

Abstract
Oxidative stress-induced decrease in tissue or systemic glutathione (GSH) and damage to the vascular endothelium of the blood-brain barrier such as occurs in diabetes or stroke will have important implications for brain homeostasis. Endothelial proliferation or repair is crucial to preserving barrier function. Cell proliferation has been associated with increased intracellular GSH, but the kinetic and distribution of GSH during cell cycle is poorly understood. Here, we determined the influence of cellular GSH status on the early dynamics of nuclear-to-cytosol (N-to-C) GSH distribution (6-h interval) during proliferation in a human brain microvascular endothelial cell line (IHEC). Control IHECs exhibited two peak S-phases of the cell cycle at 48 and 60 h post seeding that temporally corresponded to peak nuclear GSH levels and expression of cdk1, the S-to-G2-to-M checkpoint controller, suggesting a link between cell cycle progression and nuclear GSH. Sustained inhibition of GSH synthesis delayed S-to-G2/M cell transition; cell arrest in the S-phase was correlated with decreased total nuclear GSH and increased nuclear expressions of chk2/phospho-chk2 and GADPH. The temporal correspondence of nuclear chk2 activation and GAPDH expression with S-phase prolongation is consistent with enhanced DNA damage response and extended time for DNA repair. Strikingly, when GSH synthesis was restored, cell transit time through S-phase remained delayed. Significantly, total nuclear GSH remained depressed, indicating a time lag between restored cellular GSH synthetic capacity and recovery of the nuclear GSH status. Interestingly, despite a delay in cell cycle recovery, nuclear expressions of chk2/phospho-chk2 and GAPDH resembled those of control cells. This means that restoration of nuclear DNA integrity preceded normalization of the cell cycle. The current results provide important insights into GSH control of endothelial proliferation with implications for cell repair or wound healing in recovery post-oxidative damage.
Abbreviations: GSH, glutathione; GSSG, glutathione disulfide; H1, histone H1; cdk1, cyclin dependent kinase 1; ATM, ataxia telangiectasia mutated; chk2, checkpoint kinase 2; GAPDH, glyceraldehyde 3-phosphate dehydrogenase
Keywords: Nuclear-to-cytosolic GSH distribution, GSH and endothelial proliferation, GSH and cell cycle, DNA damage response, Brain microvascular endothelial cells
Graphical abstract
Highlights
► Increased nuclear-to-cytosol GSH was associated with brain endothelial cell (IHEC) cell cycle progression. ► Inhibition of GSH synthesis decreased nuclear GSH and delayed S-to-G2 transition. ► S-phase prolongation was linked to elevated DNA damage response. ► Recovery of nuclear GSH and normal cell cycle lagged behind the restoration of GSH synthesis.
Introduction
Recent findings support an intrinsic role for redox control of the cell cycle. Progression through the cell cycle at defined times is influenced by the cellular redox environment, which modulates the activity of cell cycle redox-sensitive proteins [1]. The redox environment within a cell is determined by the ratio of the concentration of the reduced and oxidized forms of various redox couples, such as glutathione (GSH), thioredoxin (Trx), and pyridine nucleotides [2,3]. Glutathione/glutathione disulfide (GSH/GSSG) is the most abundant thiol redox buffer in cells and quantitatively plays a key role in the maintenance of the cellular redox environment. GSH participates in multiple metabolic functions and redox signaling, including thiol-disulfide exchange and protein S-glutathiolation. Such redox mechanisms modulate the function of redox-sensitive protein cysteines, such as those involved in cell growth, proliferation, differentiation, or apoptosis [4,5]. Thus, GSH is recognized as a regulator of cell proliferation. GSH synthesis is a pivotal contributor to cytosolic GSH homeostasis that impacts the redox states of intracellular compartments of mitochondria, nucleus, and endoplasmic reticulum. Notably, the nuclear-to-cytosol (N-to-C) distribution of GSH is reportedly a factor in redox-based signaling in cell proliferation [6].
Vascular endothelial cells are located at the interface between the vascular lumen and underlying tissues, and as such, are in direct contact with the systemic circulation. In disease states, such as diabetes, elevated levels of systemic or locally generated mediators, free radicals, and reactive oxygen or carbonyl species can contribute to the disruption of the vascular endothelium [7]. Therefore, the preservation or post-injury restoration of endothelial barrier integrity is central to the maintenance of endothelial function. A major mechanism for the repair and/or restitution of the endothelial monolayer is proliferation of endothelial cells adjacent to the lesion or injury site. Little is known of the contribution of GSH to the control of vascular endothelial cell proliferation.
The current study is designed to examine the influence of cellular GSH status on the kinetics of N-to-C GSH distribution in a human brain microvascular endothelial cell line, and the changes in the expression of the cell cycle checkpoint controller, cdk1, and DNA damage response markers, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and phospho-chk2. Previous studies of GSH and cell growth in epithelial [8] and 3T3 fibroblasts [6] have focused on 24 h time intervals or days, which, while convenient, missed the crucial dynamic and shorter window of GSH-cell cycle changes during cell proliferation. In this study, we have selected 6-h time intervals post-seeding throughout a 72 h culture. Our results bring new insights into the early temporal changes in endothelial N-to-C GSH distribution and the relationship to S-phase responses in the cell cycle under conditions of normal, decreased, or restored GSH states following initial GSH depletion. Significantly, our results provide novel evidence that inhibition of endothelial GSH synthesis lengthened the resident time of endothelial cells in the S-phase of the cell cycle that was coincident with decreased nuclear GSH and activation of DNA damage responses. These results suggest that decreased nuclear GSH may be associated with enhanced DNA damage and that a delay in S-to-G2-to M progression allows for extended time for DNA repair and cell survival.
