

Abstract
Fibrin deposition mediated through activation of tissue factor (TF) in the airspace is central to the pathogenesis of acute lung injury. Defining the mechanisms of TF regulation in the lung is critical to understanding pulmonary fibrin formation. Tumor necrosis factor-α (TNF-α) upregulates TF in the injured lung, and there is emerging evidence that another cytokine, interferon-γ (IFN-γ) also modulates expression. The effects of TNF and IFN on regulation of TF were studied in alveolar epithelial A549 cells. In addition, potential mechanisms of modulation of TF expression by the two cytokines were analyzed with the hypothesis that IFN-γ acts synergistically with TNF-α to upregulate alveolar epithelial TF through modulation of TNFR expression. TNF-α but not IFN-γ treatment increased TF mRNA, protein and cell surface TF activity. The combination of IFN-γ and TNF-α treatment augmented the effects of TNF-α on TF upregulation and also increased release of procoagulant microparticles (MPs) from A549 cells. IFN-γ modulated expression of both TNF-α receptors, studies utilizing neutralizing antibodies against the two TNF receptors showed the TF effects were mediated primarily through augmenting TNFR1-dependent cellular responses. These findings have important implications for regulation of fibrin formation in the lung in the setting of acute inflammation.
Keywords: Coagulation, acute lung injury, acute respiratory distress syndrome, tumor necrosis factor alpha receptor, TNFR1, TNFR2, microparticles
INTRODUCTION
Activation of the coagulation cascade in the airspace is central to the pathogenesis of both acute (1) and chronic lung diseases. (2) Although TF is the major procoagulant protein in the lung in patients with acute lung injury (ALI) and acute respiratory distress syndrome (ARDS), (3) mechanisms that modulate TF activity are incompletely understood. In a baboon model of sepsis, systemic blockade of TF attenuated lung injury and improved survival. (4–6) However, in humans with severe sepsis, the major risk factor for the development of ARDS, systemic blockade of TF with recombinant tissue factor pathway inhibitor had no effect on survival or other clinical endpoints. (7) We and others have shown that TF procoagulant activity in the lung is regulated locally by the lung epithelium (8–10) but the specific factors that regulate TF transcription, translation and activity are still poorly defined.
Activation of TF is complex and involves transcriptional and translational processes (11, 12) as well as de-encryption of TF at the cell surface. (13) We recently reported that the alveolar epithelium is capable of modulating intra-alveolar fibrin deposition through upregulation of active TF on the cell surface in response to cytomix, a combination of TNF-α, IL-1β and IFN-γ, by increasing TF transcription, translation, and cell surface TF activity. (10) Although cytomix recapitulates the complex inflammatory environment in the alveolar space in ARDS, these studies did not determine the role of each individual cytokine in the upregulation and activation of TF in the alveolar epithelium. Understanding the effect of individual cytokines on TF transcription, translation and activity may be important for the development of targeted therapies to modulate TF activity in ALI/ARDS.
The effect of TNF-α and IL-1β on TF regulation in several different cell types has been previously reported. (14, 15) Both TNF-α and IL-1β upregulate TF through induction of TF gene transcription and upregulation of protein production. (16–18) However, very little is known about the role of IFN-γ on the regulation of TF. Our initial work showed that even small amounts of cytomix (1 ng/ml of each cytokine) can robustly upregulate alveolar epithelial cell TF protein and activity, (10) suggesting that there may be potential synergy between the cytokines. Interestingly, other investigators have shown TNF-α and IFN-γ act synergistically through several distinct cellular mechanisms. In human airway smooth muscle cells, TNF-α and IFN-γ synergistically upregulated the chemokine CXCL10 through recruitment of co-activators to the CXCL10 gene. (19) IFN-γ can also potentiate TNF-α induced phosphorylation and degradation of IκBα in the NF-κB transcriptional complex. (20) In addition to effects on gene transcription, IFN-γ modulates tumor necrosis factor receptor (TNFR) activity in astrocytes and intestinal epithelial cells through upregulation of TNFR2 transcription and protein expression. (21, 22) With regards to TF, there has been one small study which showed IFN-γ synergized with C reactive protein to upregulate TF on peripheral macrophages isolated from healthy human volunteers. (23) Whether these same mechanisms function to upregulate TF in the alveolar epithelium is unknown. Given the extensive body of literature detailing the synergistic effects of IFN-γ and TNF-α in several cell types, we chose to study the effects of these two cytokines on lung epithelial coagulation. We designed a series of experiments to test the hypothesis that IFN-γ acts synergistically with TNF-α to upregulate alveolar epithelial TF through modulation of TNFR expression.
METHODS
Cell culture
Alveolar epithelial cells (A549 cells, American Type Culture Collection, Manassas, VA) were grown in minimal essential media (MEM) (Cellgro, Herndon, VA) with 10% fetal bovine serum (Cellgro, Herndon, VA) and 10,000 units/mL each of penicillin and streptomycin (Sigma, St. Louis, MO). All experiments were done at 6 hours.
