

Abstract
During DNA synthesis in vitro using dNTP and rNTP concentrations present in vivo, yeast replicative DNA polymerases α, δ and ε (Pols α, δ and ε) stably incorporate rNTPs into DNA. rNTPs are also incorporated during replication in vivo, and they are repaired in an RNase H2-dependent manner. In strains encoding a mutator allele of Pol ε (pol2-M644G), failure to remove rNMPs from DNA due to deletion of the RNH201 gene encoding the catalytic subunit of RNase H2, results in deletion of 2-5 base pairs in short repetitive sequences. Deletion rates depend on the orientation of the reporter gene relative to a nearby replication origin, suggesting that mutations result from rNMPs incorporated during replication. Here we demonstrate that 2-5 base pair deletion mutagenesis also strongly increases in rnh201Δ strains encoding wild type DNA polymerases. As in the pol2-M644G strains, the deletions occur at repetitive sequences and are orientation-dependent, suggesting that mismatches involving misaligned strands arise that could be subject to mismatch repair. Unexpectedly however, 2-5 base pair deletion rates resulting from loss of RNH201 in the pol2-M644G strain are unaffected by concomitant loss of MSH3, MSH6, or both. It could be that the mismatch repair machinery is unable to repair mismatches resulting from unrepaired rNMPs incorporated into DNA by M644G Pol ε, but this possibility is belied by the observation that Msh2-Msh6 can bind to a ribonucleotide-containing mismatch. Alternatively, following incorporation of rNMPs by M644G Pol ε during replication, the conversion of unrepaired rNMPs into mutations may occur outside the context of replication, e.g., during the repair of nicks resulting from rNMPs in DNA. The results make interesting predictions that can be tested.
Keywords: ribonucleotide incorporation, RNase H2, DNA mismatch repair, deletions, genome instability
1. Introduction
Most DNA polymerases are relatively efficient at excluding ribonucleoside triphosphates (rNTPs) from being incorporated into DNA during DNA synthesis in vivo ([1–4] and references therein). Nonetheless, rNTP exclusion by DNA polymerases is not absolute, and the concentrations of rNTPs in cells are much higher than are the concentrations of dNTPs [3,5,6]. These facts imply that some rNTPs will be incorporated into DNA in vivo, and that they will eventually be removed to maintain the chemical identity of organisms whose genomes are comprised of DNA. In support of these possibilities, we recently reported [7] that rNTPs are incorporated during replication by Saccharomyces cerevisiae DNA polymerase ε (Pol ε), a putative leading strand replicase [8,9]. We also reported that these rNMPs are efficiently removed from DNA by RNase H2-dependent repair. However, in yeast strains harboring a mutator allele (pol2-M644G) encoding a form of Pol ε with a 10-fold higher than normal propensity to incorporate rNMPs into DNA, deletion of the RNH201 gene encoding the catalytic subunit of RNase H2 results in replication stress and genome instability [7]. This genome instability primarily involves the deletion of 2-5 base pairs in short repetitive DNA sequences. Interestingly, many of these deletions occurred at locations where the mutation rate is high only when the URA3 reporter gene is in one of its two possible orientations relative to the nearest origin of replication. This orientation bias, and the fact that the mutagenesis is observed in the pol2-M644G strain encoding a mutator derivative of the putative leading strand replicase, raises interesting questions about DNA replication fidelity, but in this case for discrimination against the sugar rather than discrimination against an incorrect or misaligned base.
The first studies to demonstrate that deletion of the gene encoding the catalytic subunit of RNase H2 was mutagenic [10,11] were performed in strains encoding wild type DNA polymerases. The mutagenesis observed in those studies was suggested to reflect aberrant processing of rNMPs incorporated by the Pol α–associated RNA primase that initiates Okazaki fragments, and the increase in overall mutation rate was small. In strains harboring the pol2-M644G allele, loss of RNase H2 activity was much more mutagenic, suggesting that the mutagenesis depends on unrepaired rNMPs incorporated during leading strand replication by the M644G mutator derivative of Pol ε. However, the same study also confirmed the earlier work [10,11], by demonstrating that deleting RNH201 in strains with wild type polymerases increased mutation rates by only about 2-fold at each of three different reporter loci in yeast. Thus, existing evidence leaves open the question of whether unrepaired rNMPs incorporated during replication by wild type replicative DNA polymerases, rather than rNMPs incorporated by RNA primase, are in fact mutagenic. Here we address this question by defining specific mutation rates in rnh201Δ strains encoding wild type replicases. We find that 2-5 base pair deletion rates in repetitive sequences are elevated an average of 65-fold, and the site specificity of mutagenesis recapitulates that seen in the pol2-M644G rnh201Δ strains. These data strongly support the conclusion that rNMPs incorporated during normal DNA replication are highly mutagenic if not repaired by RNase H2.
