

Abstract
Opioid substitution and antiretroviral therapies have steadily increased the life spans of AIDS patients with opioid addiction, while the adverse drug-drug interactions and persistence of HIV-associated neurocognitive disorders still require new strategies to target opioid abuse and HIV-1 comorbidities. A bivalent ligand 1 with a 21-atom spacer was thus synthesized and explicitly characterized as a novel pharmacological probe to study the underlying mechanism of opioid-enhanced NeuroAIDS. The steric hindrance generated from the spacer affected the binding affinity and Ca2+ flux inhibition function activity of bivalent ligand 1 at the chemokine receptor CCR5 more profoundly than it did at the mu opioid receptor (MOR). However, the CCR5 radioligand binding affinity and the Ca2+ flux inhibition function of the ligand seemed not necessarily to correlate with its antiviral activity given that it was at least two times more potent than maraviroc alone in reducing Tat expression upon HIV-1 infection in human astrocytes. Furthermore, the ligand was also about two times more potent than the simple mixture of maraviroc and naltrexone in the same viral entry inhibition assay. Therefore bivalent ligand 1 seemed to function more effectively by targeting specifically the putative MOR–CCR5 heterodimer in the viral invasion process. The results reported here suggest that a properly designed bivalent ligand may serve as a useful chemical probe to study the potential MOR–CCR5 interaction during the progression of NeuroAIDS.
Keywords: MOR, CCR5, bivalent ligand, functional selectivity, NeuroAIDS
Introduction
Drug abuse and acquired immunodeficiency syndrome (AIDS) are two intertwined epidemics which still pose a huge challenge in the modern society.1 Statistics showed that injecting drug use was responsible for nearly 10% of the global human immunodeficiency virus (HIV) infection while HIV infection could also increase drug addiction vulnerability. 2–3
Among the abused substances, opioids account for the third major class behind cannabis and amphetamines.4 Studies have shown that chronic opioid use and abuse have severe negative impact on the immune system.5 Furthermore, opioids may intrinsically promote the pathogenesis of AIDS directly through central nervous system (CNS).6 As a result, chronic opioid use and abuse not only compromise HIV medical treatment,7 but also exacerbate NeuroAIDS, neurodegenerative diseases caused by chronic HIV infection8. Although opioid substitution therapy (OST) has improved opioid-dependent HIV patients’ adherence to the antiretroviral therapy, the overall effect was compromised by the adverse drug-drug interactions between substituted opioids and anantiretroviral agents.9 Therefore, new strategies are still urgently desired to treat opioid abuse and HIV-1 comorbidities.
One of the pathways that opioids accelerate HIV-1 infection and AIDS progression is through upregulation of the chemokine receptor CCR5 and CXCR4 (two major HIV-1 entry co-receptors), as well as a number of proinflammatory chemokines.5 While there are at least three types of opioid receptors, opioids analgesic and dependence/addiction liability are predominantly attributed to the activation of the mu opioid receptor (MOR).10 Accumulating in vitro studies have demonstrated that the CCR5 and the MOR, two members of the G-protein coupled receptor (GPCR) super family, may undergo crosstalking through dimerization.11–15
It has been reported that GPCR dimerization may lead to a differentiated pharmacology from that of the monomers for some GPCRs.16, 17 Hence, it is tempting to speculate that under certain conditions, a ligand that could interact simultaneously with both receptors of the heterodimer might display a distinct biology profile from that of the ligands which can only interact with one of them. A number of bivalent ligands have been developed to study the biological and pharmacological process of GPCR dimerization.9 Thus, in order to study the putative MOR–CCR5 heterodimer, a bivalent ligand 1 (Figure 1) that combines the pharmacophores of naltrexone (a MOR antagonist) and maraviroc (a CCR5 antagonist) into one molecule was designed and synthesized.9 We here report the comprehensive characterization of this novel molecule for its binding affinity, Ca2+ flux functional activity, and HIV-1 inhibition potency.
Figure 1.

Chemical structures of bivalent ligand (1), monovalent ligands (2, 3, 6), and maraviroc analogues, 4 and 5.
