

Abstract
Parkinson's disease (PD) is characterized by the progressive loss of dopaminergic neurons in the substantia nigra (SN) and depletion of striatal dopamine (DA), leading to a range of motor symptoms, including resting tremor, rigidity, bradykinesia and postural abnormalities. The neurotoxin (MPTP) and its active metabolite, 1-methyl-4-phenylpyridinium (MPP+), cause dopaminergic cell loss in a variety of animal species and produce symptoms similar to those seen in PD. Our lab has shown that MPP+ activates cell stress pathways, including the unfolded protein response (UPR) in mouse primary mesencephalic cultures. The BH3-only protein, PUMA (p53 upregulated mediator of apoptosis), has been shown to be activated in response to many cellular stresses, including endoplasmic reticulum (ER) stress and UPR, and to induce cell death. Therefore, we hypothesized that PUMA may mediate MPP+ toxicity. To test this hypothesis, we compared the response of primary mesencephalic cultures from wild-type and PUMA deficient (−/−) mice to MPP+. We also utilized cultures from p53 −/− and activating transcription factor 3 (ATF3) −/− mice to further elucidate the pathways involved. These studies revealed that PUMA and p53, but not ATF3, are required for MPP+-induced cell death, suggesting that UPR activation is parallel to the induction of MPP+-induced cell death.
Keywords: Parkinson's, MPP+, UPR, p53, PUMA, ATF3
1. Introduction
PD is the second most common neurodegenerative disorder and is characterized by the progressive loss of dopaminergic neurons in the SN and the resulting loss of dopaminergic innervation to the striatum. Although the molecular mechanisms underlying the pathogenesis of PD remain unclear, oxidative stress, mitochondrial dysfunction and ER stress have all been implicated in the etiology of this disorder (Dauer and Przedborski, 2003; Vila and Przedborski, 2004).
The neurotoxin, MPTP, and its active metabolite, MPP+, are used to model dopaminergic degeneration in vivo and in vitro, respectively (Blum et al., 2001). After intraperitoneal injection of MPTP, it crosses the blood-brain barrier and is rapidly converted to its active metabolite, MPP+, by monoamine oxidase B (MAOB) in glial cells (Markey et al., 1984). In in vitro systems, direct application of MPP+ induces cell death specifically in dopaminergic neurons due to its high affinity for the dopamine transporter (DAT) and other catecholamine uptake systems. Once inside the cell, MPP+ is sequestered into vesicles by the vesicular monoamine transporter 2 (VMAT2), displacing DA in the process (Staal and Sonsalla, 2000). The toxicity of MPP+ is likely due to its actions at cytosolic and mitochondrial sites of action rather than through the displacement of DA from vesicles to cytosol as dopamine deficient animals do not show altered MPTP toxicity (Hasbani et al., 2005).
MPTP- and MPP+-induced toxicity seems to involve activation of UPR and ubiquitin-proteasome system (UPS) dysfunction. UPR can be triggered by any or all of three different gatekeeper proteins: IRE1, PERK and ATF6 (Zhang and Kaufman, 2004, 2006). In cell lines and primary dopaminergic neurons, MPP+ activates UPR; however, which branches are upregulated varies by cell type (Holtz and O'Malley, 2003; Ryu et al., 2002). For example, in PC12 cells, both the PERK and IRE1 branches are activated, while in a CNS dopaminergic cell line only the PERK pathway is activated. Whether this activation of UPR is protective or leads to cell death is not yet clear. Recent evidence suggests that it may be protective, as Xbp-1 overexpression protects against both MPTP in vivo and MPP+ in vitro (Sado et al., 2009) and ATF6α deletion accelerates MPTP toxicity (Egawa et al., 2011; Hashida et al., 2012).
Many studies have demonstrated the induction of at least some markers of apoptosis by MPTP and MPP+ (Blum et al., 2001). However, whether MPTP and MPP+ induce bona fide apoptosis or other forms of cell death depends on both the dosing paradigm and the cell type. For example, MPTP induces both caspase-3 activation and poly (ADP-ribose) polymerase (PARP) cleavage in the SN, if the toxin is given in small doses over five days (chronic model), but not if given in small doses within a single day (acute model) (Blum et al., 2001). In addition, chronic MPTP exposure leads to elevation of Bax mRNA and decreased Bcl-2 levels in the mouse SN (Vila et al., 2001). Using this same dosing paradigm, deletion of Bax is protective against MPTP (Vila et al., 2001). However, in primary dopaminergic neurons, Bax deletion did not protect against MPP+ (O'Malley et al., 2003). Thus, exactly how MPTP/MPP+ leads to cell death in dopaminergic neurons is still unclear (Blum et al., 2001).
