

Abstract
The emergence of hybrid materials combining biomacromolecules and organic polymers has received broad attention based on their potential applications in chemical, biological and materials sciences. Among different coupling strategies, the grafting of oligonucleotides to organic polymers as side chains by covalent bonds provides a novel platform whereby the properties of both oligonucleotides and polymer backbone are integrated, manipulated and optimized for various applications. In this review, we give the perspective on this specific type of DNA polymer hybrid materials , using selected examples with emphasis on bioanalysis, biomedicine and stimuli-responsive materials. It is expected the success of DNA-grafting polymers will not only impact the frabication of novel bimolecule incorporated materials, but also will influence how the properties of synthetic materials are tailored using different functional groups.
1. Introduction
A primary goal in synthetic chemistry is the ability to synthesize macromolecules with well-defined monomer composition and sequence while, at the same time, exhibiting novel properties and functions. Materials science and polymer chemistry have been extensively used in the generation of artificial macromolecules with different building blocks, among which bio-originated macromolecules, including proteins, polypeptides and nucleic acids, are of enormous importance.1-4 For decades, the combination of peptides with organic polymers has been explored in depth with a multitude of reviews on their synthesis and supramolecular organization.5, 6 However, the coupling of DNAs with organic polymers has been understudied.7, 8
Owing to the technological breakthrough, the revolutionary principle of solid phase peptide synthesis (SPPS) has been adapted for the construction of other biomacromolecules such as DNAs. The solid phase synthesis method enables scientists to generate DNAs that share similar properties with their natural counterparts. Moreover, solid phase synthesis offers a highly automated and convenient way for the incorporation of non-nucleic acid functional groups into the oligonucleotides which then exhibit exciting and unprecedented functions. DNA has been coupled with various inorganic materials, such as silica nanoparticles and gold nanoparticles, using different functional groups.9, 10 Small organic molecules, including fluorescent dyes and photoresponsive moieties, have also been incorporated into DNA chains.11, 12 Recently, advances in combinatorial chemistry have enabled widespread research in artificial oligonucleotides, particularly the introduction of a novel class of synthetic oligonucleotides known as aptamers. Aptamers are single-stranded oligonucleotides that bind to specific targets and are generated from an in vitro process known as Systematic Evolution of Ligands by Exponential Enrichment (SELEX).13, 14 With a wide range of targets, including small molecules, proteins, and even whole cells, 15-17 aptamers have found numerous applications in biosensing, molecular imaging, and targeted cancer therapy.18-20 Rapid development in the field of artificial DNA has propelled its applications in biosensing, biomedicine, and functional materials.21-23
During the last decade, a new class of hybrid materials, DNA block copolymers consisting oligonucleotides and organic polymers, has emerged as one of the newest research areas. Depending on the resulting structures of the hybrid macromolecules, they are divided into two categories: linear DNA block copolymers and DNA-grafting linear polymers. A linear DNA block copolymer consist of an oligonucleotide and a terminal organic polymer, while a DNA-grafting linear polymer is composed of a linear organic polymer with multiple strands of oligonucleotide as side chains. Different types of moieties were also modified on one single polymer chain, thus providing new ways for the construction of multifunctional materials.24-26 Furthermore, DNA block copolymers were engineered to form even more complex materials, such as multiblock structures, micelles, stimuli-responsive hydrogels.27-30 Therefore, this materials class shows promise in applications ranging from bioanalysis to biomedicine and materials science. In this review, emphasis is placed on DNA-grafting linear polymers, and selected examples are given to illustrate the most recent developments in the engineering and applications of DNA block copolymer hybrid materials. The review is split into three sections: synthesis of DNA-grafting linear polymers, direct applications of the DNA-grafting linear polymers, and functional hydrogels assembled from DNA-grafting linear polymers. Additionally, linear DNA block copolymers are excluded as this field of research has been extensively reviewed elsewhere.7, 8, 31, 32
2. Synthesis of DNA-grafting linear polymers
DNA-grafting hybrid linear polymer features a synthetic polymer backbone with multistrand DNAs as side chains to form a comb-like structure. Its synthesis can be realized by two general strategies (Figure 1). In the first strategy, DNAs are functionalized onto the pre-synthesized linear polymer backbone through various types of coupling chemistry (Figure 1A). One of the classic method includes active ester groups-mediated formation of amide bonds between polymers and DNAs. For example, amino-terminated DNAs could be coupled with carboxyl groups of poly(acrylic acid) using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) as coupling reagent.33 Alternatively, a polymer backbone with activated ester groups could directly react with amino-modified DNAs to form amide bonds. 33 Another method towards DNA modification on pre-synthesized polymers relies on covalent coupling of DNAs with polymers on a solid support. Phosphoramidite chemistry has been employed starting with the formation of organic polymers bearing multiple phosphoramidite groups. This phosphoramidite-polymer was then reacted with detritylated 5′-OH end of the DNA on the solid support followed by the cleavage of DNA from the support.25, 34 Moreover, reactive synthetic polymers were also immobilized on the solid support for the purpose of constructing DNA side-chain polymers.35 In one report, controlled pore glass (CPG) surface covered by hydroxyl groups was covalently coated with an alternating copolymer which carried maleic anhydride moieties along the backbone via ester bond formation. Afterwards, the remaining anhydride moieties were reacted to form amide bonds with 5′-dimethoxytrityl thymidine 3′-(6-aminohexyl phosphate) which initiated the oligonucleotide synthesis. The selective cleavage of DNA-grafting side-chain polymers after DNA synthesis was carried out by basic treatment due to the difference in stability between ester and amide connections. However, the strategy based on DNA coupling to presynthesized polymers generally results in conjugates with a small number of DNA strands.
Figure 1.
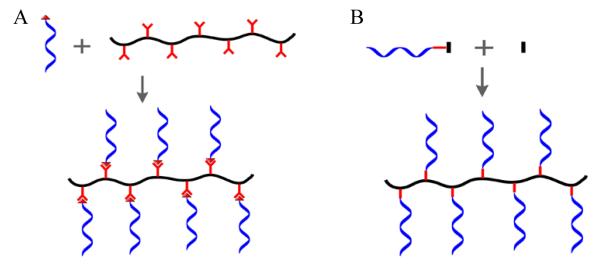
Synthesis of DNA-grafting polymers. A) Covalent modification of nucleic acids to polymers via coupling chemistry. B) Copolymerization of polymerizable group terminated nucleic acids with polymer monomers.
A fundamentally different strategy to synthesize DNA side-chain polymers is based on copolymerization of polymerizable DNA monomers with regular polymer monomers such as acrylamide (Figure 1B). This type of DNA monomer can be synthesized by modifying polymerizable moieties onto functional groups connected with DNAs. For instance, Murakami and coworkers reported the fabrication of a methacryloyl group- conjugated ssDNA using methacryloyloxy succinimide and amino-terminated ssDNA.36 Alternatively, polymerizable DNA monomers can also be generated directly by solid phase synthesis using a phosphoramidite consisting of a polymerizable group such as acrydite. Our group has been using acrydite- modified DNAs for synthesis of DNA-grafting polymer materials for different applications.26, 37 However, limitations of DNA polymers generated by this strategy include the poor solubility and high viscosity which make it difficult to handle. Another drawback is that the resulting structure cannot be fully characterized.