Results and discussion
Bivalent ligand 1 was first characterized in hMOR-expressed CHO cells in the competitive radioligand binding assay as described previously.18, 19 As shown in Table 1, bivalent ligand 1 retained moderate binding affinity to the MOR, as indicated by the two-digit nanomolar Ki value. Monovalent ligand 2 displayed relatively high MOR affinity compared to the parent compound, naltrexone. The decreased binding affinity compared to the parent pharmacophores has been reported for several series of bivalent ligands targeting the MOR.9 In our case the adoption of the 21-atom spacer was based on several reported GPCR bivalent ligands,9 while it seemed to deserve further elaboration and exploration.
Table 1.
MOR Radioligand binding assay and DAMGO stimulated Ca2+ flux assaya
| Compound | Radioligand binding | Ca2+ inhibition |
|---|---|---|
| Ki ± SEM (nM) | IC50 ± SEM (nM) | |
| naltrexone | 0.7 ± 0.1 | 8.9 ± 0.9 |
| 1 | 51.8 ± 7.9 | 40.0 ± 4.8 |
| 2 | 9.2 ± 3.4 | 37.8 ± 4.4 |
[3H]naloxone was used as the radioligand in the binding assay. The values are the means ± S.E.M. of three independent experiments.
As Ca2+ flux is associated with the activation of the MOR,20–22 the functional activity of bivalent ligand 1, monovalent ligand 2, and naltrexone was then evaluated in a Ca2+ mobilization assay in hMOR-CHO cells transfected with chimeric Gqi5 following a published protocol.23 No agonism was observed for any of the tested compounds (data not shown). Thus, they were further assessed for their antagonist properties as the ability to inhibit DAMGO (a MOR agonist) induced Ca2+ flux. As shown in Table 1, the potency of bivalent ligand 1 to antagonize DAMGO-induced Ca2+ flux was modestly reduced (less than 5-fold) compared to that of naltrexone. Nevertheless, bivalent ligand 1 did fulfill the original design as a potent MOR antagonist with moderate binding affinity.
Afterwards, the pharmacological profile of bivalent ligand 1 at the chemokine receptor CCR5 was characterized consequently. The competitive radioligand binding assay was conducted in CCR5 rhesus macaque membrane preparations from Chem-1 cells. Monovalent ligand 3 and compound 4, an analogue of maraviroc, were tested along under the same condition. Introduction of the 4-NH2 group onto the phenyl ring of maraviroc, as seen in compound 4, caused approximately 65-fold decrease in the binding affinity, compared to maraviroc. The decrease of the binding affinity was even more profound for bivalent ligand 1 and monovalent ligand 3, as their Ki values dropped to submicromolar range, respectively. Apparently a para-substitution on the phenyl ring of maraviroc, no matter its size, is detrimental for the CCR5 binding (Table 2).
Table 2.
CCR5 Radioligand binding assay and RANTES stimulated Ca2+ flux assaya
| Compound | Radioligand binding | Ca2+ inhibition |
|---|---|---|
| Ki ± SEM (nM) | IC50 ± SEM (nM) | |
| maraviroc | 0.24 ± 0.06 | 2.2 ± 0.3 |
| 1 | 239 ± 56 | 126 ± 28 |
| 3 | 151 ± 44 | 622 ± 36 |
| 4 | 15.3 ± 4.8 | 14.2 ± 1.9 |
[125I]MIP-1α was used as the radioligand in the binding assay. The values are the means ± S.E.M. of three independent experiments.
Then the Ca2+ functional activity of bivalent ligand 1 was evaluated in the Gqi5 transfected CCR5-MOLT-4 cells as described in the literature.23 As expected, no CCR5 agonism was detected for the bivalent ligand 1 (data not shown). In the RANTES induced Ca2+ flux inhibition assay (Table 2), the bivalent ligand 1 was approximately 60-fold less potent than maraviroc. A more significant potency decrease (nearly 300 times) was observed for the monovalent ligand 3, compared to maraviroc. In order to figure out the possible reasons for such a dramatic drop of their potency, two analogues (4 and 5, Figure 1) of mavaviroc carrying gradient steric hindrance characters at the same substitution position were evaluated under the same condition. Compound 4 showed a modest reduction of the potency (Table 2). However, the inhibition potency of the N-t-Boc protected analogue 5 dropped to micromolar (IC50 = 1.57 ± 0.18 μM). It thus appeared that steric hindrance may play an essential role for the reduced potency, as seen in both the bivalent ligand 1 and the monovalent ligand 3. To further test this hypothesis, a monovalent ligand 6 (Figure 1) with a 7-atom spacer was synthesized and assessed under the same conditions. The inhibition potency of the monovalent ligand 6 (IC50 = 7.91 ± 0.76 μM) was further reduced compared to analogue 5. This again suggested that steric hindrance generated through introducing substitutions onto the para-position of phenyl ring system in maraviroc is disadvantageous for its antagonism in Ca2+ mobilization. Nonetheless, being a CCR5 antagonist, bivalent ligand 1 still met the basic requirements of the original design though its relatively lower binding affinity to the CCR5 was somehow less promising.
