

Abstract
Intestinal epithelial cells (IEC) are constantly exposed to enteric microbes. Although IECs express TLRs that recognize bacterial products, activation of these TLRs is strictly controlled through poorly understood mechanisms, producing a state of hypo-responsiveness and preventing unwanted inflammation. The Single IgG IL-1 related receptor (Sigirr) is a negative regulator of TLRs that is expressed by IEC and recently shown to inhibit experimental colitis. However, the importance of Sigirr in IEC hyporesponsiveness and its distribution within the human colon is unknown. In this study, we investigated the role of Sigirr in regulating epithelial specific TLR responses and characterize its expression in colonic biopsies. Transformed and non-transformed human IEC were cultured as monolayers. Transient gene silencing and stable over-expression of Sigirr was performed to assess innate IEC responses. Sigirr expression in human colonic biopsies was examined by immunohistochemistry. Bacterial infection of IEC and exposure to flagellin transiently decreased Sigirr protein expression, concurrent with secretion of the neutrophil chemokine IL-8. Sigirr gene silencing augmented chemokine responses to bacterial flagellin, Pam3Cys and the cytokine IL-1β. Conversely, stable over-expression of Sigirr diminished NF-κB mediated IL-8 responses to TLR ligands. We also found that Sigirr expression increased as IECs differentiated in culture. This observation was confirmed in biopsy sections, where Sigirr expression within colonic crypts was prominent in IECs at the apex and diminished at the base. Our findings show that Sigirr broadly regulates innate responses in differentiated human IEC, and may therefore modulate epithelial involvement in infectious and inflammatory bowel diseases.
Keywords: Enterocytes, chemokines, flagellin, bacterial infection, differentiation
Introduction
The intestinal epithelial cells (IEC) lining the human colon undergo continuous transition from an undifferentiated and proliferative state at the base of colonic crypts to a mature and differentiated phenotype at the crypt apex (1). Throughout this transition, IEC are exposed to a multitude of commensal and pathogenic microbes, including their products such as LPS and flagellin. Under steady state conditions, IEC remain generally quiescent and tolerant of these products, as they have evolved critically important yet poorly defined mechanisms to remain hypo-responsive to these stimuli (2). In part, this tolerance is maintained by limiting the activation of innate receptors in IEC, which express a specific subset of TLR including TLR5 and TLR9 (required for detecting bacterial flagellin and CpG DNA respectively) (3, 4) as well as low levels of TLR2 and TLR4 (5). Numerous studies have shown that these innate receptors are involved in the host’s recognition of enteric pathogens such as Shigella, Salmonella typhimurium and Enteropathogenic Escherichia coli (2, 6, 7) triggering inflammation and playing a central role in host defense.
While innate receptors normally provide protection against bacterial and viral pathogens, dysregulated activation of TLRs in the gastrointestinal (GI) tract could impair host defenses as well as lead to chronic inflammation and cancer (8). It is therefore not surprising that IEC have evolved strategies to control innate sensing, including the expression of negative regulators of TLRs. Negative regulators inhibit TLR signaling by binding key adaptor proteins such as IRAK and TRAF6 thereby impeding downstream signaling to NF-κB (9). At present, only negative regulators of TLR4 such as Tollip have been identified within IEC (9), but considering that colonic IEC express relatively little TLR4 or its co-receptor MD-2 (10), the impact of such regulation on gut homeostasis is unclear. In contrast, negative regulators of the innate receptors expressed at higher levels by IECs such as TLR5 (11) have not been previously reported. However, it seems likely that such regulators do exist and they may play a significant role in controlling the innate and inflammatory responsiveness of the mucosal epithelium within the GI tract. Identifying these negative regulators and clarifying their roles in TLR responses and expression patterns in human IEC could significantly increase our understanding of gastrointestinal health and disease.
The single IgG IL-1 receptor (Sigirr) was first described as a negative regulator of IL-1β and TLR4 signaling (12). Expressed throughout the human GI tract with highest expression in the colon (13), Sigirr was recently shown to regulate inflammation as well as mucosal homeostasis in a mouse model of chemical colitis (14); however, the mechanisms involved as well as the cellular distribution of Sigirr within the intestine remain obscure. The objectives of this study were therefore to characterize the expression and elucidate the role of Sigirr in regulating the immune responsiveness of human IEC. We show that Sigirr inhibits responses to several bacterial derived TLR ligands as well as signaling due to the pro-inflammatory cytokine IL-1β. Moreover, the innate response to infection by the attaching and effacing bacterial pathogen Enteropathogenic Escherichia coli (EPEC), which causes diarrheal disease, was also modulated by Sigirr. In contrast, IEC responses to IFN gamma and PMA were unaffected by Sigirr. Interestingly, we found that Sigirr expression directly correlated with the maturation state of cultured IECs, a finding that was corroborated in the epithelium of human colonic biopsy sections. These findings designate Sigirr as a critical modulator of IEC responses in the human colon and highlight the need to address Sigirr function in the context of host responses to enteric pathogens and idiopathic intestinal diseases including inflammatory bowel disease (IBD).
Materials and Methods
Cell culture
Caco-2, HT-29 (ATCC) and TLR5 expressing HEK-293T cells (Invivogen) were grown in DMEM with 10% serum and antibiotics. NCM460 is a human non-transformed colonic mucosal IEC line (15) grown in M3 medium. Chinese hamster ovary (CHO) cells stably co-transfected with human TLR5 and NF-kB luciferase reporter were gifted by Dr. Stuart Turvey (UBC, Canada) and maintained as described (16). IEC were used for experiments 3–5 days after confluence.
Sigirr gene silencing and over-expression studies
Gene silencing was performed with Sigirr and control siRNA duplexes for transient transfection using the Hiperfect transfection reagent (Qiagen), as per manufacturer’s recommendations. IEC uptake of siRNA was confirmed by fluorochrome-tagged control siRNA. Two sets of Sigirr 27-mers siRNA (NAPS, UBC) duplexes were tested simultaneously with negative control siRNA. A single duplex that produced greater knockdown of target gene was selected for transfection. Monolayers of IEC (104 cells)/well were transfected in 24 well plates (WP) and assessed after 48–72 h. The pUNO-Sigirr mammalian expression vector and empty pUNO control vector (Invivogen) were used to transfect IECs and generate stable clones as per manufacturer’s specifications. Briefly, pUNO transformed E. coli was grown in LB and plated on blasticidin containing agar plates for selection. Bacteria were then grown in recommended culture medium and purified pUNO control and Sigirr containing plasmids were extracted for stable transfection of epithelial cells with the Effectene transfection reagent (Qiagen). Stably transfected IEC were selected and maintained in DMEM with serum and blasticidin (20–40 μg/ml). Sigirr knockdown and over-expression was analyzed by RT-PCR and western blot analysis.