How are unrepaired rNMPs in DNA converted into deletions? In the field of cancer research, the deletion of repeat units from repetitive DNA sequences is called microsatellite instability (MSI), and MSI is an established hallmark of tumors with defects in repair of errors arising by strand misalignments that occur during DNA replication (reviewed in [12–14]). The 2-5 base pair deletions in repetitive sequences observed in the pol2-M644G rnh201Δ strains [7] can be considered a specialized form of this type of misalignment mutagenesis (see further discussion below). Theoretically, the deletions could result from rNMP incorporation during replication, followed by strand slippage during the next round of replication as a DNA polymerase attempts to bypass the rNMP that remains in the template strand because RNase H2 is absent. In this model, a mismatch containing a 2-5 base loop in the template strand should arise that is stabilized by the correct base pairs possible at repetitive sequences [15,16]. Such deletion mismatches are predicted to be subject to MMR. Perhaps especially important is MMR initiated by Msh2•Msh3 (MutSβ), which has evolved to repair loop mismatches in this size range (reviewed in [17–19]). Here we test this prediction by comparing the rates of 2-5 base pair deletions in pol2-M644G rnh201Δ strains that are proficient in MMR to strains that are deficient in MMR repair. The results show that loss of MMR has little effect on the 2-5 base pair deletion rate. This suggests that either MMR does not repair deletion mismatches promoted by rNMPs incorporated into DNA by M644G Pol ε, or that the deletion mismatches are formed during a DNA transaction that is not subject to MMR, e.g., during processing of rNMPs remaining in DNA when RNase H2 is defective.
2. Materials and methods
2.1. Construction of yeast strains
S. cerevisiae strains were isogenic derivatives of strain Δ|(−2)|-7B-YUNI300 (MATa CAN1 his7-2 leu2-Δ::kanMX ura3-Δ trp1-289 ade2-1 lys2-ΔGG2899-2900) [20]. The rnh201Δ, pol2-M644G and pol2-M644G rnh201Δ strains have been described previously [7]. The msh3Δ variants were constructed by deleting and replacing MSH3 via transformation with a PCR product containing the nourseothricin resistance cassette (NAT-R) amplified from pAG25 [21] and flanked by 50 nucleotides of sequence homologous to the intergenic regions upstream and downstream of the MSH3 open reading frame. Transformants that arose by homologous recombination containing the replacement of the MSH3 open reading frame with NAT-R were isolated on YPDA plates containing 100 μg/ml NAT and verified by PCR analysis and DNA sequencing of genomic DNA. To construct msh6Δ and msh3Δ msh6Δ, strains, diploid yeast strains were first made heterozygous for MSH6 (Supplementary Table 1, strains d031 and d032). MSH6 was removed from diploid cells (ySNM1001 and ySNM1005 [9]) via transformation with a PCR product containing the TRP1 gene flanked by sequences homologous to 200 nucleotides of intergenic regions upstream and downstream of the MSH6 open reading frame. Transformants were isolated on complete synthetic media plates lacking tryptophan and verified by PCR analysis to have replaced MSH6 with TRP1 via homologous recombination. Dissection of meiotic tetrads confirmed that only one copy of MSH6 had been deleted. Mating type α (mat α) haploids resulting from these dissections (strains h031 and h032) were mated on YPDA with pol2-M644G mating type a (mat a) haploid yeast (above; ySNM70 and ySNM77 [8]). Diploids (non-maters; strains d231.5 and d232.5) were sporulated and underwent meiotic tetrad dissection. Small samples of TRP1+ spore colonies generated (lacking MSH6) were taken and the remainder of each colony was replica plated to media containing 5-fluoro-orotic acid (5-FOA). Genomic DNA was isolated from samples corresponding to colonies with elevated mutation rates and the pol2-M644G mutation was confirmed by sequencing. Mat α isolates of the pol2-M644G msh6::TRP1 haploid strains generated (strains h231 and h232) were mated with mat a pol2-M644G msh3::NAT rnh201::HYG haploids (above; ABC2001 and ABC2003). Transformants bearing all three markers were selected. The resulting diploids (strains d281 and d282) were homozygous for pol2-M644G and heterozygous for msh6Δ, msh3Δ, and rnh201Δ. These were sporulated and meiotic tetrads were dissected, resulting in pol2-M644G haploid cells with all combinations of the presence or absence of MSH6, MSH3, and RNH201, spore colonies of which (strains h271, h272, h281, and h282) were used to determine mutation rates and to generate mutation spectra.
2.2. Measurement of spontaneous mutation rates and sequence analysis
Spontaneous mutation rates were measured by fluctuation analysis as described previously [22]. For each ura3 mutant that was sequenced, an independent colony was patched to YPDA and then replica plated to media containing 5-fluoro-orotic acid (5-FOA). Genomic DNA from a single 5-FOA resistant colony from each patched colony was isolated and the ura3 gene was PCR-amplified and sequenced.