Methods
Reagents
Medium 199, L-buthionine-(S,R)-sulfoximine (BSO), insulin-transferin-sodium selenite solution, propidium iodine (PI), iodoacetic acid (IAA), 2,4-dinitrophenyl fluorobenzene (DNFB), ethanol, trichloracetic acid (TCA), imidazol, sucrose, RNAase A were purchased from Sigma-Aldrich (St. Louis, MO). Digitonin was purchased from Wako, Japan. Protease and phosphatase inhibitor cocktail (Complete Mini, EDTA and PhosphoStop, EDTA free, 1 tablet of each in 10 ml lysis buffer) were purchased from Roche Diagnosis (Indianapolis, IN). Fetal bovine serum (FBS) was obtained from Atlanta Biologicals (Lawrenceville, GA). Trypsin-EDTA and antibiotic/antimycotic were from Gibco (Carlsbad, CA). Purified mouse monoclonal anti-β actin and anti-cdk1 antibodies were purchased from BD Biosciences (San Jose, CA). Mouse monoclonal anti-GAPDH and anti- H1 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA), and mouse monoclonal anti-chk2 and rabbit monoclonal anti-phospho-chk2 antibodies were from Millipore (Temecula, CA).
Cell culture
The human brain endothelial cell (IHEC) line was obtained from Dr. Danica Stanimirovic at the National Research Council, Canada Institute for Biological Sciences and was propagated by Dr. Steven J. Alexander at LSUHSC—Shreveport, LA. IHECs were maintained in 75 cm2 culture flasks in M199 media supplemented with 10% FBS, 1% insulin–transferrin–sodium selenite solution, and 1X antibiotic/antimycotic (complete M199 media) until confluence. Cells were incubated in humidified atmosphere with 5% CO2 at 37 °C. Media was changed every 2 days.
Cell incubations
For all experiments cells from confluent IHEC monolayer were seeded at a density of 2×105/well in 6-well plates and cultured for 0–72 h with a media change at 28 h post seeding. Cells were collected at the following time points: 0 (at seeding), 30, 36, 42, 48, 55, 60, 66, and 72 h. To inhibit GSH synthesis, cells were treated with 10 μM BSO at 4 and 24 h after seeding with a change to fresh media at 28 h. Sustained decrease in cellular GSH was maintained with the addition of 2 μM BSO in the fresh media. To allow for re-synthesis of GSH, the 2 μM BSO dose was omitted from the fresh media. At designated time points, cells were harvested for flow cytometric analyses of S-phase responses in the cell cycle, for cellular fractionation and GSH determination, or for Western blot analyses.
Cell cycle analysis by flow cytometry
At each designated time point cells were trypsinized, washed with PBS and fixed in 70% cold ethanol. Fixed cells were centrifuged at 4 °C, 1200 rpm for 10 min to remove ethanol, resuspended in 1 mL PBS, and incubated with 1 mg/mL DNAse-free RNAase A for 40 min at 37 °C. Propidium iodine (PI, 50 μg/ml) was added and samples were incubated at 4 °C for 15 min in the dark. Flow cytometric analysis was performed using a BD Biosciences LSRII Flow Cytometer (San Jose, CA). A minimum of 2×106 cells per sample was counted. Data was processed using the Cell Quest software and gated on pulse-processed PI signals to exclude doublets and large aggregates, using a multiparameter gate strategy.
Cellular fractionation
Separation of cytosolic and nuclear fractions was achieved by digitonin fractionation [9]. Cells were trypsinized, counted, and fractionation was performed in 1.5 mL eppendorf tubes. Briefly, 3×106 cells were resuspended in 0.5 mL of fractionation buffer A, containing 0.25 M sucrose, 3 mM imidazol, pH 7.4, and 1 mg/mL digitonin. When samples were used for GSH measurements, cell trypsinization and fractionation buffer A also contained 8 mM iodoacetic acid. Cells were centrifuged at 14,000g (20 s, 4 °C) and the supernatant was collected as the cytosolic fraction. The pellet was resuspended in 0.5 mL fractionation buffer A, homogenized with a Dounce homogenizer (5–10 passes) and centrifuged at 14,000g, 4 °C (3 s). The resultant pellet, representing the nuclear pellet was washed twice in 0.5 mL buffer A (2 s at 14,000g, 4 °C). The final nuclear pellet was washed once with 0.5 mL PBS. For GSH measurements, cytosolic and nuclear fractions were treated with 5% ice-cold TCA prior to analyses. The purity of nuclear and cytosolic fractions was validated by the enrichment of histone H1 on western blots and lactate dehydrogenase activity, respectively.
Quantification of GSH
Cellular levels of GSH were determined by high-performance liquid chromatography (HPLC) [10] as we previously described [11,12]. TCA-soluble proteins were derivatized with 6 mM IAA and 1% 2, 4 DNFB to yield the S-carboxymethyl and 2, 4-dinitrophenyl derivative of GSH, respectively. Separation of GSH derivatives was performed on a 250×4.6 mm Alltech Lichrosorb NH2 10 μm anion-exchange column. GSH contents were quantified by comparison to standards derivatized in the same manner. Protein pellets were resuspended in 1 mL 0.1 M NaOH for protein quantitation. GSH concentrations were expressed as nmoles/mg protein.
Western blot analyses
Protein expressions of cyclin-dependent kinase, cdk-1, histone H1, DNA damage response genes (GAPDH, cell cycle checkpoint regulator, chk-2 and phospho-chk2), and β-actin were determined by western blot analyses. At designated times, cells were suspended in a reducing buffer containing 0.5 M Tris, 10% SDS, glycerol, β-mercaptoethanol, phenol red and a protease and phosphatase inhibitor cocktail, and then sonicated. 30 μg proteins of cytosolic or nuclear extracts were loaded onto 10% or 4–10% polyacrylamide gels and size separated at 110–115 V. Proteins were transferred onto PVDF membranes overnight at 30 V, 4 °C. Membranes were blocked in 5% milk or BSA in TBS-Tween at room temperature for 1 h and then incubated with the respective antibodies as follows: anti-histone H1 (1:3000), anti-chk2 (0.150 μg/mL), anti-p-chk2 (1:2000), anti-cdk1 (1:10,000), anti-β actin (1:75,000), and anti-GAPDH (1:1000). After washing, membranes were incubated in HRP-conjugated goat-anti-mouse or anti rabbit secondary antibody for 1 or 2 h at room temperature followed by washing and 5 min incubation with ECL reagents per the manufacturer's protocol. Detection was performed using radiographic films. Quantification of band intensity relative to β-actin or H1 was performed for at least 4 separate blots from 2 to 3 experiments performed in duplicates. Results are mean±SEM. Equal protein loading was assessed by Ponceau red staining which showed uniform staining in all western blots. β-actin expression was relatively uniform at the different times, but the expression levels of H1 on western blots (Figs. 2–4) were notably variable, suggesting that the purity of the nuclear fraction from one cell preparation to another can be quite varied, a recognized limitation of the fractionation procedure. However, the relative expressions of nuclear cdk1, chk2 or GAPDH paralleled the respective changes in H1 levels; therefore, quantification of their band intensities were performed relative to H1.