Stimulation and infection of IEC
Recombinant purified and endotoxin free bacterial flagellin (Salmonella Typhimurium), LPS, Pam3Cys, and TLR5 neutralizing antibody (Invivogen) were prepared in DMSO according to manufacturer’s specifications. Recombinant IL-1β, IFN gamma and Phorbol myristate acetate-PMA (Sigma) were also prepared as recommended by manufacturer. To assess innate responses, IEC were stimulated for 6 h with the TLR ligands in DMEM and serum without antibiotics, and IL-8 concentration was quantified by ELISA. TLR ligands used in this study are as follows: bacterial flagellin FliC-10–20 ng/ml, LPS -20 ng/ml and Pam3Cys (TLR2 ligand)-20 ug/ml. IECs were also stimulated with IL-1β (5ng/ml) for 3h, IFN gamma (100 ng/ml) and PMA (50 ng/ml) for 6 h. The wild-type EPEC strain 2348/69 was used to infect IECs in this study (17). Caco-2 cells cultured in 12 WP were infected by EPEC in DMEM Nutrient Mixture F12 Ham (DMEM-F12) for 3 h. At this time point, the MOI ranged from 30–40. After infection, cells were washed twice with DMEM-F12 and gentamycin (1 μg/ml) added to prevent overgrowth of extracellular bacteria, cell culture supernatant was collected 24 h later for IL-8 ELISA as described previously (7).
IL-8 ELISA and Western analysis
IECs were cultured as confluent monolayers in 6 WP were washed twice in 2 ml DMEM and exposed to various TLR ligands in 1 ml DMEM. After 6 h, cell culture supernatant was collected for IL-8 quantification by ELISA kit (BD Biosciences) as per manufacturer’s specifications. For western analysis, IECs were washed twice with 2 ml of ice-cold Hank’s Balanced Salt Solution (Sigma). Cells were then lysed in 350–500 μl of lysis buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycero-phosphate, 1 mm Na3VO4, 1 μg/ml leupeptin and 1 mm PMSF) on ice for 5–10 min and then scraped into microcentrifuge tubes. The tubes were centrifuged at 13 000 g for 5 min to pellet debris, and the supernatant was transferred to another tube for Western blots. Caco-2 proteins (30–50 μg in cleared cell lysate) were resolved by 9–10% sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to 0.2 μm polyvinylidene fluoride (PVDF) membranes. Blots were then blocked for 1 h with 5% nonfat milk in TBST. Membranes were then incubated with primary antibody in TBST overnight at 4 °C and probed with the respective secondary antibody next day for 1 h at room temperature. Rabbit polyclonal antibody to Sigirr (Proscience Incorporated) was used to detect protein expression in IEC.
Conventional semi-quantitative RT-PCR analysis
IECs were scraped in RLT lysis buffer (Qiagen) for 30 seconds in an eppendorf tube, centrifuged for 5 min at 13,000g at room temperature. 1–2 mL of cleared supernatant was used for extraction of RNA using the RNeasy Protect Minikits (Qiagen) spin columns. Total RNA from Caco-2 cells grown in 12–24 WP was extracted by RNeasy Protect Minikits as per manufacturer’s recommendations. 1–2 micrograms of RNA were reverse transcribed into complementary DNA for 60 min at 37°C in 20-μL reaction buffer containing 10 mM Tris-HCl (pH 8.3), 50 mmol/L KCl, 5.0 mmol/L MgCl2, 100 μmol/L pooled deoxynucleotide triphosphates U of Moloney murine leukemia virus reverse transcriptase (RT), 4-μg random hexamer primers and 20 U of ribonuclease inhibitor. Random hexamers were used to provide a “one-step” RT reaction that yielded complementary DNA for amplification by PCR. A semi-quantitative conventional PCR method of amplification was used because the target messenger RNA was expressed at low copy numbers. Aliquots of reverse-transcribed complementary DNA were added to PCR reaction buffer to obtain a reaction mixture containing 20 mmol/L Tris-HCl (pH 8.3), 50 mmol/L KCl, 1.5 mmol/L MgCl2, 100 μmol/L pooled deoxynucleotide triphosphates, 1–2 U of Taq DNA polymerase, and 0.25 μmol/L each of GAPDH and gene specific oligonucleotide primers (NAPS unit, UBC). PCR conditions were optimized for different genes of interest as reported previously (7). Primers in this study are listed in Table I.
Human TLR5-NF-kB luciferase assay
CHO cells were stably transfected with human TLR5 cDNA (cloned into the pEF6 V5/His TOPO vector (Invitrogen) and Elam-Luc plasmids (Promega) as reported previously (16). CHO cells expressing TLR5 and NF-B luciferase reporter were stably transfected with control and Sigirr expressing plasmids as described in materials and methods. CHO cells stably expressing Sigirr were stimulated with bacterial flagellin for 6 hr before assaying for luciferase activity (Promega).
Intestinal biopsy sampling and immunofluorescent staining
Two colonic biopsies were obtained from each patient at a single pass using standard crocodile biopsy forceps. Informed consent was obtained from all patients undergoing diagnostic colonoscopy for lower gastrointestinal symptoms such as abdominal pain and altered bowel movements. Study approval was obtained from the UBC Clinical Ethics Review Board. Colonic biopsy samples were collected in 4% formaldehyde solution and rapidly processed for Immunohistochemical analysis as described below.
Immunofluorescence staining of colon tissues
Colonic tissue samples were rapidly processed for immunofluorescence and staining was performed as described previously (19). In brief, tissues were rinsed in ice-cold PBS, embedded in optimal cutting temperature compound (OCT, Sakura, Finetech), frozen with isopentane (Sigma) and liquid N2, and stored at −70°C. Serial sections were cut at a thickness of 6–8 μm and fixed in ice-cold acetone for 10 min. Tissue sections were directly blocked with 1% BSA followed by the addition of mouse monoclonal and goat polyclonal antibodies to Sigirr (R&D), mouse monoclonal antibodies to Claudin-3 (Invitrogen) and rabbit polyclonal antibodies to Indian Hedgehog (Santa Cruz Biotechnology), all at 1:200 dilution. These were followed by secondary Alexa 568/488-conjugated goat anti-rabbit IgG antibodies (Molecular Probes) and Prolong® Gold antifade reagent containing DAPI (Invitrogen). Cells and tissues were visualized at 350 and 594 nm using a Leica DM 4000B microscope (Leica Microsystems).
Data presentation and statistical analysis
All the results are expressed as the mean value ± standard error of the mean and analyzed with Graphpad prism software (La Jolla, USA). Results are from one representative experiment out of at least three. Statistical analysis was performed using the one-way ANOVA test and student t-test with p < 0.05 considered significant. Quantity-one software (Chemidoc XRS-Bio-Rad, USA) was used for densitometry.
Results
Bacterial flagellin induces Sigirr expression in human IEC
While Sigirr has been shown to regulate the inflammatory response to LPS, it is unclear if it regulates the responses to other TLR ligands such as flagellin. We exposed human NCM (non-transformed colonic mucosal) IEC to flagellin (FliC), which transiently decreased Sigirr protein levels by 50% after 12h. However, prolonged exposure to flagellin (24–48h) significantly increased Sigirr protein levels by 50–100% over baseline levels in unstimulated cells (Figure 1A). To assess if enteric bacterial infection also alters Sigirr expression, we infected Caco-2 colonic IEC with the diarrheal pathogen EPEC. We recently showed that the inflammatory response to EPEC is triggered by flagellin activation of TLR5 in IEC (7). Through RT-PCR analysis, we observed transient down-regulation of Sigirr message by approximately 50% after 6h, which subsequently increased by 50–60% over baseline 24–48 h after EPEC infection (Figure 1B). These alterations in Sigirr expression were found to inversely correlate with the induction of IL-8 mRNA in Caco-2 IEC infected with EPEC (Figure 1C). These results indicate that Sigirr expression is temporally regulated in response to flagellin and bacterial stimulation of IEC, and this regulation occurs within the same time point as the IL-8 response.
