

Abstract
Chronic periodontitis (CP) is a common oral disease that confers substantial systemic inflammatory and microbial burden and is a major cause of tooth loss. Here, we present the results of a genome-wide association study of CP that was carried out in a cohort of 4504 European Americans (EA) participating in the Atherosclerosis Risk in Communities (ARIC) Study (mean age—62 years, moderate CP—43% and severe CP—17%). We detected no genome-wide significant association signals for CP; however, we found suggestive evidence of association (P < 5 × 10−6) for six loci, including NIN, NPY, WNT5A for severe CP and NCR2, EMR1, 10p15 for moderate CP. Three of these loci had concordant effect size and direction in an independent sample of 656 adult EA participants of the Health, Aging, and Body Composition (Health ABC) Study. Meta-analysis pooled estimates were severe CP (n = 958 versus health: n = 1909)—NPY, rs2521634 [G]: odds ratio [OR = 1.49 (95% confidence interval (CI = 1.28–1.73, P = 3.5 × 10−7))]; moderate CP (n = 2293)—NCR2, rs7762544 [G]: OR = 1.40 (95% CI = 1.24–1.59, P = 7.5 × 10−8), EMR1, rs3826782 [A]: OR = 2.01 (95% CI = 1.52–2.65, P = 8.2 × 10−7). Canonical pathway analysis indicated significant enrichment of nervous system signaling, cellular immune response and cytokine signaling pathways. A significant interaction of NUAK1 (rs11112872, interaction P = 2.9 × 10−9) with smoking in ARIC was not replicated in Health ABC, although estimates of heritable variance in severe CP explained by all single nucleotide polymorphisms increased from 18 to 52% with the inclusion of a genome-wide interaction term with smoking. These genome-wide association results provide information on multiple candidate regions and pathways for interrogation in future genetic studies of CP.
INTRODUCTION
Chronic periodontitis (CP) is a common complex disease of the oral cavity that is characterized by an inflammatory response to commensal and pathogenic oral bacteria (1) and is found in about 20% of the adult US population. It manifests with gingival pocket formation and clinical attachment loss (CAL) and results in gradual destruction of periodontal tissues and tooth-supporting alveolar bone. CP is considered the main cause of tooth loss among adults and is associated with severe quality of life impacts (2). Moreover, a growing body of evidence links the disease with increased risk of systemic conditions, including coronary heart disease (CHD) (3), pregnancy outcomes (4), poor diabetes mellitus (DM) control (5) and others. Although definitive mechanistic evidence is lacking, it has been suggested that these links may be mediated to some degree via both the microbial component and the inflammatory burden that characterize the disease (6–8).
Risk factors for CP have been well studied and include smoking and DM. In addition, age, race and obesity have also been shown to be important risk indicators (9). A genetic component of CP risk was supported by early reports of familial aggregation of severe forms of the disease (10), as well as twin studies (11), but the magnitude of risk conferred by genetics and the role of specific genes has been under debate. Recent candidate gene studies for CP have focused on genes related to host immunity and inflammatory response such as cytokines, cell-surface receptors, chemokines, enzymes and antigen recognition. Most of these studies have examined polymorphisms in the interleukin (IL)-1, IL-6, Fc gamma receptor (FCGR2A), tumor necrosis factor (TNF) alpha, human vitamin D receptor (VDR), cluster of differentiation (CD)-14, matrix metalloproteinase-1, toll-like receptor (TLR), cyclo-oxygenase-2 (COX-2) and C-reactive protein genes with mixed results (12,13). A recent meta-analysis of studies investigating the association between CP and IL1 polymorphisms reported significantly elevated summary estimates for two IL1A polymorphisms [rs1800587 and rs17561, in tight linkage disequilibrium (LD)—r2 = 1.0; odds ratios (OR = 1.48; 95% confidence interval (CI = 1.17–1.86))] and one IL1B polymorphism (rs1143634; OR = 1.54; 95% CI = 1.03–2.30); the authors, however, noted an underlying heterogeneity in the published estimates (14). Whereas most of what is known or hypothesized with regard to genetic risk loci for CP has been based on candidate gene studies, to date no genome-wide association study (GWAS) has been performed. To add to the knowledge base of the genetic etiology of CP, this study aimed to investigate genetic risk loci for CP using a genome-wide association (GWA) approach in the context of a well-defined cohort.
RESULTS
GWA analysis results in the Atherosclerosis Risk in Communities (ARIC) cohort
The Atherosclerosis Risk in Communities (ARIC) participants had a mean age of 62 years, with a balanced sex distribution. Twelve percent were current smokers, and 11% had DM. In terms of Centers of Disease Control [CDC/American Academy of Periodontology (AAP)] periodontal diagnoses, 17% (n = 761) were classified as severe, 43% (n = 1920) as moderate and 40% (n = 1823) as healthy. The descriptive characteristics of the European American Dental ARIC cohort participants that were included in this analysis are presented in the Supplementary Material, Table S1. After exclusions that are described in the Materials and Methods section, there were 2 135 236 single nucleotide polymorphisms (SNPs) included in the GWA analysis. Genomic inflation factors were low, 1.019 for moderate and 1.024 for severe CP (Fig. 1). No genome-wide significant signals were noted; however, 26 SNPs in 6 loci (3 for moderate and 3 for severe CP) emerged below a P < 5 × 10−6 threshold (Fig. 2). The SNP with the lowest P-value in each of these six loci was, thus, prioritized for further annotation and follow-up (Table 1). The quality scores and proxies of these prioritized SNPs are presented in the Supplementary Material, Table S2. Adjustment for smoking and diabetes did not result in any important change in estimate (less than 10%) of association for these SNPs (Supplementary Material, Table S3). Similarly, sex-stratified analyses did not reveal any departures from homogeneity (Supplementary Material, Table S4). Consistent with the current trends in reporting of GWAS (15), we have made available the results of the entire set of SNPs that we investigated in ascending P-value order at: http://genomewide.net/public/aric/dental/periodontitis/CDC/cdc1vs0_full.txt and http://genomewide.net/public/aric/dental/periodontitis/CDC/cdc2vs0_full.txt.
Figure 1.
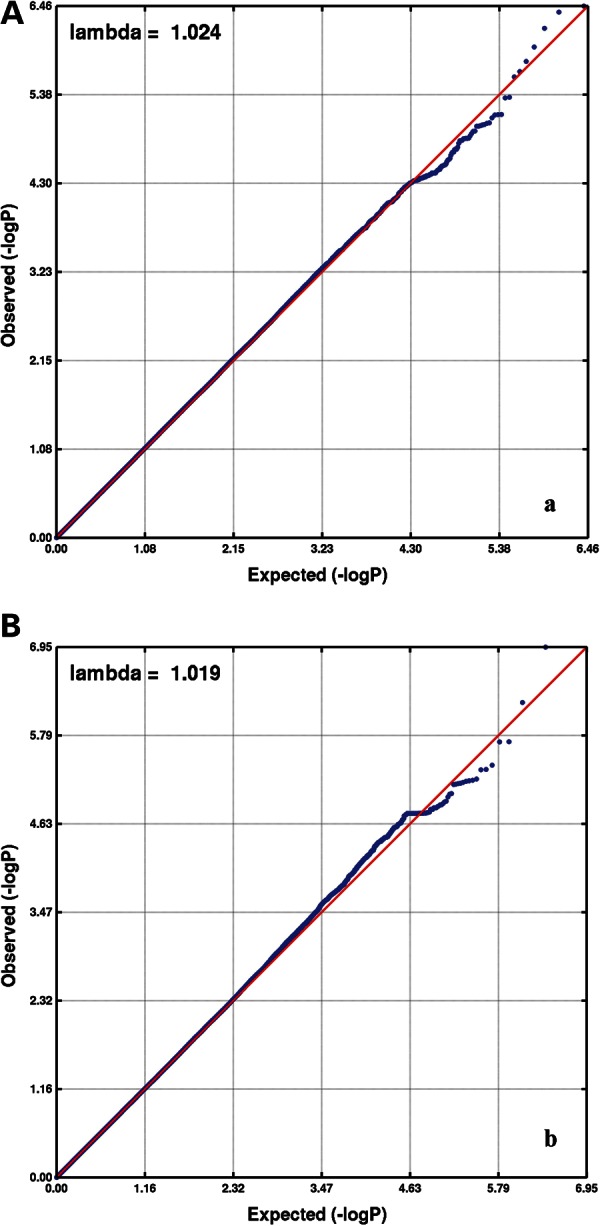
Quantile–quantile plots of genome-wide association analysis results of severe CP (A) and moderate CP (B) complex in the ARIC cohort.