Tissue factor activity assay
A549 cells were treated with TNF-α and/or IFN-γ (R&D Systems, Minneapolis, MN), washed twice with serum-free MEM and incubated at 37°C for 60 minutes with 25 μL of 40 nM factor VIIa and 25μL of 1μM factor X. (factors VIIa, X, Xa Enzyme Research Laboratories, South Bend, IN) 100μL from each sample well was transferred to the new plate and 5μL of 100mM EDTA was added to stop the reaction. Xa was quantified by adding 100μL of the chromogenic substrate S-2222. (Chromogenix, Milano, Italy) to each well followed by measurement of OD at 405 nm and comparing to a Xa standard curve.
Cell viability
Cell viability was measured using a chromogenic electron coupling reagent (Promega, Madison, WI).
Cell supernates and lysates
A549 cells were grown to confluence in 24 well plates and exposed to cytokines or control medium, conditioned media was removed, centrifuged at 1500g and stored at −70°C. For measurement of TF protein, cells were lysed with 250 μL of 50 mM tris, 100 mM NaCl, 0.1% Triton X-100, pH 7.45, centrifuged at 1500g then stored at −70°C. For measurement of TNFRs, the adherent cells were incubated with RIPA buffer (50 mM Tris, 1 mM EDTA, 150 mM NaCl, sodium deoxydodecholate, NP-40) protease inhibitor cocktail tablet (Roche, Mannheim, Germany) and stored at −70°C.
Isolation of mRNA
Cells were trypsinized and mRNA was extracted (Qiagen, Valencia, CA) according to manufacturer’s instructions and stored at −70°C.
Real time PCR
Semi-quantitative PCR was done using the Applied Biosystems Method (Carlsbad, CA). cDNA was prepared using Superscript RT (Invitrogen, Carlsbad, CA). Both TF and GAPDH primers and probe sets were obtained from Applied Biosystems. Semi-quantitation was established using the ΔΔCt method. (24)
Fixed cell-surface ELISAs
As previously described, (25–27) Cells were rinsed with phosphate-buffered saline (PBS), fixed with 4% formaldehyde (Thermoscientific, Rockford, IL), then rinsed with PBS. Cells were blocked for 20 min (5% nonfat dried milk in PBS), incubated for 2 h with blocking buffer and either goat-antihuman TF (R&D Systems, Cat. AF2339) at 0.1μg/ml, mouse-antihuman TNFR1 (R&D Systems, Cat. MAB625) a 0.5μg/ml, or mouse-antihuman TNFR2 (R&D Systems, Cat MAB726) at 0.5μg/ml. Cells were washed (PBS) then incubated with blocking buffer and 1:1000 horseradish peroxidase–linked rabbit anti-goat secondary antibody (R&D Systems, Cat. HAF017) for TF ELISA or 1:1000 mouse-HRP secondary (R&D systems, cat number HAF007) for TNFR ELISAs for 45 min. Cells were washed (PBS) and then incubated for 5–20 min with TMB (Sigma, St. Louis, MO). The reaction was terminated with 5% sulfuric acid and absorbance was read at 450 nm (Synergy HT spectrophotometer (Biotech)).
Quantification of microparticles
Microparticles were quantified in cell free conditioned media using a microparticle capture assay (Hyphen Biomed, Neuville sur Oise, France).
Clot time measurements
Clot time was measured using a mechanical clot detector (STart4 Coagulometer; Diagnostica Stago, Asnieres, France). 50 μl conditioned media was warmed to 37°C. Samples were incubated with 50 μl of pooled normal human plasma (Fisher Diagnostics, Middletown, VA). Clot time was determined following the addition of 50 μl of 50 mM calcium chloride.
Measurement of TF, TNFR1 and TNFR2
Proteins were measured by ELISA (TF-American diagnostica, Stamford, CT; TNFRs-R&D Systems).
TNFR blocking antibodies
A549 cells were incubated with cytokines and TNFR1 or TNFR2 blocking antibody (10 μg/ml each antibody) (R&D Systems) for 6 hours prior to TF activity assay.
RESULTS
IFN-γ augments TNF-α induced upregulation of cell surface TF activity
Cell surface TF activity (TF index) was measured by generation of factor Xa in A549 cells treated with increasing concentrations of cytokines for 6 hours. (Figure 1) There were no changes in cell viability with any of the treatments (Supplementary Data Figure 1). Treatment of A549 cells with IFN-γ at any concentration did not alter TF activity compared to control-treated cells, however exposure to 20–80 ng/ml TNF-α significantly increased the cell surface TF index. This TNF-α mediated increase in TF activity was further augmented by co-treatment with 20 ng/ml IFN-γ. Interestingly, there was no dose response with either TNF-α alone or the combination of TNF and IFN suggesting a threshold response of A549 cells to TNF.
Figure 1. Cell surface TF activity in response to increasing doses of TNF-α and IFN-γ.
