [Google Scholar]
- 405.Cusack M, Fraser AC, Stachel T. Comparative Biochemistry and Physiology B-Biochemistry & Molecular Biology. 2003;134:63. doi: 10.1016/s1096-4959(02)00185-9. [DOI] [PubMed] [Google Scholar]
- 406.Blake DF, Peacor DR, Allard LF. Micron and Microscopica Acta. 1984;15:85. [Google Scholar]
- 407.Tsipursky SJ, Buseck PR. American Mineralogist. 1993;78:775. [Google Scholar]
- 408.Rosenberg GD. In: Skeletal Biomineralization: Patterns, Processes and Evolutionary Trends. Carter JG, editor. Vol. 1 Van Nostrand Reinhold; N.Y: 1990. [Google Scholar]
- 409.Meibom A, Yurimoto H, Cuif JP, Domart-Coulon I, Houlbreque F, Constantz B, Dauphin Y, Tambutte E, Tambutte S, Allemand D, Wooden J, Dunbar R. Geophysical Research Letters. 2006:33. [Google Scholar]
- 410.Clode PL, Marshall AT. Protoplasma. 2003;220:153. doi: 10.1007/s00709-002-0046-3. [DOI] [PubMed] [Google Scholar]
- 411.Clode PL, Marshall AT. Tissue & Cell. 2002;34:187. doi: 10.1016/s0040-8166(02)00031-9. [DOI] [PubMed] [Google Scholar]
- 412.STOLARSKI J, MAZUR M. Acta Palaeontol Pol. 2005;50(4):847. [Google Scholar]
- 413.Emlet RB. Biological Bulletin. 1982;163:264. doi: 10.1086/BBLv216n2p163. [DOI] [PubMed] [Google Scholar]
- 414.Yang HG, Zeng HC. Angew Chem-Int Edit. 2004;43:5930. doi: 10.1002/anie.200461129. [DOI] [PubMed] [Google Scholar]
- 415.Gehrke N, Cölfen H, Pinna N, Antonietti M, Nassif N. Crystal Growth & Design. 2005;5:1317. [Google Scholar]
- 416.Nassif N, Gehrke N, Pinna N, Shirshova N, Tauer K, Antonietti M, Cölfen H. Angew Chem-Int Edit. 2005;44:6004. doi: 10.1002/anie.200500081. [DOI] [PubMed] [Google Scholar]
- 417.Zolotoyabko E, Pokroy B. CrystEngComm. 2007;9(12):1156. [Google Scholar]
- 418.Bolze J, Peng B, Dingenouts N, Panine P, Narayanan T, Ballauff M. Langmuir. 2002;18:8364. [Google Scholar]
- 419.Kurc M, Farina M, Lins U, Kachar B. Hearing Research. 1999;131:11. doi: 10.1016/s0378-5955(99)00007-6. [DOI] [PubMed] [Google Scholar]
- 420.Lins U, Farina M, Kurc M, Riordan G, Thalmann R, Thalmann I, Kachar B. J Struct Biol. 2000;131:67. doi: 10.1006/jsbi.2000.4260. [DOI] [PubMed] [Google Scholar]
- 421.Hetherington NBJ, Kulak AN, Sheard K, Meldrum FC. Langmuir. 2006;22:1955. doi: 10.1021/la0523356. [DOI] [PubMed] [Google Scholar]
- 422.Allemand D, Cuif J-P, editors. Biomineralization 93. Vol. 14 Musee Oceanographique; Monaco: 1994. [Google Scholar]
- 423.Janofske D. In: Biomineralization 93- 7th International Symposium on Biomineralization. 4 Allemand D, Cuif J-P, editors. Vol. 14. Monaco Musee Oceanographique; Monaco: 1993. [Google Scholar]
- 424.Patel VM, Sheth P, Kurz A, Ossenbeck M, Shah DO, Gower LB. Concentrated Dispersions: Theory, Experiments, and Applications. Washington, DC: 2002. p. 15. [Google Scholar]
- 425.Olszta MJ, Douglas EP, Gower LB. Calcif Tissue Int. 2003;72:583. doi: 10.1007/s00223-002-1032-7. [DOI] [PubMed] [Google Scholar]
- 426.Wustman BA, Morse DE, Evans JS. Biopolymers. 2004;74:363. doi: 10.1002/bip.20086. [DOI] [PubMed] [Google Scholar]
- 427.Kim IW, Morse DE, Evans JS. Langmuir. 2004;20:11664. doi: 10.1021/la0481400. [DOI] [PubMed] [Google Scholar]
- 428.Weiner S, Addadi L. Science. 2002;298:375. doi: 10.1126/science.1078093. [DOI] [PubMed] [Google Scholar]
- 429.Mann S, Sparks NHC, Blakemore RP. Proceedings of the Royal Society of London Series B, Biological Sciences. 1987;231:469. [Google Scholar]
- 430.Scheffel A, Gruska M, Faivre D, Linaroudis A, Plitzko JM, Schüler D. Nature. 2006;440:110. doi: 10.1038/nature04382. [DOI] [PubMed] [Google Scholar]
- 431.Kroger N, Deutzmann R, Bergsdorf C, Sumper M. Proc Natl Acad Sci U S A. 2000;97:14133. doi: 10.1073/pnas.260496497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Kroger N, Deutzmann R, Sumper M. Science. 1999;286:1129. doi: 10.1126/science.286.5442.1129. [DOI] [PubMed] [Google Scholar]
- 433.Kroger N, Rainer D, Bergsdorf C, Sumper M. PNAS. 2000;97:14133. doi: 10.1073/pnas.260496497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Zhang XD, Sun YY, Zhuang JQ, Yang WS. Chemical Research in Chinese Universities. 2006;22:368. [Google Scholar]
- 435.Patwardhan SV, Clarson SJ. Mater Sci Eng C-Biomimetic Supramol Syst. 2003;23:495. [Google Scholar]
- 436.Sumper M, Kröger N. J Mater Chem. 2004;14:2059. [Google Scholar]
- 437.Cha Jn, Shimizu K, Zhou Y, Christiansen SC, Chmelka BF, Stucky GD, Morse DE. Proc Natl Acad Sci USA. 1999;96:361. doi: 10.1073/pnas.96.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Jodaikin A, Weiner S, Traub W. Journal of Ultrastructure Research. 1984;89:324. doi: 10.1016/s0022-5320(84)80048-9. [DOI] [PubMed] [Google Scholar]
- 439.Mann S, Sparks NHC, Blakemore RP. Proc R Soc Lond B. 1987;231:469. [Google Scholar]
- 440.Khan SR. The Journal of Urology. 1997;157:376. [PubMed] [Google Scholar]
- 441.Kroger N, Lorenz S, Brunner E, Sumper M. Science. 2002;298:584. doi: 10.1126/science.1076221. [DOI] [PubMed] [Google Scholar]
- 442.Uriz MJ. Canadian Journal of Zoology-Revue Canadienne De Zoologie. 2006;84:322. [Google Scholar]