Figure 2.

Manhattan plots of genome-wide association analysis results of severe CP (A) and moderate CP (B) complex in the ARIC cohort.
Table 1.
Genome-wide association analysis results of the CDC/AAP CP classification traits (severe CP versus healthy and moderate CP versus healthy) among the European American participants of the Dental ARIC study cohort (n = 4504), including single nucleotide polymorphisms (SNPs) with MAF-HapMap II CEU ≥ 5% and associated P < 5 × 10−6; the SNP with the lowest P-value per locus is presented; additional prioritized SNPs in each locus are presented in the footnote, along with corresponding r2 (based on 1000 genomes pilot 1 release) with the top SNPs
| Locus | SNP | Position Build36 | Strand | Imputed | Major allele | MAFa (HapMap II-CEU) | Closest gene(s) and position or distance | Pb | Risk allele frequency disease/healthy | OR (95% CI) |
|---|---|---|---|---|---|---|---|---|---|---|
| Severe CP | ||||||||||
| 14q21c | rs12883458 | 50349129 | + | Yes | T | [C] 0.10 | NIN (Intronic) | 3.5 × 10−7 | [C] 0.13/0.09 | 1.89 (1.48, 2.41) |
| 7p15d | rs2521634 | 24344565 | + | Yes | G | [A] 0.25 | NPY (47Kb) | 1.6 × 10−6 | [G] 0.80/0.74 | 1.47 (1.25, 1.73) |
| 3p21e | rs11925054 | 55365926 | + | No | G | [T] 0.14 | WNT5A (109Kb); ERC2 (151Kb) | 6.5 × 10−7 | [G] 0.90/0.86 | 1.69 (1.37, 2.10) |
| Moderate CP | ||||||||||
| 6p21.1f | rs7762544 | 41487293 | + | Yes | A | [G] 0.18 | NCR2 (61Kb) | 1.1 × 10−7 | [G] 0.21/0.16 | 1.41 (1.24, 1.60) |
| 19p13.3g | rs3826782 | 6838736 | + | Yes | G | [A] 0.07 | EMR1 (Intronic); VAV1 (30Kb) | 4.0 × 10−6 | [A] 0.05/0.04 | 2.00 (1.48, 2.70) |
| 10p15 | rs12260727 | 10378335 | + | Yes | G | [A] 0.15 | CELF2 (709Kb) | 6.0 × 10−7 | [G] 0.89/0.85 | 1.54 (1.30, 1.82) |
CI, confidence interval.
aMinor allele frequency.
bBased on logistic regression, log-additive models, including terms for age, sex, study center and ancestry (10 first PCs).
cAdditional SNPs in locus with P < 5 × 10−6: rs1004832 (r2 = 1.00), rs8009874 (r2 = 0.84) and rs12893300 (r2 = 0.49).
dAdditional SNP in locus with P < 5 × 10−6: rs11771124 (r2 = 1.00).
eAdditional SNP in locus with P < 5 × 10−6: rs503022 (r2 = 0.52).
fAdditional SNPs in locus with P < 5 × 10−6: rs9357360 (r2 = 0.89), rs1853406 (r2 = 0.89) and rs1535582 (r2 = 0.33).
gAdditional SNP in locus with P < 5 × 10−6: rs12610529 (r2 = 0.79).
Visualizations of the six loci of interest, along with nearby genes and recombination rates, are presented in Figure 3. For severe CP, the strongest (with respect to P-value) association in the 14q21 locus was produced by rs12883458. This SNP is located in an intron of Ninein (GSK3B interacting protein; NIN). The minor allele [C] showed a 3.5% enrichment among severe CP patients and was associated with an OR = 1.89 (95% CI = 1.48–2.41; P = 3.5 × 10−7). In the 7p15 locus, the major allele [G] of an intergenic SNP, rs2521634 [minor allele frequency (MAF) [A] = 0.25; 47 Kb downstream from Neuropeptide Y (NPY)], produced an OR = 1.47 (95% CI = 1.25–1.73; P = 1.6 × 10−6). Similarly, the major allele [G] of rs11925054 (MAF [T] = 0.14) in the 3p21 locus, downstream from wingless-type mouse mammary tumor virus integration site family, member 5A (WNT5A) and ELKS/RAB6-interacting/CAST family member 2 (ERC2), produced the strongest signal in the region (OR = 1.69; 95% CI = 1.37–2.10; P = 6.5 × 10−7), showing 4% enrichment among severe CP patients.
Figure 3.

Visualization of the six loci that were prioritized based on a P < 5 × 10−6 criterion in the ARIC cohort. Three loci were prioritized based on their association with severe CP (A: 14q21, NIN, rs12883458; B: 7p15, NPY, rs2521634; C: 3p21, WNT5A/ERC2, rs11925054) and three for their association with moderate CP (D: 6p21.1, NCR2, rs7762544; E: 19p13.3, EMR1, rs3826782; F: 10p15, rs12260727). The vertical axis corresponds to each locus' (index) SNP−log10 P-value; the SNP associated with the lowest P-value is labeled. The overlaid recombination rate plot and the color-coded pairwise linkage disequilibrium values with index SNPs were calculated based on HapMap II–CEU (human genome 18, build 36).
With regard to moderate CP, the SNP with the lowest P-value in the 6p21.1 locus was rs7762544 (MAF [G] = 0.18; OR = 1.41; 95% CI = 1.24–1.60; P = 1.1 × 10−7). This variant is 61 Kb downstream from natural cytotoxicity triggering receptor 2 (NCR2), and its minor (risk) allele showed 4.4% enrichment among moderate CP patients when compared with healthy participants. SNP rs3826782 (MAF [A] = 0.07), located in an intronic region of Egf-like module containing, mucin-like, hormone receptor-like 1(EMR1) and 30 Kb downstream from Vav 1 guanine nucleotide exchange factor (VAV1), provided the strongest signal in that locus (OR = 2.00; 95% CI = 1.48–2.70; P = 4.0 × 10−6), showing a small 1% enrichment among ARIC ‘cases’. Examination of the SCAN database for expression-associated data (Supplementary Material, Table S5) revealed that this SNP has been associated (P = 2 × 10−5) with the expression of GPR113 on lymphoblastoid cell lines (LCLs). GPR113 has been found to be expressed intraorally, by taste receptor cells (16), and has functions on the neuropeptide signaling pathway [Gene Ontology (GO) accession: 0007218] and signal transducer activity (GO accession: 0004871)—pathways relevant to NPY. Finally, the major allele [G] of rs12260727 (MAF [A] = 0.15) in the 10p15 locus was associated with OR = 1.54 (95% CI = 1.30–1.82; P = 6.0 × 10−7). SNP rs12260727 is located distant (700 Kb upstream) from the closest gene CUGBP, Elav-like family member 2 (CELF2). As expected, all SNPs in LD (r2 > 0.80) displayed directionally consistent and of a similar magnitude effect estimates as the index SNP at each locus.
Examination of the prioritized SNPs in the subset of 1020 European American Dental ARIC participants previously reported in a GWAS of periodontal pathogen colonization by Divaris et al. (17) showed no important association with high colonization with periodontal pathogens of the ‘red’ or ‘orange’ complex, or individual bacteria such as Aggregatibacter actinomycetemcomitans and Porphyromonas gingivalis (Supplementary Material, Table S6). The number of concordant effect estimates (n = 13) that we detected in this additional ‘look-up’ was not statistically different from what would be expected by chance alone, among 24 examined effect estimates (binomial test P = 0.5). However, NPY and NCR2 were two loci that showed concordant effect estimates with all four bacterial traits. Examination of additional polymorphisms in the CD14, FCGR2A, IL1A, IL1B, IL1RN, IL4, IL6, IL10, TLR4, TNF and VDR genes that have previously been reported (13) as associated with CP in at least one candidate gene study among whites of European descent did not show any evidence of association (Table 2).