Figure 1.
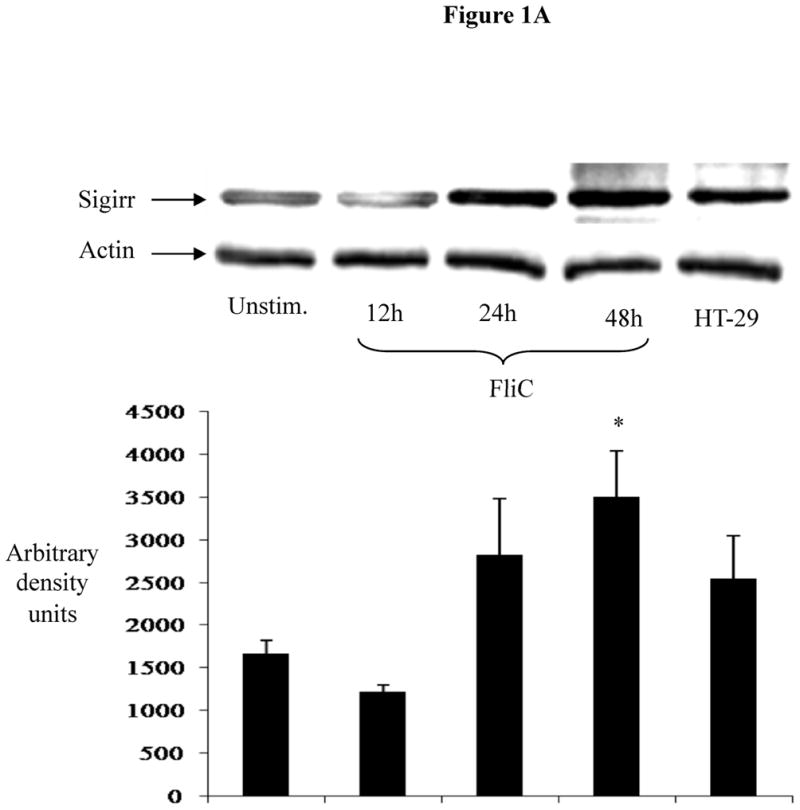
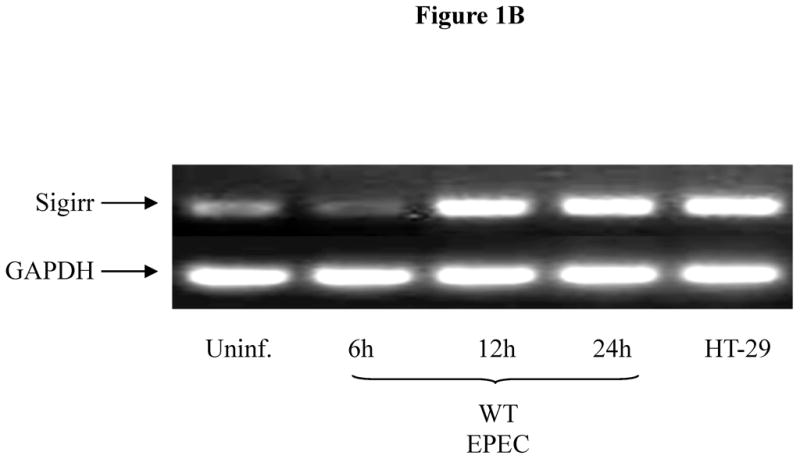

Figure 1A: Exposure to bacterial flagellin induces Sigirr protein expression in non-transformed human colonic (NCM) IEC. NCM were grown in M3 growth media and exposed to 10 ng/ml purified flagellin (FliC). Cell lysates were prepared as described in materials and methods. 30–50μg of cleared cell lysates were subjected to SDS-PAGE and probed with rabbit polyclonal antibody to Sigirr. Blots were developed in ECL and bands analyzed by densitometry. Result representative of three separate experiments.
Figure 1B: EPEC infection increases Sigirr message levels in Caco-2 IEC. Cells were grown to confluence in 12 WP and infected with WT EPEC for 3 h. After infection, cells were washed twice and fresh media added with gentamycin to prevent bacterial growth. Total RNA extracted at different time-points was subjected to semi-quantitative RT-PCR to measure gene expression as described in Materials and Methods. GAPDH was included as internal control. Blot representative of three independent experiments.
Figure 1C: EPEC infection increases IL-8 expression in Caco-2 IEC. Cells were grown to confluence in 12 WP and infected with WT EPEC for 3 h as above. Afterwards, cells were washed twice and fresh media added with gentamycin. Total RNA was extracted and subjected to semi-quantitative IL-8 RT-PCR. GAPDH was included as internal control. Blot representative of two separate experiments.
Sigirr gene silencing augments responses to bacterial flagellin and heat killed bacteria
The transient downregulation of Sigirr gene expression and its correlation with IL-8 release suggests a possible intrinsic mechanism in IECs, in which a temporary reduction of Sigirr protein facilitates TLR activation leading to chemokine responses. We therefore assessed if Sigirr gene silencing would lead to augmentation of TLR responses in IEC. Transient gene silencing reduced Sigirr protein levels by 50% in Caco-2 IEC (Figure 2A). Following exposure to FliC at 48–72 hours post transfection, these cells produced a two-fold increase in IL-8 secretion compared to cells transfected with non-silencing control siRNA (NSC) (1280.25 ± 68.6 vs. 615 ± 152.9 pg/ml, p < 0.05) (Figure 2B). Time-course analysis indicated that this effect was transient, but confirmed that the effect of Sigirr knockdown was maximal at 48–72 hours post-transfection (not shown), so we continued the remainder of the gene-silencing studies within these timepoints. Thus, Sigirr deficiency also exaggerated the IL-8 response to pro-inflammatory IL-1β used as control (1492.6 ± 223.3 vs. 721.6 ± 107.4 pg/ml, p < 0.05). In contrast, the IL-8 response to IFNg stimulation, which involves the JAK-STAT pathway instead of IRAK and TRAF6, was unaffected by Sigirr knockdown (Figure 2B). We also exposed these cells to heat-killed (HK) EPEC to examine whether the observed effect would occur in response to multiple TLR ligands from a bacterial pathogen. Similar to the results obtained from purified flagellin, HK-EPEC-treated Sigirr-deficient Caco-2 cells released three-fold more IL-8 compared to cells transfected with NSC, suggesting Sigirr modulates the responses to multiple TLR ligands? (Figure 2C).
Figure 2.