2.3. MutSα binding to a ribo-containing mismatch
Enzyme mobility shift assays were performed as described, using yeast MutSα purified as previously described [23]. Oligonucleotide heteroduplexes were created by annealing radiolabeled dTCGTTTTACAACGTCGTGAATGAGAAAACCCTGGCGTTACC with unlabeled dGGTAACGCCAGGGTTTTCTCGTTCACGACGTTGTAAAACGA or dGGTAACGCCAGGGTTTTCTCrGTTCACGACGTTGTAAAACGA (Dharmacon). Bold and underlined bases define the residues creating the rG•dT and dG•dT mispairs. Binding reaction mixtures (20 μl) containing 1 nM heteroduplex and 250, 50 or 10 nM of MutSα were incubated at 25°C for 10 minutes. Bound substrate was separated from unbound substrate using a nondenaturing 5% polyacrylamide TBE gel (BioRad) run at at 100V with 0.5% TBE buffer at 4°C. Following electrophoresis, gels were dried and exposed to a phosphor screen.
3. Results
3.1. Mutator effect of unrepaired rNMPs in strains with wild type DNA polymerases
As previously reported for rnh201Δ strains encoding wild type DNA polymerases [10,11], the spontaneous mutation rate at the URA3 locus is only slightly increased upon deletion of RNH201 (Table 1). This is the case when URA3 is in either of two orientations relative to ARS306, the closest origin of replication. To determine if this reflects larger increases for specific types of mutations, the URA3 coding sequences of independent FOAr mutants were determined (Table 1, Fig. 1), and rates were calculated for specific types of mutations. The results (Fig. 2A) indicate that deleting RNH201 (i.e., the rnh201Δ strains, designated with a “−” on the X axis) does not increase the rate of single base substitutions or single base deletions beyond the rates in the corresponding RNH201 strains (designated with a “+” on the X axis). In contrast, the overall rate of 2-5 base pair deletions is increased by 20-fold in orientation 1 (OR1) and by 65-fold in orientation 2 (OR2). These are all deletions of one repeat unit from repetitive sequences that are usually perfect but occasionally imperfect, and comprised of two to four repeat units (Fig. 1). The deletions preferentially occur at certain locations, e.g., base pairs 178-181, 216-219 and 688-694. They are largely, but not exclusively, orientation-dependent. At these locations, deletion rates are increased from ≥5-fold up to ≥90-fold. There is also a 3-fold, rnh201Δ-dependent increase in G•C to A•T substitution rate at base pair 768.
Table 1.
Mutation rates and sequencing data for RNH201 and rnh201Δ strains
| Strain | RNH201 | rnh201Δ | ||
|---|---|---|---|---|
| URA3 Orientation | OR1 | OR2 | OR1 | OR2 |
| Mutation rate (x 10−7) | 0.34 | 0.48 | 0.36 | 0.75 |
| 95% CI | 0.23 – 0.79 | 0.21 – 0.47 | 0.34–0.95 | 0.68 – 1.2 |
| ura3 mutants sequenced | 191 | 239 | 251 | 273 |
| Base substitutions | 119 | 137 | 86 | 89 |
| ±1 frameshifts | 11 | 21 | 27 | 14 |
| 2 - 5 base deletions | 1 | 2 | 25 | 96 |
| Others* | 9 | 15 | 19 | 13 |
Others include mutations involving multiple bases (deletions, duplications and complex mutations; see Supplementary Table 2). A number of FOAr mutants had no sequence change in the 804 base pair URA3 open reading frame. These mutants were not investigated further, but they may contain sequence changes in the promoter or 5′ untranslated region of URA3, or in another gene that affects resistance to 5-FOA.
Figure 1. Mutational spectra in rnh201Δ strains.

The coding strand of the 804 base pair URA3 open reading frame is shown. The sequence changes observed upon sequence analysis of independent ura3 mutants are depicted above the coding sequence for URA3 orientation 1, and below the coding sequence for URA3 orientation 2. Letters indicate single base substitutions, closed triangles indicate single base additions, open triangles indicate single base deletions, and short lines above or below the coding sequence indicate 2-5 base deletions. Large insertions and deletion mutations and complex mutations are listed in Supplementary Table 2. Solid boxes enclose perfect direct repeat sequences, and dashed boxes enclose imperfect direct repeat sequences.
Figure 2. Mutation rates for RNH201 and rnh201Δ in WT and pol2-M644G strains.

(A). Results for wild type DNA polymerase strains that are either RNH201 (+) or rnh201Δ (−). Individual rates, shown above the columns, were calculated from the results in Table 1 and Figure 1, as the proportion of each type of event among the total mutants sequenced, multiplied by the total mutation rates for each strain. Columns are shaded for POL2 RNH201 URA3 in orientation 1 (□), POL2 rnh201Δ URA3 in orientation 1 (
 ), POL2 RNH201 URA3 in orientation 2 (
), POL2 RNH201 URA3 in orientation 2 (
 ), POL2 rnh201Δ URA3 in orientation 2 (■). (B). Results for pol2-M644G strains that are either RNH201 (+) or rnh201Δ (−). Individual rates, above the columns, were calculated as in (A) from results in Table 1 and Figure 4 in [7]. Columns are shaded for pol2-M644G RNH201 URA3 in orientation 1 (□), pol2-M644G rnh201Δ URA3 in orientation 1 (
), pol2-M644G RNH201 URA3 in orientation 2 (
), pol2-M644G rnh201Δ URA3 in orientation 2 (■).