Many studies have suggested that prolonged and severe UPR can lead to cell death, possibly via apoptosis (Zhang and Kaufman, 2004, 2006). Amongst other mechanisms, BH3-only proteins, such as PUMA, have been hypothesized to serve as links between ER stress pathways and apoptosis (Nakano and Vousden, 2001; Reimertz et al., 2003; Yu et al., 2001). We have previously demonstrated that PUMA mediates cell death induced by another parkinsonian mimetic, 6-hydroxydopamine (Bernstein et al., 2011). Therefore, we tested whether PUMA also mediates cell death in response to MPP+ exposure. Here, we show that PUMA is required for MPP+-induced cell death in primary mesencephalic cultures. However, further analysis using animals deficient in a key UPR pathway (ATF3) suggests that MPP+-mediated cell death is independent of the upregulation of UPR. Instead, the DNA damage pathway (p53) may play a more direct role in mediating cell death in this model of PD.
2. Materials and Methods
2.1 Animals
Animals were treated in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Wild-type C57/Bl6 mice were from Charles River Laboratories (Wilmington, MA). Puma knockout mice were previously generated and characterized (Jeffers et al., 2003). ATF3 knockout mice were generated and provided by Dr. Tsonwin Hai (Ohio State University) (Hartman et al., 2004). p53 knockout mice were provided by Dr. Helen Pwinica-Worms (Washington University Medical School) (Jacks et al., 1994).
2.2 Cell cultures and toxin treatment
For RT-PCR experiments, embryonic day 14 (E14) C57Bl/6 murine midbrains (Charles River Laboratories, Wilmington, MA, USA) were pooled and prepared as described previously (Lotharius et al., 1999). To assess survival of neurons after MPP+ treatment, mice heterozygous for Puma, ATF3 or p53 were mated to produce wild type, heterozygous, and homozygous deficient embryos. Cultures were derived from individual pups and pups were individually genotyped using cortical tissue as described previously (Antenor-Dorsey and O'Malley, 2012; Bernstein et al., 2011). After seven days in vitro (DIV7), cells were treated with 1 μM MPP+, a dose that produces 50% loss of dopaminergic neurons 48 hours after treatment. Previous experiments from the lab have established this exposure paradigm (Antenor-Dorsey and O'Malley, 2012; Holtz and O'Malley, 2003; Kim-Han et al., 2011; Lotharius et al., 1999; O'Malley et al., 2003).
2.3 Reverse Transcription PCR
Dissociated midbrain neurons were plated in 12-well plates and treated with MPP+ at DIV7. Cultures were washed with PBS, total RNA was extracted (RNeasy Mini Kit; Qiagen, Valencia, CA) and then reverse transcribed (High Capacity cDNA Reverse Transcription Kit; Applied Biosystems, Foster City, CA). Levels of Puma and 18S rRNA were analyzed by semi-quantitative reverse transcription PCR (RT-PCR) using primers specific for the gene of interest. All reactions were performed in triplicate. PCR products were resolved with polyacrylamide gel electrophoresis, visualized (SYBR Safe DNA; Invitrogen) and image (Storm PhosphorImager; Molecular Dynamics, Piscataway, NJ). Band intensities were measured (ImageQuant; Amersham Biosciences, Piscataway, NJ) and Puma levels were normalized to 18S rRNA levels and then compared to levels in untreated samples. Data was pooled from 3 independent experiments. The following primer sequences were used:
18S rRNA (5′-GGGAACGCGTGCATTTATCAG-3′, 5′-CGCTATTGGAGCTGGAATTAC-3′)
PUMA (5′-ACGACCTCAACGCGCAGTA-3′, 5′-CTAGTTGGGCTCCATTTCTGG-3′)
2.4 Immunocytochemistry
Primary cultures were plated in 7 mm microwell plates (MatTek Corp., Ashland, MA). Cells were treated with MPP+ and fixed with 4% paraformaldehyde in PBS after the appropriate incubation time. Cultures were stained with sheep polyclonal anti-tyrosine hydroxylase (TH) (Novus Biologicals, Littleton, CO) and Alexa488 α-sheep (Molecular Probes, Carlsbad, CA). Cultures were co-stained for NeuN where with mouse monoclonal α-NeuN (Chemicon, Billerica, MA). Cy3 α-mouse secondary antibody was purchased from Jackson Labs (Bar Harbor, ME). Cells were counted using unbiased stereological methods modified for use in a cell culture dish (Stereo Investigator, MicroBrightField, Williston, VT) (Antenor-Dorsey and O'Malley, 2012; Bernstein et al., 2011; Kim-Han et al., 2011). The estimated total number of TH neurons in the culture dish was calculated based on the following formula: N = Q- ×1/ssf × 1/asf, where N is the estimate of the total number of cells, Q- is the number of objects counted, ssf is the section sampling fraction and asf is the area sampling fraction. Gundersen (m=1) coefficients of error were less than 0.1. TH-positive neurite length was estimated by an unbiased stereological method (Petrimetrics, Stereo Investigator, MicroBrightField). Cell counts and neurite length were normalized to untreated control for each pup. Images were acquired by confocal microscopy (Olympus Fluoview 500, Olympus, Center Valley, PA) and processed in ImageJ.