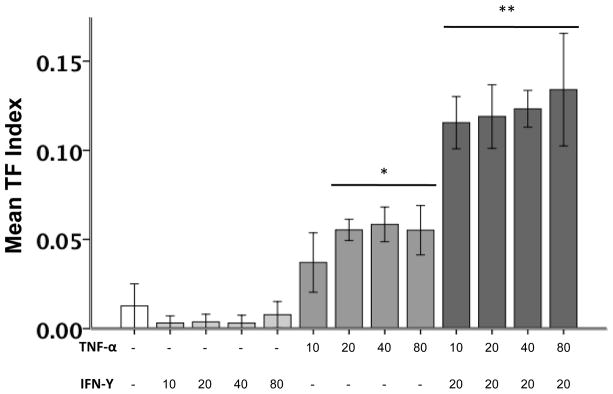
Bar graph depicting mean cell surface TF activity (N=6 for each condition) as measured by factor Xa generation corrected for cell viability (TF index). Cytokine concentrations are in ng/ml. IFN-γ treatment alone does not increase cell surface TF activity while TNF-α treatment alone increases cell surface TF activity. The combination if IFN-γ and TNF-α increases TF activity in an dose-independent manner. * p≤0.003 vs all other treatments, **p<0.001 vs all other treatments by ANOVA with post hoc Tukey test.
IFN-γ and TNF-α synergistically upregulate TF mRNA
To determine if the increase in cell surface TF activity was mediated transcriptionally, TF mRNA levels were measured by semi-quantitative real time PCR. (Figure 2) IFN-γ (20 ng/ml) had no effect on TF mRNA while TNF-α (20 ng/ml) increased TF mRNA 4.9 fold over control (although this increase was not statistically significant). The combination of cytokines increased TF mRNA by 16.5 fold over control (p < 0.01).
Figure 2. Semi-quantitative RT-PCR for TF mRNA in A549 cells.
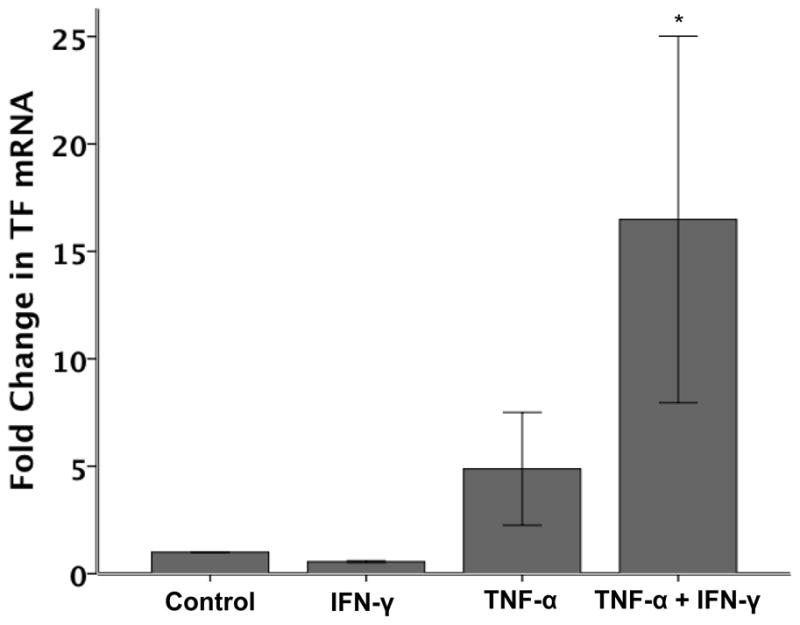
After 6 hours of cytokine treatment, A549 cells have increased TF mRNA in response to TNF-α (4.9 fold increase, p=NS versus control) but not IFN-γ (N=5 per group). The combination of TNF-α (20 ng/ml) and IFN-γ (20ng/ml) increases TF mRNA 16.5 fold over control. *p ≤ 0.010 versus all other groups by ANOVA with post hoc Tukey Test.
IFN-γ and TNF-α synergistically upregulate total cellular TF protein and release
To determine whether the cytokine-mediated increase in TF mRNA was paralleled by increased protein levels, total cellular protein from A549 lysates as well as conditioned media were collected and analyzed by ELISA for TF. Although total cellular protein measured in A549 cell lysates was increased, after treatment for 6 hours with 20 ng/ml of TNF-α, soluble TF levels were unchanged compared to control-treated cells. Interestingly, whereas IFN-γ alone had no effect on either total cellular TF (Figure 3A) or TF released from cells (Figure 3B), the combination of IFN-γ and TNF-α significantly increased both total cellular TF (Figure 3A) and TF released into the conditioned media (Figure 3B). These results suggest that IFN co-treatment not only augments TNF-induced cellular TF levels, but also modulates the mechanisms whereby TNF- augments TF release in lung epithelial cells.
Figure 3. TF protein concentration in A549 cells.