), POL2 rnh201Δ URA3 in orientation 2 (■). (B). Results for pol2-M644G strains that are either RNH201 (+) or rnh201Δ (−). Individual rates, above the columns, were calculated as in (A) from results in Table 1 and Figure 4 in [7]. Columns are shaded for pol2-M644G RNH201 URA3 in orientation 1 (□), pol2-M644G rnh201Δ URA3 in orientation 1 (
), pol2-M644G RNH201 URA3 in orientation 2 (
), pol2-M644G rnh201Δ URA3 in orientation 2 (■).
The rnh201Δ-dependent mutational specificity observed in strains encoding wild type DNA polymerases (Fig. 1) is similar to the specificity previously seen in pol2-M644G rnh201Δ strains (Fig. 4 in [7]). In fact, when the data from the earlier study [7] were used to calculate rates for 2-5 base pair deletions and the G•C to A•T substitution at base pair 768, the rates in the pol2-M644G rnh201Δ strains (Fig. 2B) were an amplified reflection of the mutagenesis observed in the rnh201Δ single mutant strains.
3.2. Mutator effect of unrepaired rNMPs in DNA in strains defective in mismatch repair
To determine if 2-5 base deletions are subjected to mismatch repair, we then compared mutation rates in pol2-M644G rnh201Δ double mutants to mutation rates in isogenic strains also deleted for MSH3, MSH6, or both (Table 2). Overall spontaneous mutation rates in pol2-M644G rnh201Δ msh3Δ triple mutants deficient in Msh2-Msh3-dependent mismatch repair (9.8 × 10−7 in OR1 and 9.9 × 10−7 in OR2) were similar to those in the mismatch repair-proficient pol2-M644G rnh201Δ double mutants (5.7 × 10−7 in OR1 and 11 × 10−7 in OR2). Mutation rates in pol2-M644G rnh201Δ msh6Δ triple mutants deficient in Msh2-Msh6-dependent mismatch repair were much higher, 200 × 10−7 in OR1 and 81 × 10−7 in OR2. As predicted by the well established role for Msh2-Msh6 in correcting single base-base mismatches (reviewed in [17–19]), the vast majority of ura3 mutants in these strains contained single base substitutions. Only a few single base deletions were observed, as expected because these strains retain the capacity to repair single base deletion mismatches using Msh2-Msh3 [24]. More importantly, only six 2-base deletions were observed in the pol2-M644G rnh201Δ msh6Δ triple mutants, and all were deletions of a CA dinucleotide at base pairs 216-219, the orientation 2-dependent hotspot. Four G•C to A•T substitutions at base pair 768 were also observed in the pol2-M644G rnh201Δ msh6Δ triple mutants, all in OR1. Mutation rates in the pol2-M644G rnh201Δ msh3Δ msh6Δ quadruple mutants were also high, 270 × 10−7 in OR1 and 280 × 10−7 in OR2. As predicted by deficiencies in both Msh2-Msh6 and Msh2-Msh3, the vast majority of ura3 mutants in these strains contained single base changes that were about equally distributed between substitutions and deletions. Only one 2-base deletion was observed in the pol2-M644G rnh201Δ msh3Δ msh6Δ quadruple mutants, the deletion of a CA dinucleotide at base pairs 216-219.
Table 2.
URA3 mutation rates and sequencing data for MMR-proficient and MMR-deficient pol2 M644G rnh201Δ strains.
| MMR Status | MSH+ | msh3Δ | msh6Δ | msh3Δ/msh6Δ | ||||
|---|---|---|---|---|---|---|---|---|
| OR1 | OR2 | OR1 | OR2 | OR1 | OR2 | OR1 | OR2 | |
| Mutation rate (x 10−7) | 5.7 | 11 | 9.8 | 9.9 | 200 | 81 | 270 | 280 |
| 95% Confidence Intervals | 5.1 – 44 | 8.3 – 20 | 7.3 – 17 | 8.4 – 17 | 150 – 270 | 58 – 110 | 190 – 370 | 230 – 350 |
| ura3 Mutants Sequenced | 126 | 140 | 87 | 95 | 141 | 134 | 157 | 116 |
| Base Pair Substitutions | 56 | 9 | 39 | 9 | 126 | 116 | 83 | 44 |
| One Base Deletions | 3 | 7 | 17 | 29 | 7 | 3 | 64 | 52 |
| Others * | 2 | 0 | 3 | 4 | 0 | 6 | 0 | 2 |
| Δ2-5 Base Pair -Total | 61 | 124 | 26 | 48 | 0 | 6 | 0 | 1 |
| ΔTG 95-100 | 0 | 6 | 0 | 3 | 0 | 0 | 0 | 0 |
| ΔAC 178-181 | 8 | 0 | 3 | 0 | 0 | 0 | 0 | 0 |
| ΔCA 216-219 | 1 | 91 | 0 | 28 | 0 | 6 | 0 | 1 |
| ΔATT 688-694 | 21 | 6 | 7 | 5 | 0 | 0 | 0 | 0 |
| G•C to A•T 768 | 21 | 0 | 8 | 0 | 4 | 0 | 0 | 0 |
Measurements were performed as described in Methods. In those cases where the total number of mutations does not equal the total number of FOAr mutants sequenced, some mutants had no sequence change and others had two sequence changes in the URA3 coding sequence.