As the bivalent ligand 1 has been designed as a molecular probe to study the underlying mechanisms of opioid-enhanced NeuroAIDS by targeting the MOR-CCR5 heterodimerization, a luciferase-based HIV-1SF162 infection assay measuring the relative transactivator of transcription (Tat) protein expression in human astrocytes was performed (Figure 2). Tat is encoded by HIV-1 and is the first protein to be produced after HIV-1 infection.24 Thus, Tat expression level is directly proportioned to the extent of HIV-1 invasion. Astrocytes were chosen based on the following facts: first, astrocytes express both the MOR and the CCR5;25, 26 Second, the human blood-brain barrier (BBB) is composed of interacting adjacent cerebral endothelial cells (CECs) and astrocytes,8 which makes astrocytes a readily accessible target during the course of HIV infection; Thirdly, during NeuroAIDS development astroglia are important cellular sites within the CNS where opioids synergistically potentiate the pathophysiological effects of HIV-1 infection.27–29
Figure 2.
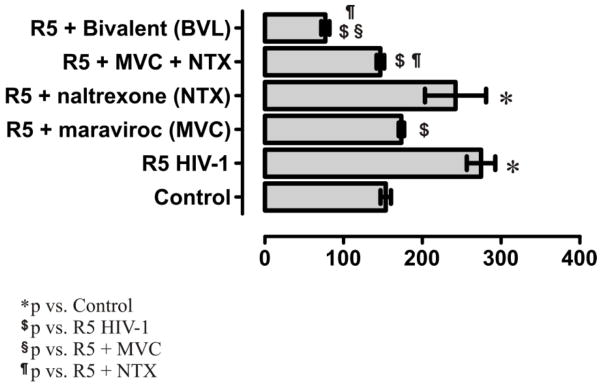
HIV-1SF162 infectivity in human astrocytes (HA) was determined based on the relative amount of Tat protein expressed by the virus using a luciferase based assay. Dose response effect was first studied for each compound (See Supporting Information). Values are absorbance ± SEM (x-axis) of three independent experiments at 18 h post-infection [Maraviroc (MVC), 100 nM; naltrexone (NTX), 1.5 μM; bivalent ligand 1, 100 nM; *p < 0.005 vs. uninfected cells (control); $p < 0.05 vs. R5 HIV-1; §p < 0.05 vs. R5 + MVC; ¶p < 0.05 vs. R5 + NTX].
In Figure 2 it showed that the relative Tat expression was significantly increased in astrocytes after infection with R5 HIV-1SF162 isolated from the CNS of AIDS patients. As expected, exposure to maraviroc (MVC, 100 nM) considerably inhibited viral entry by interrupting the role of CCR5 as a co-receptor of HIV-1. In contrast, naltrexone (NTX, 1.5 μM) did not show significant effect on Tat production. Not surprisingly, the blockage of virus infection by a simple mixture of maraviroc (100 nM) and naltrexone (1.5 μM) was similar to the effect from maraviroc alone. However, in a dose response screening (see Supplementary Information) bivalent ligand 1 (100 nM) was significantly effective in inhibiting viral entry into human astrocytes, causing at least a two-fold decrease in Tat expression when compared to maraviroc alone, or the combination of maraviroc and naltrexone, respectively. In a separate study the bivalent ligand 1 (100 nM) did not affect the viability of astroglial cells using dual (green/red) fluorescence labeling with AOPI (data not shown). The results thus indicated that the bivalent ligand 1 might function as a potent HIV-1 inhibitory agent in human astrocytes through interacting specifically with the putative MOR–CCR5 heterodimer.