2.5 Statistical analysis
GraphPad Prism software (San Diego, CA) was used for statistical analysis. All data were collected from a minimum of three independent experiments. The significance of effects between control and drug treatment conditions was determined by one-way ANOVA with Bonferroni Multiple Comparisons tests. The significance of effects between genotypes and drug treatment conditions was determined by two-way ANOVA with Bonferroni post-tests.
3 Results
3.1 MPP+ induces upregulation of PUMA mRNA
Previously, we used an unbiased, bioinformatics approach to survey genes responding to MPP+ treatment. This analysis did not detect changes in known BH3-only proteins; however, PUMA was not represented on the chip surveyed (Holtz and O'Malley, 2003). Since PUMA has been shown to be induced by ER stress and to trigger mitochondrial events leading to cell death in a variety of cell types, we sought to determine if PUMA is transcriptionally upregulated in response to MPP+. Primary mesencephalic cultures were treated with MPP+ and PUMA mRNA levels were analyzed by RT-PCR and normalized to 18S rRNA mRNA levels. Levels of PUMA mRNA were significantly increased by 12 hours after treatment with MPP+ (Figure 1A,B). These results demonstrate that MPP+ induces the upregulation of PUMA mRNA, suggesting that PUMA does play a role in MPP+-mediated cell death.
Figure 1. Loss of PUMA attenuates MPP+-mediated cell death.

A) Cultures derived from C57Bl/6 mice were treated with 1 μM MPP+ and total RNA was collected at the indicated times. Levels of PUMA and 18S rRNA were analyzed by semi-quantitative RT-PCR. All reactions were performed in triplicate and three independent experiments were performed. A representative gel is shown. B) Gels were quantitated in ImageQuant and analyzed by one-way ANOVA (***, p < 0.001) with Bonferroni post-tests to compare each time point to untreated (12 hr and 24 hr: ***, p < 0.001). C) Cultures derived from PUMA +/+ and PUMA −/− cultures were treated with 1 μM MPP+ for 48 hours. Cells were fixed and stained for TH and NeuN. D) TH-positive and NeuN-positive cells were counted, the percentage of TH-positive cells was calculated and survival expressed as a percentage of untreated control. Data were analyzed by Student's t-test (*, p < 0.05).
3.2 Loss of PUMA protects against MPP+ toxicity
To determine if PUMA plays an essential role in MPP+ toxicity, primary mesencephalic cultures from PUMA +/+ and −/− mice were treated with or without MPP+. After 48 hours, cells were fixed and stained for TH, a marker of dopaminergic neurons, and NeuN, a marker of neuronal nuclei. TH-positive and NeuN-positive cells were counted by a non-biased stereological method and the percentage of TH-positive cells was calculated to determine the level of cell survival. In wild-type cultures, MPP+ induced a 50% loss of TH neurons, while only about 20% of TH neurons were lost in PUMA −/− cultures (Figure 1C,D). Neurite length was also estimated and loss of PUMA did not prevent loss of neurites (data not shown). These data indicate that loss of PUMA protects cells bodies against MPP+-induced cell death.