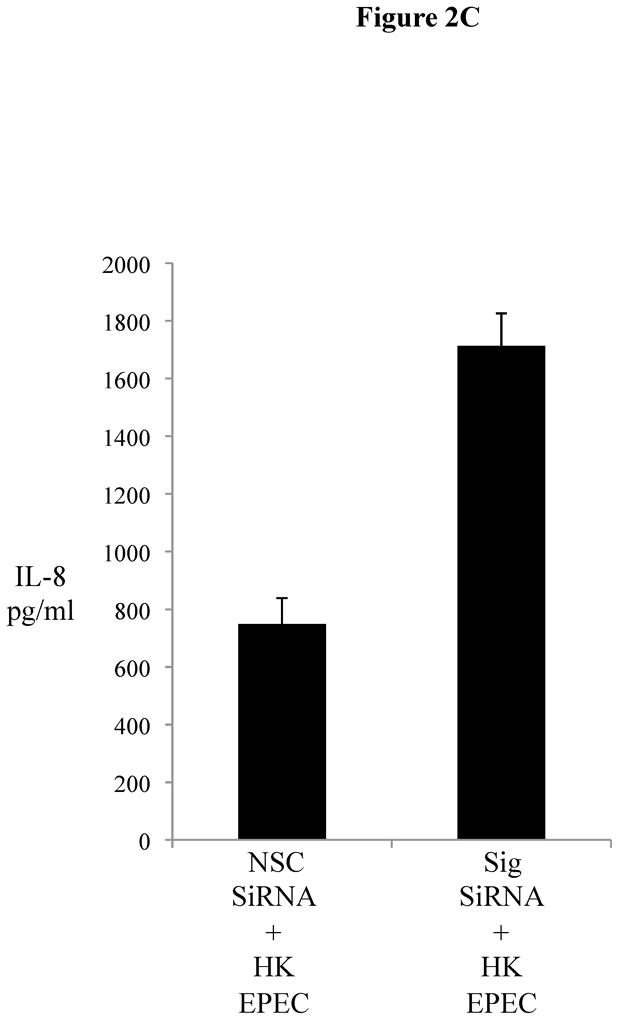
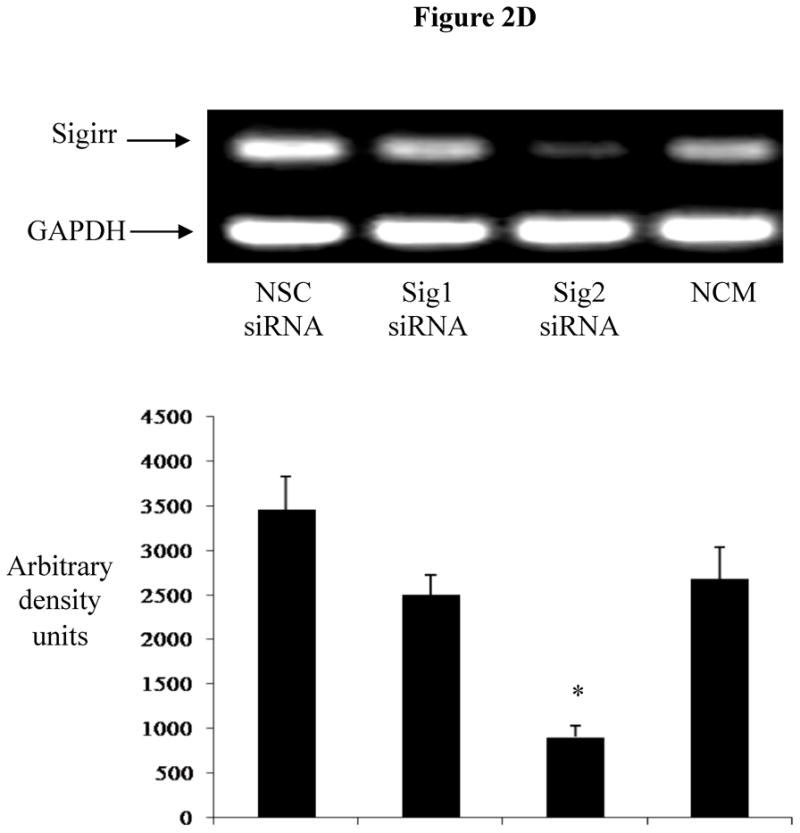



Figure 2A: Transient gene silencing reduces Sigirr protein in Caco-2 cells. IEC (2 X 105/well) were seeded in 24 WP. After 48h, cells were transfected with Sigirr and non-silencing control (NSC) siRNA in Hiperfect transfection reagent. Sigirr protein was analyzed by SDS-PAGE as described in materials and methods and developed in ECL. Bands were quantified by densitometry with BioRad quantity one software program.
Figure 2B: Sigirr gene silencing augments flagellin induced IL-8 chemokine secretion from Caco-2 cells. Sigirr and NSC siRNA transfected Caco-2 cells were exposed to 10 ng/ml purified flagellin (FliC) and Interferon gamma (IFNg) (100 ng/ml) in DMEM with serum and antibiotics. IL-1β (5 ng/ml) was included as a positive control. Cell culture supernatant was collected after 6h and IL-8 quantified by ELISA. Error bars and * p<0.05 vs. NSC siRNA + FliC.
Figure 2C: Heat-killed (HK)-EPEC-induced IL-8 production is enhanced upon Sigirr gene silencing in Caco2 cells. . Sigirr and NSC siRNA transfected Caco-2 cells were exposed to HK-EPEC in DMEM with serum and antibiotics. Cell culture supernatant was collected after Xhr and IL-8 quantified by ELISA. Error bars = ?.
Figure 2C 2D: Transient gene Silencing decreases Sigirr gene expression in HT-29 cells. IEC (2 X 105/well) were seeded in 24 WP and transfected after 48 h with Sigirr and non-silencing control (NSC) siRNA. Total RNA was extracted and Sigirr gene expression measured by semi-quantitative RT-PCR. GAPDH was included as internal control. Blot is representative of three experiments and bands were analyzed by densitometry
Figure 2D 2E: Sigirr gene silencing augments TLR5 mediated IL-8 secretion. siRNA transfected HT-29 cells were exposed to FliC (10 ng/ml), in presence and absence of a specific TLR5 neutralizing antibody. Cell culture supernatant was collected after 6 h and IL-8 quantified by ELISA. IL-1β (5 ng/ml) was included as positive control. Error bars and * p<0.05 vs. NSC siRNA + FliC and Sig siRNA + FliC.
Figure 2E 2F: Sigirr gene silencing augments diverse TLR responses in IEC. siRNA transfected HT-29 cells were exposed to FliC (10 ng/ml), LPS (20 ng/ml), Pam3Cys (Pam-20 ug/ml) and Phorbol myristate acetate (PMA-50 ng/ml) for 6 hours in DMEM with serum and antibiotics. Cell culture supernatant was collected and IL-8 quantified by ELISA. Error bars and * p<0.05 vs. NSC siRNA + FliC, NSC siRNA + LPS and NSC siRNA + Pam.
Figure 2F 2G: EPEC infection up-regulates chemokine response in Sigirr deficient HT-29 cells. IEC were grown to confluence and infected with wild type EPEC for 3 h. After infection, cells were washed twice and fresh media added with gentamycin treatment. Cell culture supernatant was collected after 12 h for IL-8 ELISA. Error bars and * p<0.05 vs. NSC siRNA + EPEC.
Sigirr deficiency enhances IL-8 responses to diverse TLR ligands
To ensure the actions of Sigirr were not cell line specific, we silenced Sigirr in the enterocytic HT-29 IEC cell line. After 70% knockdown of Sigirr mRNA (Figure 2C 2D), we noted that IL-8 release doubled in response to FliC (971 ± 143.8 vs. 423.67 ± 85.5 pg/ml, p < 0.05) as shown in Figure 2D(2E). To confirm TLR5 involvement, we examined the FliC response in the presence of a neutralizing antibody to TLR5 which abolished IL-8 release in Sigirr deficient HT-29 IEC (971 ± 143.8 vs. 153 ± 52 pg/ml, p < 0.05) Figure 2D (2E), but did not affect IL-1β responses.