During the development of CCR5 antagonists as anti-HIV agents, Lemoine et al. found that there was no definite correlation between the radioligand binding affinity and the antiviral activity.30 Meanwhile, a number of GPCR ligands have been proposed to show “functional selectivity: differences in ligand-induced intermediate conformational states, diversity of G proteins, scaffolding and signaling partners, and receptor oligomers may lead to different functions”.31 Based on the results obtained from the current study for bivalent ligand 1 and maraviroc, a possible mechanism of their “functional selectivity” from the receptor dimerization prospective was proposed (Figure 3). In the radioligand binding assay from the Chem-1 cell membrane and Ca2+ functional assay in MOLT-4 cells, the CCR5 receptor may exist as monomer or homodimers. The conformation of maraviroc may fit the binding pocket of the CCR5 monomer and/or the homodimer nicely to induce its function. Whereas in the HIV-1 invasion assay, CCR5 and MOR may form heterodimers in the human astrocytes, and the subsequent conformation change might enable the binding pocket of CCR5 to accommodate the bivalent ligand (the conformation of the CCR5 antagonist pharmacophore portion might also be changed due to the introduction of the spacer and the MOR antagonist portion) to induce the inhibition of viral entry. On the contrary, the bivalent molecule may not fit as well as maraviroc could into the binding pocket of the CCR5 monomer and/or homodimer. Similarly maraviroc may not recognize the CCR5 heterodimer binding pocket as well as the bivalent ligand could. As a result, maraviroc was observed to be much more potent than bivalent ligand 1 in the monocloned receptor competition binding assay and the Ca2+ flux inhibition assay while bivalent ligand 1 apparently was more potent than maraviroc to inhibit HIV-1 infection in the invasion assay. A similar scenario could be speculated for the MOR binding pocket recognition process. Meanwhile such speculation does not exclude the existence of functional CCR5 monomers or homodimers in astrocytes, either other possible mechanisms (e.g. lipid raft32 involvement in the receptor dimerization).
Figure 3.

Schematic illustration of the proposed mechanism for the “functional selectivity” of bivalent ligand 1 and maraviroc in the radioligand binding assay, Ca2+ flux inhibition assay, and HIV-1 invasion assay. The CCR5 receptor may exist as monomer or form homodimers in the monocloned receptor expressed assays while the CCR5 and the MOR may exist as heterodimers in the human astrocytes and the CCR5 binding pocket in the heterodimer might accommodate the bivalent ligand preferably.
Conclusions
In conclusion, the bivalent ligand 1 acted as a CCR5 and a MOR dual antagonist with moderate binding affinity to both receptors compared to the parent pharmacophores. The steric hindrance generated from the introduction of the spacer to the molecule affected its binding and Ca2+ function activity at the CCR5 more profoundly than it did at the MOR, whereas it was more potent than maraviroc in reducing Tat expression upon HIV-1 infection in human astrocytes. It thus seemed that small molecule maraviroc and bivalent ligand 1 displayed a “functional selectivity” profile upon binding to the CCR5 under different circumstances. More importantly, bivalent ligand 1 was two times more potent than the mixture of maraviroc and naltrexone in HIV-1 entry inhibition. The results reported here thus support the hypothesis that a properly designed bivalent ligand could block HIV-1 invasion into host cells by targeting specifically the putative MOR–CCR5 heterodimer, which makes the bivalent ligand 1 a useful chemical entity to study the mechanism of opioid abuse-enhanced NeuroAIDS.
Supplementary Material
Acknowledgments
We thank EMD Millipore for conducting the CCR5 radioligand binding assay and the funds from the National Institutes of Health (Y.Z., DA024022; N.E., DA026744; K.F.H., DA019398 and DA027374).
Footnotes
Synthetic procedure and spectral characterization of monovalent ligand 6, and experimental procedures for the biological assays. This material is available free of charge via the Internet.
Notes and references
- 1.National Institute of Drug Abuse. [Accessed on May 12, 2012];DrugFacts: HIV/AIDS and Drug Abuse: Intertwined Epidemics. http://www.drugabuse.gov/publications/drugfacts/hivaids-drug-abuse-intertwined-epidemics.
- 2.Mathers BM, Degenhardt L, Phillips B, Wiessing L, Hickman M, Strathdee SA, Wodak A, Panda S, Tyndall M, Toufik A, Mattick RP. Lancet. 2008;372:1733–1745. doi: 10.1016/S0140-6736(08)61311-2. [DOI] [PubMed] [Google Scholar]
- 3.Chang SL, Vigorito M. Am J Infect Dis. 2006;2:98–106. [Google Scholar]
- 4.United Nations Office on Drugs and Crime. [Accessed on May 12, 2012];World Drug Report. 2011 http://www.unodc.org/unodc/en/data-and-analysis/WDR-2011.html.