Others include rare mutants containing duplications or deletions of more than five base pairs or more complex sequence changes, e.g. tandem double base substitutions or substitution-deletions.
The results in Table 2 were then used to calculate mutation rates for the types of mutations characteristic of loss of RNase H2. The results (Fig. 3) demonstrate that the overall rates of 2-5 base pair deletions and deletions at the four major orientation-dependent hotspots were not increased by deficiencies in Msh3, Msh6 or both. Likewise, the rate of G•C to A•T substitutions at base pair 768 in OR1 was not increased by loss of Msh3 or loss of both Msh3 and Msh6 (Fig. 3). Msh6 deficiency did reveal a higher rate of G•C to A•T substitutions at base pair 768 in OR1 (Fig. 3). This leaves open the possibility of some Msh6-dependent repair of the responsible base-base mismatch. However, the increase is small, and is based on four occurrences (Table 2), so it might reflect experimental fluctuation, or repair of base-base mismatches at this location that do not depend on rnh201Δ
Figure 3. Effect of msh3Δ, msh6Δ or msh3Δ msh6Δ on rnh201Δ-dependent mutation rates in pol2 M644G strains.

Site-specific rates, shown to the right of the bars, were calculated as in Figure 2. Columns are shaded for pol2-M644G rnh201Δ (□), pol2-M644G rnh201Δ msh3Δ(
), pol2-M644G rnh201Δ msh6Δ (
), pol2-M644G rnh201Δ msh3Δ msh6Δ (■). Orientation of URA3 is designated as OR1 or OR2.
3.3. Msh2-Msh6 binding to a mismatch containing a ribonucleotide
To determine if the mismatch repair system can recognize a mismatch when one of the mismatched bases is a ribonucleotide, we used an electrophoretic mobility shift assay to measure yMsh2-Msh6 binding to DNA substrates containing a rG•dT mismatch or a corresponding dG•dT mismatch. The results (Fig. 4) demonstrate that yMsh2-Msh6 binds to both mismatches, thus providing initial evidence that mismatches containing ribonucleotides do not escape detection by the mismatch repair machinery.
Figure 4. Binding of yMsh2•Msh6 to heteroduplexes containing a mismatched ribonucleotide.
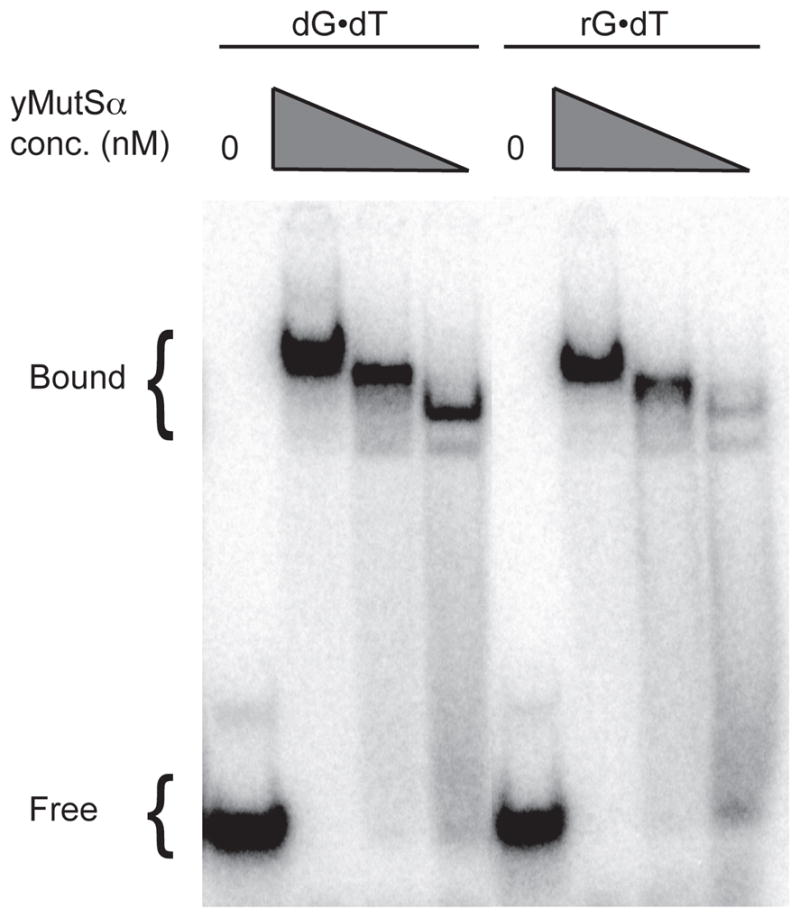
Binding reactions contained 1 nM heteroduplex with a dG•dT or a rG•dT mismatch and 250, 50 or 10 nM concentration of yMsh2•Msh6. Protein bound heteroduplexes were separated from free heteroduplexes by PAGE using a 5% polyacrylamide TBE gel.