3.3 Loss of ATF3 does not protect against MPP+ toxicity
The transcription factor ATF3, which is rapidly induced in response to ER stress, is a key mediator of the PERK branch of the UPR pathway (Jiang et al., 2004). Since we previously showed that ATF3 levels were significantly upregulated in response to MPP+, we sought to determine if MPP+-induced UPR was playing a direct role in cell death by utilizing ATF3-deficient mice (Holtz and O'Malley, 2003). Primary cultures from ATF3 +/+ and −/− mice were treated with or without MPP+ for 48 hours before being evaluated for surviving TH-positive cells. Surprisingly, loss of ATF3 did not protect cells from MPP+; 40% of TH neurons in wild-type cultures and 50% of TH neurons in ATF3 −/− cultures survived (Figure 2). Furthermore, loss of ATF3 did not affect the loss of neurites induced by MPP+ (data not shown). These results indicate that ATF3 is not required for cell death induced by MPP+ and suggest that UPR does not play a direct role in MPP+-induced dopaminergic degeneration.
Figure 2. ATF3 is not required for MPP+-induced cell death.
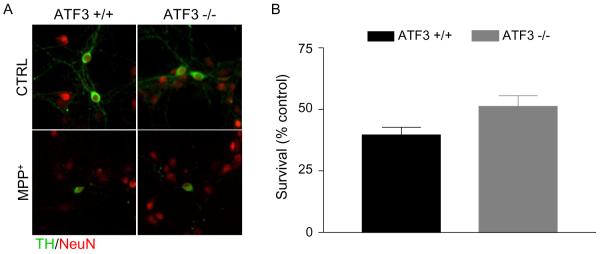
A) Cultures prepared from ATF3 +/+ and −/− mice were treated with 1 μM MPP+. 48 hours after treatment, cultures were fixed and stained for TH and NeuN. B) TH-positive and NeuN-positive cells were counted, the percentage of TH neurons was calculated and survival expressed as a percent of untreated control. Data were analyzed by Student's t-test (ns).
3.4 Loss of p53 protects against MPP+
Since activation of UPR did not appear to be mediating cell death in response to MPP+, we sought to determine how PUMA is regulated in this model. PUMA is a known transcriptional target of p53 and we have previously shown that p53 and PUMA are required for 6-OHDA-induced cell death in this system (Bernstein et al., 2011). In addition, MPP+ is known to increase levels of p53 and to induce p53-dependent cell death in the peripheral neuronal cell line, PC12 (Duan et al., 2002; Kook et al., 2011). In vivo, DNA damage and p53 pathways have been implicated in acute MPTP toxicity, although this paradigm primarily exhibits hallmarks of necrosis (Duan et al., 2002; Karunakaran et al., 2008; Mandavilli et al., 2000; Mandir et al., 2002; Trimmer et al., 1996; Wang et al., 2003). Therefore, we sought to test whether this upregulation of p53 seen in cell lines mediates MPP+-induced toxicity.
To test this hypothesis, we assessed the survival of TH neurons in cultures derived from mice deficient for p53. Primary cultures were prepared from p53 +/+ and −/−mice, treated with or without MPP+ for 48 hours and subsequently fixed and stained for TH and NeuN. Cultures from p53 −/− mice were significantly protected (20% cell death) against MPP+ compared to p53 +/+ cultures (60% cell death) (Figure 3). However, as with the PUMA −/− cultures, neurite loss was not prevented by loss of p53 (data not shown). These results indicate that loss of p53 protects against MPP+ induced cell body loss and that p53 is required for MPP+-induced cell death.
Figure 3. p53 is required for MPP+-induced cell death.
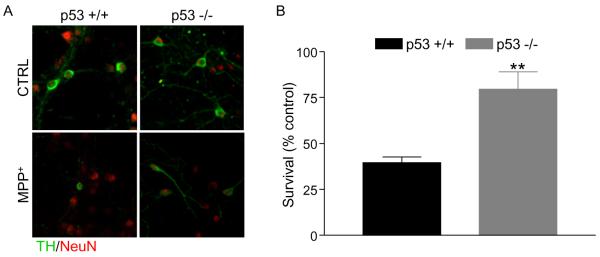
A) Cultures prepared from p53 +/+ and −/− mice were treated with 1 μM MPP+. 48 hours after treatment, cultures were fixed and stained for TH and NeuN. B) TH-positive and NeuN-positive cells were counted, the percentage of TH neurons was calculated and survival expressed as a percent of untreated control. Data were analyzed by Student's t-test (**, p < 0.01).