Fig. 2.
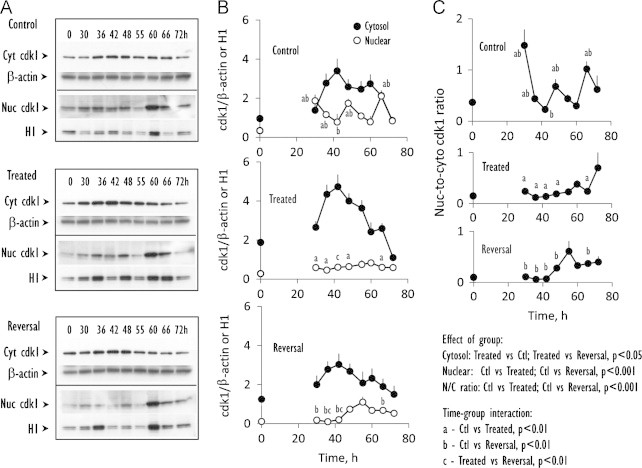
Time course of protein expression of cdk1, a cell cycle checkpoint controller of S-to-G2-to-M progression. (A) Western blot analyses of cdk1 expression from 0–to–72 h in the cytosolic or nuclear fractions prepared from control IHECs (Control), cells treated with BSO to maintain low GSH (Treated) or cells treated with BSO that was subsequently removed to allow for GSH recovery (Reversal). (B) shows the quantitative data of band intensities of cytosolic (closed circles) or nuclear (open circles) cdk1 expression relative to β-actin or H1, respectively for the 3 treatment groups. Results are mean±SEM for 4 separate blots. C illustrates the N-to-C ratios of cdk1. Statistical differences for time-group interactions among the 3 groups in (B and C) are denoted by letters a, b, c that are explained in the statistical analyses on the figure.
Fig. 3.
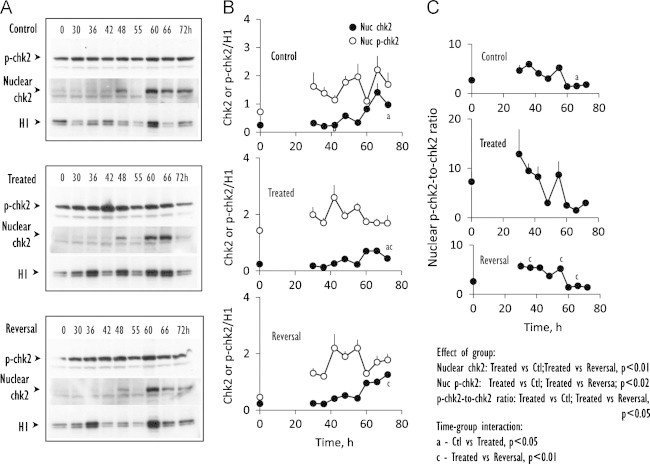
Kinetics of expression of nuclear chk2 and its phosphorylation status. (A) Western blot analyses of nuclear expression of chk2 and phospho-chk2 from 0–to–72 h in nuclear fractions prepared from control IHECs, cells treated with BSO to maintain low GSH (Treated) or cells treated with BSO that was subsequently removed to allow for GSH recovery (Reversal). (B) shows the quantitative data of band intensities of nuclear chk2 (closed circles) or phospho-chk2 (open circles) expression relative to H1 for the 3 treatment groups. Results are mean±SEM for 4 separate blots. (C) illustrates the N-to-C ratios of p-chk2-to-chk2. Statistical differences for time-group interactions among the 3 groups in (B and C) are denoted by letters a, c that are explained in the statistical analyses on the figure.
Fig. 4.
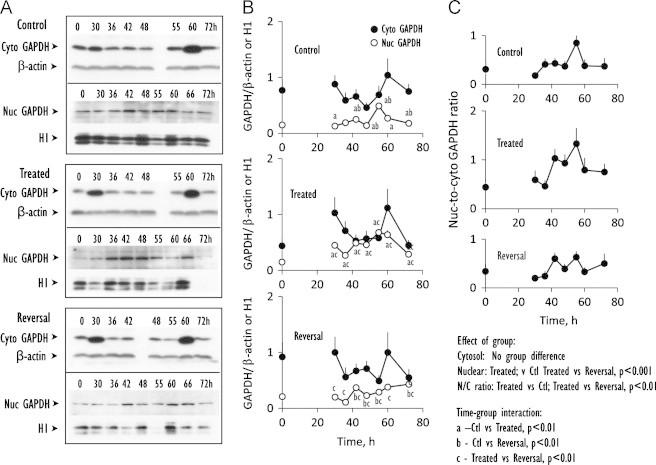
Time course of cytosolic and nuclear expression of GAPDH, an index of DNA damage response. (A) Western blot analyses of expression of GAPDH from 0 to 72 h in cytosolic and nuclear fractions prepared from control IHECs, cells treated with BSO to maintain low GSH (Treated) or cells treated with BSO that was subsequently removed to allow for GSH recovery (Reversal). (B) shows the quantitative data of band intensities of cytosolic GAPDH (closed circles) or nuclear GAPDH (open circles) expression relative to β-actin or H1, respectively, for the 3 treatment groups. Results are mean+SEM for 4 separate blots. (C) illustrates the N-to-C ratios of nuclear-to-cytosolic GAPDH. Statistical differences for time-group interactions among the 3 groups in (B and C) are denoted by letters a, b, c that are explained in the statistical analyses on the figure.
Protein assay
Protein contents were measured using the Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Hercules, CA), according to manufacturers' protocol.