In addition to the conventional coupling chemistry for generation of DNA-grafting polymer, it is also worthy of attention that click chemistry based strategies have shown great promise in construction of complex macromolecular architectures using nucleic acids and other biomolecules. By far the coppercatalyzed azide–alkyne cycloaddition (CuAAC) reaction is one of the most widely used click chemistry approaches because of the high efficiency and specificity. CuAAC has been extensively employed to facilitate the engineering of functional macromolecular structures, such as brush and graft copolymers, shell or core cross-linked micelles, and hyperbranched materials.38 DNA has also been modified to organic polymers by this method to create novel macromolecules or assemblies such as star polymer and micelles.39, 40 It is believed that click chemistry will extend the methodology for preparation of sophisticated DNA-polymer conjugates including DNA side-chain polymers. However, click chemistry-based coupling schemes also have limitations. Excess copper may lead to drastic consequences if products are used for drug delivery.
3. Applications of DNA-grafting hybrid linear polymer
DNA side-chain copolymers are generated by grafting DNAs onto the organic polymers such that the attractive features of both materials are maintained and integrated. For example, both increased melting point and sharp transition are achieved when DNA block copolymer hybridizes with complementary DNA block copolymer because of Neighboring-Duplex Cooperativity.41 Compared with mono-strand DNA materials, the assembly of multi-strand DNAs along a single polymer chain introduces significant offers important advantages in various applications, such as signal-amplified bioanalysis and enhanced therapeutic effect.
3.1. DNA-grafting hybrid linear polymers as sensors for DNA detection
DNA detection is important in the practice of clinical diagnostics, gene therapy, forensic science, and biomedical research. Considerable effort has been put into developing novel materials and methodologies for highly sensitive and selective DNA analysis. To achieve highly enhanced fluorescence signals, various nanomaterials, such as quantum dots, silica, and gold nanoparticles, have been investigated as sensing platforms for bioanalysis.42-44 Similar to DNA-nanoparticle conjugates, multiple DNA strands are attached to one single polymer chain. It has been observed that hybridization between two complementary DNA-grafting hybrid linear polymers has a sharper melting profile and higher melting temperature than that observed between two individual complementary strands. 41, 45 It is well known that sharp melting curve is an essential feature of DNA-modified gold nanoparticles and accounts for the enhanced discriminatory power of even single-base mismatches.46 Therefore, because of the arrangement of several DNAs along the polymer backbone, the cooperative behavior of DNA hybridization and coupling flexibility with multiple signaling groups have led to significant improvements in the selectivity and sensitivity of DNA block copolymer-based assays.
DNA detection depends on the vital factors of molecular recognition and signal readout. In one attempt, Mori and coworkers reported a strategy using DNA-grafting Poly(N-isopropylacrylamide) (PNIPAAm) colloidal particles.47 These particles aggregated in the presence of complementary DNA by the destabilization caused by DNA hybridization on the particle’s surface, observed as a turbidity increase of the dispersion. However, this method fell short of sensitivity with a detection limit in the micromolar range. A second example, as demonstrated by Mirkin and coworkers, took advantage of the numerous functional groups on DNA block copolymers by adding a number of ferrocene groups as signaling moieties (Figure 2A).24 The newly constructed triblock conjugates were applied in an electrochemical assay, where the target DNA served as a bridge to bring the conjugates onto the electrode. Consequently, a redox signal from ferrocenyl moieties was measured. Both pM-level limit of detection and single-base mismatch discrimination were achieved. Moreover, dual-channel electrochemical detection of two different DNA targets was enabled using two distinct block copolymers. Conjugated polymers were also employed in the generation of DNA block copolymers for DNA analysis (Figure 2B).48 On-chip DNA synthesis was carried out on a poly (oxadiazole-fluorene) derivative polymer-coated glass slide to fabricate a signal-amplifying DNA chip. Upon the hybridization of dye-labeled target DNA with the DNA grafted on the conjugated polymer, fluorescence resonance energy transfer (FRET) from the conjugated polymer to the dye on the target yielded a fluorescence readout for detection. Furthermore, the chip-based assay can be performed with ease in a parallel fashion, which has the potential for developing a high-throughput method. However, up to now, methods based on DNA-grafting polymers generally have a relatively high limit of detection. To further improve the performance, it is necessary to introduce signal amplification via approaches such as enzymatic reactions. Moreover, the capability for single-base mismatch discrimination is still lack. More sophisticated probe designs are expected in the future.
Figure 2.
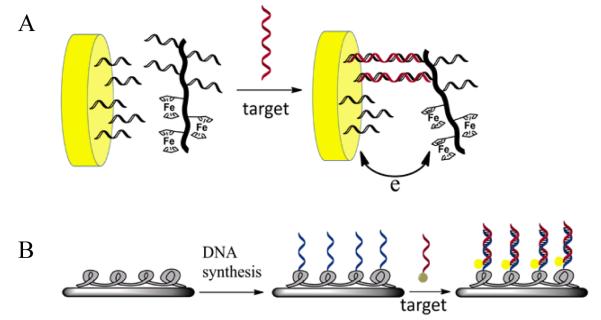
Examples of biosensors for DNA detection based on DNA-grafting polymers. A) Electrochemical detection of DNA employing a DNA–ferrocene–polymer hybrid (adapted from Ref. [24]. B) DNA detection using a light-emitting polymer immobilized on a chip surface (adapted from Ref. [48]).
3.2. DNA-grafting hybrid linear polymers used in DNA delivery
DNA not only provides the molecular basis for the understanding of the biology of human life but also serves as a fundamental building block for the construction of novel therapeutics. Functional DNA-based therapeutics include plasmids for gene therapy, antisense oligonucleotides, DNAzymes and aptamers. However, the therapeutic applications of DNA has been limited due to the poor cellular uptake and rapid enzymatic degradation. Different methods and delivery systems have been developed to facilitate cellular internalization of DNA-based therapeutics with maintained activity. 49, 50
DNA-grafting polymers also find their unique applications in this area. For a few decades, polymer-based therapies have gained attention as a result of their potential advantages over conventional drugs, including biocompatibility, effectiveness in transmembrane delivery and flexible integration with chemotherapies.51, 52 It is well known that positively charged polymers can be effectively internalized into cells, thus serving as an effective carrier for various anticancer drugs. Lebleu and coworkers engineered a DNA-modified poly(L-lysine) (PLL) polymer (Figure3A).53 This type of DNA polymer hybrid can be more easily transported into cells than pristine DNAs. More interestingly, a 15-mer antisense DNA-grafting PLL polymer targeting the initiation region of the mRNA of vesicular stomatitis virus (VSV) N-protein specifically inhibited expression of VSV proteins at a dosage as low as 100 nM. Moveover, a similar approach was employed to deliver antisense oligonucleotides into human immunodeficiency virus type 1 (HIV-1) for inhibition of virus reverse transcription.54
Figure 3.
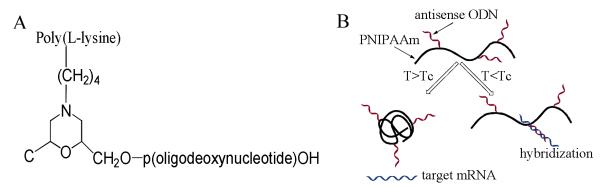
A) Structure of DNA-poly(L-lysine) conjugate. (adapted from Ref. [53]) B) Temperature controlled binding of DNA-PNIPAM conjugate with target mRNA. (adapted from Ref. [55])
Poly(N-isopropylacrylamide) (PNIPAM) is a temperature-responsive polymer regarded as one of the most well-know intelligent polymer materials. Maeda and coworkers reported a antisense DNA-PNIPAM side chain polymer of which the antisense activity can be controlled by temperature (Figure 3B).55 This DNA-PNIPAM conjugate could specifically bind complementary RNA sequence at 27°C below the lower critical solution temperature (TLCST, 33°C) when the conjugate stayed in random coil structure. However, as the temperature was raised to 37°C above TLCST but still below the DNA melting temperature, coil-to-globule transition of PNIPAM backbone significantly inhibited the binding of antisense DNA with the target. The conjugate was later applied in in-vitro study, gene expression at 27°C was inhibited at a similar level to that when pristine DNA was used, while at 37°C, no significant inhibition effect was observed.56 Overall, DNA delivery using DNA-grafting polymers possesses biological potentials, an systematic understanding is still needed to interpret the behavior of the DNA-grafting polymers on the transmembrane delivery, intracellular fate, biocompatibility, etc.