To address whether the inhibition of innate signaling by Sigirr was limited to TLR5, we also tested other TLR ligands in the LPS responsive HT-29 cells (20). In these IEC (Figure 2E 2F), we saw more than two-fold increase in their FliC response (1108 ± 149.9 vs. 392.3 ± 72.5 pg/ml, p < 0.05), a two-fold increase in their LPS response (870.6 ± 66.9 vs. 435.3 ± 57.7 pg/ml, p < 0.05), and a four-fold increase in response to Pam3Cys (833.3 ± 123.5 vs. 154.6 ± 20.4 pg/ml, p < 0.05). We also tested TLR9 responses and found similar elevations in IL-8 release (data not shown). Notably, while Sigirr gene silencing amplified inflammatory responses to TLR ligands, it had no effect on IL-8 responses to PMA (Figure 2E 2F). To assay the potential impact of Sigirr in regulating the IL-8 response to an enteric bacterial pathogen, we infected HT-29 cells with EPEC. We noted that EPEC induced a significantly higher IL-8 response in Sigirr deficient cells (609.79 ± 163 vs. 281.5 ± 67.3 pg/ml, p < 0.05) (Figure 2F 2G), demonstrating that loss of Sigirr enhances the chemokine response to this bacterial pathogen.
Immunofluorescence analysis of Sigirr expression in human IEC
While IEC express Sigirr its localization within these cells has not been shown previously. In order to visualize Sigirr, we first assessed Sigirr staining in transfected HEK293T cells. While no signal was detected in cells transfected with control vector (Figure 3A), immunostaining of cells stably overexpressing Sigirr revealed a homogenous green signal corresponding to Sigirr protein, present in the cytoplasm and the cell membrane (Figure 3B). Specificity was confirmed by incubation with the Sigirr peptide, which abolished Sigirr immunoreactivity (Figure 3C vs. Figure 3D). Next, we analyzed the pattern of native Sigirr expression by immunocytochemistry in Caco-2 IECs as well as Caco-2 and HT-29 cell lines stably over-expressing Sigirr. Interestingly, unlike in HEK293T cells, Sigirr expression in native spontaneously differentiating Caco-2 cells was found to be patchy and clustered in cells growing in smaller colonies (Figure 3E and 3F), with staining localized to the cell membrane as well as in a punctate pattern within the cytoplasm. We observed increased Sigirr immunoreactivity in Caco-2 cells seeded at higher cell densities (Figure 3F vs. 3E), in Sigirr over-expressing Caco-2 (Figure 3H vs. 3G) and in HT-29 (Figure 3J vs. 3I) cells relative to their controls.
Figure 3.


Immunofluorescent staining for Sigirr expression in HEK293T cells and human intestinal epithelial cells. HEK293T cells grown on coverslips were stably transfected with pUNO Sigirr over-expression vector (b) and the control vector (b). After formalin fixation, Sigirr expression was analyzed using a rabbit polyclonal antibody and Alexafluor 468 (green) conjugated secondary antibody. Sigirr antibody was pre-incubated with full length Sigirr peptide and used for staining Sigirr over-expressing cells (d) vs. Sigirr antibody alone (c), nuclei (blue) stained with ProlongGold dapi. Native Caco-2 cells grown on coverslips were assessed for Sigirr expression with a goat polyclonal primary Sigirr antibody and Alexa fluor 568 conjugated (34) and dapi nuclear stain (blue). Caco-2 cells seeded at lower density (e) and higher density (f). Immunostaining in Caco-2 cells over-expressing Sigirr (h) and control (g). HT-29 cells over-expressing Sigirr (j) and control vector (i). Magnification 20X.
Over-expression of Sigirr dampens NF-kB mediated TLR responses
We next assessed the impact of increased Sigirr levels on TLR and cytokine induced responses, using our stably Sigirr overexpressing Caco-2 and HT-29 IEC (Figure 4A), as well as Sigirr over-expressing CHO cells (transfected with NF-κB luciferase reporter) and TLR5 expressing HEK293T cells. NF-κB luciferase activity in response to FliC decreased by approximately 25% in CHO cells over-expressing Sigirr (Figure 4B), while in Caco-2 IEC, IL-8 release in response to FliC significantly declined by 30% (Figure 4C-left panel) compared to control (pUNO) transfected cells (870.33 ± 150 vs. 581.6 ± 103 pg/ml, p < 0.05). These data were reproduced in HEK293T cells stably co-expressing TLR5 and Sigirr (571.6 ± 109.4 vs. 295.8 ± 90.3pg/ml, p < 0.05) (Figure 4C-right panel). Over-expression of Sigirr in HT-29 IEC also significantly reduced IL-8 responses to FliC, LPS and Pam3Cys (Figure 4D) relative to control cells. Moreover, consistent with our results observed with Sigirr –deficient Caco2 cells, over-expressing Sigirr in Caco-2 cells also produced less IL-8 when exposed to HK-EPEC compared to pUNO-transfected cells (Figure 4E). These results confirm that increasing Sigirr expression attenuates TLR signaling and depresses IEC responses to bacterial derived products.
Figure 4.





(A, B, C, D and E): Sigirr over-expression depresses NF-kB mediated diverse TLR responses. (a) HT-29 and Caco-2 IEC were stably transfected with pUNO Sigirr mammalian expression vector or empty pUNO vector with Effectene transfection reagent. Clones over-expressing Sigirr were maintained in blastocidin and subsequently assessed for Sigirr by RT-PCR and immunoblotting. Blots are representative of two experiments. (b) CHO cells containing a NF-kB luciferase reporter were stably transfected with Sigirr over-expressing pUNO vector and control vector. Clones were maintained in blastocidin and subsequently tested for flagellin (FliC) responses. Whole cells lysate was collected after 6 h and relative luciferase activity measured by luminometry. * p<0.05 vs. pUNO + FliC and unstimulated cells. (c) Over-expression of Sigirr blunts FliC induced chemokine response. HT-29 IEC (left panel) and HEK293T cells (right panel) overexpressing Sigirr were exposed to 10 ng/ml of flagellin (FliC) for 6 h and IL-8 quantified by ELISA in cell culture supernatant. Error bars and * p<0.05 vs. pUNO + FliC. (d) Over-expression of Sigirr blunts IL-8 responses to diverse TLRs and inflammatory cytokines. Stably transfected HT-29 cells over-expressing Sigirr were exposed to 10 ng/ml Flagellin (FliC), 20 ng/ml of LPS and 20ug/ml of Pam3Cys (Pam) for 6 hours in DMEM, and IL-8 secretion quantified by ELISA. Error bars and * p<0.05 vs. pUNO + FliC, pUNO + LPS and pUNO + Pam. (e) HK-EPEC induced IL-8 production is abrogated upon Sigirr overexpression in Caco-2 cells. Stably transfected Caco-2 cells over-expressing Sigirr were treated with HK-EPEC for Xhrs in DMEM, and IL-8 secretion was quantified by ELISA as above. Error bars = ?