Table 2.
P-values for the association with severe and moderate CP in the Dental ARIC GWAS of polymorphisms that have been examined and reported as associated in at least one study among whites of European descent in the review of Laine et al. (13)
| ARIC GWAS Pb |
|||||
|---|---|---|---|---|---|
| Gene | SNP | Position (build 36) | MAFa (HapMap II-CEU) | Severe CP | Moderate CP |
| CD14 | rs2569190 | 139993100 | [A] 0.47 | 0.57 | 0.75 |
| FCGR2A | rs1801274 | 159746369 | [A] 0.50 | 0.66 | 0.37 |
| IL10 | rs1800872 | 205013030 | [T] 0.22 | 0.89 | 0.56 |
| IL1A | rs1800587 | 113259431 | [A] 0.31 | 0.20 | 0.51 |
| rs17561 | 113253694 | [A] 0.31 | 0.20 | 0.50 | |
| IL1B | rs1143634 | 113306861 | [A] 0.26 | 0.21 | 0.27 |
| IL1RN | rs419598 | 113603678 | [C] 0.27 | 0.31 | 0.97 |
| IL4 | rs2070874 | 132037609 | [T] 0.15 | 0.17 | 0.62 |
| rs2243250 | 132037053 | [T] 0.15 | 0.17 | 0.63 | |
| IL6 | rs1800795 | 22733170 | [C] 0.43 | 0.87 | 0.18 |
| rs2069827 | 22731981 | [T] 0.10 | 0.31 | 0.49 | |
| MMP1 | rs475007 | 102174522 | [T] 0.44 | 0.90 | 0.12 |
| TLR4 | rs4986791 | 119515423 | [T] 0.06 | 0.93 | 0.27 |
| rs4986790 | 119515123 | [G] 0.06 | 0.93 | 0.27 | |
| TNF | rs1800629 | 31651010 | [A] 0.17 | 0.68 | 0.77 |
| VDR | rs731236 | 46525024 | [G] 0.41 | 0.69 | 0.59 |
| rs1544410 | 46526102 | [T] 0.41 | 0.68 | 0.59 | |
aMinor allele frequency.
bBased on logistic regression, log-additive models, including terms for age, sex, study center and ancestry (10 first PCs).
Replication and meta-analysis in the Health, Aging and Body Composition (Health ABC) Study
Three SNPs associated with severe CP (rs12883458, rs2521634 and rs11925054) and three SNPs associated with moderate CP (rs7762544, rs3826782, rs12260727) in the ARIC Study (P < 5 × 10−6) were investigated using the next largest cohort available to us, the Heath ABC Study (n = 686). The descriptive characteristics of the Health, Aging, and Body Composition (Health ABC) Study participants included in this analysis are presented in Supplementary Material, Table S1.
Of the three SNPs investigated with severe CP, one SNP (NPY locus: rs2521634) was nominally associated with severe CP in the Health ABC Study (OR = 1.64; 95% CI = 1.01–2.65; P = 0.046), with a concordant effect direction and magnitude as in ARIC (Table 3). Similarly, the EMR1 locus (rs3826782) showed concordant effect direction and magnitude (OR = 2.06; 95% CI = 1.03–4.60; P = 0.06). The NCR2 (rs7762544) locus showed similar effect direction for moderate CP risk in Health ABC (OR = 1.32, 95% CI = 0.81–2.14; P = 0.27). None of these SNPs met statistical significance criteria based on a Bonferroni-corrected (assuming α = 0.05 and six independent tests) P-value criterion of 0.0083. No significant effect was observed with rs12260727 at the 10p15 locus (OR = 0.93; 95% CI = 0.56–1.57; P = 0.8). The remaining three loci showed non-significant effects of opposite direction in Health ABC. Allele frequencies of these SNPs in the Health ABC sample are presented in the Supplementary Material, Table S7.
Table 3.
Meta-analysis of GWAS results (meta-analysis P-values and effect estimates) of prioritized SNPs for severe and moderate CP in the ARIC and the Health ABC studies
| ARIC |
Health ABC |
Meta-analysis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pooled estimatesa | Heterogeneity assessmentb | ||||||||||
| Locus | SNP | Pc | OR (95% CI) | Pc | OR (95% CI) | P | OR (95% CI) | Diff. direction | I2 | X2 | P < 0.2 |
| Severe CP | |||||||||||
| NIN | rs12883458 | 3.5 × 10−7 | 1.89 (1.48, 2.41) | 0.25 | 0.65 (0.32, 1.35) | * | 86.6 | 7.5 | * | ||
| NPY | rs2521634 | 1.6 × 10−6 | 1.47 (1.25, 1.73) | 0.05 | 1.64 (1.01, 2.65) | 3.5 × 10−7 | 1.49 (1.28, 1.73) | 0 | 0.2 | ||
| WNT5A/ERC2 | rs11925054 | 6.5 × 10−7 | 1.69 (1.37, 2.10) | 0.06 | 0.51 (0.25, 1.00) | * | 90.1 | 10.1 | * | ||
| Moderate CP | |||||||||||
| NCR2 | rs7762544 | 1.1 × 10−7 | 1.41 (1.24, 1.60) | 0.27 | 1.32 (0.81, 2.14) | 7.5 × 10−8 | 1.40 (1.24, 1.59) | 0 | 0.6 | ||
| EMR1/VAV1 | rs3826782 | 4.0 × 10−6 | 2.00 (1.48, 2.70) | 0.06 | 2.06 (1.03, 4.60) | 8.2 × 10−7 | 2.01 (1.52, 2.65) | 0 | 0.9 | ||
| 10p15 | rs12260727 | 6.0 × 10−7 | 1.54 (1.30, 1.82) | 0.80 | 0.93 (0.56, 1.57) | * | 67.7 | 3.1 | * | ||
Reported effect estimates correspond to the ‘risk’ allele in the ARIC cohort. Pooled estimates based on inverse-variance weighted meta-analysis are presented for results that did not demonstrate substantial heterogeneity (homogeneity X2 P < 0.2 criterion)
OR, odds ratio; CI, confidence interval.
aBased on inverse-variance weighted meta-analysis.
bAsterisks (*) correspond to effects of different direction in the ARIC and Health ABC cohorts and homogeneity X2 P-values of less than 0.2.
cBased on logistic regression, log-additive models, including terms for age, sex, study center and ancestry (10 first PCs).
The three SNPs (rs2521634, rs7762544 and rs3826782) that showed concordant effect direction and magnitude in the discovery and replication cohorts were carried forward for meta-analysis, resulting in the following summary estimates: severe CP—NPY, rs2521634 [G]: ORpooled = 1.49 (95% CI = 1.28–1.73; P = 3.5 × 10−7); moderate CP—NCR2, rs7762544 [G]: ORpooled = 1.40 (95% CI = 1.24–1.59; P = 7.5 × 10−8), EMR1, rs3826782 [A]: ORpooled = 2.01 (95% CI = 1.52–2.65; P = 8.2 × 10−7).
Genome-wide interactions with smoking
The exploratory genome-wide interaction analysis between all SNPs and smoking history (ever versus never smoker) revealed one genome-wide significant locus for moderate CP at NUAK1 (12q23.3), where the intronic rs11112872 (MAF [G] = 0.42) produced an interaction term P = 2.9 × 10−9. Smoking-stratified estimates of moderate CP for the minor allele were never smokers (n = 1825)—OR = 0.77 (95% CI = 0.67–0.88; P = 2.2 × 10−4) and ever smokers (n = 1838)—OR = 1.42 (95% CI = 1.23–1.64; P = 2.8 × 10−6). This interaction for moderate CP was not replicated in Health ABC (never smokers—n = 227, ever smokers—n = 232 in the moderate CP versus health analysis), where the stratified estimates were of discordant direction when compared with ARIC. All other interaction estimates for moderate and severe CP were of substantially smaller magnitude (P-values around 10−6 or greater) and are available as Supplementary Material at http://genomewide.net/public/aric/dental/periodontitis/CDC/cdc1vs0_smoke.txt and http://genomewide.net/public/aric/dental/periodontitis/CDC/cdc2vs0_smoke.txt.