3.3. DNA-grafting hybrid linear polymers used in cancer therapy
Anticancer therapeutic drugs to inhibit cellular proliferation are limited by their general toxicity. Although anticancer drugs work more intensely on a particular type of cancer cell, rather than on healthy cells, some side effects always occur throughout the systematic dosing process. Consequently, different types of novel therapeutic agents have been developed with distinct mechanisms. However, specific selectivity of these therapeutics toward cancer cells is still lacking, and none has been clinically useful. Monoclonal antibodies offer an alternative strategy for selective cancer therapy with less toxicity; however, they fall short owing to availability problems and their modest effects. Similarly, the development of multidrug resistance, another challenge in the field of cancer therapeutics, is driving advances in target-specific drugs with enhanced efficacy.57, 58
In the pursuit of better anticancer therapeutics, much attention has been focusing on the engineering of new therapeutic hybrid materials using anticell aptamers selected using the method known as cell-based systematic evolution of ligands by exponential enrichment (Cell-SELEX).17, 59 Aptamers with special properties have been identified, such as Sgc8, which can be internalized by specific cancer cells. Owing to the flexibility of solid phase synthesis, different types of functional groups can be modified on aptamers. Combining aptamers and polymers, a targeted anticancer system was developed by using the targeting property of aptamers and the intrinsic toxicity of polymers (Figure 4A).26 The hybrid polymer was synthesized by copolymerization of acrylamide, acrydite-modified Sgc8 aptamer, and acrydite/FITC-modified reporter DNA monomer. The resulting polymers exhibited high specificity and internalization ability based on the coupling of Sgc8 aptamer. Furthermore, the DNA polymer hybrid induced selective cytotoxicity in target cell lines, while having little effect on nontarget cells (Figure 4B). Even more striking, when the drug-resistant K562/D cell line was tested for therapeutic effect of this type of aptamer-polymer conjugate, significant cytotoxicity similar to that of the corresponding nondrug-resistant K562 cell line was observed (Figure 4C). The results indicated that the aptamer-polymer conjugate could bypass the P-glycoprotein (P-gp), a drug efflux transporter, on the cell membrane of the drug-resistant K562/D cells and interrupt the cellular metabolism. Therefore, this approach might find potential applications in the development and improvement of anticancer drugs, as well as targeted delivery of cancer therapies. However, the exact mechanism of the therapeutic effect remains unclear although further studies showed that the significant cytotoxicity of DNA-grafting polyacrylamide conjugates is likely rooted in their intrinsic properties of polyacrylamide backbone, including physical size, primary amine groups, and flexibility.
Figure 4.

Aptamer-grafting acrylamide polymers for selective cytotoxicity. A) Scheme of polymeric aptamers. Selective cytotoxicity achieved with B) non-drug-resistant cancer cells and C) drug-resistant cells. (adapted from Ref. [26])
4. DNA-cross-linked dynamic polymer hydrogels
A hydrogel is a network of cross-linked hydrophilic polymers. Hydrogels can hold large amounts of water, up to more than 99% of their weight. Stimuli-responsive dynamic hydrogels have shown great potential in various applications, such as biosensing, drug delivery and microfluidics. Based on the different designs, several classes of hydrogels have been developed with sensitivities towards temperature, pH, biomolecules, and other stimuli.60-63 These dynamic hydrogels can change such properties as volume, color and transparency in response to environmental stimuli.64, 65
Towards the development of novel functional hydrogel materials, various types of molecular interactions have been introduced into the polymeric hydrogel systems, including antibody/antigen binding and conformational change of proteins.66, 67 The DNA-grafting polymers can also be further assembled to form hydrogels, mainly through two different strategies. The first involves addition of permanent linker groups to induce the covalent-bond crosslinking of polymer backbones where the DNAs are also modified. In this strategy, either a dual-functionalized ssDNA, such as ssDNA with amino groups on both ends, or other types of molecules, such as N,N’-Methylenebisacrylamide (MBAm), serve as crosslinkers.36, 68 In the second approach, duplex DNA formed by DNA hybridization serves as the cross-linking groups for the hydrogels. The reversible nature of DNA hybridization in response to external stimulus causes this type of DNA polymer hydrogel to have special properties, such as sol-gel phase transition and responsive releasing capability. As a result, a number of systems with different applications have been developed based on this class of hydrogels.
4.1. DNA-grafting polymer hydrogels for sensor applications
Stimuli-sensitive hydrogels that can sense environmental changes and undergo structural changes hold great promise in the development of chemical sensors.69, 70 For example, some sensitive hydrogels can swell in response to specific biomolecules, such as proteins and glucose, and have become important in the development and application of responsive biomaterials and smart drug delivery systems.66, 71 A ratiometric fluorescent pH sensor was also developed based on resonance energy transfer caused by the volume change of chitosan hydrogels.72
DNA sensing hydrogels have been fabricated using volume change as the output. Maeda and Murakami synthesized a polyacrylamide hydrogel containing DNA-grafting polymer conjugates in a semi-interpenetrating network (semi-IPN) manner (Figure 5A).68 The addition of target DNA caused DNA hybridization resulting in shrinking of the hydrogel. Although the exact mechanism was still unclear, one possible explanation was proposed that dehydration took place upon the formation of duplex DNA and water molecules diffused out of the hydrogel. Using DNA as crosslinkers, the same group designed a new DNA-responsive hydrogel structure (Figure 5B).36 A DNA-polymer hybrid hydrogel was prepared by copolymerization of acrylamide and ssDNA modified with methacryloyl groups on both 3′ and 5′ ends. When the DNA crosslinker was designed as a hairpin, the hydrogel swelled upon the addition of complementary DNA. This volume increase was explained by the hybridization induced elimination of stem-loop structure and longitudinal extension of the DNA crosslinkers. In contrast, when the DNA crosslinker was designed with no secondary structure, a volume shrinkage was obtained due to the addition of the complementary ssDNA. As a summary of the DNA crosslinked hydrogel, on the one hand, the volume change behavior largely depends on the structure of the DNA crosslinkers which can lead to the rational design of hydrogels with desired responsiveness for DNA sensing applications. On the other hand, the application is limited by the slow response rate and small percentage volume change. Further efforts are necessary to optimize the structural properties of this type of DNA-crosslinked hydrogel.
Figure 5.