Sigirr expression depends on the maturation state of IEC
As Caco-2 cells grow in culture they form colonies where cell to cell contact induces their differentiation from an immature state towards a mature enterocytic phenotype (21). As these cells form monolayers, they acquire a cylindrical polarized morphology with microvilli on their apical surface, tight junction between adjacent cells and express intestinal hydrolase enzymes such as alkaline phosphatase, sucrose isomaltase and dipeptidyldipeptidase (22, 23). In this context, the patchy staining for Sigirr observed in the midst of cultured Caco-2 cell colonies in Figures 3E–G suggested that Sigirr expression was possibly related to the maturation state of IEC. We examined this possibility using sodium butyrate, an agent known to induce IEC differentiation (24). Exposure to sodium butyrate progressively increased Sigirr protein levels in Caco-2 cells after 24 h and 72 h compared to pre-confluent untreated IEC (Figure 5A). Since PI-3-Kinase-pathway is known to play a role differentiation of Caco-2 cells(*), we tested whether inhibition of this pathway after sodium-butyrate treatment would abrogate Sigirr expression. Indeed, the PI-3K inhibition with LY294002 confirmed that interference with sodium butyrate- mediated Caco-2 cell maturation resulted in reduced Sigirr protein production (Figure 5A). Sigirr expression also increased significantly in untreated spontaneously differentiating Caco-2 cells. Therefore, we tested whether the seeding density of Caco-2 cells affected Sigirr expression as cells seeded at higher density reach maturation sooner than cells seeded at lower density. As seen in Figure 5B, cells seeded at 3 × 106 cells/well have higher Sigirr expression compared to cells seeded at 1 × 105 cells/well. These results indicate that the level of Sigirr expression in IEC is linked to their differentiation state. To verify this, we investigated dipeptidyl dipeptidase expression (DPP), a brush border associated marker of differentiation in colonic IEC (25). Immunocytochemical analysis revealed that DPP was patchy and confined to small clusters of cells (Figure 5C), remarkably similar to Sigirr staining pattern seen in Figure 3E and 3F. Further, Sigirr staining co-localized with DPP expression (Figure 5D), suggesting that Sigirr expression by Caco-2 cells coincides with expression of this brush border enzyme.
Figure 5.

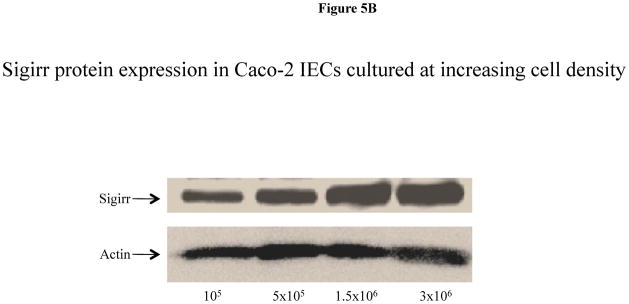
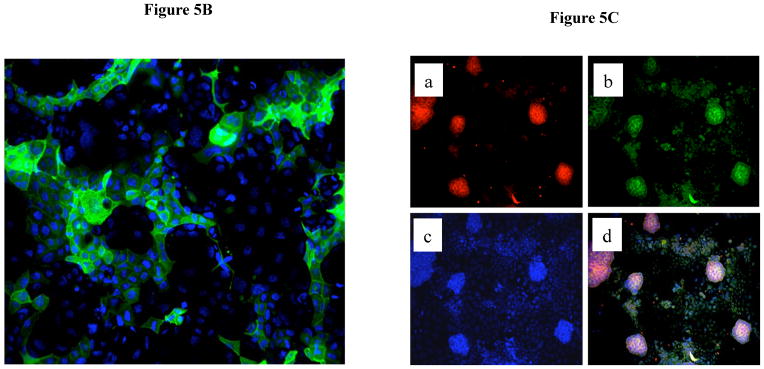
Sigirr expression in Caco-2 cells is dependent on the differentiation state. (A) Caco-2 monolayers were exposed to sodium butyrate (2mM) for 24–72 h in DMEM supplemented with serum and antibiotics. 30–50μg of total protein from pre-confluent, sodium butyrate treated and spontaneously differentiating post-confluent cells were probed with Sigirr rabbit polyclonal antibody in western blots. Result representative of two experiments. (B) Caco-2 cells were seeded at increasing densities as indicated, and xug of total protein from post-confluent cells were probed with Sigirr antibody in western blots as above. (C) Caco-2 cells express the differentiation marker Dipeptidyl dipeptidase (DPP) in IEC monolayers. Formalin fixed Caco-2 monolayers grown on coverslips were incubated with mouse monoclonal DPP antibody conjugated with Alexa fluor-488 (green). Cell nuclei stained with dapi. Magnification 40X. Result representative of three independent experiments. (D) Sigirr co-localizes with the differentiation marker DPP in Caco-2 cells. Cells grown higher density (5 × 105/well) were fixed in 4% formalin and incubated simultaneously with goat polyclonal Sigirr antibody (a-red) with mouse monoclonal DPP antibody conjugated to Alexa fluor 488 (b-green) and nuclear dapi stain (c-blue). Images taken with Zeiss fluorescent microscope were merged to show co-localization (d). Result representative of two independent experiments. Magnification 20X.
Sigirr is expressed by differentiated epithelial cells in the human colon
To examine the in vivo relevance of our findings we proceeded to stain for Sigirr expression in human colonic biopsy sections. By immunofluorescence, we detected maximal Sigirr expression on the apical surface of colonic crypts (Figure 6A), with the signal diminishing in cells at their base. Additional sporadic staining was also observed in cells within the colonic lamina propria which likely emanates from immune cells resident in lamina propria. To address whether Sigirr expression was mostly localized to IECs, we stained for the tight junction protein claudin-3 (26). As expected, claudin-3 staining was maximal on the lateral intercellular junctions of the IEC, while Sigirr staining was most intense on the apical surface of these IEC (Figure 6B). To confirm that Sigirr was predominantly expressed by differentiated epithelial cells, we further stained for Indian hedgehog (Ihh), a marker of differentiated apical IECs in both small intestine and colonic epithelial cells (27–29). We found abundant Ihh expression in epithelial cells that were also expressing Sigirr (Figure 6C). These findings are consistent with our data in cultured IEC, and confirm that within the human colon, Sigirr is predominantly expressed by mature and differentiated IEC present on the luminal surface of the crypts.
Figure 6.

Immunohistochemical analysis of Sigirr expression in human colonic tissues. (A) Colonic tissues obtained at biopsy were fixed in formalin and prepared as described in materials and methods. 8–10 uM sections were mounted on slides and incubated with either control antibody (a), or goat polyclonal antibody to Sigirr (b and c). Immunoreactivity to epithelial Sigirr (34) is prominent on apical epithelial surface (arrows). (B) Epithelial Sigirr expression co-localizes with Claudin-3 tight junction protein in human colon. Colonic tissues from biopsy were prepared as described. 8–10 microns sections were mounted on slide and double immunostaining performed with goat polyclonal Sigirr and mouse monoclonal Claudin-3 antibody. Sigirr immunoreactivity (a-red) co-localized with Claudin-3 (b-green) in epithelial cells on crypt surface (d-arrow). Nuclei stained with dapi (blue) stain. (C) Indian Hedgehog (Ihh) expression by IEC coincides with Sigirr expression in human colon. Colonic tissues from biopsy were prepared as described. Double immunostaining was performed with a goat polyclonal Sigirr antibody and a rabbit polyclonal antibody to Ihh. Sigirr immunoreactivity (a-red) co-localized with Ihh (b-green) in epithelial cells on crypt surface (d-arrow). Nuclei stained with dapi (blue) stain. Magnification 20X.