Estimation of variance explained by all SNPs
The results of the examination of heritable variance explained by all GWAS SNPs are presented in Table 4. Variance explained (h2) estimates were consistently higher for severe CP when compared with moderate CP. Some variations in the h2 estimates were also noted with regard to the SNP sets that were used for analyses. Interestingly, higher variance in moderate disease was explained with the use of imputed versus genotyped SNPs, whereas the inverse was true for severe CP. After adjustment for population stratification and using imputed SNPs, h2 was 7% for moderate CP and 8% for severe CP. Higher phenotypic variance (18%) was explained in severe disease with the use of genotyped-only SNPs. However, standard errors were large for all these estimates. Notably, the inclusion of a G × E interaction term with smoking history produced high estimates of variance jointly explained by SNPs and their interaction with smoking, ranging between 36 and 41% for moderate CP and 42 and 52% for severe CP.
Table 4.
Phenotypic variance explained for moderate and severe CP by all genotyped and imputed autosomal SNPs available and their interaction with smoking history (ever/never smoker) using REML analysis implemented with GCTA [Yang et al. (56)], among the European American participants of the Dental ARIC study (n = 4504)
| Genotyped SNPs | Imputeda SNPs |
||
|---|---|---|---|
| Exclusion filters: | MAF < 0.05 | MAF < 0.05, R2b < 0.6 | MAF < 0.05, R2b < 0.3 |
| n (SNPs)c: | 656 292 | 2 104 905 | 2 131 070 |
| Varianced explained (SE) | Varianced explained (SE) | Varianced explained (SE) | |
| Moderate CP | |||
| Only SNPs considered | 0.057 (0.13) | 0.097 (0.11) | 0.099 (0.11) |
| +10 PCs for population structure | 0.006 (0.14) | 0.068 (0.12) | 0.069 (0.12) |
| +10 PCs, sex | 0.000 (0.14) | 0.048 (0.12) | 0.049 (0.12) |
| +10 PCs, age | 0.008 (0.14) | 0.082 (0.12) | 0.083 (0.12) |
| +10 PCs, sex, age | 0.000 (0.14) | 0.066 (0.12) | 0.067 (0.12) |
| +10 PCs, sex, age, [G] × [smoking history]e | 0.358 (0.26) | 0.405 (0.23) | 0.391 (0.24) |
| [G] × [E] term (LRT X2) P | 0.1 | 0.04 | 0.04 |
| Severe CP | |||
| Only SNPs considered | 0.298 (0.18) | 0.214 (0.15) | 0.212 (0.15) |
| +10 PCs for population structure | 0.175 (0.19) | 0.083 (0.16) | 0.080 (0.16) |
| +10 PCs, sex | 0.221 (0.19) | 0.110 (0.16) | 0.109 (0.16) |
| +10 PCs, age | 0.171 (0.19) | 0.097 (0.16) | 0.094 (0.16) |
| + 10 PCs, sex, age | 0.220 (0.19) | 0.127 (0.16) | 0.127 (0.16) |
| +10 PCs, sex, age, [G] × [smoking history]e | 0.515 (0.35) | 0.425 (0.31) | 0.418 (0.31) |
| [G] × [E] term (LRT X2) P | 0.04 | 0.06 | 0.06 |
GCTA, genome-wide complex trait analysis; MAF, minor allele frequency; SE, standard error; PCs, principal components; LRT, likelihood ratio test comparing models, including the [G] × [E] term against ‘reduced’ models without the interaction term.
aImputed using HapMap II-CEU.
bImputation quality score.
cNumber of SNPs that were used to estimate the GRM after exclusions, among the study participants as a first step in the GCTA prior to conducting REML.
dAdjusted to the prevalence of CP in the Dental ARIC cohort, moderate CP—0.43 and severe CP—0.17.
eSmoking history was defined as a binary variable, where 0: never smoker (47% of participants) and 1: ever smoker (53% of participants).
Canonical signaling pathway analysis
Of the 147 Ingenuity Pathway Analysis (IPA) canonical signaling pathways that were tested in the moderate and severe CP results data, 11 were significantly enriched (P < 3.4 × 10−4) (Supplementary Material, Figs S1 and S2). Contrarily, none of the metabolic pathways that we tested as controls was associated with the data. Two of the identified pathways (axonal guidance and neuropathic pain signaling) were common for severe and moderate CP, eight (i.e. synaptic long-term potentiation, cAMP responsive element binding protein signaling in neurons, N-formyl-methionyl-leucyl-phenylalanine (fMLP) signaling in neutrophils, neuronal nitric oxide synthase 1 signaling in neurons and virus entry via endocytic pathways) were unique to severe CP and one (amyotrophic lateral sclerosis signaling) was unique to moderate CP. Overall, neurotransmitters and nervous signaling pathways were the majority (9 out of 12) of those enriched. Axonal guidance signaling was the most significant (severe CP—P = 2.1 × 10−7, ratio = 0.194; moderate CP—P = 1.4 × 10−5, ratio = 0.173) and included WNT5A, the only prioritized locus that was represented in the top pathways. Axonal guidance signaling remained the top canonical pathway for both moderate and severe CP when the pathway analysis was repeated using a smaller set of loci that met more stringent association criteria (P < 10−3), but did not reach statistical significance. The remaining three identified pathways were cellular immune response related, namely fMLP signaling in neutrophils, CXCR4 signaling and virus entry via endocytic pathways.
DISCUSSION
In this work, we present the results of the first genome-wide investigation of CP. In our GWAS, we identified six loci providing suggestive evidence of association with CP. Although no signals met genome-wide significance criteria, we found that three of these loci (NPY, NCR2 and EMR1) had concordant effect direction in a replication sample, with NPY being nominally associated with severe CP. Similarly, a genome-wide significant interaction of NUAK1 with smoking was not replicated. Nevertheless, our estimates of phenotypic heritability explained by all SNPs were significantly increased with the inclusion of a G × E interaction term with smoking. Furthermore, our findings suggest an important role of neurotransmitter and nervous system signaling pathways in the risk of CP, complementary to immune response-related ones.
Although limited by its sample size of about 5000 subjects, this study was conducted using a well-defined cohort with detailed characterization of CP, using full-mouth periodontal examinations and the latest consensus taxonomy of CP. These results provide a wealth of new information on potential candidate genes that will require further exploration, replication and validation in future studies. Our GWA scan did not identify any SNP that met strict genome-wide statistical significance criteria (P < 5 × 10−8). However, many true signals, elements of the ‘missing heritability’ may be found below this threshold (18–22). Therefore, we used a more lenient P-value threshold for prioritization and annotation of a set of ‘promising’ SNPs, reported estimates of variance explained by all GWAS SNPs and made available the complete set of our GWAS results.
The fact that virtually all prioritized loci are related to host immunity is consistent with the current understanding of the etiology and pathogenesis of periodontal diseases (1,8,23). The preponderance of neurotransmitter and nervous system signaling pathways among those significantly associated with our data echo reports that emphasize the role of the nervous system in the pathophysiology of peripheral inflammation and suggest a neurogenic inflammatory component for periodontitis (24). In fact, a recent study found differences in gingival crevicular fluid levels of NPY between healthy and periodontitis-affected sites (25). Nevertheless, CP is a complex, polygenic disease, wherein multiple genes are likely to confer risk or protection by influencing the host inflammatory response and the qualitative and quantitative composition of the oral microbiome. Noteworthy, two of the six prioritized loci (NPY and NCR2) showed concordant effect estimates for all examined ‘high periodontal pathogen colonization’ traits in a previous GWAS (17), an observation that parallels the pathogenic oral microbial shift that is characteristic of periodontitis (8).
It is well established that ‘environmental’ and behavioral factors are important in CP, with the major lifestyle risk factor being smoking (26,27). Although smoking is an unlikely confounder of genetic effects on CP risk, synergistic effects are likely. Tomar and Asma (28) used 1988–1994 NHANES data to estimate that up to 52% of periodontitis cases in the USA were attributable to current or former smoking, whereas among current smokers, 75% of periodontitis cases were attributable to smoking. Although our study was under-powered to efficiently examine G × E interactions, we detected one significant interaction of NUAK1 with smoking that was not replicated in the Health ABC sample. Although some level of misclassification in the environmental exposure cannot be ruled out, we support that further investigation of candidate gene interactions with smoking, possibly involving more sensitive measures such as serum cotinine levels and smoking pack years, may lead to the discovery of novel risk loci for CP. The overall strong effect of smoking was confirmed in our data, where the inclusion of a smoking interaction term with the genetic heritable variance component improved significantly the phenotypic variance explained in both periodontitis traits, reaching up to 52% for severe periodontitis. Although these results were imprecise and should be interpreted with caution, they provide support for a strong interaction of smoking with common variation that should be explored in detail in future genetic and mechanistic studies.