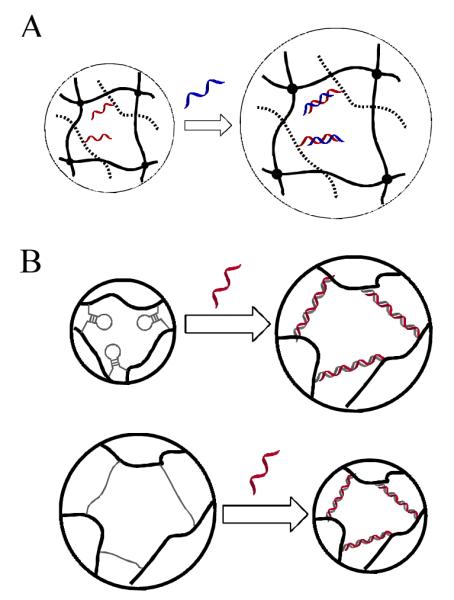
A) Target ssDNA induced swelling of semi-IPN DNA-grafting polymer hydrogel (adapted from Ref. [68]) B). Target ssDNA induced volume change of hairpin DNA (top) and DNA without secondary structure crosslinked polymer hydrogels. (adapted from Ref. [36])
Beside the DNA responsive hydrogels, small molecule responsive hydrogels were also engineered based on aptamer-target interactions (Figure 6A).73 This dynamic hydrogel underwent a gel-to-sol transition in the presence of a specific target. The basic principle of operation relies on target-induced decrease in duplex DNA-crosslinker density of the hydrogel. In order to construct this hydrogel, two acrydite-modified DNA strands, Strand A and Strand B, are separately copolymerized with acrylamide to generate DNA-grafting polymer A and B in solution. A linker ssDNA containing an aptamer segment was then added to form the hydrogel. The sequences were designed such that the three-strand DNA complex would dissociate in the presence of target due to the target induced structural change of aptamer. Consequently, the hydrogel converted back to the solution state if a significant amount of targets is added. In this detection approach, the target-responsive sol/gel phase transition could be visually detected, and the detection of adenosine and thrombin as model targets was demonstrated.
Figure 6.

A) Detection of adenosine using aptamer crosslinked hydrogels by sol-gel transition. (adapted from Ref. [73]) B) Signal amplified detection of targets based on colorimetric reaction catalyzed by enzymes released from target-responsive hydrogels (adapted from Ref. [77]). C) Detection of mersury ion using thymine-rich DNA-grafting polymer hydrogels. (adapted from Ref. [78])
Direct visual detection based on gel-to-sol conversion of DNA polymer hydrogels was made possible for biosensing. However, the detection limit is poor, and quantitative analysis is difficult to achieve. Moreover, most biosensing systems based on the structural changes of hydrogels are unsuitable for practical applications.74-76 Recently, colorimetric assay-based visual detection methods have provided a simple and rapid, yet cost-effective, way for sensitive biosensing as a result of the emergence of aptamers. In order to improve detection using the target-responsive hydrogel, a novel visual detection system using colorimetric agent-caging hydrogels was developed (Figure 6B).28 Similar to the system described above, two DNA-grafting polymers were synthesized separately. Before the addition of aptamers to induce gelation, agents for signal generation were mixed with the DNA polymers. To demonstrate the colorimetric detection, a first attempt was made to use gold nanoparticles, and the upper solution turned red when target was added. As a further step, better analytical performance was reached when an enzyme, amylase, was introduced instead. It is well known that amylose induces a color change of iodine solution from yellow to dark blue and that amylase converts amylose into sugar. In this hydrogel system, the presence of targets caused the release of amylase into the dark blue solution containing amylose and iodine which changed color to yellow as a readout. Overall, this colorimetric agent-caging hydrogel system showed good sensitivity and high specificity.
Additionally, Liu and coworkers described a strategy using a thymine-rich DNA modified polyacrylamide hydrogel to detect and remove mercury ions from water simultaneously (Figure 6C).77 Detection relied on the formation of hairpin structures induced by Hg2+ mediated T-T DNA base-pairing in the hydrogel. The T-Hg2+-T base pair has a higher stability than the classical T-A pair and cannot be exist with ions other than Hg2+.78 After the formation of hairpin in the present of Hg2+, SYBR Green I, a cyanine dye that emits green fluorescence when binding to duplex DNA, is added to the hydogel. Green fluorescence was observed. However, in the absence of Hg2+, yellow fluorescence was obtained. A detection limit of 10 nM Hg2+ by visual detection was achieved with a 50 mL water sample. Moreover, the adsorbed Hg2+ could be removed by acid treatment for the regeneration of the hydrogel.
The above example demonstrated the applications of DNA-grafting hydrogels for detection. Currently, the performance of this class of hydrogels are limited by some intrinsic properties. First, response rate is much slower compared with solution based detection methods, which is due to the restricted diffusion of molecules in the hydrogel network. Second, quantitation by this strategy is hard to achieve. For example, the volume change or sol-gel conversion cannot be directly related to the target concentration. Although the introduction of release of signaling groups provides well-measurable signals, the accurate quantitation still remains as a problem due to the experimental variations.
4.2. DNA-cross-linked polymer hydrogels designed for logic gates
Aside from the utilization of DNA cross-linked polymer hydrogels for biosensor development, other new functions are being explored. The engineering of molecular logic gate systems that generate output signals in response to chemical and physical inputs has attracted extensive attention. Recently, a new class of DNA-based molecular logic gates has shown special features and is considered an excellent platform for in vitro computation.79, 80 DNA-grafting polymer hydrogels have emerged as one of the promising platform for logic gate systems.
An example of this type of logic gate systems involved the synthesis of a polyacrylamide hydrogels with both permanent and duplex DNA crosslinkers. Depending on DNA hybridization, the hydrogel transformed DNA based logic operations into the volume change of the hydrogel.81 As illustrated in Figure 7, two DNA strands with two isolated complementary segments were modified on the hydrogel polymers two form the duplex DNA crosslinkers. The swelling behavior of the hydrogel could be adjusted by adding different complementary DNAs which induced partial or complete dissociation of the duplex DNA crosslinkers through toe-hold displacement reactions. The volume change degree was evaluated by interferometric technique using height as the output. Logical AND and OR operations were realized based on the volume swelling levels. When a single complementary DNA (P1 or P2) was added breaking one of the two duplex segments of the DNA crosslinker, the hydrogel swelled to a level represented by the height L1. At the level L1, the OR gate was realized with the signal “1” when either P1 or P2 was present. Furthermore, a more pronounced signal (L2) was achieved when both P1 and P2 were added to the hydrogel to completely disintegrate the duplex DNA crosslinkers. Therefore, at the level L2, the AND gate was realized with the signal “1” in the presence of both P1 and P2. The reason for the different swelling behaviors upon addition of different complementary DNAs was attributed to the corresponding Donnan effect on the polyelectrolyte hydrogel swelling balance and change in crosslinker density.
Figure 7.
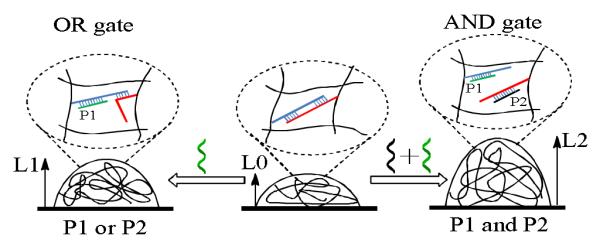
Illustration of hydrogel swelling behaviors in response to complementary sequences monitored by the interferometric technique. (adapted from Ref. [82])
Inspired by the target-responsive DNA polymer hydrogels, extra target recognition functions were introduced to the system in order to construct colorimetric logic gates that respond to chemical stimuli.82 Three ssDNA strands containing aptamer segments were involved in the self-assembly of responsive DNA nanostructures. Two logic gates (AND and OR) for cocaine and ATP were fabricated. In the AND gate (Figure 8A), a hydrogel loaded with output gold nanoparticles (AuNPs) was generated by crosslinking two DNA-grafting polymers through a three-way DNA junction when a linker DNA was added. The DNA sequences were designed such that the presence of a single target only could disintegrate one arm of the three-way junction. Therefore, the DNA polymers were still crosslinked to maintain the hydrogel structure resulting in no release of AuNPs (output “0”). However, the hydrogel AND gate yielded an output “1” only when both targets were added to destroy the three-way junction structure.