Discussion
This study is aimed at elucidating the inhibitory functions of Sigirr in the human colonic epithelium. Our results indicate that Sigirr plays a major role in maintaining the hypo-responsiveness of IEC to a variety of bacterial and pro-inflammatory stimuli. We show for the first time that bacterial flagellin as well as direct infection of IEC with the enteric bacterial pathogen EPEC causes a transient decrease followed by a substantial and prolonged increase in Sigirr expression that inversely correlates with the expression and release of the neutrophil chemokine IL-8. Sigirr gene silencing in IEC caused a significant and selective enhancement of IL-8 secretion in response to TLR ligands and augmented responses to the pro-inflammatory cytokine IL-1β. Conversely, stable over-expression of Sigirr diminished the same TLR and cytokine responses. While exploring different IEC densities, we determined that Sigirr expression increases with the differentiation/maturation state of these cells in culture. Correspondingly, Sigirr expression was found to be minimal in cells at the base of human colonic crypts and maximal in the differentiated epithelial cells at the crypt apex, providing a basis for the previously identified hypo-responsiveness of mature IECs to bacterial products (1, 20).
The recent demonstration of exaggerated colitis and tumorigenesis in Sigirr deficient mice designated this receptor as potentially important in gastrointestinal inflammation; however the underlying mechanism remained unclear. Although Sigirr is known to suppress TLR4 and IL-1β signaling, whether exaggerated signaling through these receptors led to the resulting pathology was unknown. In part, this is because the potential role of Sigirr in regulating other innate receptors expressed at higher levels within the gut epithelium, such as TLR5, has not been examined. It is now evident that the inflammatory responses mediated through several innate and cytokine receptors are regulated by Sigirr within IEC. Based on the broad actions of Sigirr, we speculate that it may affect responses to other members of the IL-1 superfamily such as IL-26 (30) and IL-22 (31) and were recently described to act on IEC to mediate inflammation and host defense respectively.
While a tumor derived intestinal epithelial cell line (HT-29) was previously shown to express Sigirr mRNA (12), its protein expression and function had not been examined in non-transformed human IEC or in human colonic tissues. Moreover, although IECs are known to differentiate while migrating apically in colonic crypts, becoming less responsive to bacterial products as they mature (20), the impact of differentiation on the expression of Sigirr by IEC was previously unknown. Interestingly, we found that the differentiation agent sodium butyrate (32) induced expression of Sigirr in IEC. We noted a similar increase in Sigirr expression in spontaneously differentiating Caco-2 monolayers, indicating that the maturation of IEC promoted Sigirr expression. Immunohistochemical analysis of human colonic tissues confirmed that Sigirr protein levels were maximal in differentiated epithelial cells on the surface of the crypts.
These data are consistent with earlier studies in which IEC differentiation down-regulated IL-1β induced chemokine secretion (1). Moreover, previous immunohistochemical analysis of TLR4 and TLR5 in human colonic sections also demonstrated that these TLRs were preferentially expressed by mature IEC, therefore requiring the increased expression of negative regulators like Sigirr in these type of cells (33, 34). Taken together, these studies support our findings that Sigirr expression within IEC is related to their differentiation state. We propose that in differentiated IEC, innate responses are controlled to prevent unwanted inflammatory responses against commensal microbes, whereas in less mature cells, limited Sigirr expression leads to heightened inflammatory responses, potentially contributing to the more sterile environment found at the base of crypts.
In spite of the regulatory role of Sigirr, limited activation of specific epithelial TLRs may be required for the maintenance of colonic mucosal integrity (19, 35). In fact, TLR2, TLR4 and TLR9 signaling have all been shown to contribute to normal IEC function (4, 8, 35), at least in mice, and this TLR stimulation may in fact be regulated by Sigirr, as epithelial homeostasis appears dysregulated in the absence of Sigirr (14). While TLR5 is not known to play a role in intestinal homeostasis, IEC expression of TLR5 have been shown to play a critical role in recognizing and responding to flagellated enteric pathogens for protection of the host (11, 36). Thus Sigirr’s regulation of TLR5 and other innate receptors may play a role in enteric host defense. The observed transient down-regulation of Sigirr in response to flagellin and EPEC followed by its prolonged up-regulation likely reflects a means to transiently recruit neutrophils to clear enteric bacterial pathogens, while attenuating collateral tissue damage and suppressing colonic inflammation after infection.
Based on our findings, Sigirr dysregulation could dramatically impact IEC responsiveness, potentially contributing to the development of chronic inflammation, as seen in IBD, not only in response to microbes, but possibly to other endogenous cytokines such as IL-22 required for host defense against enteric pathogens (31). At present it is unclear if single nucleotide polymorphisms within the Sigirr gene would alter its function and/or distribution, but such assessment in human subjects should be a priority. These findings also indicate that modulation of Sigirr expression may prove to be of therapeutic benefit. In steady state conditions, IEC are exposed to minimal amounts of flagellin and butyrate derived as a fermentation product from commensal bacteria in the gut. Both factors may facilitate the expression of Sigirr and perhaps other negative regulators and thereby offer attractive targets in treatment of inflammatory disorders of the GI tract.
Table 1.
Oligonucleotide primers used in this study
| Target Gene | Forward | Reverse |
|---|---|---|
| GAPDH | 5′–ATGACCTTGCCCACAGCC–3′ | 5′–CCCATCACCATCTTCCAG–3′ (7) |
| Sigirr | 5′–GCTGACTGCAAGGACAGAGA–3′ | 5′–ACTCGTGGAGGCTGTAGTGG–3′ (13) |
| 1L-8 | 5′-TCTGCAGCTCTGTGTGAAGGTGCAGTT-3′ | 5′-TTCCTTTGACCCACGTCTCCCAA-3′ (18) |
References
- 1.Bocker U, Schottelius A, Watson JM, Holt L, Licato LL, Brenner DA, Sartor RB, Jobin C. Cellular differentiation causes a selective down-regulation of interleukin (IL)-1beta-mediated NF-kappaB activation and IL-8 gene expression in intestinal epithelial cells. J Biol Chem. 2000;275:12207–12213. doi: 10.1074/jbc.275.16.12207. [DOI] [PubMed] [Google Scholar]
- 2.Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat Rev Immunol. 2008;8:411–420. doi: 10.1038/nri2316. [DOI] [PubMed] [Google Scholar]
- 3.Abreu MT, Fukata M, Arditi M. TLR signaling in the gut in health and disease. J Immunol. 2005;174:4453–4460. doi: 10.4049/jimmunol.174.8.4453. [DOI] [PubMed] [Google Scholar]