The lack of an overlap of risk loci for the moderate and severe disease traits is not surprising. Although these results are preliminary, the lack of overlap is consistent with our approach of examining the two CP diagnoses separately: moderate and severe CP are considered largely distinct entities, rather than disease progression stages. Since the first population-based studies of periodontal disease natural history, the most severe forms of disease were distinguished into moderate and mild forms; those in the top 15–18% of the distribution have different rates of clinical progression, microbial composition, host response and molecular (inflammatory) characteristics (29–31). Interestingly, the heritability estimates that we obtained for the severe CP trait in this GWAS were consistently higher when compared with those obtained for the moderate trait. Also, from a statistical standpoint, the lack of overlap is not surprising because small stochastic variations can have a big impact on the tails of the test statistic distribution. However, because these traits share a common pathogenetic underpinning, and in our analyses we used the same controls for both contrasts, some overlap in GWA signals should be anticipated. The discovery of the same nervous system signaling pathway as the most significantly associated with both traits may indeed be an indication of a shared pathogenetic framework. Explorations at lower P-value thresholds may reveal more ‘good signals’ and common risk loci.
The heritability estimates attributable to common variation that we obtained are lower when compared with previous estimates reported by Michalowicz et al. (11,32) from studies among twins. A more recent study used an animal model to estimate 35% heritability in alveolar bone loss, after adjustment for age and sex (33). As noted by Yang et al. (34), heritability estimates obtained via the use of GWAS SNPs are likely underestimates of the true heritability due to common variation because of incomplete LD between causal variants and available SNPs and the fact that causal variants may have lower MAF when compared with all other SNPs examined. We anticipate that the precision and validity of these estimates will improve as higher density imputations and whole genome sequencing are becoming more common and larger GWAS samples more feasible.
A recent GWAS of generalized aggressive periodontitis (gAgP) among a sample of whites of European descent identified associations with a susceptibility locus on 9p34.3 intronic to the glycosyltransferase 6 domain containing 1 (GLT6D1) gene and a shared susceptibility locus on 9p21.3 for both gAgP and CHD (35,36). However, gAgP is a rare form of periodontitis, found in less than 1% of adults and is a distinct entity from CP. Noteworthy, none of the previously reported CP risk candidate gene polymorphisms met nominal statistical significance criteria in our study. This finding requires further study, nevertheless it may be reflective of the unavoidable systematic biases that may affect small sample size case-control candidate gene studies; in this respect, the ‘agnostic’ genome-wide approach that we employed using two population-based samples constitutes an improvement.
Our examination of two dichotomous disease traits, including shared ‘healthy’ controls in both cohorts can be considered as one of the study limitations. Examinations of quantitative traits such as extent of attachment loss, probing depth and/or tooth loss that capture a continuum in disease severity might offer a more powerful approach when conducting GWAS; however, we suggest that using current diagnostic criteria for CP is a valuable first step in exploring the genetic basis of the disease. We are aware of the ongoing debate on CP definition and classification (37,38). Although the CDC/AAP classification is the consensus taxonomy implemented in epidemiologic studies and surveillance, it has several limitations. First, the case definitions include a reversible clinical marker (probing depth) and second, disease ascertainment is influenced by tooth loss (sites with severe disease are lost and unobservable). Although the latter issue is common to all CP classifications, improvements have been suggested. For example, Offenbacher et al. (31) introduced a refined CP classification characterizing the disease's biological (versus clinical-only) phenotype, a feature that may be advantageous in the exploration of genetic effects. Future research directions may include the interrogation of CP ‘endophenotypes’ (39), pleiotropic effects (40) in the context of phenome-wide association studies (41) and composite phenotypes involving clinical, molecular and oral microbiome characteristics. Such investigations may help shed light on the complex CP etiology, pathogenesis and variable clinical manifestation, as well as clarify possible shared genetic underpinnings with other systemic conditions.
In summary, we present the results of the first GWAS of CP among EA. Although none of the reported loci reached genome-wide significance levels, our data provide support for further investigation for the role of several loci in the risk of CP. Acknowledging that interpretation of these results should be made with caution, we support that the suggestive evidence on the six prioritized loci, the heritability estimates, including the significant interaction with smoking, the associated canonical signaling pathways involving the nervous system and immunity and the full set of GWAS results, may serve as a rich hypothesis-generating resource. Future research, including larger GWA, replication and fine-mapping studies, as well as mechanistic and experimental investigations will be required to further our understanding of the pathogenesis of the disease and may lead to novel preventive and therapeutic approaches.
MATERIALS AND METHODS
Study population
Our discovery GWA analysis was performed in a sample of European American participants of the ARIC study (42) age 53–74. The ARIC is a prospective cohort study of atherosclerosis, cardiovascular disease risk factors and outcomes that enrolled 15 792 community-dwelling residents in 4 US communities (Jackson, MS; Washington County, MD; suburban Minneapolis, MN; and Forsyth County, NC) between 1987 and 1989. The Dental ARIC is a National Institute of Dental and Craniofacial Research-funded ancillary study that took place during the fourth ARIC visit (1996–1998) and included complete oral-dental examinations in a subset (n = 6017) of dentate ARIC participants. The Health ABC Study is a National Institute on Aging-sponsored longitudinal investigation examining factors that contribute to incident disability and functional decline of healthier older persons, with a particular emphasis on changes in body composition in old age (43). Between April 1997 and June 1998, the Health ABC study had recruited 3075 70–79-year-old well-functioning community-dwelling adults. Study participants were recruited from a random sample of Medicare beneficiaries in Pittsburgh, Pennsylvania and Memphis, Tennessee. As part of the study year 2 and 3 follow-up clinical visits (1998–2000), a total of 1133 EA and AfricanAmerican participants received complete dental and periodontal examinations (44,45).
Phenotype measurement and definition
As part of the Dental ARIC, ancillary study participants underwent detailed oral-periodontal examinations that recorded the number of missing teeth, probing depths, attachment loss measurements and bleeding upon probing at six sites per tooth, including third molars. Dental ARIC clinical examiners were trained and calibrated against a standard examiner, with kappas indicating excellent to outstanding level of agreement (46). During the Health ABC oral examinations, trained and calibrated examiners obtained clinical plaque index, gingival index, PD and CAL measurements. An a priori minimum level of 90% agreement on all measures was set as a benchmark and was achieved by all examiners (45). We used the CDC and AAP consensus three-level classification for the disease trait definition in both cohorts. The CDC/AAP taxonomy uses CAL and PD criteria to define three CP categories as, healthy-mild (n = 1864), moderate (n = 1961) and severe (n = 785) (47). Based on this definition, we created two dichotomous traits: severe CP (severe versus healthy) and moderate CP (moderate versus healthy). To fully describe the clinical features of these traits, we present the distribution of the following clinical periodontal measures, overall and stratified by periodontitis diagnosis in each cohort: plaque score, gingival index, extent of bleeding on probing, mean probing depth (mm), mean attachment loss (mm), extent of probing depth (≥4 mm), extent of attachment loss (≥3 mm) and number of natural teeth.
Genotyping, imputation and quality control
In the ARIC study, population DNA was extracted from blood samples drawn from an antecubital vein into tubes containing serum separator gel. Blood samples were analyzed at a central ARIC laboratory in Houston, TX, USA. Genotyping was performed using the Affymetrix Genome-Wide Human SNP Array 6.0 chip that offers 906 600 markers for SNPs. The quality control procedures included initial blind duplicate genotyping and identification/flagging of SNPs with κ < 0.95 and reconciliation of unintentional duplicate samples (17 duplicates and 1 triplicate). Imputation to 2.5 million markers was performed using 669 450 SNPs and the MACH program version 1.0.16 (48), based on HapMap Phase II CEU build 36. The SNPs used for imputation were selected from 839 048 autosomal SNPs restricted to those with MAF > 0.01 (129 543 excluded), Hardy–Weinberg equilibrium (HWE) P > 10−5 (12 432 excluded) and call rate >95% (1693 excluded). We used the following criteria for exclusion of SNPs from further analyses: quality score < 0.8 and missing data rate > 10% after imputation and MAF of <5%.