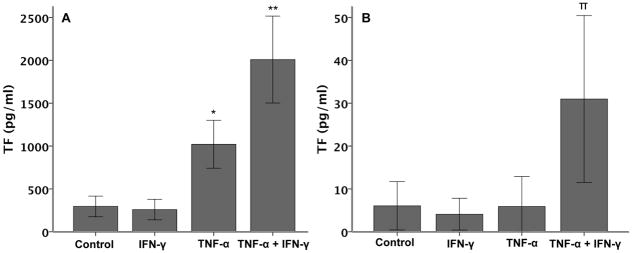
Panel A shows TF protein concentration in A549 cell lysates after 6 hours of treatment with cytokines (20 ng/ml each). (*p≤0.014 versus all other groups, **p≤0.001 versus all other groups both by ANOVA with post hoc Tukey test). Panel B shows TF released into the conditioned media of A549 cells after 6 hours of treatment with cytokines (N=6 per group). (π p≤0.020 versus all other groups by ANOVA with post hoc Tukey test).
TNF-α-induced cell surface TF is increased by co-treatment of cells with IFN-γ
To determine if the increase in total cellular TF is accompanied by an increase in cell surface, and thus potentially functional TF, we measured cell surface TF protein by cell surface ELISA (Figure 4). TNF-α treatment alone (20 ng/ml, 6 h) significantly increased cell surface TF protein. This effect was significantly augmented by concomitant treatment with IFN-γ (20 ng/ml, 6 h).
Figure 4. Cell surface TF protein expression.

A549 cell surface TF protein was measured by ELISA after 6 hours of stimulation with cytokines (20 ng/ml each, N=6 per group). TNF-α treatment increases cell surface TF protein (*p≤0.002 versus all other treatments by ANOVA with post hoc Tukey test). This is significantly augmented by the addition of IFN-γ (**p≤0.002 versus all other treatments by ANOVA with post hoc Tukey test).
IFN-γ and TNF-α synergistically increase the release of procoagulant microparticles from A549 cells
To determine if the TF released by the co-treatment of cells with IFN-γ and TNF-α was contained in procoagulant microparticles, we measured plasma recalcification time (clot time) (Figure 5A) and microparticle concentration (Figure 5B) in conditioned media from A549 cells treated with cytokines for 6 hours. Similar to the soluble TF levels, neither cytokine alone augmented procoagulant MP release compared to control-treated cells; however, the combination of IFN-γ and TNF-α synergistically reduced clot time which was concomitant with increased microparticle release. Since IFN enhanced TNF-α–induced mRNA expression, microparticle release and total cell-associated protein levels of TF in A549 cells, subsequent experiments examined the involvement of the two TNF receptor types in modulation of IFN-mediated upregulation of TNF-induced TF.
Figure 5. A549 cells release procoagulant microparticles.
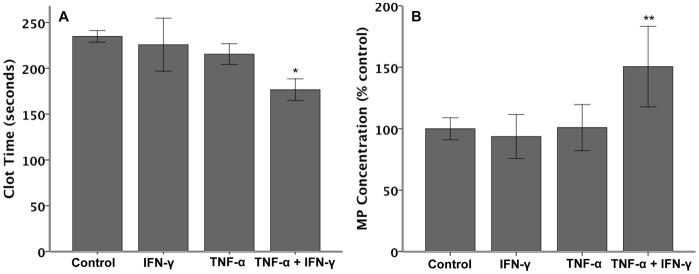
Clot time was measured in conditioned media from A549 cells treated with cytokines (20 ng/ml each) for 6 hours (Panel A). Although neither cytokine alone significantly reduced clot time, the combination of cytokines significantly reduced clot time (*p≤0.019 versus all other treatments by ANOVA with post hoc Tukey test). Panel B shows microparticle (MP) released from A549 cells after 6 hours of cytokine treatment (N=6 per group). Again, the combination of cytokines significantly increased MP release (**p≤0.013 versus all other treatments by ANOVA with post hoc Tukey test).
IFN-γ attenuates the effects of TNF-α on TNFR1 and augments the effects on TNFR2
To further define the mechanism of synergy between IFN-γ and TNF-α we studied the effects of cytokine treatment on modulation of the expression of the two TNF-α receptors, TNFR1 and TNFR2 (Figure 6). TNF-α downregulated TNFR1 mRNA (Figure 6A) and total cellular protein levels as measured by semi-quantitative RT-PCR and ELISA respectively (Figure 6B). Although IFN-γ significantly attenuated the TNF-mediated downregulation of TNFR1 mRNA levels, cotreatment only resulted in a trend towards increased total and cell surface TNFR1 (Figures 6A, 6B and 6C). Similar to TNFR1, exposure of A549 cells to IFN did not significantly modulate mRNA or protein levels of TNFR2. However, although TNF-α treatment trended to increase TNFR2 mRNA expression, (Figure 6D; 58 fold increase compared to control, p=0.624 by ANOVA with post hoc Tukey test) total cellular TNFR2 protein was significantly increased (Figure 6E). Cell surface TNFR2 expression was variable with individual cytokine treatment and only yielded consistently measureable protein with co-treatment (data not shown). Interestingly, both TNFR2 mRNA and protein were significantly increased by the combination of IFN-γ and TNF-α, once again demonstrating synergy between the two cytokines in modulating expression of alveolar epithelial cell-derived proteins.