- 5.Roy S, Ninkovic J, Banerjee S, Charboneau RG, Das S, Dutta R, Kirchner VA, Koodie L, Ma J, Meng J, Barke RA. J Neuroimmune Pharmacol. 2011;6:442–465. doi: 10.1007/s11481-011-9292-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hauser KF, El-Hage N, Buch S, Berger JR, Tyor WR, Nath A, Bruce-Keller AJ, Knapp PE. Neurotoxicity Res. 2005;8:63–80. doi: 10.1007/BF03033820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gonzalez A, Barinas J, O’Cleirigh C. Curr HIV/AIDS Rep. 2011;8:223–234. doi: 10.1007/s11904-011-0093-5. [DOI] [PubMed] [Google Scholar]
- 8.Shapshak P, Kangueane P, Fujimura RK, Commins D, Chiappelli F, Singer E, Levine AJ, Minagar A, Novembre FJ, Somboonwit C, Nath A, Sinnott JT. AIDS. 2011;25:123–141. doi: 10.1097/QAD.0b013e328340fd42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Liu Z, Zhang J, Zhang A. Design of multivalent ligand targeting G-protein-coupled receptors. Curr Pharm Des. 2009;15:682–718. doi: 10.2174/138161209787315639. [DOI] [PubMed] [Google Scholar]; b) Lenhart JA, Ling X, Gandhi R, Guo TL, Gerk PM, Brunzell DH, Zhang S. “Clicked” bivalent ligands containing curcumin and cholesterol as multifunctional Aβ oligomerization inhibitors: design, synthesis, and biological characterization. J Med Chem. 2010;53:6198–6209. doi: 10.1021/jm100601q. [DOI] [PubMed] [Google Scholar]; c) Li F, Liu J, Jas GS, Zhang J, Qin G, Xing J, Cotes C, Zhao H, Wang X, Diaz LA, Shi ZZ, Lee DY, Li KCP, Li Z. Synthesis and evaluation of a near-infrared fluorescent non-peptidic bivalent integrin αvβ3 antagonist for cancer imaging. Bioconjugate Chem. 2010;21:270–278. doi: 10.1021/bc900313d. [DOI] [PubMed] [Google Scholar]; d) Yuan Y, Arnatt CK, Li G, Haney KM, Ding D, Jacob JC, Selley DE, Zhang Y. Org Biomol Chem. 2012;10:2633–2646. doi: 10.1039/c2ob06801j. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zheng Y, Akgün E, Harikumar KG, Hopson J, Powers MD, Lunzer MM, Miller LJ, Portoghese PS. Induced association of μ opioid (MOP) and type 2 cholecystokinin (CCK2) receptors by novel bivalent ligands. J Med Chem. 2009;52:247–258. doi: 10.1021/jm800174p. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Xie Z, Bhushan RG, Daniels DJ, Portoghese PS. Interaction of bivalent ligand KDN21 with heterodimeric δ-κ opioid receptors in human embryonic kidney 293 cells. Mol Pharmacol. 2005;68:1079–1086. doi: 10.1124/mol.105.012070. [DOI] [PubMed] [Google Scholar]; g) Peng X, Knapp BI, Bidlack JM, Neumeyer JL. Synthesis and preliminary in vitro investigation of bivalent ligands containing homo- and heterodimeric pharmacophores at μ, δ, and κ opioid receptors. J Med Chem. 2006;49:256–262. doi: 10.1021/jm050577x. [DOI] [PubMed] [Google Scholar]; h) Neumeyer JL, Zhang A, Xiong W, Gu XH, Hilbert JE, Knapp BI, Negus SS, Mello NK, Bidlack JM. Design and synthesis of novel dimeric morphinan ligands for κ and μ opioid receptors. J Med Chem. 2003;46:5162–5170. doi: 10.1021/jm030139v. [DOI] [PubMed] [Google Scholar]
- 10.Schmidhammer H. Prog Med Chem. 1998;35:83–132. [PubMed] [Google Scholar]
- 11.Song C, Rahim RT, Davey PC, Bednar F, Bardi G, Zhang L, Zhang N, Oppenheim JJ, Rogers TJ. J Biol Chem. 2011;286:20354–20365. doi: 10.1074/jbc.M110.177303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen C, Li J, Bot G, Szabo I, Rogers TJ, Liu-Chen LY. Eur J Pharmacol. 2004;483:175–186. doi: 10.1016/j.ejphar.2003.10.033. [DOI] [PubMed] [Google Scholar]