4 Discussion
Many lines of evidence suggest a role of ER stress in the pathogenesis of PD. Genetic mutations linked to PD, in genes such as α-synuclein, parkin and UCH-L1, as well as evidence from toxin models, implicate dysfunction of the UPS in this disorder (Eriksen et al., 2005; Snyder and Wolozin, 2004). Although previous results have established that MPP+ leads to upregulation of UPR and cell death, whether these two events are sequential or parallel events was not determined (Holtz and O'Malley, 2003; Ryu et al., 2002). In the present study, we demonstrate that PUMA is required for MPP+-induced cell death (Figure 1). Surprisingly, loss of ATF3, a critical transcription factor in UPR, had no effect on MPP+ toxicity, while loss of p53 is required (Figures 2 and 3). These data suggest that UPR and cell death are parallel, rather than sequential events. Taken together, the data suggest that a p53-dependent pathway mediates PUMA upregulation and cell death in response to MPP+ (Figure 4).
Figure 4. Model of PUMA action in response to MPP+.
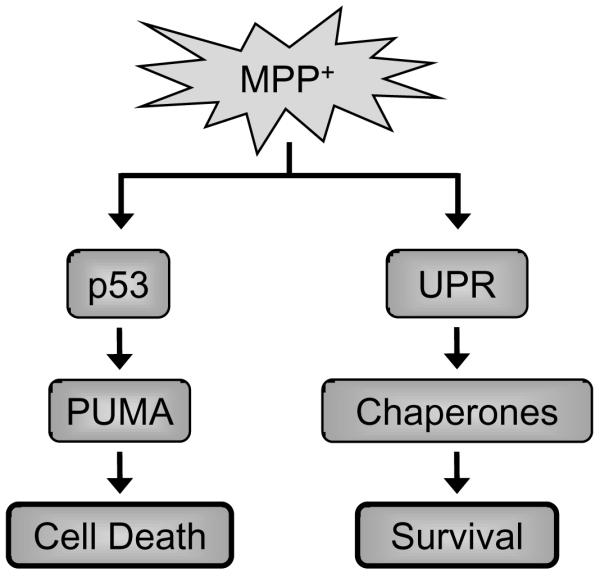
In this model, MPP+ activates both UPR and p53. UPR pathways lead to the upregulation of adaptive mechanisms, while p53 leads to PUMA upregulation and eventual cell death.
4.1 PUMA and p53 mediated MPP+-induced toxicity
We have demonstrated that PUMA and p53 are required for MPP+-induced cell death. (Figures 1 and 3) However, the protection provided by deletion of each of these genes is only partial, indicating that an additional pathway not examined here also contributes to cell death. The results reported here are consistent with work from other labs demonstrating that MPP+ can increase p53 levels and induce p53-dependent Bax upregulation and apoptosis in cell lines derived from the peripheral nervous system (Duan et al., 2002). It has also been demonstrated that MPP+ treatment in the same cell lines leads to upregulation of PUMA (Kook et al., 2011). In addition, acute administration of MPTP has been shown to induce both DNA damage pathways and activation of p53 in vivo (Mandavilli et al., 2000; Mandir et al., 2002; Wang et al., 2003). Furthermore, loss of p53 by pharmacological or genetic means can attenuate cell body loss, but not metabolites or terminal fields after acute administration of MPTP (Duan et al., 2002; Trimmer et al., 1996). Finally, phosphorylation and nuclear localization of p53, as well as upregulation of PUMA, in dopaminergic neurons of the SN after a single MPTP dose or chronic administration of MPTP was reported (Karunakaran et al., 2008). We have confirmed that p53 is a critical mediator of cell death in this model and demonstrated that PUMA also plays an important role. While other groups have shown upregulation of PUMA in response to MPP+ or MPTP, they have not shown that this upregulation mediates cell death (Karunakaran et al., 2008; Kook et al., 2011). By utilizing cultures generated from PUMA knockout animals, we demonstrate here that PUMA is a mediator of cell death, extending the previous findings. These findings also confirm the data from the peripherally derived PC12 cells (Duan et al., 2002; Kook et al., 2011). Together, these results implicate a p53- and PUMA-dependent pathway, possibly triggered by DNA damage, in MPP+-toxicity.