Figure 8.

Colorimetric logic gates based on aptamer-crosslinked hydrogels: A) AND gate and B) OR gate using cocaine and ATP as stimuli. (adapted from Ref. [83])
In the design of the OR gate (Figure 8B), the linker DNA bridged the two DNA-grafting polymers to form the hydrogel. Moreover, the sequence of the linker DNA was designed to contain two toe-hold aptamer segments on both termini for binding with their respective targets. The existence of either cocaine or ATP could cause the breakage of the crosslinkers by the aptamer-target induced dissociation of duplex DNA. Therefore, the logic AND gate was realized to yield an output “1” upon the addition of either target. Since the logic gate hydrogel system can perform Boolean operations in response to different molecular inputs, molecular computation is a practical outcome. Moreover, this type of multi-input hydrogel system has potential applications in diagnostics and biomedicine, especially when medical conditions are determined based on combinations of biomarkers. On the other hand, the described hydrogels are synthesized at macroscopic level which is not sensitive to the logic input under a micro-environment. In addition, the slow response of the hydrogels may also limit their applications. Future studies are need to scale down the logic DNA hydrogels and improve the diffusion properties within the network.
4.3. DNA-cross-linked polymer hydrogels for controlled drug delivery
Drug delivery systems with temporal and spatial control are highly desired in the development of next-generation therapeutics with excellent efficacy and specificity, but minimal side effects. Responsive polymer materials have been intensively investigated for controlled drug delivery, and smart polymer hydrogels have received enormous attention.83, 84 The DNA-grafting polymer hydrogel provides a versatile platform with the potential for different biomedical applications.7, 8 Integrated with different functional groups, the structures, as well as functions, of nucleic acids can be easily tuned with various stimuli, such as chemicals, pH, or light, which forms the basis of stimulus-responsive DNA hydrogels for controlled drug delivery and release.85-87
In one report, Simmel and co-workers demonstrated a DNA-controlled hydrogel release system using quantums dot (QD) as a model drug (Figure 9A).88 The DNA hydrogel was synthesized using a similar method described above.73 The DNA crosslinker strand had a toehold which could recognize the release DNA. When complementary release DNA strands were added to the system, toehold initiated strand displacement caused the hydrogel to dissovle followed by the release of QDs from the network. Afterwards, the hydrogel could be regenerated by adding crosslinker DNA strands again. This type of duplex DNA crosslinked hydrogel is biocompatible and programmable. The properties such as mechanical strength, pore size and melting temperature can be adjusted by the length, sequence design and concentration of DNA strands employed during the hydrogel synthesis. Furthermore, the integration of aptamers will facilitate the development of controlled delivery systems responsive to a broad range of targets.
Figure 9.

Controlled release from DNA-crosslinked hydrogels. A) DNA induced release of QDs. (adapted from Ref. [89]) B) Light controlled release from photoresponsive DNA-crosslinked hydrogel. (adapted from Ref. [29] C) NIR light triggered thermal release from DNA nanogels coated on gold nanorods. (adapted from Ref. [37])
In addition, stimuli other than chemicals were also applied to trigger the cargo release from the DNA hydrogels. A photoresponsive DNA-cross-linked hydrogel that undergoes reversible sol–gel conversion upon the irradiation of UV and visible light was created as shown in Figure 9B.29 In the synthesis of the hydrogel, a azobenzene modified DNA crosslinker which hybridized simultaneously with both DNA strands grafted on the polymers was added instead of a regular DNA crosslinker. The azobenzene-regulated DNA hybridization was responsible for the reversible sol-gel conversion. Specifically, the UV/visible light irradiation drove the isomerization of azobenzene moieties between the trans and cis states, thereby regulating the association and dissociation of complementary ssDNA.89, 90 The encapsulation and release of small molecules, proteins, and nanoparticles were tested in a light-controlled fashion. The results showed that significant release happened only upon the irradiation of UV light. Biocompatibility of the hydrogels was also evaluated in cell experiments, which exhibited limited toxicity. Furthermore, the biomedical applications were demonstrated using the photoresponsive DNA hydrogel for controlled release of an anticancer drug, doxorubicin, to kill cancer cells.
As shown in the previous example of controlled release, DNA-cross-linked hydrogels showed impressive capability. However, they may not be optimal when the targeting region is deep in the human body or when the stimuli are subtle, i.e., weak biochemical signals or subnanomolar-level biomarkers. One feasible approach is to design hydrogels that respond to external stimuli, such as light or magnetic and electrical fields. Another approach is to engineer nanometer-sized delivery systems that are sensitive to localized weak stimuli. Gold nanoparticles can absorb light energy in the near-infrared (NIR) range and generate heat to cause an increase of temperature in the surrounding area which can be used to manipulate heat-sensitive events, such as DNA hybridization and cell destruction.91, 92 Based the photothermal nanoparticles and the DNA-cross-linked hydrogel, a NIR light-responsive core-shell nanogel for targeted drug delivery (Figure 9C).37 This nanostructured hydrogel particle was constructed from a nanosized DNA-cross-linked polymer hydrogel and an embedded gold/silver nanoparticle with drug molecules trapped in the hydrogel layer. Cell-specific aptamers were also modified on the hydrogel network to guide the therapeutic nanogels to the targets. Upon irradiation of NIR light, elevated temperature caused the DNA-cross-linked hydrogel layer to undergo a rapid melting process by the dissociation of DNA duplex. Meanwhile, the payload drugs were released to generate therapeutic effect. The NIR light-controlled release of therapeutics was investigated using doxorubicin on cancer cells. The results showed that the drug-loaded targeted NIR light-responsive nanogel possessed high therapeutic effect with specificity toward target cancer cells.
Same as the hydrogels mentioned in the previous sections sharing the same gel-to-sol conversion, this class of stimulus-responsive hydrogel delivery systems may suffer from the large sizes which results in problems including insensitivity to stimulus, lack of accurate control over release and nonspecific leakage of drugs. The azobenzene functionalized DNA hydrogel offers a way of using light as the external stimulus of which the dosage and position can be better administered. But the required UV light has limited tissue penetration capability and is harmful to human body. The NIR light responsive nanogel partially solves the problem with extra targeting function. Future research may include the optimization of hydrogel properties and the development of DNA hydrogel release systems responsive to other stimulus such as pH, electrical field and magnetic field.
4.4. DNA-cross-linked polymer hydrogels exhibiting photoreversible volume change
On one hand, stimuli-responsive hydrogels as platforms for encapsulation of sensing materials and therapeutic drugs have significant applications in bioanalysis, diagnostics and biomedicine. On the other hand, the physical changes of hydrogels responding to external stimuli can also be utilized in the engineering of novel dynamic devices, such as actuators and microlenses. This type of hydrogel can perform mechanical operations, including volume change and deformation.93, 94 Depending on the design, the dynamic hydrogel devices are able convert different types of energy, including chemical energy, electrical energy and heat, into mechanical movement.