Statistical analysis
Two-Factor Analysis of Variance (ANOVA) was used to determine significant effects of group namely, Control cells, GSH depleted cells (Treated) and GSH restored cells (Reversal), times (30, 36, 42, 48, 55, 60, 66 and 72 h), and interaction between group and time. For each of the three variables, multiple (pair wise) comparisons among the 3 groups and among the 8 time points were performed using the Bonferoni method. Results are expressed as mean±SEM. The relationships between % cell in S-phase and nuclear GSH were analyzed by linear regression analysis. The slope of the fitted line is significant for control.
Results
Cellular GSH manipulation on cell cycle responses and cytosolic and nuclear GSH contents
The relationship between IHEC proliferation and GSH was examined by cell cycle analyses (Fig. 1, Table 1). Regardless of treatment conditions, media change elicited rapid entry of IHECs into cell cycle with a peak S-phase at 30 h. However, significant differences in cell cycle response among groups were observed between 36 and 66 h. Over this time period, % cells in S-phase exhibited two distinct peaks at 48 h and 60 h (peak S-phases, Fig. 1A). Specifically, peak S-phases at these times were significantly higher than at 42, 55, 66, and 72 h (Table 1B), indicating that under normal conditions, control cells underwent two rounds of cell cycle. In contrast, GSH-depleted (Treated) cells exhibited a single S-phase peak between 36 h and 66 h (Fig. 1A), consistent with a dramatic increase in the resident time of cells in the S-phase in conjunction with cellular GSH depletion. Remarkably, the S-phase profile in the reversal state was identical to that of GSH-depleted cells, indicating that cell transition from S-to-G2 remained delayed.
Fig. 1.
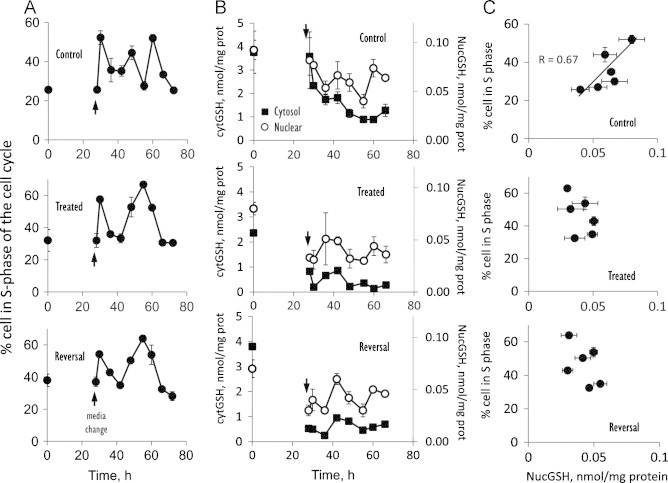
Temporal relationship between cell cycle S phase and nuclear-to-cytosolic (N-to-C) GSH distribution. Cells were cultured in complete M199 media at a density of 2×105/well in 6 well plates for 72 h. Media was changed at 28 h (arrow) and cell samples were harvested at every 6-h interval starting at 30 h post seeding for cell cycle analysis and cell fractionation and GSH measurements. Details on achievement of sustained GSH depletion and GSH recovery were described in the Methods section. (A) shows the percent of cells in the S-phase of the cell cycle for control, GSH-depleted (Treated), and GSH-restored (Reversal) cells. Results are mean±SEM and statistical analyses for effects of Group, times and group-time interactions are summarized in Table 1. (B) shows the concentrations of GSH in the cytosol (filled squares) or in the nucleus (open circle) for controls, treated or reversal. Results are mean±SEM and statistical analyses for effects of Group and Group means for cytosolic and nuclear GSH are summarized in Table 2. (C) illustrates the relationship between % of cell in the S-phase and nuclear GSH levels. Horizontal and vertical error bars represent mean±SEM of GSH and % cells in S-phase, respectively at times 36, 42, 48, 55, 60 and 66 h.
Table 1.
A Effects of Group, Time and Group-Time Interaction and Group Means for S-Phase among control cells, GSH-depleted (Treated) cells and GSH-restored (Reversal) cells.
| Effect | Na | Mean±SD b | p-value |
|---|---|---|---|
| Group | <0.01⁎⁎ | ||
| Control | 36 | 35.8±10.6 c | |
| Treated | 48 | 44.4±14.7 | |
| Reversal | 36 | 43.9±14.7 | |
| Time point | <0.01⁎⁎ | ||
| 42 h | 20 | 33.1±3.6 | |
| 48 h | 20 | 49.3±6.3 d | |
| 55 h | 20 | 53.9±17.9 d | |
| 60 h | 20 | 52.7±7.5 d | |
| 66 h | 20 | 32.2±2.3 | |
| 72 hour | 20 | 28.6±5.1 | |
| Group–time interaction | <0.01⁎⁎ | ||
| 42 h—Control | 6 | 30.3±4.6 | |
| Treated | 8 | 33.4±3.1 | |
| Reversal | 6 | 34.9±1.7 | |
| 48 h—Control | 6 | 44.5±9.4 | |
| Treated | 8 | 52.8±1.5 | |
| Reversal | 6 | 50.3±3.3 | |
| 55 h—Control | 6 | 27.7±4.3 e | |
| Treated | 8 | 66.7±4.4 | |
| Reversal | 6 | 63.9±1.5 | |
| 60 h—Control | 6 | 51.6±5.4 | |
| Treated | 8 | 52.4±9.4 | |
| Reversal | 6 | 53.8±8.1 | |
| 66 h—Control | 6 | 33.4±3.4 | |
| Treated | 8 | 30.7±2.0 | |
| Reversal | 6 | 32.5±0.6 | |
| 72 h—Control | 6 | 27.4±4.7 | |
| Treated | 8 | 30.5±4.8 | |
| Reversal | 6 | 28.1±5.9 | |
| B Comparisons among six time points for control cells. | |||
|---|---|---|---|
| Time point | N | Mean±SD | p-value |
| 42 h | 6 | 30.3±4.6 | <0.1⁎⁎ |
| 48 h | 6 | 44.5±9.4 f | |
| 55 h | 6 | 27.7±4.3 | |
| 60 h | 6 | 51.6±5.4 f | |
| 66 h | 6 | 33.4±3.4 | |
| 72 h | 6 | 27.4±4.7 | |
N—number of observations.