- 13.Szabo I, Wetzel MA, Zhang N, Steele AD, Kaminsky DE, Chen C, Liu-Chen LY, Bednar F, Henderson EE, Howard OM, Oppenheim JJ, Rogers TJ. J Leukoc Biol. 2003;74:1074–1082. doi: 10.1189/jlb.0203067. [DOI] [PubMed] [Google Scholar]
- 14.Szabo I, Chen XH, Xin L, Adler MW, Howard OM, Oppenheim JJ, Rogers TJ. Proc Natl Acad Sci USA. 2002;99:10276–10281. doi: 10.1073/pnas.102327699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki S, Chuang LF, Yau P, Doi RH, Chuang RY. Exp Cell Res. 2002;280:192–200. doi: 10.1006/excr.2002.5638. [DOI] [PubMed] [Google Scholar]
- 16.Devi LA. Trends Pharmacol Sci. 2001;22:532–537. doi: 10.1016/s0165-6147(00)01799-5. [DOI] [PubMed] [Google Scholar]
- 17.Milligan G. Br J Pharmacol. 2009;158:5–14. doi: 10.1111/j.1476-5381.2009.00169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li G, Aschenbach LC, Chen J, Cassidy MP, Stevens DL, Gabra BH, Selley DE, Dewey WL, Westkaemper RB, Zhang Y. J Med Chem. 2009;52:1416–1427. doi: 10.1021/jm801272c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan Y, Li G, He H, Stevens DL, Kozak P, Scoggins KL, Mitra P, Gerk PM, Selley DE, Dewey WL, Zhang Y. ACS Chem Neurosci. 2011;2:346–351. doi: 10.1021/cn2000348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hauser KF, Stiene-Martin A, Mattson MP, Elde RP, Ryan SE, Godleske CC. Brain Res. 1996;720:191–123. doi: 10.1016/0006-8993(96)00103-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stiene-Martin A, Mattson MP, Hauser KF. Dec Brain Res. 1993;76:189–196. doi: 10.1016/0165-3806(93)90207-q. [DOI] [PubMed] [Google Scholar]
- 22.Kaneko S, Yuasa J, Takahashi H, Satoh M. Mol Brain Res. 1994;22:69–75. doi: 10.1016/0169-328x(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 23.Conklin BR, Farfel Z, Lustig KD, Julius D, Bourne HR. Nature. 1993;363:274–276. doi: 10.1038/363274a0. [DOI] [PubMed] [Google Scholar]
- 24.Johri MK, Mishra R, Chhatbar C, Unni SK, Singh SK. Expert Opin Biol Ther. 2011;11:269–283. doi: 10.1517/14712598.2011.546339. [DOI] [PubMed] [Google Scholar]
- 25.Khurdayan VK, Buch S, El-Hage N, Lutz SE, Goebel SM, Singh IN, Knapp PE, Turchan-Cholewo J, Nath A, Hauser KF. Eur J Neurosci. 2004;19:3171–3182. doi: 10.1111/j.0953-816X.2004.03461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashraf T, Ronaldson PT, Persidsky Y, Bendayan R. J Neurosci Res. 2011;89:1773–1782. doi: 10.1002/jnr.22720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou S, Fitting S, Hahn YK, Welch SP, El-Hage N, Hauser KF, Knapp PE. Brain. 2011;134:3613–3628. doi: 10.1093/brain/awr281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.El-Hage N, Bruce-Keller AJ, Yakovleva T, Bazov I, Bakalkin G, Knapp PE, Hauser KF. PLoS ONE. 2008;3:e4093. doi: 10.1371/journal.pone.0004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.El-Hage N, Gurwell JA, Singh IN, Knapp PE, Nath A, Hauser KF. Glia. 2005;50:91–106. doi: 10.1002/glia.20148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lemoine RC, Petersen AC, Setti L, Baldinger T, Wanner J, Jekle A, Heilek G, deRosier A, Ji C, Rotstein DM. Bioorg Med Chem Lett. 2010;20:1674–1676. doi: 10.1016/j.bmcl.2010.01.080. [DOI] [PubMed] [Google Scholar]
- 31.Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki KGN. Lipid rafts generate digital-like signal transduction in cell plasma membranes. Biotechnol J. 2012;7:753–761. doi: 10.1002/biot.201100360. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.