Our group has engineered a novel hydrogel with reversible volume changes based on light-regulated DNA hybridization (Figure 10). 30 Photoresponsive hydrogels have attracted enormous attention.64, 95 Light is one of the most attractive stimuli for hydrogels since it can be delivered remotely with controlled intensity and high accuracy. Moreover, light-responsive materials are increasingly studied because of their potential in solar energy harvesting and utilization.96, 97 In our design, azobenzene-regulated photoswitchable DNA duplex complexes were grafted into the hydrogel network as reversible crosslinkers. A two-step polymerization method was used, resulting in a hydrogel where an azobenzene-modified DNA-grafting linear polymer was entrapped in a complementary DNA-grafting cross-linked polymer network. The hydrogel was maintained by the permanent N,N’-methylenebisacrylamide (MBAA) crosslinkers, while the azobenzene-modified DNA duplex complexes served as reversible crosslinkers. Therefore, the overall hydrogel crosslinker density could be reversibly manipulated by UV and visible light. Upon UV light irradiation, the DNA crosslinkers were dissociated, yielding a larger volume of the hydrogel, while the irradiation of visible light caused the recovery of the DNA crosslinkers, resulting in a smaller volume. The process of volume transition was reversible when UV and visible light were applied alternately. The volume change of the hydrogel could potentially be 1) converted into mechanical or electrical energy if proper device designs are used or 2) extended to similar photoresponsive systems, such as azobenzene/α-cyclodextrin complexation. On the other hand, there is still plenty room for improvement of this type of hydrogels. The response rate is relatively slow due to the large size and the restricted diffusion. Possible strategies to further enhance the responsiveness include the utilization of a porous structure and the fabrication of micro-sized hydrogel particles.
Figure 10.
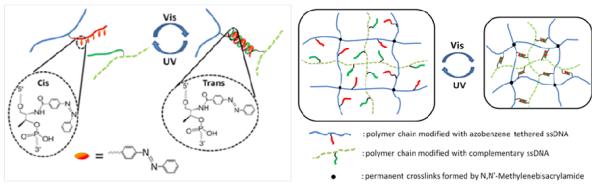
A) Light-controlled DNA hybridization. (B) Reversible volume transition of the DNA-crosslinked hydrogel regulated by UV and visible light. (adapted from Ref. [30])
5. Summary and outlook
Synthetic polymers have been the focus of materials science and chemistry ever since their invention because of their major impact on scientific research, as well as our daily life. Nucleic acids, known as the molecular foundation of all life forms, have found new applications in areas other than genetics, such as materials science, nanoscience and combinatorial science. Thanks to developments in the field of artificial nucleic acids, very different functions can be integrated into one system, which expands our toolbox of functional nucleic acids. Specifically, hybrid materials combining nucleic acids and synthetic polymers provide us with special features that are not seen in either material alone. Various methods have been developed to synthesize DNA polymer hybrid materials with novel properties and applications. As discussed in this review, DNA-grafting linear polymers have been successfully applied in bioanalysis and biomedicine. Furthermore, this class of polymers supports the formation of more complicated structures, such as cross-linked hydrogels to assist DNA hybridization. Applications in biosensor development, logic gate fabrication, controlled drug release and novel photodynamic materials have been explored. However, their physical and chemical properties need more intensive investigation in order to extend their applications. Moreover, to understand the mechanisms underlying their therapeutic effects, the interaction of DNA-grafting polymers with cells also requires systematic study. Similarly, the stimuli-responsive release process of DNA hydrogels should be elucidated. As our knowledge of the DNA-grafting polymers deepens and broadens, we believe that they will be better controlled for more practical applications.
Acknowledgement
The authors would like to thank Dr. Kathryn R. Williams for manuscript review. This work was also supported by grants awarded by the National Institutes of Health (GM066137, GM079359 and CA133086).
References
- 1.Caruthers MH. Acc. Chem. Res. 1991;24:278–284. [Google Scholar]
- 2.Dawson PE, Kent SBH. Annu. Rev. Biochem. 2000;69:923–960. doi: 10.1146/annurev.biochem.69.1.923. [DOI] [PubMed] [Google Scholar]
- 3.Seeberger PH, Werz DB. Nat. Rev. Drug Discovery. 2005;4:751–763. doi: 10.1038/nrd1823. [DOI] [PubMed] [Google Scholar]
- 4.Seeberger PH, Werz DB. Nature. 2007;446:1046–1051. doi: 10.1038/nature05819. [DOI] [PubMed] [Google Scholar]
- 5.Deming TJ. Adv. Drug Deliv. Rev. 2002;54:1145–1155. doi: 10.1016/s0169-409x(02)00062-5. [DOI] [PubMed] [Google Scholar]
- 6.Klok HA. Angew. Chem., Int. Ed. 2002;41:1509–1513. doi: 10.1002/1521-3773(20020503)41:9<1509::aid-anie1509>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 7.Alemdaroglu FE, Herrmann A. Org. Biomol. Chem. 2006;5:1311–1320. doi: 10.1039/b617941j. [DOI] [PubMed] [Google Scholar]
- 8.Kwak M, Herrmann A. Angew. Chem. , Int. Ed. 2010;49:8574–8587. doi: 10.1002/anie.200906820. [DOI] [PubMed] [Google Scholar]
- 9.Zhao X, Tapec-Dytioco R, Tan W. J. Am. Chem. Soc. 2003;125:11474–11475. doi: 10.1021/ja0358854. [DOI] [PubMed] [Google Scholar]
- 10.Elghanian R, Storhoff JJ, Mucic RC, Letsinger RL, Mirkin CA. Science. 1997;277:1078–1081. doi: 10.1126/science.277.5329.1078. [DOI] [PubMed] [Google Scholar]
- 11.Tyagi S, Kramer FR. Nat. Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 12.Asanuma H, Ito T, Yoshida T, Liang X, Komiyama M. Angew. Chem. , Int. Ed. 1999;38:2393–2395. doi: 10.1002/(sici)1521-3773(19990816)38:16<2393::aid-anie2393>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 13.Ellington AD, Szostak JW. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 14.Tuerk C, Gold L. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 15.Huizenga DE, Szostak JW. Biochemistry (N. Y.) 1995;34:656–665. doi: 10.1021/bi00002a033. [DOI] [PubMed] [Google Scholar]