We then attempted to determine if loss of PUMA is protective against MPTP in vivo. However, using the lowest dose that we have used to produce striatal terminal loss and nigral cell loss (4 × 15 mg/kg, 1 hour apart), all of the PUMA knockout mice, but none of the wild-type, died within the first 24 hours, suggesting enhanced peripheral toxicity in these knockouts (Hasbani et al., 2005). Therefore, we were unable to test this in vivo. A targeted deletion of PUMA that does not affect its peripheral expression may provide a better system for testing whether loss of PUMA protects against MPTP.
4.2 ATF3 does not mediate MPP+-induced cell loss
UPR is thought to activate both adaptive and apoptotic pathways, depending on the severity and duration of ER stress (Malhotra and Kaufman, 2007a, b; Wu and Kaufman, 2006). Pro- and anti-apoptotic proteins also directly regulate UPR proteins, reinforcing the idea that UPR is a highly regulated process intimately tied to cellular homeostasis, as well as cell death pathways (Hetz and Glimcher, 2008). UPR is regulated by three interconnected pathways mediated by PERK, IRE1 and ATF6. Activation of PERK triggers activation of a cascade of transcription factors, including ATF4, ATF3 and CHOP. If PERK signaling were contributing to cell death in this system, loss of ATF3 would have provided protection against MPP+. However, loss of ATF3 did not prevent cell loss (Figure 2). In this paradigm of MPP+-induced toxicity, the PERK-ATF3- branch of the UPR does not appear to contribute to cell death. It is possible that, over a longer exposure time, the prolonged activation of UPR could lead to cell loss. However, within the 48 hours tested here, UPR, specifically the PERK-ATF3 branch, does not mediate cell death. It is possible that one or both of the other UPR pathways (IRE1 or ATF6) are more important for mediating UPR-induced cell death and we did not see protection when impairing only this one branch of this complex network. However, recent data suggest that activation of UPR is protective in these cellular models of PD (Egawa et al., 2011; Hashida et al., 2012; Sun et al., 2013). In fact, deletion of ATF6α has been shown to exacerbate MPTP toxicity, suggesting that that this pathway not only does not mediate cell loss, but also promotes cell survival (Egawa et al., 2011; Hashida et al., 2012). Additionally, in a PC12 model, ATF4 also protects against MPP+-toxicity (Sun et al., 2013). Our data, taken together with this work form other groups, suggest that UPR is a protective mechanism employed after MPP+ exposure rather than a mediator of cell death.
4.3 Conclusions
The results of this study suggest that that activation of UPR does not mediate cell death in response to MPP+. Instead, the p53-dependent activation of PUMA induces cell death. It is possible that UPR represents a protective mechanism employed by neurons in response to protein damage and UPS dysfunction. Multiple lines of evidence support a role for both the activation of UPR pathways and p53-dependent pathways in toxin models of PD and PD itself. Therefore, understanding which of these pathways is protective and which lead to neuronal degeneration will help in the development of new interventions for PD.
Highlights
PUMA and p53 mediate MPP+-induced toxicity.
ATF3 does not mediate cell loss in response to MPP+.