SD—Standard deviation.
Control significantly lower than the other 2 groups.
Significantly higher than 42 h, 66 h, &and 72 h.
Control significantly lower than the other 2 groups.
Significantly higher than 42 h, 55 h, 66 h and 72 h; troughs at 42 h, 55 h, 66 h, and 72 h; peaks at 48 h and 60 h.
Significant at 1% level (p-value<0.01).
Cytosolic and nuclear GSH contents were examined in parallel with cell cycle changes. In control cells, cytosolic GSH time-dependently decreased from 30 to 60 h, while nuclear GSH was cyclical, exhibiting two peaks between 42 and 48 h and at 60 h (Fig. 1B) that temporally correlated with the two peak S-phases (Fig. 1A). Significantly, there was a positive correlation of % cells in S-phase with nuclear GSH (R=0.67, Fig. 1C). BSO treatment decreased cytosolic GSH that was <50% lower than controls (Table 2). Nuclear GSH contents were similarly decreased post BSO-treatment; interestingly, a cyclical pattern of nuclear GSH remained evident even though the levels at various times were not statistically different (Fig. 1B). Importantly, there was no positive linear correlation of % cells in S-phase with nuclear GSH under these GSH-depleted conditions (Fig. 1C). During reversal, the contents of cytosolic GSH, nuclear GSH and its cyclical pattern, as well as the relationship between % cells in S-phase with nuclear GSH resembled those of GSH-depleted cells (Fig. 1, Table 2).
Table 2.
Effect of group and group means for cytosolic and nuclear GSH among control cells, GSH-depleted (treated) cells and GSH-restored (reversal) cells.
| Effect | Na | Mean±SD b | p-value | Effect | Na | Mean±SD b | p-value |
|---|---|---|---|---|---|---|---|
| Cytosolic GSH | Nuclear GSH | ||||||
| Group | <0.01⁎⁎ | Group | <0.01⁎⁎ | ||||
| Control | 54 | 1.69±1.18 c | Control | 54 | 0.07±0.03 c | ||
| Treated | 54 | 0.43±0.33 | Treated | 54 | 0.037±0.01 | ||
| Reversal | 54 | 0.64±0.28 | Reversal | 54 | 0.042±0.02 |
N—number of observations.
SD—Standard deviation.
Control significantly higher than the other 2 groups.
Significant at 1% level (p-value<0.01).
GSH status and cytosolic and nuclear expression of cdk1
The expression kinetics of the cell cycle checkpoint controller, cdk1, was determined to further investigate the relationship of cell cycle and GSH. The results revealed distinct patterns of cdk1 expression in the cytosolic and nuclear fractions among the treatment groups. In control cells, cytosolic cdk1 was elevated between 36 h and 60 h while nuclear cdk1 exhibited peaks and troughs, statistically higher values occurring at 48 and 66 h (Fig. 2A, B). These peak nuclear cdk1 responses temporally correlated with that of peak S-phase responses (within 6 h, see Fig. 1A), consistent with the function of cdk1 in S-to-G2-to-M transition [13]. Over this time period, GSH-depleted cells elicited high expression of cytosolic cdk1, but uniformly low expression of nuclear cdk1 (Fig. 2B). During reversal, nuclear cdk1 expression remained depressed even though cytosolic cdk1 returned to near control levels (Fig. 2B). Of note, the nuclear-to-cytosolic cdk1 ratios in GSH-depleted and GSH-restored cells were significantly lower (as a group) when compared to control cells, in accordance with a delay of S-to-G2-to-M transition in the cell cycle as reflected in a prolongation of the S-phase (see Fig. 1A).
GSH status on chk2 activation status and GAPDH expression
A significant decrease in nuclear GSH in BSO-treated cells suggested the possibility of enhanced DNA damage which would elicit a DNA damage response. To address this, we determine the nuclear activation of chk2 and the nuclear-to-cytosol distribution of GAPDH, indices of DNA damage response and repair, respectively. Chk2 is a downstream effector of the ataxia telangiectasia mutated (ATM) signaling pathway that is often associated with the DNA damage signaling network [14], while GAPDH is a pleiotropic enzyme known to function in mRNA stability, DNA repair and cell cycle progression [15–18]. In control cells, nuclear chk2 was notably low between 0 h and 42 h and at 55 h; elevated nuclear chk2 was evident at 48 h and was strongly expressed between 60 h and 72 h (Fig. 3A, B). The phosphorylation of nuclear chk2 was strong at all times (Fig. 3). In BSO-treated cells, the expression profile of nuclear chk2 resembled that of controls but quantitatively lower; however, as a group the proportion of phospho-chk2-to-chk2 was significantly higher especially at times between 30 h and 42 h and at 55 h (Fig. 3C), consistent with early increase in chk2 phosphorylation in conjunction with GSH depletion. It is notable that this increase in chk2 activation occurred over a 6 h time period prior to cell entry into the prolonged S-phase. During reversal, nuclear chk2 expression and phospho-chk2-to-chk2 ratios resembled those in control cells that was statistically lower than BSO-depleted cells (Fig. 3), suggesting that GSH recovery was associated with chk2 de-phosphorylation.
Cytosolic GAPDH expressions were not different among groups and were characteristically higher at 30 and 60 h post-seeding (Fig. 4A, B). However, statistical differences were observed in group and time-group effects among control and treatment groups in that GSH synthesis inhibition elicited higher nuclear GAPDH expression and N-to-C ratios than control conditions and during reversal. Significantly higher nuclear GAPDH levels in GSH-depleted cells were evident at all time points except at 66 h (Fig. 4B). Overall, these results suggest that GSH deficiency is linked to DNA damage responses, which could contribute to a delay in cell transition from S-to-G2 to favor DNA repair.