Figure 6. Effects of IFN-γ and TNF-α on TNF Receptors.

Following 6 hours of cytokine treatment, TNF-α (20 ng/ml) reduces both TNFR1 mRNA (A), total cellular protein (B) and cell surface protein (C). Co-stimulation with IFN-γ (20 ng/ml) prevents the reduction in TNFR1 mRNA but has minimal effects on TNFR1 protein. *p<0.001 versus all other treatments, **p≤0.006 versus control and IFN-γ, πp≤0.001 versus TNF-α and co-treatment and p=0.059 versus control by ANOVA with post hoc Tukey test. Following 6 hours of cytokine treatment, TNF-α and IFN-γ increase both TNFR2 mRNA (D) and protein (E) (N=6 per group). *p<0.001 versus all other treatments, **p≤0.007 versus all other groups, πp≤0.02 versus control and TNF-α + IFN-γ by ANOVA with post hoc Tukey test.
The effects of IFN-γ on cell surface TF activity are mediated through TNFR1
To define the functional effects of TNFR1 and -2 on cytokine-mediated TF upregulation, we measured TF activity in response to cytokine treatment following neutralization of either TNFR1 or TNFR2 (Figure 7). In the presence of a TNFR1 neutralizing antibody, the effects of both TNF-α alone and the synergistic effects of IFN-γ and TNF-α on cell surface TF activity were significantly attenuated compared to control-treated and control, IgG antibody-treated cells whereas neutralization of TNFR2 did not alter TNF–mediated TF activity and only slightly modulated IFN and TNF-induced effects. However, TF activity induced by the cotreatment of both cytokines was not completely inhibited by neutralization of either TNFR1 or TNFR2 suggesting regulation of cytokine-induced TF activity is complex and may involve other mechanisms in addition to activation of TNFR1.
Figure 7. TF activity in cytokine stimulated A549 cells treated with TNFR blocking antibodies.

TNFR1 inhibition (*p=0.004), but not TNFR2 inhibition (p=1.0) decreases the TNF-α induced upregulation of TF (p<0.001 TNF-γ versus control). TNFR1 inhibition (**p<0.001) also significantly reduced TNF-α+IFN-γ upregulation of TF activity (p<0.001 versus control) while TNFR2 inhibition had no effect on the combination of cytokines (p=0.108 versus TNF-α+IFN-γ). All comparisons by ANOVA with post hoc Tukey test (N=6 per group).
DISCUSSION
The major findings of this study are summarized as follows. First, IFN-γ treatment alone had no effect on TF transcription, translation, activity or microparticle in alveolar epithelial cells. Second, IFN-γ treatment significantly augmented the effects of TNF-α on upregulation of TF protein and activity and induced the release of procoagulant MPs from lung alveolar epithelial cells. Third, this synergistic effect of IFN-γ could be attenuated by inhibition of TNFR1 but not TNFR2 signaling suggesting that the synergistic effects are mediated in part by augmenting TNFR1-dependent cellular responses. These findings are novel and deepen our understanding of TF regulation in the airspace in the setting of inflammation.
We have previously reported that inflammatory stimuli (cytomix, a mixture of proinflammatory cytokines and pulmonary edema fluid from patients with ARDS upregulate lung epithelial TF protein and activity. (10) However, multiple studies have shown that broad inhibition of inflammation with corticosteroids (28) does not substantially alter the course of ARDS; thus leading to the conclusion that specific, targeted therapies will be needed to improve outcomes in ARDS. Therefore, understanding the mechanisms that govern lung-derived TF regulation are an important first step in developing strategies to modulate its activity.
Interferon-γ is an important cytokine for both innate and adaptive immunity and is critical for host defense against viral and intracellular bacterial pathogens. (29) Although its role in TF regulation is poorly understood, it has been shown to upregulate TF activity in endometrial stromal cells (30) and to augment LPS induced TF expression in macrophages. (31) In the lung, one can postulate that IFN-γ may serve to augment TF activity and thus fibrin deposition in the setting of infection which could help to contain the infection in the airspace. In this study, we identified a unique and as yet undescribed role for IFN-γ in the regulation of TF activity in a lung A549 epithelial cell line. Although it had no effect on its own, treatment of A549 cells with IFN-γ in the presence of another cytokine, TNF, resulted in an augmented TNF-α-response. Specifically, IFN-γ significantly increased TNF-mediated upregulation of TF mRNA and protein, cell surface expression and activity and release of TF into the culture media which correlated with an increase in procoagulant MPs. However, although our data are interesting and identify a novel pathway of TF modulation in the lung, further studies into the functional significance of IFN-γ in the in vivo setting are necessary.