MPP+-induced UPR may represent a protective mechanism in response to protein damage
Acknowledgments
Dr. Gerard Zambetti (St. Jude's Children's Research Hospital) generously provided the PUMA knockout mice. Dr. Tsonwin Hai at Ohio State University provided us with the ATF3 knockout mice. Dr. Helen Pwinica-Worms provided us with the p53 mice. Ms. Lynn White in the Pwinica-Worms lab performed all mating and genotyping of the p53 mice. All images were acquired at the Bakewell Neuroimaging Facility, which is supported by National Institutes of Health Neuroscience Blueprint Core Grant NS057105 to Washington University and the Bakewell Family Foundation. This work was supported by National Institutes of Health grant NS39084 and MH45330.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antenor-Dorsey JA, O'Malley KL. WldS but not Nmnat1 protects dopaminergic neurites from MPP+ neurotoxicity. Mol Neurodegener. 2012;7:5. doi: 10.1186/1750-1326-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein AI, Garrison SP, Zambetti GP, O'Malley KL. 6-OHDA generated ROS induces DNA damage and p53- and PUMA-dependent cell death. Mol Neurodegener. 2011;6:2. doi: 10.1186/1750-1326-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum D, Torch S, Lambeng N, Nissou M, Benabid AL, Sadoul R, Verna JM. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson's disease. Prog Neurobiol. 2001;65:135–172. doi: 10.1016/s0301-0082(01)00003-x. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Duan W, Zhu X, Ladenheim B, Yu QS, Guo Z, Oyler J, Cutler RG, Cadet JL, Greig NH, Mattson MP. p53 inhibitors preserve dopamine neurons and motor function in experimental parkinsonism. Ann Neurol. 2002;52:597–606. doi: 10.1002/ana.10350. [DOI] [PubMed] [Google Scholar]
- Egawa N, Yamamoto K, Inoue H, Hikawa R, Nishi K, Mori K, Takahashi R. The endoplasmic reticulum stress sensor, ATF6alpha, protects against neurotoxin-induced dopaminergic neuronal death. J Biol Chem. 2011;286:7947–7957. doi: 10.1074/jbc.M110.156430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen JL, Wszolek Z, Petrucelli L. Molecular pathogenesis of Parkinson disease. Arch Neurol. 2005;62:353–357. doi: 10.1001/archneur.62.3.353. [DOI] [PubMed] [Google Scholar]
- Hartman MG, Lu D, Kim ML, Kociba GJ, Shukri T, Buteau J, Wang X, Frankel WL, Guttridge D, Prentki M, Grey ST, Ron D, Hai T. Role for activating transcription factor 3 in stress-induced beta-cell apoptosis. Mol Cell Biol. 2004;24:5721–5732. doi: 10.1128/MCB.24.13.5721-5732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasbani DM, Perez FA, Palmiter RD, O'Malley KL. Dopamine depletion does not protect against acute 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity in vivo. J Neurosci. 2005;25:9428–9433. doi: 10.1523/JNEUROSCI.0130-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashida K, Kitao Y, Sudo H, Awa Y, Maeda S, Mori K, Takahashi R, Iinuma M, Hori O. ATF6alpha Promotes Astroglial Activation and Neuronal Survival in a Chronic Mouse Model of Parkinson's Disease. PLoS One. 2012;7:e47950. doi: 10.1371/journal.pone.0047950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Glimcher L. The daily job of night killers: alternative roles of the BCL-2 family in organelle physiology. Trends Cell Biol. 2008;18:38–44. doi: 10.1016/j.tcb.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Holtz WA, O'Malley KL. Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem. 2003;278:19367–19377. doi: 10.1074/jbc.M211821200. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland JL, Zambetti GP. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, Wang X, Ron D, Cavener DR, Wek RC. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol. 2004;24:1365–1377. doi: 10.1128/MCB.24.3.1365-1377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunakaran S, Saeed U, Mishra M, Valli RK, Joshi SD, Meka DP, Seth P, Ravindranath V. Selective activation of p38 mitogen-activated protein kinase in dopaminergic neurons of substantia nigra leads to nuclear translocation of p53 in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice. J Neurosci. 2008;28:12500–12509. doi: 10.1523/JNEUROSCI.4511-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim-Han JS, Antenor-Dorsey JA, O'Malley KL. The parkinsonian mimetic, MPP+, specifically impairs mitochondrial transport in dopamine axons. J Neurosci. 