Discussion
The dynamic relationship between nuclear and cytosolic GSH compartmentation and cell cycle responses at the early time points (6 h intervals) during proliferation of endothelial cells is poorly understood. Our current study shows for the first time that brain microvascular endothelial cells (IHECs) exhibit distinct cell cycle characteristics post-seeding after an initial 30-h quiescent period, depending on the cellular GSH status. Under normal conditions, control IHECs completed two cycles of cell division with peak S-phases at 48 h and 60 h (Fig. 1A) which temporally associate with peak nuclear expressions of cdk1 and nuclear GSH at these times. These results suggest a link between cell cycle progression and nuclear GSH [19], a suggestion that was supported by the finding of a positive correlation between % cells in S-phase of cell cycle with nuclear GSH levels. The cyclical pattern of nuclear GSH over the 12-h period is an interesting observation that could reflect an impact of circadian rhythm that is separate from cell cycle. However, the relationship between biological rhythms and cell cycle is not easily resolved in cell culture studies given our lack of understanding of the regulation of biological clocks in vitro. Indeed, most of the studies on circadian biology are conducted in animal studies wherein clock genes were mutated to determine their impact on GSH rhythms [20,21].
A novel observation in our study is that decreased nuclear GSH is associated with a delay in cell exit from S-phase in the cell cycle. Strikingly, only one peak S-phase was evident in BSO-treated cells as compared to control cells that typically proceeded with two rounds of cell cycle over the same time period (Fig. 1A). This slowed S-to-G2 progression in GSH-depleted cells was preceded by higher localization of the checkpoint controller, cdk1 in the cytosol (Fig. 2), suggesting that cytosol-to-nuclear cdk1 translocation was decreased by a disruption in the GSH status [22,23]. Importantly, under GSH disruption, a positive linear correlation of % cell in S-phase with nuclear GSH was no longer evident; rather it appears that the arrest of cells in the S-phase corresponded to a significant decrease in the total nuclear GSH content (Table 2). It is notable that a >50% decrease in baseline cytosolic GSH was reflected in 50% decrease in nuclear GSH; however the nuclear GSH pool appeared to retain an inherent cyclical profile and was relatively stable over 72 h [24]. This means that the GSH status within the cytosolic and nuclear compartments is differentially controlled during cell cycle in favor of nuclear GSH accumulation. The results are also consistent with a slower turnover of the nuclear GSH pool which would ensure the preservation of critical cysteine residues of nuclear proteins, such as histones, telomerase, and polyADP ribose [25], and the maintenance of a reducing nuclear environment that promotes DNA binding and gene expression [26].
The correspondence of decreased nuclear GSH with a prolonged S-phase in BSO-treated cells is consistent with the following scenario during active DNA synthesis: increased oxidative DNA damage at low nuclear GSH would result in a DNA damage response and induce S-phase arrest, thus providing an extended time for DNA repair. Our current results support the suggestion that inhibition of GSH synthesis elicited DNA damage response and repair as evidenced by elevated nuclear chk2 phosphorylation (activation) and increased N-to-C GAPDH distribution, prior to peak cell arrest in S-phase. Enhanced cytosol-to-nuclear GAPDH translocation [27] is evidenced by an increase in nuclear GAPDH in conjunction with decreased cytosolic GAPDH.
Recent studies demonstrated that GAPDH is a substrate for the ATM/ATR pathway [28], implicating a role for nuclear chk2. The presence of phosphorylated chk2 in the nucleus of quiescent cells means that DNA replication is not an error free process under physiological conditions, and that a basal activity for DNA repair exists to maintain the integrity of nuclear DNA. Moreover, chk2-mediated phosphorylation was shown to be essential in accurate spindle assembly in normal mitosis [29,30]. However, the extent of chk2 phosphorylation relative to chk2 is lower in quiescent and proliferating control cells and increased markedly during GSH deficiency. An enhanced nuclear phospho-chk2-to-chk2 ratio between 30 h and 55 h in GSH-compromised cells is consistent with activation of the chk2/ATM/ATR pathway for DNA repair, likely in response to elevated DNA damage secondary to decreased nuclear GSH. Since phospho-chk2 is an inhibitor of Cdc 25C that is required for cyclin B-cdk1 complex activation and G2M transition [31], the delay in S-to-G2 transition (Fig. 1A) and greater retention of cdk1 in the cytosol of GSH-depleted cells (Fig. 2A) would correlate with an increase in chk2 activation in these cells.
It is remarkable that the reversal of GSH inhibition and restored GSH synthetic capacity did not restore endothelial cell cycle vis-à-vis S-to-G2 progression over 72 h post BSO removal. A possible explanation is the temporal delay in recovery of nuclear GSH which remained depressed over this time frame (Fig. 1B). Low nuclear GSH was reflective of decreased cytosolic GSH (Table 2); presumably, during reversal and active proliferation, amino acids (including cysteine, glutamate, glycine) were preferentially utilized for protein synthesis rather than GSH synthesis. However, despite a delay in cell cycle recovery, there was evidence that IHECs were transitioning to the control phenotype, as evidenced by the expressions of nuclear chk2 and GAPDH which resembled control cells. The attenuated DNA damage responses would be consistent with restored nuclear DNA integrity such that cells can begin to exit the S-phase and proceed with normal cell cycle. A lagging time line for normalization of S-phase progression behind that of decreased DNA damage responses is consistent with this interpretation.
To our knowledge, the current findings are the first to establish a temporal link between nuclear GSH, DNA damage response, and the S-phase of the cell cycle in brain microvascular endothelial cells in the normal and GSH-deficient states (Fig. 5). These findings complement and extend recent studies of GSH changes and epithelial cell proliferation [32]. Importantly, our results provide insights into the relationship between cellular GSH disruption and recovery and cell cycle progression at 6-h intervals during endothelial proliferation in the first three days post emergence from quiescence. This approach allowed us to better describe the influence of nuclear GSH changes on up-or-down expressions of cdk1, GAPDH, chk2 and its activation state between the times of 36h and 60 h during active cell proliferation. At present, the mechanistic relationships that govern nuclear GSH, proliferation-associated responses in the cell cycle, DNA damage response, and activation of DNA repair are unknown and are the subjects of current investigation in the laboratory.
Fig. 5.