In the Health ABC Study, population genomic DNA was extracted from buffy coat collected using PUREGENE® DNA Purification Kit during the baseline examination. Genotyping was performed by the Center for Inherited Disease Research using the Illumina Human1M-Duo BeadChip system. Illumina BeadStudio was used to call genotypes. Samples were excluded from the dataset for the reasons of sample failure, sex mismatch and first-degree relative of an included individual based on genotype data. Genotyping was successful for 1 151 215 SNPs in 1663 European American participants. Imputation for autosomal SNPs was done using MACH version 1.0.16. SNPs with MAF ≥ 1%, call rate ≥ 97% and HWE P ≥ 10−6 were used for imputation using HapMap Phase II CEU build 36 resulting in a final set of 2 543 887 SNPs.
Population stratification
To obtain estimates of relatedness and population stratification in ARIC, a subset of 85 947 ‘high quality’ SNPs was selected. These SNPs met the following criteria: MAF ≥ 0.1, call rate > 99.5%, HWE P ≥ 10−3, autosomal, with annotation in the platform annotation file, not labeled ‘AFFX’ or ‘chromosome 0’ and not monomorphic. Using these SNPs, identity-by-state (IBS) allele sharing distance (DST values) was computed using PLINK (49), as such: DST = IBS distance (IBS2 + 0.5 × IBS1)/(n SNP pairs). First-degree relative status was assigned to pairs of individuals with DST ≥ 0.8, and second-degree relatives were considered those with 0.763 ≤ DST < 0.8. There were 380 pairs of first degree and 207 pairs of second-degree relatives identified among the ARIC white participants of European descent. To minimize exclusions, related pairs were broken by iterative selection of individuals with most relatives using a custom-written program.
Population stratification was further evaluated with principal component (PC) analysis using the EIGENSTRAT program (50). The previously chosen set of SNPs was used for the computation of 10 PCs. Genetic outliers were considered those that were farther than 8 standard deviations away from any of 10 PCs over 10 runs of PC computation. Based on DST and PC criteria, there were 716 subjects flagged for removal from the analysis [206 as genetic outliers based on PCs and 16 based on average DST values (‘too little IBS sharing’ with the rest of the sample), 351 first-degree relatives and 143 second-degree relatives. All but 10 second-degree relatives (whose relatives were excluded as genetic outliers) were reentered in the dataset and were assigned PCs. After exclusion of 364 individuals (4%), there were 9349 European Americans who were included in the GWA analysis, and of those, 4504 had periodontal phenotype data available as dental ARIC participants.
Analytical strategy
The association between SNPs and the two disease traits (severe and moderate CP) was tested using logistic regression models assuming log-additive allelic effects, adjusting for age, sex, examination center and ancestry (10 first PCs). Per allele ORs and 95% CIs and associated P-values were estimated for each CP trait. Consistent with the current trends in reporting of GWAS (15), we have made available the results of the entire set of SNPs that we investigated. Because no confounding of the association between SNPs and the examined traits is expected by CP risk factors, the main analysis models were not adjusted for smoking or diabetic status. However, in the Supplementary Material, we present the results (effect estimates and percentage of change in estimate) of additional exploratory analyses, adjusting for smoking status and diabetes. In the ARIC ‘discovery’ cohort, we applied a multiple-test correction assuming 1 million independent tests, resulting to a genome-wide significance level of P < 5 × 10−8. All genetic analyses were performed with the ProbABEL software (51). An a priori threshold of P < 5 × 10−6 was set for prioritizing SNPs for further investigation and replication in the Health ABC Study. To inspect for any substantial differences in the effects of prioritized SNPs between males and females in ARIC, we conducted additional exploratory analyses stratified by sex. The departure from between-sexes homogeneity was assessed by inspection of non-overlapping CIs. To provide a more comprehensive view of the potential role of these SNPs in periodontitis-related traits, we obtained and empirically examined their effect size, direction, and P-values for four additional ‘high periodontal pathogen colonization’ traits that were available for a subset of participants in the European American Dental ARIC cohort (n = 1020) and have been previously studied in the context of a GWAS of the oral-periodontal microbiome (17).
Post-analysis procedures included the generation of quantile–quantile (Q–Q) and Manhattan plots and detailed annotations of gene context. We also examined the prioritized SNPs association with gene expression (as expression quantitative trait loci) using the SCAN (http://www.scandb.org) database and reported the P-values associated with gene expression on LCLs. Furthermore, P-values and effect estimates [OR (95% CI)] of the prioritized variants (one ‘top’ SNP per prioritized locus, as determined by the lowest P-value) in the Health ABC Study were obtained and examined for nominal significance, as well as for effect direction and magnitude concordance. ‘Replication’ was determined as concordant effect direction and nominal significance (P < 0.05) in the Health ABC Study. Effect estimates of SNPs that did not show substantial heterogeneity and ‘generalized’ to the Health ABC Study were combined in inverse-variance weighted meta-analysis to produce pooled (summary) ORs.
The prioritized SNPs were annotated using WGAViewer ver.1.26l (52) and Snipper ver. 1.2 (http://csg.sph.umich.edu/boehnke/snipper/), and regions of interest were viewed using LocusZoom ver.1.1 (53). We used PolyPhen-2 for the prediction of potentially damaging missense changes (54) and METAL (55) for the summarization and meta-analysis of estimates derived from the two cohorts. We used additional online resources of the National Center for Biotechnology Information (NCBI http://www.ncbi.nlm.nih.gov/). Reporting of genes was based on the ‘HUGO Gene Nomenclature’ naming convention (http://www.genenames.org). The full names and genomic locations of the reported genes are presented in the Supplementary Material, Table S8.
The estimation of heritable variance (h2) in the two periodontitis traits explained by all available SNPs in the present GWAS was performed with the use of GCTA (56). GCTA is based on a two-step method, where in the first step, all available SNPs are used to estimate a genetic relationship matrix (GRM) among all study participants. Subsequently, the GRM is used in restricted maximum likelihood (REML) analyses to estimate the proportion of variance explained by all SNPs. Adjustment for population stratification and the inclusion of additional covariates, including gene–environment interaction (G × E) terms are feasible in the REML step. Because the variance explained may be influenced by the unknown LD structure of available SNPs with causal variants, we examined three different sets of SNPs for the computation of the GRM: genotyped SNPs with MAF ≥0.05, as well as combinations of imputed SNPs with MAF ≥ 0.05 and imputation quality score (R2) ≥ 0.3 and ≥0.6. The maximum number of SNPs used was 2 131 070. Additionally, the inclusion of an interaction term with smoking was examined using a binary definition of smoking history (ever/never smoker) and inspecting the variance explained and a likelihood ratio test P-value (using a P < 0.2 criterion, as the examination of interaction effects is based on de facto underpowered analyses).
For the conduct of pathway analyses, we first identified a set of ‘independent’ signals providing nominal evidence association (using an arbitrary P-value criterion of <10−2) for each of the two disease traits using LD pruning. This step was implemented with PLINK, filtering SNPs that were correlated (r2 ≥ 0.1) with the index SNP (lowest P-value) in each locus and within a distance of 400 Kb. Next, the closest or harboring gene was assigned to each locus, if one was found within 100 Kb of the index SNP. Thus, 2 sets of 2328 loci and genes nominally associated with moderate CP and 2425 nominally associated with severe CP were carried forward to pathway analysis that was conducted using IPA (Ingenuity® Systems, www.ingenuity.com, Redwood City, CA, USA). As a sensitivity analysis, we explored the use of a more stringent criterion (P < 10−3) to carry forward association signals in pathway analyses that resulted in 2 additional sets of loci, enumerating 361 genes for each disease trait.