- 16.Macaya RF, Schultze P, Smith FW, Roe JA, Feigon J. Proc. Natl. Acad. Sci. U. S. A. 1993;90:3745. doi: 10.1073/pnas.90.8.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shangguan D, Li Y, Tang Z, Cao ZC, Chen HW, Mallikaratchy P, Sefah K, Yang CJ, Tan W. Proc. Natl. Acad. Sci. U. S. A. 2006;103:11838–11843. doi: 10.1073/pnas.0602615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Potyrailo RA, Conrad RC, Ellington AD, Hieftje GM. Anal. Chem. 1998;70:3419–3425. doi: 10.1021/ac9802325. [DOI] [PubMed] [Google Scholar]
- 19.Zheng D, Seferos DS, Giljohann DA, Patel PC, Mirkin CA. Nano Lett. 2009;9:3258–3261. doi: 10.1021/nl901517b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang YF, Shangguan D, Liu H, Phillips JA, Zhang X, Chen Y, Tan W. Chembiochem. 2009;10:862–868. doi: 10.1002/cbic.200800805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X, Tan W. Anal. Chem. 1999;71:5054–5059. doi: 10.1021/ac990561c. [DOI] [PubMed] [Google Scholar]
- 22.Smith JE, Medley CD, Tang Z, Shangguan D, Lofton C, Tan W. Anal. Chem. 2007;79:3075–3082. doi: 10.1021/ac062151b. [DOI] [PubMed] [Google Scholar]
- 23.Park SY, Lytton-Jean AKR, Lee B, Weigand S, Schatz GC, Mirkin CA. Nature. 2008;451:553–556. doi: 10.1038/nature06508. [DOI] [PubMed] [Google Scholar]
- 24.Gibbs JM, Park SJ, Anderson DR, Watson KJ, Mirkin CA, Nguyen SBT. J. Am. Chem. Soc. 2005;127:1170–1178. doi: 10.1021/ja046931i. [DOI] [PubMed] [Google Scholar]
- 25.Watson KJ, Park SJ, Im JH, Nguyen SBT, Mirkin CA. J. Am. Chem. Soc. 2001;123:5592–5593. doi: 10.1021/ja0156845. [DOI] [PubMed] [Google Scholar]
- 26.Yang L, Meng L, Zhang X, Chen Y, Zhu G, Liu H, Xiong X, Sefah K, Tan W. J. Am. Chem. Soc. 2011;133:13380–13386. doi: 10.1021/ja201285y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang H, Liu H, Kang H, Tan W. J. Am. Chem. Soc. 2008;130:6320–6321. doi: 10.1021/ja801339w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu Z, Wu C, Liu H, Zou Y, Zhang X, Kang H, Yang CJ, Tan W. Angew. Chem. , Int. Ed. 2010;49:1052–1056. doi: 10.1002/anie.200905570. [DOI] [PubMed] [Google Scholar]
- 29.Kang H, Liu H, Zhang X, Yan J, Zhu Z, Peng L, Yang H, Kim Y, Tan W. Langmuir. 2011;27:399–408. doi: 10.1021/la1037553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng L, You M, Yuan Q, Wu C, Han D, Chen Y, Zhong Z, Xue J, Tan W. J. Am. Chem. Soc. 2012;134:12302–12307. doi: 10.1021/ja305109n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu J, Zhang G, Liu S. Chem. Soc. Rev. 2012;41:5933–5949. doi: 10.1039/c2cs35103j. [DOI] [PubMed] [Google Scholar]
- 32.Randolph LM, Chien MP, Gianneschi NC. Chem. Sci. 2012;3:1363–1380. doi: 10.1039/C2SC00857B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taira S, Yokoyama K. Biotechnol. Bioeng. 2004;88:35–41. doi: 10.1002/bit.20205. [DOI] [PubMed] [Google Scholar]
- 34.Nagahara S, Matsuda T. Polym. Gels Networks. 1996;4:111–127. [Google Scholar]
- 35.Chaix C. J. Appl. Polym. Sci. 1998;70:2487–2497. [Google Scholar]
- 36.Murakami Y, Maeda M. Biomacromolecules. 2005;6:2927–2929. doi: 10.1021/bm0504330. [DOI] [PubMed] [Google Scholar]
- 37.Kang H, Trondoli AC, Zhu G, Chen Y, Chang YJ, Liu H, Huang YF, Zhang X, Tan W. ACS Nano. 2011;5:5094–5099. doi: 10.1021/nn201171r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Golas PL, Matyjaszewski K. Chem. Soc. Rev. 2010;39:1338–1354. doi: 10.1039/b901978m. [DOI] [PubMed] [Google Scholar]
- 39.Averick S, Paredes E, Li W, Matyjaszewski K, Das SR. Bioconjug. Chem. 2011;22:2030–2037. doi: 10.1021/bc200240q. [DOI] [PubMed] [Google Scholar]
- 40.Pan P, Fujita M, Ooi WY, Sudesh K, Takarada T, Goto A, Maeda M. Polymer. 2011;52:895–900. [Google Scholar]
- 41.Gibbs-Davis JM, Schatz GC, Nguyen SBT. J. Am. Chem. Soc. 2007;129:15535–15540. doi: 10.1021/ja073034g. [DOI] [PubMed] [Google Scholar]
- 42.Chan WCW, Nie S. Science. 1998;281:2016–2018. doi: 10.1126/science.281.5385.2016. [DOI] [PubMed] [Google Scholar]
- 43.Santra S, Zhang P, Wang K, Tapec R, Tan W. Anal. Chem. 2001;73:4988–4993. doi: 10.1021/ac010406+. [DOI] [PubMed] [Google Scholar]
- 44.Taton TA, Mirkin CA, Letsinger RL. Science. 2000;289:1757–1760. doi: 10.1126/science.289.5485.1757. [DOI] [PubMed] [Google Scholar]
- 45.Kudlay A, Gibbs JM, Schatz GC, Nguyen SBT, de la Cruz MO. J. Phys. Chem. B. 2007;111:1610–1619. doi: 10.1021/jp0664667. [DOI] [PubMed] [Google Scholar]
- 46.Rosi NL, Mirkin CA. Chem. Rev. 2005;105:1547–1562. doi: 10.1021/cr030067f. [DOI] [PubMed] [Google Scholar]
- 47.Mori T, Maeda M. Polym. J. 2002;34:624–628. [Google Scholar]
- 48.Lee K, Rouillard JM, Pham T, Gulari E, Kim J. Angew. Chem. , Int. Ed. 2007;46:4667–4670. doi: 10.1002/anie.200700419. [DOI] [PubMed] [Google Scholar]
- 49.Patil SD, Rhodes DG, Burgess DJ. The AAPS journal. 2005;7:61–77. doi: 10.1208/aapsj070109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luo D, Saltzman WM. Nat. Biotechnol. 2000;18:33–37. doi: 10.1038/71889. [DOI] [PubMed] [Google Scholar]
- 51.Dong X, Mattingly CA, Tseng MT, Cho MJ, Liu Y, Adams VR, Mumper RJ. Cancer Res. 2009;69:3918–3926. doi: 10.1158/0008-5472.CAN-08-2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Farokhzad OC, Jon S, Khademhosseini A, Tran TNT, LaVan DA, Langer R. Cancer Res. 2004;64:7668. doi: 10.1158/0008-5472.CAN-04-2550. [DOI] [PubMed] [Google Scholar]