4. Discussion
Previous work revealed that in yeast strains encoding a Pol ε derivative with an increased capacity to incorporate rNTPs, deletion of the gene encoding the catalytic subunit of RNase H2 results in orientation-dependent deletions of 2-5 base pairs in repetitive sequences [7]. Here we report a similar pattern of mutagenesis in strains encoding wild type DNA polymerases (Figs. 1 and 2). This similarity indicates that the genome can be destabilized by unrepaired rNMPs incorporated even by wild type DNA polymerases, and possibly by primase, during normal DNA replication. The deletion of 2-5 base pair repeat units from short repetitive sequences is somewhat reminiscent of the infamous “microsatellite instability” (MSI) resulting from failure of MMR to correct mismatches resulting from strand slippage during DNA replication. For cancer diagnosis [13,14], MSI is assessed at loci containing much greater numbers of repeat units than are present in the URA3 coding sequence. Thus, it will be interesting to test if an RNase H2 defect results in even greater instability in longer repetitive sequences than those present in the URA3 coding sequence. MSI in tumors has been categorized in two general forms [13]. “MSI-high” represents a high degree of instability in tumors defective in core MMR gene products like MSH2 and MLH1. “MSI-low” is a lesser degree of instability that can result from defects in other MMR genes, and perhaps in other, yet to be identified, genes. The results presented here suggest that one source of MSI-low in humans could be independent of MMR, i.e., defective RNase H2-dependent repair of rNMPs incorporated into DNA during replication. It should be possible to examine this possibility using RNase H2-defective cells obtained from patients suffering from Acardi Goutiéres Syndrome [25], a severe autoimmune disease.
During DNA synthesis in vitro using cellular concentrations of the four dNTPs and rNTPs, Pols α and δ readily incorporate rNTPs into DNA, but they do so with different site specificity than Pol ε [3]. If this were also the case in vivo, then one might anticipate that the pattern of mutagenesis due to unrepaired rNMPs incorporated during replication would differ between the pol2-M644G strains, where mutagenesis is primarily due to rNMP incorporation by M644G Pol ε, and strains with wild type polymerases, where mutagenesis could be due to rNMP incorporation by wild type Pols α, δ and ε. However, this is not what is observed. Instead, the mutagenesis due to the RNase H2 defect in the pol2-M644G strain (Fig. 2B) is largely an amplified version of the specificity in the strains with wild type polymerases (Fig. 2A). This implies that the main culprit for incorporating mutagenic rNMPs is Pol ε. If so, our currently favored model wherein Pol ε participates in replicating the leading strand template [8,9] can be used to predict which particular rNMPs might be responsible for the mutagenesis. As one example, the strongest hotspot involves deleting a dinucleotide from the CACA repeat at nucleotides 216-219. The orientation 2 bias for this deletion suggests that incorporation of rNTPs (possibly rUTP and/or rGTP) during Pol ε-mediated leading strand replication of the 5′-CACA-containing template is responsible for the mutagenesis, rather than incorporation of rNTPs during Pol ε-mediated leading strand replication of the complementary 5′-TGTG-containing template in orientation 1. We are currently attempting to test this prediction. The mutational specificity data also lead one to wonder if unrepaired rNMPs incorporated by Pols α and δ also contribute to genome instability. If so, it is possible that the mismatches responsible for any such mutations may be subject to correction by MMR. We are investigating these possibilities using mutant alleles of Pols α and δ. However, the similar mutational patterns in Figure 2A and 2B suggest that unrepaired rNMPs incorporated by Pols α and δ may not strongly contribute to genome instability. If not, then perhaps when RNase H2 is defective, rNMPs incorporated into DNA by Pol ε are processed differently than rNMPs incorporated into DNA by Pols α and δ, which participate in replicating the lagging strand template [9,26]. Perhaps like rNMPs incorporated by RNA primase to initiate replication, rNMPs introduced into DNA by lagging strand DNA polymerases can be removed during Okazaki fragment maturation. This mechanism would rarely be available to the leading strand replication machinery, e.g., only at replication origins or following replication restart.