Consistent with the current understanding of pathogenesis of periodontitis that entails dysbiotic host–pathogen interactions, detrimental immune response (1,8) and the emerging role of the nervous system (24,57), we investigated the enrichment of ingenuity canonical signaling pathways in the following categories: cellular immune response, cytokine signaling, humoral immune response, neurotransmitters and other nervous system signaling and pathogen-influenced signaling. There were 147 canonical pathways listed in these categories (http://genomewide.net/public/aric/dental/periodontitis/CDC/CP_candidate_canonical_pathways.xls). To serve as control, we examined the enrichment of the IPA metabolic group of pathways (n = 126). The association between pathways and the data was determined with Fisher's exact test and a Bonferroni-corrected P-value threshold for multiple pathways tested (critical P < 3.4 × 10−4). For each pathway, we also present the ratio of the number of molecules in the data that mapped to each canonical pathway over the number of molecules in the pathway.
SUPPLEMENTARY MATERIAL
FUNDING
ARIC Study: The Atherosclerosis Risk in Communities Study was carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C and HSN268201100012C), R01HL087641, R01HL59367 and R01HL086694; National Human Genome Research Institute contract U01HG004402; National Institutes of Health contract HHSN268200625226C; National Institute of Environmental Health Sciences grant P30ES010126; and National Institute of Dental and Craniofacial Research grants R01DE11551 and R01DE021418. Infrastructure was partly supported by Grant Number UL1RR025005, a component of the National Institutes of Health and NIH Roadmap for Medical Research.
Health ABC Study: This research was supported by NIA contracts N01AG62101, N01AG62103 and N01AG62106. The genome-wide association study was funded by NIA grant 1R01AG032098-01A1. Wake Forest University Health Sciences and genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR was fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number HHSN268200782096C. This research was additionally supported by National Institute of Health grants N01AG6210 and R01HL74104. Funding to pay the Open Access publication charges for this article was provided by the National Institute of Dental and Craniofacial Research grant number R01DE021418.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the staff and participants of the ARIC and Health ABC studies for their important contributions.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Berezow A.B., Darveau R.P. Microbial shift and periodontitis. Periodontol. 2000. 2011;55:36–47. doi: 10.1111/j.1600-0757.2010.00350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petersen P.E., Ogawa H. Strengthening the prevention of periodontal disease: the WHO approach. J. Periodontol. 2005;76:2187–2193. doi: 10.1902/jop.2005.76.12.2187. [DOI] [PubMed] [Google Scholar]
- 3.Beck J.D., Offenbacher S., Williams R., Gibbs P., Garcia R. Periodontitis: a risk factor for coronary heart disease? Ann. Periodontol. 1998;3:127–141. doi: 10.1902/annals.1998.3.1.127. [DOI] [PubMed] [Google Scholar]
- 4.Xiong X., Buekens P., Fraser W.D., Beck J., Offenbacher S. Periodontal disease and adverse pregnancy outcomes: a systematic review. BJOG. 2006;113:135–143. doi: 10.1111/j.1471-0528.2005.00827.x. [DOI] [PubMed] [Google Scholar]
- 5.Lalla E., Papapanou P.N. Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat. Rev. Endocrinol. 2011;28:738–748. doi: 10.1038/nrendo.2011.106. [DOI] [PubMed] [Google Scholar]
- 6.Loos B.G., Craandijk J., Hoek F.J., Wertheim-van Dillen P.M., van der Velden U. Elevation of systemic markers related to cardiovascular diseases in the peripheral blood of periodontitis patients. J. Periodontol. 2000;71:1528–1534. doi: 10.1902/jop.2000.71.10.1528. [DOI] [PubMed] [Google Scholar]
- 7.Desvarieux M., Demmer R.T., Rundek T., Boden-Albala B., Jacobs D.R., Jr, Sacco R.L., Papapanou P.N. Periodontal microbiota and carotid intima-media thickness: the Oral Infections and Vascular Disease Epidemiology Study (INVEST) Circulation. 2005;111:576–582. doi: 10.1161/01.CIR.0000154582.37101.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Darveau R.P. Periodontitis: a polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 2010;8:481–490. doi: 10.1038/nrmicro2337. [DOI] [PubMed] [Google Scholar]
- 9.Saito T., Shimazaki Y., Sakamoto M. Obesity and periodontitis. N. Engl. J. Med. 1998;339:482–483. doi: 10.1056/NEJM199808133390717. [DOI] [PubMed] [Google Scholar]
- 10.Saxén L. Heredity of juvenile periodontitis. J. Clin. Periodontol. 1980;7:276–288. doi: 10.1111/j.1600-051x.1980.tb01970.x. [DOI] [PubMed] [Google Scholar]
- 11.Michalowicz B.S., Aeppli D., Virag J.G., Klump D.G., Hinrichs J.E., Segal N.L., Bouchard T.J., Jr, Pihlstrom B.L. Periodontal findings in adult twins. J. Periodontol. 1991;62:293–299. doi: 10.1902/jop.1991.62.5.293. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J., Sun X., Xiao L., Xie C., Xuan D., Luo G. Gene polymorphisms and periodontitis. Periodontol. 2000. 2011;56:102–124. doi: 10.1111/j.1600-0757.2010.00371.x. [DOI] [PubMed] [Google Scholar]
- 13.Laine M.L., Crielaard W., Loos B.G. Genetic susceptibility to periodontitis. Periodontol. 2000. 2012;58:37–68. doi: 10.1111/j.1600-0757.2011.00415.x. [DOI] [PubMed] [Google Scholar]
- 14.Karimbux N.Y., Saraiya V.M., Elangovan S., Allareddy V., Kinnunen T., Kornman K.S., Duff G.W. Interleukin-1 gene polymorphisms and chronic periodontitis in adult Caucasians: a systematic review and meta-analysis. J. Periodontol. 2012;83:1407–1419. doi: 10.1902/jop.2012.110655. [DOI] [PubMed] [Google Scholar]
- 15.Asking for more. Nat. Genet. 2012;44:733. doi: 10.1038/ng.2345. Editorial. [DOI] [PubMed] [Google Scholar]
- 16.LopezJimenez N.D., Sainz E., Cavenagh M.M., Cruz-Ithier M.A., Blackwood C.A., Battey J.F., Sullivan S.L. Two novel genes, Gpr113, which encodes a family 2 G-protein-coupled receptor, and Trcg1, are selectively expressed in taste receptor cells. Genomics. 2005;85:472–482. doi: 10.1016/j.ygeno.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Divaris K., Monda K.L., North K.E., Olshan A.F., Lange E.M., Moss K., Barros S.P., Beck J.D., Offenbacher S. Genome-wide association study of periodontal pathogen colonization. J. Dent. Res. 2012;91:21S–28S. doi: 10.1177/0022034512447951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J., McCarthy M.I., Ramos E.M., Cardon L.R., Chakravarti A., et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cantor R.M., Lange K., Sinsheimer J.S. Prioritizing GWAS results: a review of statistical methods and recommendations for their application. Am. J. Hum. Genet. 2010;86:6–22. doi: 10.1016/j.ajhg.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi G., Boerwinkle E., Morrison A.C., Gu C.C., Chakravarti A., Rao D.C. Mining gold dust under the genome wide significance level: a two-stage approach to analysis of GWAS. Genet. Epidemiol. 2011;35:111–118. doi: 10.1002/gepi.20556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tong S., Neale R.E., Shen X., Olsen J. Challenges for epidemiologic research on the verge of a new era. Eur. J. Epidemiol. 2011;26:689–694. doi: 10.1007/s10654-011-9615-0. [DOI] [PubMed] [Google Scholar]
- 22.Panagiotou O.A., Ioannidis J.P. for the Genome-Wide Significance Project. What should the genome-wide significance threshold be? Empirical replication of borderline genetic associations. Int. J. Epidemiol. 2012;41:273–286. doi: 10.1093/ije/dyr178. [DOI] [PubMed] [Google Scholar]