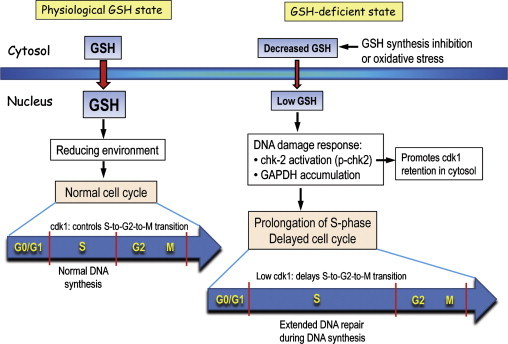
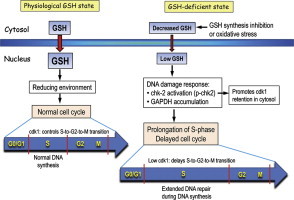
Endothelial cell cycle responses under physiological and GSH-deficient states. During cell proliferation, cytosol-to-nuclear GSH transport is increased under physiological GSH conditions. An increase in intra-nuclear reducing environment promotes gene transcription that brings about normal cell cycle progression wherein DNA synthesis occurs during the S-phase. Normal nuclear cdk1expression controls S-to-G2-to-M cell transition. Decreased cytosolic GSH due to inhibition of synthesis or enhanced oxidative stress results in decreased nuclear GSH import. Low nuclear GSH induces a DNA damage response, presumably due to increased oxidative DNA damage. An increase in chk-2 activation (increased p-chk2) and nuclear GAPDH accumulation contributes to a prolongation of the S-phase and a delay in cell cycle progression. In part, increased p-chk2 promotes the retention of cdk1 in the cytosol; a decrease in nuclear cdk1 delays the S-to-G2-to-M transition. Significantly, the lengthening of the S-phase allows for extended time for DNA repair.
In the current study, the IHEC cell line was used as a surrogate of the blood-brain barrier endothelium in vivo. The capability of the cerebral microvascular endothelial monolayer to repair itself after wounding is critical to preserving blood-brain barrier function against fluctuating systemic influences to maintain brain homeostasis [7]. Furthermore, an understanding of the intrinsic role for GSH control of endothelial proliferation and restitution will have key implications for endothelial integrity in various microvascular beds under conditions of oxidative stress or associated vascular pathologies. Future therapeutic approaches that target endothelial restoration post oxidative insult would have significant clinical implications for the neurovascular disorders of diabetes and stroke and more broadly, for other neurodegenerative and neurological disorders as well.
Acknowledgment
We thank Deborah Chervenak for technical assistance in flow cytometry. This study was supported by NIH Grant DK44510 (TYA) and by an LSUHSC Malcolm Feist Cardiovascular Fellowship (WL).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Tu B.P., Kudlicki A., Rowicka M., McKnight S.L. Logic of the yeast metabolic cycle: temporal compartmentalization of cellular processes. Science (New York, NY) 2005;310(5751):1152–1158. doi: 10.1126/science.1120499. [DOI] [PubMed] [Google Scholar]
- 2.Jones D.P., Go Y.M., Anderson C.L., Ziegler T.R., Kinkade J.M., Jr., Kirlin W.G. Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. FASEB Journal. 2004;18(11):1246–1248. doi: 10.1096/fj.03-0971fje. [DOI] [PubMed] [Google Scholar]
- 3.Okouchi M., Ekshyyan O., Maracine M., Aw T.Y. Neuronal apoptosis in neurodegeneration. Antioxidants and Redox Signaling. 2007;9(8):1059–1096. doi: 10.1089/ars.2007.1511. [DOI] [PubMed] [Google Scholar]
- 4.Dalle-Donne I., Rossi R., Giustarini D., Colombo R., Milzani A. S-glutathionylation in protein redox regulation. Free Radical Biology and Medicine. 2007;43(6):883–898. doi: 10.1016/j.freeradbiomed.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 5.Fratelli M., Gianazza E., Ghezzi P. Redox proteomics: identification and functional role of glutathionylated proteins. Expert Review of Proteomics. 2004;1(3):365–376. doi: 10.1586/14789450.1.3.365. [DOI] [PubMed] [Google Scholar]
- 6.Markovic J., Borras C., Ortega A., Sastre J., Vina J., Pallardo F.V. Glutathione is recruited into the nucleus in early phases of cell proliferation. The Journal of Biological Chemistry. 2007;282(28):20416–20424. doi: 10.1074/jbc.M609582200. [DOI] [PubMed] [Google Scholar]
- 7.Bradbury M.W. The blood-brain barrier. Experimental Physiology. 1993;78(4):453–472. doi: 10.1113/expphysiol.1993.sp003698. [DOI] [PubMed] [Google Scholar]
- 8.Attene-Ramos M.S., Kitiphongspattana K., Ishii-Schrade K., Gaskins H.R. Temporal changes of multiple redox couples from proliferation to growth arrest in IEC-6 intestinal epithelial cells. American Journal of Physiology—Cell Physiology. 2005;289(5):C1220–1228. doi: 10.1152/ajpcell.00164.2005. [DOI] [PubMed] [Google Scholar]
- 9.Bronfman M., Loyola G., Koenig C.S. Isolation of intact organelles by differential centrifugation of digitonin-treated hepatocytes using a table Eppendorf centrifuge. Analytical Biochemistry. 1998;255(2):252–256. doi: 10.1006/abio.1997.2453. [DOI] [PubMed] [Google Scholar]
- 10.Reed D.J., Babson J.R., Beatty P.W., Brodie A.E., Ellis W.W., Potter D.W. High-performance liquid chromatography analysis of nanomole levels of glutathione, glutathione disulfide, and related thiols and disulfides. Analytical Biochemistry. 1980;106(1):55–62. doi: 10.1016/0003-2697(80)90118-9. [DOI] [PubMed] [Google Scholar]
- 11.Noda T., Iwakiri R., Fujimoto K., Aw T.Y. Induction of mild intracellular redox imbalance inhibits proliferation of CaCo-2 cells. FASEB Journal. 2001;15(12):2131–2139. doi: 10.1096/fj.01-0131com. [DOI] [PubMed] [Google Scholar]
- 12.Okouchi M., Okayama N., Aw T.Y. Hyperglycemia potentiates carbonyl stress-induced apoptosis in naive PC-12 cells: relationship to cellular redox and activator protease factor-1 expression. Current Neurovascular Research. 2005;2(5):375–386. doi: 10.2174/156720205774962665. [DOI] [PubMed] [Google Scholar]