The mechanism of the observed effects of IFN-γ were found to be mediated in part through TNFR1 since neutralization of TNFR1 signaling with a receptor-specific neutralizing antibody significantly reduced, but did not ablate, the synergistic effect of cotreating A549 cells with both cytokines on TF expression and activity. (Figure 7A). It is possible that the concentration of blocking antibody used were insufficient however, the dose of 10 μg/ml was chosen because it has been shown to completely block TNFR1 activity in a mouse fibrobalst cell line (R&D Systems) but this concentration may have been insufficient to completely block TNFR1 signaling. This lack of complete inhibition suggests that some of the effects of IFN-γ are independent of TNFR1 signaling and/or expression. Specifically, our data showed exposure of A549 cells to IFN-γ inhibited the TNF-α induced downregulation of TNFR1 mRNA without altering total cellular or cell surface TNFR1 protein compared to TNF-treated cells alone. However, prior studies have shown that TNFR1 is shed from the cell surface by metalloproteases (32) thus down-regulating cell responsiveness to TNF-α. It is possible that in the presence of both cytokines, cellular expression of TNFR1 was increased compared to cognate ligand alone yet was offset by increased release from the plasma membrane.
Interestingly, IFN-γ significantly augmented TNF-induced TNFR2 mRNA and protein although this did not translate into a functional effect on TF activity as TNFR2 inhibition had no significant effect on TNF-α + IFN-γ synergy. However, in A549 cells TNFR2 is not expressed constitutively, instead it is induced by cognate ligand in a time dependent manner. Therefore, experiments utilizing the TNFR2 specific neutralizing antibody at different time points may show a role for TNFR2. However, our data suggest that in alveolar epithelial cells TNFR2 is not critical for TF regulation and may serve some other purpose.
There are several potential mechansism by which IFN-γ may potentiate the effects of TNF-α in our system. Both cytokines have been shown to synergistically activate the mitogen-activated protein (MAP) kinase JNK in pancreatic β-cells and induce apoptosis. (33) Interferon can also augment TNF-α receptor signaling by through signal transducers and activators of transcription (STAT) signaling. In the absence of IFN-γ STAT-1 is recruited to TNFR1 and limits it’s signaling in response to TNF-α. Upon treatment with IFN-γ, STAT-1 translocates to the nucleus and allows optimal NF-κB activity. (34) Interferon can also potentiate TNF-α mediated NF-κB activation through phosphorylation and degradation of the NF-κB repressor Iκ-Bα. (20) Others have shown that IFN-γ can act synergistically with TNF-α to increase the transcriptional coactivators CREB-binding protein (CBP) and RNA polymerase II to target genes (19). Whether these mechanisms also contribute to IFN-γ synergy with TNF-α in the lung epithelium is unknown.
Our study has some limitations. First, we studied TF regulation in a cancer cell line, A549 alveolar epithelial type II-like cells, which may not replicate the in vivo conditions in the human lung. However, we have previously used this cell line to study TF regulation and have found that A549 cell –derived TF regulation correlates both with histologic analysis of lungs from patients with ARDS and with studies in primary lung epithelial cells such as distal small airway epithelial cells. (10, 35) Second, the importance of IFN-γ in the pathogenesis of ARDS is unknown. As there is no published human data on levels of IFN-γ in clinical ARDS it is difficult to determine if the findings are clinically important. However, this study identifies IFN-γ as a novel mediator of inflammation and coagulation in ARDS and should fuel future studies in animal models and humans. Finally, although we identified the TNFR1 receptor signaling as one potential mechanism to explain the IFN-γ effects, we found that there are also TNFR1 independent effects of IFN-γ. The relative contribution of TNFR1 dependent and independent effects of IFN-γ on TNF-α induced TF upregulation are thus unknown.
In summary, we have identified a novel regulator of TF activity and microparticle release in the lung epithelium and provided insight into the mechanism of TF regulation in the airspace. This regulatory pathway represents a unique target for modulating coagulation and fibrin deposition in the lung.
Supplementary Material
Acknowledgments
Support:
NIH/NICHD 5 K12 HD 043483-05 and HL090785 to JAB, NIH HL103836 and NIH HL088263 and an American Heart Association Established Investigator Award to LBW and NIHLBI T32 HL087738 to SCS
Footnotes
Declaration of interest:
The authors report no conflicts of interest.