- 23.Hernández M., Dutzan N., García-Sesnich J., Abusleme L., Dezerega A., Silva N., González F.E., Vernal R., Sorsa T., Gamonal J. Host-pathogen interactions in progressive chronic periodontitis. J. Dent. Res. 2011;90:1164–1170. doi: 10.1177/0022034511401405. [DOI] [PubMed] [Google Scholar]
- 24.Lundy F.T., Linden G.J. Neuropeptides and neurogenic mechanisms in oral and periodontal inflammation. Crit. Rev. Oral Biol. Med. 2004;15:82–98. doi: 10.1177/154411130401500203. [DOI] [PubMed] [Google Scholar]
- 25.Lundy F.T., El Karim I.A., Linden G.J. Neuropeptide Y (NPY) and NPY Y1 receptor in periodontal health and disease. Arch. Oral Biol. 2009;54:258–262. doi: 10.1016/j.archoralbio.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 26.van Winkelhoff A.J., Bosch-Tijhof C.J., Winkel E.G., van der Reijden W.A. Smoking affects the subgingival microflora in periodontitis. J. Periodontol. 2001;72:666–671. doi: 10.1902/jop.2001.72.5.666. [DOI] [PubMed] [Google Scholar]
- 27.Haber J. Smoking is a major risk factor for periodontitis. Curr. Opin. Periodontol. 1994;1994:12–18. [PubMed] [Google Scholar]
- 28.Tomar S.L., Asma S. Smoking-attributable periodontitis in the United States: findings from NHANES III. National Health and Nutrition Examination Survey. J. Periodontol. 2000;71:743–751. doi: 10.1902/jop.2000.71.5.743. [DOI] [PubMed] [Google Scholar]
- 29.Löe H., Anerud A., Boysen H., Morrison E. Natural history of periodontal disease in man. Rapid, moderate and no loss of attachment in Sri Lankan laborers 14–46 years of age. J. Clin. Periodontol. 1986;13:431–445. doi: 10.1111/j.1600-051x.1986.tb01487.x. [DOI] [PubMed] [Google Scholar]
- 30.Elter J.R., Beck J.D., Slade G.D., Offenbacher S. Etiologic models for incident periodontal attachment loss in older adults. J. Clin. Periodontol. 1999;26:113–123. doi: 10.1034/j.1600-051x.1999.260209.x. [DOI] [PubMed] [Google Scholar]
- 31.Offenbacher S., Barros S.P., Singer R.E., Moss K., Williams R.C., Beck J.D. Periodontal disease at the biofilm-gingival interface. J. Periodontol. 2007;78:1911–1925. doi: 10.1902/jop.2007.060465. [DOI] [PubMed] [Google Scholar]
- 32.Michalowicz B.S., Diehl S.R., Gunsolley J.C., Sparks B.S., Brooks C.N., Koertge T.E., Califano J.V., Burmeister J.A., Schenkein H.A. Evidence of a substantial genetic basis for risk of adult periodontitis. J. Periodontol. 2000;71:1699–1707. doi: 10.1902/jop.2000.71.11.1699. [DOI] [PubMed] [Google Scholar]
- 33.Miley D.D., Baumgartner M.H., Cheverud J.M., Roseman C.C., Rogers J., McLeod D.E., Reyes E., Hildebolt C.F. Heritability of alveolar bone loss from periodontal disease in a baboon population: a pilot study. J. Periodontol. 2011;82:575–580. doi: 10.1902/jop.2010.100189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang J., Benyamin B., McEvoy B.P., Gordon S., Henders A.K., Nyholt D.R., Madden P.A., Heath A.C., Martin N.G., Montgomery G.W., et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schaefer A.S., Richter G.M., Groessner-Schreiber B., Noack B., Nothnagel M., El Mokhtari N.E., Loos B.G., Jepsen S., Schreiber S. Identification of a shared genetic susceptibility locus for coronary heart disease and periodontitis. PLoS Genet. 2009;5:e1000378. doi: 10.1371/journal.pgen.1000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaefer A.S., Richter G.M., Nothnagel M., Manke T., Dommisch H., Jacobs G., Arlt A., Rosenstiel P., Noack B., Groessner-Schreiber B., et al. A genome-wide association study identifies GLT6D1 as a susceptibility locus for periodontitis. Hum. Mol. Genet. 2010;19:553–562. doi: 10.1093/hmg/ddp508. [DOI] [PubMed] [Google Scholar]
- 37.Baelum V., López R. Defining a periodontitis case: analysis of a never-treated adult population. J. Clin. Periodontol. 2012;39:10–19. doi: 10.1111/j.1600-051X.2011.01812.x. [DOI] [PubMed] [Google Scholar]
- 38.López R., Baelum V. Contesting conventional periodontal wisdom: implications for periodontal classifications. Community Dent. Oral Epidemiol. 2012;40:385–395. doi: 10.1111/j.1600-0528.2012.00677.x. [DOI] [PubMed] [Google Scholar]
- 39.Gottesman I.I., Gould T.D. The endophenotype concept in psychiatry: etymology and strategic intentions. Am. J. Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- 40.He X., Zhang J. Toward a molecular understanding of pleiotropy. Genetics. 2006;173:1885–1891. doi: 10.1534/genetics.106.060269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pendergrass S.A., Brown-Gentry K., Dudek S.M., Torstenson E.S., Ambite J.L., Avery C.L., Buyske S., Cai C., Fesinmeyer M.D., Haiman C., et al. The use of phenome-wide association studies (PheWAS) for exploration of novel genotype-phenotype relationships and pleiotropy discovery. Genet. Epidemiol. 2011;35:410–422. doi: 10.1002/gepi.20589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.The ARIC investigators. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. Am. J. Epidemiol. 1989;129:687–702. [PubMed] [Google Scholar]
- 43.Pahor M., Kritchevsky S. Research hypotheses on muscle wasting, aging, loss of function and disability. J. Nutr. Health Aging. 1998;2:97–100. [PubMed] [Google Scholar]
- 44.Weyant R.J., Newman A.B., Kritchevsky S.B., Bretz W.A., Corby P.M., Ren D., Weissfeld L., Rubin S.M., Harris T. Periodontal disease and weight loss in older adults. J. Am. Geriatr. Soc. 2004;52:547–553. doi: 10.1111/j.1532-5415.2004.52160.x. [DOI] [PubMed] [Google Scholar]
- 45.Katancik J.A., Kritchevsky S., Weyant R.J., Corby P., Bretz W., Crapo R.O., Jensen R., Waterer G., Rubin S.M., Newman A.B. Periodontitis and airway obstruction. J. Periodontol. 2005;76:2161–2167. doi: 10.1902/jop.2005.76.11-S.2161. [DOI] [PubMed] [Google Scholar]
- 46.Beck J.D., Elter J.R., Heiss G., Couper D., Mauriello S.M., Offenbacher S. Relationship of periodontal disease to carotid artery intima-media wall thickness: the atherosclerosis risk in communities (ARIC) study. Arterioscler. Thromb. Vasc. Biol. 2001;21:1816–1822. doi: 10.1161/hq1101.097803. [DOI] [PubMed] [Google Scholar]
- 47.Page R.C., Eke P.I. Case definitions for use in population-based surveillance of periodontitis. J. Periodontol. 2007;78:1387–1399. doi: 10.1902/jop.2007.060264. [DOI] [PubMed] [Google Scholar]
- 48.Li Y., Willer C.J., Ding J., Scheet P., Abecasis G.R. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 51.Aulchenko Y.S., Struchalin M.V., van Duijn C.M. ProbABEL package for genome-wide association analysis of imputed data. BMC Bioinformatics. 2010;11:134. doi: 10.1186/1471-2105-11-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ge D., Zhang K., Need A.C., Martin O., Fellay J., Urban T.J., Telenti A., Goldstein D.B. WGAViewer: software for genomic annotation of whole genome association studies. Genome Res. 2008;18:640–643. doi: 10.1101/gr.071571.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pruim R.J., Welch R.P., Sanna S., Teslovich T.M., Chines P.S., Gliedt T.P., Boehnke M., Abecasis G.R., Willer C.J. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Willer C.J., Li Y., Abecasis G.R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang J., Lee S.H., Goddard M.E., Visscher P.M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Offenbacher S., Barros S.P., Paquette D.W., Winston J.L., Biesbrock A.R., Thomason R.G., Gibb R.D., Fulmer A.W., Tiesman J.P., Juhlin K.D., et al. Gingival transcriptome patterns during induction and resolution of experimental gingivitis in humans. J. Periodontol. 2009;80:1963–1982. doi: 10.1902/jop.2009.080645. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.