- 53.Lemaitre M, Bayard B, Lebleu B. Proc. Natl. Acad. Sci. U. S. A. 1987;84:648. doi: 10.1073/pnas.84.3.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bordier B, Perala-Heape M, Degols G, Lebleu B, Litvak S, Sarih-Cottin L, Helene C. Proc. Natl. Acad. Sci. U. S. A. 1995;92:9383. doi: 10.1073/pnas.92.20.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murata M, Kaku W, Anada T, Soh N, Katayama Y, Maeda M. Chem. Lett. 2003:266–267. doi: 10.1016/j.bmcl.2003.08.062. [DOI] [PubMed] [Google Scholar]
- 56.Murata M, Kaku W, Anada T, Sato Y, MACDA M, Katayama Y. Chem. Lett. 2003:986–987. doi: 10.1016/j.bmcl.2003.08.062. [DOI] [PubMed] [Google Scholar]
- 57.Frei E, Elias A, Wheeler C, Richardson P, Hryniuk W. Clin. Cancer Res. 1998;4:2027–2037. [PubMed] [Google Scholar]
- 58.Chari RVJ. Acc. Chem. Res. 2007;41:98–107. doi: 10.1021/ar700108g. [DOI] [PubMed] [Google Scholar]
- 59.Fang X, Tan W. Acc. Chem. Res. 2009;43:48–57. doi: 10.1021/ar900101s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tanaka T. Phys. Rev. Lett. 1978;40:820–823. [Google Scholar]
- 61.Suzuki A, Tanaka T. Nature. 1990;346:345–347. [Google Scholar]
- 62.Miyata T, Asami N, Uragami T. Nature. 1999;399:766–769. doi: 10.1038/21619. [DOI] [PubMed] [Google Scholar]
- 63.Beebe DJ, Moore JS, Bauer JM, Yu Q, Liu RH, Devadoss C, Jo BH. Nature. 2000;404:588–590. doi: 10.1038/35007047. [DOI] [PubMed] [Google Scholar]
- 64.Matsubara K, Watanabe M, Takeoka Y. Angew. Chem. , Int. Ed. 2007;46:1688–1692. doi: 10.1002/anie.200603554. [DOI] [PubMed] [Google Scholar]
- 65.Sui Z, King WJ, Murphy WL. Adv. Funct. Mater. 2008;18:1824–1831. doi: 10.1002/adfm.200800218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miyata T, Asami N, Uragami T. Nature. 1999;399:766–769. doi: 10.1038/21619. [DOI] [PubMed] [Google Scholar]
- 67.Murphy WL, Dillmore WS, Modica J, Mrksich M. Angew. Chem. , Int. Ed. 2007;46:3066–3069. doi: 10.1002/anie.200604808. [DOI] [PubMed] [Google Scholar]
- 68.Murakami Y, Maeda M. Macromolecules. 2005;38:1535–1537. [Google Scholar]
- 69.Gerlach G, Arndt KF. Hydrogel sensors and actuators: engineering and technology. Springer Verlag; 2010. [Google Scholar]
- 70.Miyata T, Uragami T, Nakamae K. Adv. Drug Deliv. Rev. 2002;54:79–98. doi: 10.1016/s0169-409x(01)00241-1. [DOI] [PubMed] [Google Scholar]
- 71.Ehrick JD, Luckett MR, Khatwani S, Wei Y, Deo SK, Bachas LG, Daunert S. Macromol. Biosci. 2009;9:864–868. doi: 10.1002/mabi.200800337. [DOI] [PubMed] [Google Scholar]
- 72.Mao J, McShane MJ. Adv. Mater. 2006;18:2289–2293. [Google Scholar]
- 73.Yang H, Liu H, Kang H, Tan W. J. Am. Chem. Soc. 2008;130:6320–6321. doi: 10.1021/ja801339w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shakhsher ZM, Odeh I, Jabr S, Seitz W. Rudolf. Microchim. Acta. 2004;144:147–153. [Google Scholar]
- 75.DeLouise LA, Fauchet PM, Miller BL, Pentland AA. Adv. Mater. 2005;17:2199–2203. [Google Scholar]
- 76.Ruan C, Zeng K, Grimes CA. Anal. Chim. Acta. 2003;497:123–131. [Google Scholar]
- 77.Dave N, Chan MY, Huang PJJ, Smith BD, Liu J. J. Am. Chem. Soc. 2010;132:12668–12673. doi: 10.1021/ja106098j. [DOI] [PubMed] [Google Scholar]
- 78.Miyake Y, Togashi H, Tashiro M, Yamaguchi H, Oda S, Kudo M, Tanaka Y, Kondo Y, Sawa R, Fujimoto T. J. Am. Chem. Soc. 2006;128:2172–2173. doi: 10.1021/ja056354d. [DOI] [PubMed] [Google Scholar]
- 79.Krishnan Y, Simmel FC. Angew. Chem. , Int. Ed. 2011;50:3124–3156. doi: 10.1002/anie.200907223. [DOI] [PubMed] [Google Scholar]
- 80.Adleman LM. Science. 1994;266:1021–1024. doi: 10.1126/science.7973651. [DOI] [PubMed] [Google Scholar]
- 81.Gawel K, Stokke BT. Soft Matter. 2011;7:4615–4618. [Google Scholar]
- 82.Yin BC, Ye BC, Wang H, Zhu Z, Tan W. Chem. Commun. 2012;48:1248–1250. doi: 10.1039/c1cc15639j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hoffman AS. Adv. Drug Deliv. Rev. 2002;54:3–12. doi: 10.1016/s0169-409x(01)00239-3. [DOI] [PubMed] [Google Scholar]
- 84.Stuart MAC, Huck WTS, Genzer J, Müller M, Ober C, Stamm M, Sukhorukov GB, Szleifer I, Tsukruk VV, Urban M. Nat. Mater. 2010;9:101–113. doi: 10.1038/nmat2614. [DOI] [PubMed] [Google Scholar]
- 85.Lu Y, Liu J. Curr. Opin. Biotechnol. 2006;17:580–588. doi: 10.1016/j.copbio.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 86.Liu H, Xu Y, Li F, Yang Y, Wang W, Song Y, Liu D. Angew. Chem. , Int. Ed. 2007;46:2515–2517. doi: 10.1002/anie.200604589. [DOI] [PubMed] [Google Scholar]
- 87.Modi S, Swetha M, Goswami D, Gupta GD, Mayor S, Krishnan Y. Nat. Nanotechnol. 2009;4:325–330. doi: 10.1038/nnano.2009.83. [DOI] [PubMed] [Google Scholar]
- 88.Liedl T, Dietz H, Yurke B, Simmel F. small. 2007;3:1688–1693. doi: 10.1002/smll.200700366. [DOI] [PubMed] [Google Scholar]
- 89.Asanuma H, Ito T, Yoshida T, Liang X, Komiyama M. Angew. Chem. , Int. Ed. 1999;38:2393–2395. doi: 10.1002/(sici)1521-3773(19990816)38:16<2393::aid-anie2393>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 90.Asanuma H, Liang X, Nishioka H, Matsunaga D, Liu M, Komiyama M. Nat. Protoc. 2007;2:203–212. doi: 10.1038/nprot.2006.465. [DOI] [PubMed] [Google Scholar]
- 91.Reismann M, Bretschneider JC, Plessen G, Simon U. Small. 2008;4:607–610. doi: 10.1002/smll.200701317. [DOI] [PubMed] [Google Scholar]
- 92.Huang X, El-Sayed IH, Qian W, El-Sayed MA. J. Am. Chem. Soc. 2006;128:2115–2120. doi: 10.1021/ja057254a. [DOI] [PubMed] [Google Scholar]
- 93.Shiga T, Hirose Y, Okada A, Kurauchi T. J Appl Polym Sci. 1992;44:249–253. [Google Scholar]
- 94.Beebe DJ, Moore JS, Bauer JM, Yu Q, Liu RH, Devadoss C, Jo BH. Nature. 2000;404:588–590. doi: 10.1038/35007047. [DOI] [PubMed] [Google Scholar]
- 95.Tomatsu I, Hashidzume A, Harada A. Macromolecules. 2005;38:5223–5227. [Google Scholar]
- 96.Kanai Y, Srinivasan V, Meier SK, Vollhardt KPC, Grossman JC. Angew. Chem. , Int. Ed. 2010;49:8926–8929. doi: 10.1002/anie.201002994. [DOI] [PubMed] [Google Scholar]
- 97.Cho J, Berbil-Bautista L, Pechenezhskiy IV, Levy N, Meier SK, Srinivasan V, Kanai Y, Grossman JC, Vollhardt KPC, Crommie MF. ACS Nano. 2011;5:3701–3706. doi: 10.1021/nn2000367. [DOI] [PubMed] [Google Scholar]