References
- 1.Hofstra JJ, Haitsma JJ, Juffermans NP, Levi M, Schultz MJ. The role of bronchoalveolar hemostasis in the pathogenesis of acute lung injury. Semin Thromb Hemost. 2008 Jul;34(5):475–84. doi: 10.1055/s-0028-1092878. [DOI] [PubMed] [Google Scholar]
- 2.Chambers RC. Procoagulant signalling mechanisms in lung inflammation and fibrosis: novel opportunities for pharmacological intervention? Br J Pharmacol. 2008 Mar;153( Suppl 1):S367–78. doi: 10.1038/sj.bjp.0707603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Idell S, Gonzalez K, Bradford H, MacArthur CK, Fein AM, Maunder RJ, et al. Procoagulant activity in bronchoalveolar lavage in the adult respiratory distress syndrome. Contribution of tissue factor associated with factor VII. Am Rev Respir Dis. 1987 Dec;136(6):1466–74. doi: 10.1164/ajrccm/136.6.1466. [DOI] [PubMed] [Google Scholar]
- 4.Carraway MS, Welty-Wolf KE, Miller DL, Ortel TL, Idell S, Ghio AJ, et al. Blockade of tissue factor: treatment for organ injury in established sepsis. Am J Respir Crit Care Med. 2003 May 1;167(9):1200–9. doi: 10.1164/rccm.200204-287OC. [DOI] [PubMed] [Google Scholar]
- 5.Welty-Wolf KE, Carraway MS, Miller DL, Ortel TL, Ezban M, Ghio AJ, et al. Coagulation blockade prevents sepsis-induced respiratory and renal failure in baboons. Am J Respir Crit Care Med. 2001 Nov 15;164(10 Pt 1):1988–96. doi: 10.1164/ajrccm.164.10.2105027. [DOI] [PubMed] [Google Scholar]
- 6.Welty-Wolf KE, Carraway MS, Ortel TL, Ghio AJ, Idell S, Egan J, et al. Blockade of Tissue Factor-Factor X binding attenuates sepsis-induced respiratory and renal failure. Am J Physiol Lung Cell Mol Physiol. 2005 Aug 12; doi: 10.1152/ajplung.00155.2005. [DOI] [PubMed] [Google Scholar]
- 7.Abraham E, Reinhart K, Opal S, Demeyer I, Doig C, Rodriguez AL, et al. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. JAMA. 2003 Jul 9;290(2):238–47. doi: 10.1001/jama.290.2.238. [DOI] [PubMed] [Google Scholar]
- 8.Perrio MJ, Ewen D, Trevethick MA, Salmon GP, Shute JK. Fibrin formation by wounded bronchial epithelial cell layers in vitro is essential for normal epithelial repair and independent of plasma proteins. Clin Exp Allergy. 2007 Nov;37(11):1688–700. doi: 10.1111/j.1365-2222.2007.02829.x. [DOI] [PubMed] [Google Scholar]
- 9.Jesmin S, Gando S, Matsuda N, Sakuma I, Kobayashi S, Sakuraya F, et al. Temporal changes in pulmonary expression of key procoagulant molecules in rabbits with endotoxin-induced acute lung injury: elevated expression levels of protease-activated receptors. Thromb Haemost. 2004 Nov;92(5):966–79. doi: 10.1160/TH04-03-0160. [DOI] [PubMed] [Google Scholar]
- 10.Bastarache JA, Wang L, Geiser T, Wang Z, Albertine KH, Matthay MA, et al. The alveolar epithelium can initiate the extrinsic coagulation cascade through expression of tissue factor. Thorax. 2007 Jul;62(7):608–16. doi: 10.1136/thx.2006.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mackman N. Regulation of the tissue factor gene. Thromb Haemost. 1997 Jul;78(1):747–54. [PubMed] [Google Scholar]
- 12.Rao LV, Pendurthi UR. Tissue factor on cells. Blood Coagul Fibrinolysis. 1998 Mar;9(Suppl 1):S27–35. [PubMed] [Google Scholar]
- 13.Bach RR. Tissue factor encryption. Arterioscler Thromb Vasc Biol. 2006 Mar;26(3):456–61. doi: 10.1161/01.ATV.0000202656.53964.04. [DOI] [PubMed] [Google Scholar]
- 14.Pendurthi UR, Williams JT, Rao LV. Inhibition of tissue factor gene activation in cultured endothelial cells by curcumin. Suppression of activation of transcription factors Egr-1, AP-1, and NF-kappa B. Arterioscler Thromb Vasc Biol. 1997 Dec;17(12):3406–13. doi: 10.1161/01.atv.17.12.3406. [DOI] [PubMed] [Google Scholar]
- 15.Napoleone E, Di Santo A, Lorenzet R. Monocytes upregulate endothelial cell expression of tissue factor: a role for cell-cell contact and cross-talk. Blood. 1997 Jan 15;89(2):541–9. [PubMed] [Google Scholar]
- 16.Bierhaus A, Zhang Y, Deng Y, Mackman N, Quehenberger P, Haase M, et al. Mechanism of the tumor necrosis factor alpha-mediated induction of endothelial tissue factor. J Biol Chem. 1995 Nov 3;270(44):26419–32. doi: 10.1074/jbc.270.44.26419. [DOI] [PubMed] [Google Scholar]
- 17.Schorer AE, Kaplan ME, Rao GH, Moldow CF. Interleukin 1 stimulates endothelial cell tissue factor production and expression by a prostaglandin-independent mechanism. Thromb Haemost. 1986 Dec 15;56(3):256–9. [PubMed] [Google Scholar]
- 18.Parry GC, Mackman N. Transcriptional regulation of tissue factor expression in human endothelial cells. Arterioscler Thromb Vasc Biol. 1995 May;15(5):612–21. doi: 10.1161/01.atv.15.5.612. [DOI] [PubMed] [Google Scholar]