2011;31:7212–7221. doi: 10.1523/JNEUROSCI.0711-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kook YH, Ka M, Um M. Neuroprotective cytokines repress PUMA induction in the 1-methyl-4-phenylpyridinium (MPP(+)) model of Parkinson's disease. Biochem Biophys Res Commun. 2011;411:370–374. doi: 10.1016/j.bbrc.2011.06.151. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Dugan LL, O'Malley KL. Distinct mechanisms underlie neurotoxin-mediated cell death in cultured dopaminergic neurons. J Neurosci. 1999;19:1284–1293. doi: 10.1523/JNEUROSCI.19-04-01284.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007a;18:716–731. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007b;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- Mandavilli BS, Ali SF, Van Houten B. DNA damage in brain mitochondria caused by aging and MPTP treatment. Brain Res. 2000;885:45–52. doi: 10.1016/s0006-8993(00)02926-7. [DOI] [PubMed] [Google Scholar]
- Mandir AS, Simbulan-Rosenthal CM, Poitras MF, Lumpkin JR, Dawson VL, Smulson ME, Dawson TM. A novel in vivo post-translational modification of p53 by PARP-1 in MPTP-induced parkinsonism. J Neurochem. 2002;83:186–192. doi: 10.1046/j.1471-4159.2002.01144.x. [DOI] [PubMed] [Google Scholar]
- Markey SP, Johannessen JN, Chiueh CC, Burns RS, Herkenham MA. Intraneuronal generation of a pyridinium metabolite may cause drug-induced parkinsonism. Nature. 1984;311:464–467. doi: 10.1038/311464a0. [DOI] [PubMed] [Google Scholar]
- Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- O'Malley KL, Liu J, Lotharius J, Holtz W. Targeted expression of BCL-2 attenuates MPP+ but not 6-OHDA induced cell death in dopaminergic neurons. Neurobiol Dis. 2003;14:43–51. doi: 10.1016/s0969-9961(03)00013-5. [DOI] [PubMed] [Google Scholar]
- Reimertz C, Kogel D, Rami A, Chittenden T, Prehn JH. Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J Cell Biol. 2003;162:587–597. doi: 10.1083/jcb.200305149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J Neurosci. 2002;22:10690–10698. doi: 10.1523/JNEUROSCI.22-24-10690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sado M, Yamasaki Y, Iwanaga T, Onaka Y, Ibuki T, Nishihara S, Mizuguchi H, Momota H, Kishibuchi R, Hashimoto T, Wada D, Kitagawa H, Watanabe TK. Protective effect against Parkinson's disease-related insults through the activation of XBP1. Brain Res. 2009;1257:16–24. doi: 10.1016/j.brainres.2008.11.104. [DOI] [PubMed] [Google Scholar]
- Snyder H, Wolozin B. Pathological proteins in Parkinson's disease: focus on the proteasome. J Mol Neurosci. 2004;24:425–442. doi: 10.1385/JMN:24:3:425. [DOI] [PubMed] [Google Scholar]
- Staal RG, Sonsalla PK. Inhibition of brain vesicular monoamine transporter (VMAT2) enhances 1-methyl-4-phenylpyridinium neurotoxicity in vivo in rat striata. J Pharmacol Exp Ther. 2000;293:336–342. [PubMed] [Google Scholar]
- Sun X, Liu J, Crary JF, Malagelada C, Sulzer D, Greene LA, Levy OA. ATF4 Protects Against Neuronal Death in Cellular Parkinson's Disease Models by Maintaining Levels of Parkin. J Neurosci. 2013;33:2398–2407. doi: 10.1523/JNEUROSCI.2292-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer PA, Smith TS, Jung AB, Bennett JP., Jr. Dopamine neurons from transgenic mice with a knockout of the p53 gene resist MPTP neurotoxicity. Neurodegeneration. 1996;5:233–239. doi: 10.1006/neur.1996.0031. [DOI] [PubMed] [Google Scholar]
- Vila M, Jackson-Lewis V, Vukosavic S, Djaldetti R, Liberatore G, Offen D, Korsmeyer SJ, Przedborski S. Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Proc Natl Acad Sci U S A. 2001;98:2837–2842. doi: 10.1073/pnas.051633998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila M, Przedborski S. Genetic clues to the pathogenesis of Parkinson's disease. Nat Med. 2004;10(Suppl):S58–62. doi: 10.1038/nm1068. [DOI] [PubMed] [Google Scholar]
- Wang H, Shimoji M, Yu SW, Dawson TM, Dawson VL. Apoptosis inducing factor and PARP-mediated injury in the MPTP mouse model of Parkinson's disease. Ann N Y Acad Sci. 2003;991:132–139. doi: 10.1111/j.1749-6632.2003.tb07471.x. [DOI] [PubMed] [Google Scholar]
- Wu J, Kaufman RJ. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ. 2006;13:374–384. doi: 10.1038/sj.cdd.4401840. [DOI] [PubMed] [Google Scholar]
- Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. Protein folding in the endoplasmic reticulum and the unfolded protein response. Journal of Biological Chemistry. 2004;279:25935–25938. doi: 10.1074/jbc.R400008200. [DOI] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;66:S102–109. doi: 10.1212/01.wnl.0000192306.98198.ec. [DOI] [PubMed] [Google Scholar]