In our previous study [7], we discussed a model wherein unrepaired rNMPs incorporated into repetitive DNA sequences during replication promote strand slippage during a subsequent round of replication. rNMP-dependent slippage during DNA replication would create 2-5 base deletion loop mismatches that are potentially subject to MMR, perhaps especially Msh2-Msh3-dependent MMR that has evolved to repair mismatches with loops in this size range. If this were the case, then the rate of 2-5 base deletions in the pol2-M644G rnh201Δ strains would be strongly elevated by MMR defects. This is not the case. The results in Table 2 and Figure 3 clearly reveal that deletion of MSH3, MSH6, or both, does not increase deletion rates resulting from unrepaired rNMPs in DNA. In fact, several rates were actually lower in MMR defective strains (Fig. 3). This is best exemplified by a 4-fold reduction in the overall rate of 2-5 base pair deletions in URA3 (orientation 2) in the pol2-M644G rnh201Δ msh3/6Δ strain (24 × 10−8) as compared to the pol2-M644G rnh201Δ strain (97 × 10−8). This difference is statistically significant (p ≤0.008, see Supplementary Information for calculation), consistent with the possibility that MMR proteins might actually promote rather than prevent rNMP-dependent mutagenesis. Precedent for this idea comes from studies indicating the Msh2-Msh3 promotes triplet repeat expansion (see [27] for review), and from studies showing that MMR proteins modulate somatic hypermutation of immunoglobulin genes (see [28] and references therein).
The fact that loss of MMR does not elevate mutagenesis due to unrepaired rNMPs incorporated into DNA by M644G Pol ε might be explained if MMR cannot correct mismatches that contain, or are near to, rNMPs in the DNA. Arguing against this possibility is an experiment showing that yMsh2-Msh6 binds to DNA with a mismatch containing an rNMP (Fig. 4). This initial result is consistent with the possibility that MMR can correct rNMP-containing mismatches. This possibility deserves further attention in the future, since our present results in the pol2-M644G strains (Fig. 3) do not yet exclude that unrepaired rNMPs incorporated into DNA by other polymerases and/or by primase may give rise to mismatches that can be corrected by MMR. Nonetheless, the present study does imply that MMR may not have the opportunity to correct mismatches resulting from unrepaired rNMPs incorporated into DNA by M644G Pol ε. This could happen if such mismatches arise during a process operating outside the context of a normal replication fork (see Fig. 1 in [29]). Possibilities here involve mutagenic processing of nicks generated by spontaneous hydrolysis of the ribose-containing DNA backbone, creating DNA ends that have a 5′-OH and either a 3′- or 2′-phosphate that would need to be processed to allow ligation. Structural studies [30–32] have also shown that rNMPs in DNA distorts the double helix, which could promote binding of proteins that results in nicking to initiate mutagenic processing.
Supplementary Material
Supplementary Table 1. Strains used in this study for measuring mutation rates or for strain construction.
Supplementary Table 2. Large indels and complex mutations observed in rnh201Δ strains
Acknowledgments
We thank Shay Covo and Jessica Williams for thoughtful comments on the manuscript, and the NIEHS Molecular Genetics Core for technical support in DNA sequence analysis of ura3 mutants. This work was funded by the Division of Intramural Research of the NIH, NIEHS, Project Z01 ES065089.
Footnotes
Conflict of interest statement
The authors declare that there are no conflicts of interest.
References
- 1.Joyce CM. Choosing the right sugar: how polymerases select a nucleotide substrate. Proc Natl Acad Sci U S A. 1997;94:1619–1622. doi: 10.1073/pnas.94.5.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeLucia AM, Grindley ND, Joyce CM. An error-prone family Y DNA polymerase (DinB homolog from Sulfolobus solfataricus) uses a ‘steric gate’ residue for discrimination against ribonucleotides. Nucleic Acids Res. 2003;31:4129–4137. doi: 10.1093/nar/gkg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nick McElhinny SA, Watts B, Kumar D, Watt DL, Lundström EB, Burgers PMJ, Johansson E, Chabes A, Kunkel TA. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc Natl Acad Sci U S A. 2010;107:4949–4954. doi: 10.1073/pnas.0914857107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavanaugh NA, Beard WA, Wilson SH. DNA polymerase beta ribonucleotide discrimination: Insertion, misinsertion, extension, and coding. Jorrnal of Biological Chemistry. 2010 doi: 10.1074/jbc.M110.132407. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 6.Chabes A, Georgieva B, Domkin V, Zhao X, Rothstein R, Thelander L. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell. 2003;112:391–401. doi: 10.1016/s0092-8674(03)00075-8. [DOI] [PubMed] [Google Scholar]