- 19.Clarke DL, Clifford RL, Jindarat S, Proud D, Pang L, Belvisi M, et al. TNFalpha and IFNgamma synergistically enhance transcriptional activation of CXCL10 in human airway smooth muscle cells via STAT-1, NF-kappaB, and the transcriptional coactivator CREB-binding protein. J Biol Chem. 2010 Sep 17;285(38):29101–10. doi: 10.1074/jbc.M109.0999952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qi XF, Kim DH, Yoon YS, Jin D, Huang XZ, Li JH, et al. Essential involvement of cross-talk between IFN-gamma and TNF-alpha in CXCL10 production in human THP-1 monocytes. J Cell Physiol. 2009 Sep;220(3):690–7. doi: 10.1002/jcp.21815. [DOI] [PubMed] [Google Scholar]
- 21.Choi SJ, Lee KH, Park HS, Kim SK, Koh CM, Park JY. Differential expression, shedding, cytokine regulation and function of TNFR1 and TNFR2 in human fetal astrocytes. Yonsei Med J. 2005 Dec 31;46(6):818–26. doi: 10.3349/ymj.2005.46.6.818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang F, Schwarz BT, Graham WV, Wang Y, Su L, Clayburgh DR, et al. IFN-gamma-induced TNFR2 expression is required for TNF-dependent intestinal epithelial barrier dysfunction. Gastroenterology. 2006 Oct;131(4):1153–63. doi: 10.1053/j.gastro.2006.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakagomi A, Freedman SB, Geczy CL. Interferon-gamma and lipopolysaccharide potentiate monocyte tissue factor induction by C-reactive protein: relationship with age, sex, and hormone replacement treatment. Circulation. 2000 Apr 18;101(15):1785–91. doi: 10.1161/01.cir.101.15.1785. [DOI] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001 Dec;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Sebag JA, Hinkle PM. Melanocortin-2 receptor accessory protein MRAP forms antiparallel homodimers. Proc Natl Acad Sci U S A. 2007 Dec 18;104(51):20244–9. doi: 10.1073/pnas.0708916105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saperstein S, Huyck H, Kimball E, Johnston C, Finkelstein J, Pryhuber G. The effects of interleukin-1beta in tumor necrosis factor-alpha-induced acute pulmonary inflammation in mice. Mediators Inflamm. 2009;2009:958658. doi: 10.1155/2009/958658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones BW, Song GJ, Greuber EK, Hinkle PM. Phosphorylation of the endogenous thyrotropin-releasing hormone receptor in pituitary GH3 cells and pituitary tissue revealed by phosphosite-specific antibodies. J Biol Chem. 2007 Apr 27;282(17):12893–906. doi: 10.1074/jbc.M610854200. [DOI] [PubMed] [Google Scholar]
- 28.Thompson BT. Glucocorticoids and acute lung injury. Crit Care Med. 2003 Apr;31(4 Suppl):S253–7. doi: 10.1097/01.CCM.0000057900.19201.55. [DOI] [PubMed] [Google Scholar]
- 29.Murray HW. Interferon-gamma, the activated macrophage, and host defense against microbial challenge. Ann Intern Med. 1988 Apr;108(4):595–608. doi: 10.7326/0003-4819-108-4-595. [DOI] [PubMed] [Google Scholar]
- 30.Christian M, Marangos P, Mak I, McVey J, Barker F, White J, et al. Interferon-gamma modulates prolactin and tissue factor expression in differentiating human endometrial stromal cells. Endocrinology. 2001 Jul;142(7):3142–51. doi: 10.1210/endo.142.7.8231. [DOI] [PubMed] [Google Scholar]
- 31.Scheibenbogen C, Moser H, Krause S, Andreesen R. Interferon-gamma-induced expression of tissue factor activity during human monocyte to macrophage maturation. Haemostasis. 1992;22(4):173–8. doi: 10.1159/000216316. [DOI] [PubMed] [Google Scholar]
- 32.Saperstein S, Chen L, Oakes D, Pryhuber G, Finkelstein J. IL-1beta augments TNF-alpha-mediated inflammatory responses from lung epithelial cells. J Interferon Cytokine Res. 2009 May;29(5):273–84. doi: 10.1089/jir.2008.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim WH, Lee JW, Gao B, Jung MH. Synergistic activation of JNK/SAPK induced by TNF-alpha and IFN-gamma: apoptosis of pancreatic beta-cells via the p53 and ROS pathway. Cell Signal. 2005 Dec;17(12):1516–32. doi: 10.1016/j.cellsig.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 34.Wesemann DR, Benveniste EN. STAT-1 alpha and IFN-gamma as modulators of TNF-alpha signaling in macrophages: regulation and functional implications of the TNF receptor 1:STAT-1 alpha complex. J Immunol. 2003 Nov 15;171(10):5313–9. doi: 10.4049/jimmunol.171.10.5313. [DOI] [PubMed] [Google Scholar]
- 35.Bastarache JA, Wang L, Wang Z, Albertine KH, Matthay MA, Ware LB. Intra-alveolar tissue factor pathway inhibitor is not sufficient to block tissue factor procoagulant activity. Am J Physiol Lung Cell Mol Physiol. 2008 May;294(5):L874–81. doi: 10.1152/ajplung.00372.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.