- 13.Hochegger H., Takeda S., Hunt T. Cyclin-dependent kinases and cell-cycle transitions: does one fit all? Nature Reviews Molecular Cell Biology. 2008;9(11):910–916. doi: 10.1038/nrm2510. [DOI] [PubMed] [Google Scholar]
- 14.Matsuoka S., Rotman G., Ogawa A., Shiloh Y., Tamai K., Elledge S.J. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proceedings of the National Academy of Sciences of the USA. 2000;97(19):10389–10394. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez-Pascual F., Redondo-Horcajo M., Magan-Marchal N., Lagares D., Martinez-Ruiz A., Kleinert H. Glyceraldehyde-3-phosphate dehydrogenase regulates endothelin-1 expression by a novel, redox-sensitive mechanism involving mRNA stability. Molecular and Cellular Biology. 2008;28:7139–7155. doi: 10.1128/MCB.01145-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Azam S., Jouvet N., Jilani A., Vongsamphanh R., Yang X., Yang S., Ramotar D. Human glyceraldehyde-3-phosphate dehydrogenase plays a direct role in reactivating oxidized forms of the DNA repair enzyme APE1. Journal of Biological Chemistry. 2008;283(45):30632–30641. doi: 10.1074/jbc.M801401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carujo S., Estanyol J.M., Ejarque A., Agell N., Bachs O., Pujol M.J. Glyceraldehyde 3-phosphate dehydrogenase is a SET-binding protein and regulates cyclin B-cdk1 activity. Oncogene. 2006;25(29):4033–4042. doi: 10.1038/sj.onc.1209433. [DOI] [PubMed] [Google Scholar]
- 18.Walsh J.L., Keith T.J., Knull H.R. Glycolytic enzyme interactions with tubulin and microtubules. Biochimica Biophysica Acta. 1989;999(1):64–70. doi: 10.1016/0167-4838(89)90031-9. [DOI] [PubMed] [Google Scholar]
- 19.Menon S.G., Goswami P.C. A redox cycle within the cell cycle: ring in the old with the new. Oncogene. 2007;26(8):1101–1109. doi: 10.1038/sj.onc.1209895. [DOI] [PubMed] [Google Scholar]
- 20.Laura M. Beaver, Vladimir I. Klichk, Eileen S. Chow, Joanna Kotwica-Rolinska, Marisa Williamson. Circadian regulation of glutathione levels and biosynthesis in drosophila melanogaster. Plos One. 2012;7(11):1–9. doi: 10.1371/journal.pone.0050454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tongfei A. Wang. Circadian rhythm of redox state regulates excitability in suprachiasmatic nucleus neurons. Science. 2012;337:839. doi: 10.1126/science.1222826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin P., Hardy S., Morgan D.O. Nuclear localization of cyclin B1 controls mitotic entry after DNA damage. Journal of Cellular Biology. 1998;141(4):875–885. doi: 10.1083/jcb.141.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J., Song H., Walsh S., Bardes E.S., Kornbluth S. Combinatorial control of cyclin B1 nuclear trafficking through phosphorylation at multiple sites. Journal of Biological Chemistry. 2001;276(5):3604–3609. doi: 10.1074/jbc.M008151200. [DOI] [PubMed] [Google Scholar]
- 24.Markovic J., Mora N.J., Broseta A.M., Gimeno A., de-la-Concepcion N., Vina J., Pallardo F.V. The depletion of nuclear glutathione impairs cell proliferation in 3t3 fibroblasts. PloS One. 2009;4(7):e6413. doi: 10.1371/journal.pone.0006413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pallardo F.V., Markovic J., Garcia J.L., Vina J. Role of nuclear glutathione as a key regulator of cell proliferation. Molecular Aspects of Medicine. 2009;30(1–2):77–85. doi: 10.1016/j.mam.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 26.Smith C.V., Jones D.P., Guenthner T.M., Lash L.H., Lauterburg B.H. Compartmentation of glutathione: implications for the study of toxicity and disease. Toxicology and Applied Pharmacology. 1996;140(1):1–12. doi: 10.1006/taap.1996.0191. [DOI] [PubMed] [Google Scholar]
- 27.Sundararaj K.P., Wood R.E., Ponnusamy S., Salas A.M., Szulc Z., Bielawska A., Obeid L.M., Hannun Y.A., Ogretmen B. Rapid shortening of telomere length in response to ceramide involves the inhibition of telomere binding activity of nuclear glyceraldehyde-3-phosphate dehydrogenase. Journal of Biological Chemistry. 2004;279(7):6152–6162. doi: 10.1074/jbc.M310549200. [DOI] [PubMed] [Google Scholar]
- 28.Cosentino C., Grieco D., Costanzo V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO Journal. 2011;30(3):546–555. doi: 10.1038/emboj.2010.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato K. A mitotic role for the DNA damage-response CHK2 kinase. Nature Cell Biology. 2010;12:424–425. doi: 10.1038/ncb0510-424. [DOI] [PubMed] [Google Scholar]
- 30.Stolz A., Ertych A., Kienitz A., Vogel C., Schneider V., Fritz B., Jacob R., Dittmar G., Weichert W., Petersen I., Bastians H. The CHK2-BRCA1 tumour suppressor pathway ensures chromosomal stability in human somatic cells. Nature Cell Biology. 2010;12:492–499. doi: 10.1038/ncb2051. [DOI] [PubMed] [Google Scholar]
- 31.Taylor W.R., Stark G.R. Regulation of the G2/M transition by p53. Oncogene. 2001;20(15):1803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- 32.Diaz Vivancos P., Wolff T., Markovic J., Pallardo F.V., Foyer C.H. A nuclear glutathione cycle within the cell cycle. Biochemical Journal. 2010;431(2):169–178. doi: 10.1042/BJ20100409. [DOI] [PubMed] [Google Scholar]