- 7.Nick McElhinny SA, Kumar D, Clark AB, Watt DL, Watts BE, Lundstrom EB, Johansson E, Chabes A, Kunkel TA. Genome instability due to ribonucleotide incorporation into DNA. Nat Chem Biol. 2010;6:774–781. doi: 10.1038/nchembio.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–130. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nick McElhinny SA, Gordenin DA, Stith CM, Burgers PM, Kunkel TA. Division of labor at the eukaryotic replication fork. Mol Cell. 2008;30:137–144. doi: 10.1016/j.molcel.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiu J, Qian Y, Frank P, Wintersberger U, Shen B. Saccharomyces cerevisiae RNase H(35) functions in RNA primer removal during lagging-strand DNA synthesis, most efficiently in cooperation with Rad27 nuclease. Mol Cell Biol. 1999;19:8361–8371. doi: 10.1128/mcb.19.12.8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen JZ, Qiu J, Shen B, Holmquist GP. Mutational spectrum analysis of RNase H(35) deficient Saccharomyces cerevisiae using fluorescence-based directed termination PCR. Nucleic Acids Res. 2000;28:3649–3656. doi: 10.1093/nar/28.18.3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 13.Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, Lindblom A, Lynch HT, Peltomaki P, Ramsey SD, Rodriguez-Bigas MA, Vasen HF, Hawk ET, Barrett JC, Freedman AN, Srivastava S. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet. 2009;76:1–18. doi: 10.1111/j.1399-0004.2009.01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Streisinger G, Okada Y, Emrich J, Newton J, Tsugita A, Terzaghi E, Inouye M. Frameshift mutations and the genetic code. Cold Spring Harbor Symposia on Quantitative Biology. 1966;31:77–84. doi: 10.1101/sqb.1966.031.01.014. [DOI] [PubMed] [Google Scholar]
- 16.Garcia-Diaz M, Kunkel TA. Mechanism of a genetic glissando: structural biology of indel mutations. Trends Biochem Sci. 2006;31:206–214. doi: 10.1016/j.tibs.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 17.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 18.Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem Rev. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 19.Hsieh P, Yamane K. DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech Ageing Dev. 2008;129:391–407. doi: 10.1016/j.mad.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pavlov YI, Rogozin IB, Galkin AP, Aksenova AY, Hanaoka F, Rada C, Kunkel TA. Correlation of somatic hypermutation specificity and A-T base pair substitution errors by DNA polymerase eta during copying of a mouse immunoglobulin kappa light chain transgene. Proc Natl Acad Sci U S A. 2002;99:9954–9959. doi: 10.1073/pnas.152126799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldstein AL, McCusker JH. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast. 1999;15:1541–1553. doi: 10.1002/(SICI)1097-0061(199910)15:14<1541::AID-YEA476>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 22.Shcherbakova PV, Kunkel TA. Mutator phenotypes conferred by MLH1 overexpression and by heterozygosity for mlh1 mutations. Mol Cell Biol. 1999;19:3177–3183. doi: 10.1128/mcb.19.4.3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clark AB, Kunkel TA. Cadmium inhibits the functions of eukaryotic MutS complexes. J Biol Chem. 2004;279:53903–53906. doi: 10.1074/jbc.C400495200. [DOI] [PubMed] [Google Scholar]
- 24.Marsischky GT, Filosi N, Kane MF, Kolodner R. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 1996;10:407–420. doi: 10.1101/gad.10.4.407. [DOI] [PubMed] [Google Scholar]
- 25.Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, Baumann C, Baxter P, Bertini E, Chandler KE, Chitayat D, Cau D, Dery C, Fazzi E, Goizet C, King MD, Klepper J, Lacombe D, Lanzi G, Lyall H, Martinez-Frias ML, Mathieu M, McKeown C, Monier A, Oade Y, Quarrell OW, Rittey CD, Rogers RC, Sanchis A, Stephenson JB, Tacke U, Till M, Tolmie JL, Tomlin P, Voit T, Weschke B, Woods CG, Lebon P, Bonthron DT, Ponting CP, Jackson AP. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38:910–916. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- 26.Larrea AA, Lujan SA, Nick McElhinny SA, Mieczkowski PA, Resnick MA, Gordenin DA, Kunkel TA. Genome-wide model for the normal eukaryotic DNA replication fork. Proc Natl Acad Sci U S A. 2010 doi: 10.1073/pnas.1010178107. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet. 2010;11:786–799. doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roa S, Li Z, Peled JU, Zhao C, Edelmann W, Scharff MD. MSH2/MSH6 complex promotes error-free repair of AID-induced dU:G mispairs as well as error-prone hypermutation of A:T sites. PLoS One. 2010;5:e11182. doi: 10.1371/journal.pone.0011182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clark AB, Kunkel TA. The importance of being DNA. Cell Cycle. 2010 doi: 10.4161/cc.9.22.14052. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Egli M, Usman N, Rich A. X-ray crystal structures of RNA-DNA hybrids. FASEB Journal. 1992;6:A150. [Google Scholar]
- 31.Ban C, Ramakrishnan B, Sundaralingam M. A single 2′-hydroxyl group converts B-DNA to A-DNA. Crystal structure of the DNA-RNA chimeric decamer duplex d(CCGGC)r(G)d(CCGG) with a novel intermolecular G-C base-paired quadruplet. J Mol Biol. 1994;236:275–285. doi: 10.1006/jmbi.1994.1134. [DOI] [PubMed] [Google Scholar]
- 32.Wahl MC, Sundaralingam M. B-form to A-form conversion by a 3′-terminal ribose: crystal structure of the chimera d(CCACTAGTG)r(G) Nucleic Acids Res. 2000;28:4356–4363. doi: 10.1093/nar/28.21.4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Strains used in this study for measuring mutation rates or for strain construction.
Supplementary Table 2. Large indels and complex mutations observed in rnh201Δ strains