- 4.Lee J, Mo JH, Katakura K, Alkalay I, Rucker AN, Liu YT, Lee HK, Shen C, Cojocaru G, Shenouda S, Kagnoff M, Eckmann L, Ben-Neriah Y, Raz E. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat Cell Biol. 2006;8:1327–1336. doi: 10.1038/ncb1500. [DOI] [PubMed] [Google Scholar]
- 5.Abreu MT, Vora P, Faure E, Thomas LS, Arnold ET, Arditi M. Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J Immunol. 2001;167:1609–1616. doi: 10.4049/jimmunol.167.3.1609. [DOI] [PubMed] [Google Scholar]
- 6.Fritz JH, Ferrero RL, Philpott DJ, Girardin SE. Nod-like proteins in immunity, inflammation and disease. Nat Immunol. 2006;7:1250–1257. doi: 10.1038/ni1412. [DOI] [PubMed] [Google Scholar]
- 7.Khan MA, Bouzari S, Ma C, Rosenberger CM, Bergstrom KS, Gibson DL, Steiner TS, Vallance BA. Flagellin-dependent and -independent inflammatory responses following infection by enteropathogenic Escherichia coli and Citrobacter rodentium. Infect Immun. 2008;76:1410–1422. doi: 10.1128/IAI.01141-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fukata M, Abreu MT. TLR4 signalling in the intestine in health and disease. Biochem Soc Trans. 2007;35:1473–1478. doi: 10.1042/BST0351473. [DOI] [PubMed] [Google Scholar]
- 9.Shibolet O, Podolsky DK. TLRs in the Gut. IV. Negative regulation of Toll-like receptors and intestinal homeostasis: addition by subtraction. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1469–1473. doi: 10.1152/ajpgi.00531.2006. [DOI] [PubMed] [Google Scholar]
- 10.Lenoir C, Sapin C, Broquet AH, Jouniaux AM, Bardin S, Gasnereau I, Thomas G, Seksik P, Trugnan G, Masliah J, Bachelet M. MD-2 controls bacterial lipopolysaccharide hyporesponsiveness in human intestinal epithelial cells. Life Sci. 2008;82:519–528. doi: 10.1016/j.lfs.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 11.Steiner TS. How flagellin and toll-like receptor 5 contribute to enteric infection. Infect Immun. 2007;75:545–552. doi: 10.1128/IAI.01506-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wald D, Qin J, Zhao Z, Qian Y, Naramura M, Tian L, Towne J, Sims JE, Stark GR, Li X. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2003;4:920–927. doi: 10.1038/ni968. [DOI] [PubMed] [Google Scholar]
- 13.Thomassen E, Renshaw BR, Sims JE. Identification and characterization of SIGIRR, a molecule representing a novel subtype of the IL-1R superfamily. Cytokine. 1999;11:389–399. doi: 10.1006/cyto.1998.0452. [DOI] [PubMed] [Google Scholar]
- 14.Xiao H, Gulen MF, Qin J, Yao J, Bulek K, Kish D, Altuntas CZ, Wald D, Ma C, Zhou H, Tuohy VK, Fairchild RL, de la Motte C, Cua D, Vallance BA, Li X. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity. 2007;26:461–475. doi: 10.1016/j.immuni.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 15.Moyer MP, Manzano LA, Merriman RL, Stauffer JS, Tanzer LR. NCM460, a normal human colon mucosal epithelial cell line. In Vitro Cell Dev Biol Anim. 1996;32:315–317. doi: 10.1007/BF02722955. [DOI] [PubMed] [Google Scholar]
- 16.Blohmke CJ, Victor RE, Hirschfeld AF, Elias IM, Hancock DG, Lane CR, Davidson AG, Wilcox PG, Smith KD, Overhage J, Hancock RE, Turvey SE. Innate immunity mediated by TLR5 as a novel antiinflammatory target for cystic fibrosis lung disease. J Immunol. 2008;180:7764–7773. doi: 10.4049/jimmunol.180.11.7764. [DOI] [PubMed] [Google Scholar]
- 17.de Grado M, Rosenberger CM, Gauthier A, Vallance BA, Finlay BB. Enteropathogenic Escherichia coli infection induces expression of the early growth response factor by activating mitogen-activated protein kinase cascades in epithelial cells. Infect Immun. 2001;69:6217–6224. doi: 10.1128/IAI.69.10.6217-6224.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parhar K, Ray A, Steinbrecher U, Nelson C, Salh B. The p38 mitogen-activated protein kinase regulates interleukin-1beta-induced IL-8 expression via an effect on the IL-8 promoter in intestinal epithelial cells. Immunology. 2003;108:502–512. doi: 10.1046/j.1365-2567.2003.01603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibson DL, Ma C, Rosenberger CM, Bergstrom KS, Valdez Y, Huang JT, Khan MA, Vallance BA. Toll-like receptor 2 plays a critical role in maintaining mucosal integrity during Citrobacter rodentium-induced colitis. Cell Microbiol. 2008;10:388–403. doi: 10.1111/j.1462-5822.2007.01052.x. [DOI] [PubMed] [Google Scholar]
- 20.Lee SK, Il Kim T, Kim YK, Choi CH, Yang KM, Chae B, Kim WH. Cellular differentiation-induced attenuation of LPS response in HT-29 cells is related to the down-regulation of TLR4 expression. Biochem Biophys Res Commun. 2005;337:457–463. doi: 10.1016/j.bbrc.2005.09.071. [DOI] [PubMed] [Google Scholar]
- 21.Li X, Leu S, Cheong A, Zhang H, Baibakov B, Shih C, Birnbaum MJ, Donowitz M. Akt2, phosphatidylinositol 3-kinase, and PTEN are in lipid rafts of intestinal cells: role in absorption and differentiation. Gastroenterology. 2004;126:122–135. doi: 10.1053/j.gastro.2003.10.061. [DOI] [PubMed] [Google Scholar]
- 22.Basson MD, Emenaker NJ, Sanders MA. Alpha integrin subunits regulate human (Caco-2) intestinal epithelial proliferation and phenotype. Cell Physiol Biochem. 2000;10:27–36. doi: 10.1159/000016332. [DOI] [PubMed] [Google Scholar]
- 23.Laprise P, Chailler P, Houde M, Beaulieu JF, Boucher MJ, Rivard N. Phosphatidylinositol 3-kinase controls human intestinal epithelial cell differentiation by promoting adherens junction assembly and p38 MAPK activation. J Biol Chem. 2002;277:8226–8234. doi: 10.1074/jbc.M110235200. [DOI] [PubMed] [Google Scholar]
- 24.Hase K, Eckmann L, Leopard JD, Varki N, Kagnoff MF. Cell differentiation is a key determinant of cathelicidin LL-37/human cationic antimicrobial protein 18 expression by human colon epithelium. Infect Immun. 2002;70:953–963. doi: 10.1128/iai.70.2.953-963.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peiffer I, Bernet-Camard MF, Rousset M, Servin AL. Impairments in enzyme activity and biosynthesis of brush border-associated hydrolases in human intestinal Caco-2/TC7 cells infected by members of the Afa/Dr family of diffusely adhering Escherichia coli. Cell Microbiol. 2001;3:341–357. doi: 10.1046/j.1462-5822.2001.00121.x. [DOI] [PubMed] [Google Scholar]
- 26.Prasad S, Mingrino R, Kaukinen K, Hayes KL, Powell RM, MacDonald TT, Collins JE. Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Lab Invest. 2005;85:1139–1162. doi: 10.1038/labinvest.3700316. [DOI] [PubMed] [Google Scholar]
- 27.Madison BB, Braunstein K, Kuizon E, Portman K, Qiao XT, Gumucio DL. Epithelial hedgehog signals pattern the intestinal crypt-villus axis. Development. 2005;132:279–289. doi: 10.1242/dev.01576. [DOI] [PubMed] [Google Scholar]
- 28.van den Brink GR, Bleuming SA, Hardwick JC, Schepman BL, Offerhaus GJ, Keller JJ, Nielsen C, Gaffield W, van Deventer SJ, Roberts DJ, Peppelenbosch MP. Indian Hedgehog is an antagonist of Wnt signaling in colonic epithelial cell differentiation. Nat Genet. 2004;36:277–282. doi: 10.1038/ng1304. [DOI] [PubMed] [Google Scholar]
- 29.Watt FM. Unexpected Hedgehog-Wnt interactions in epithelial differentiation. Trends Mol Med. 2004;10:577–580. doi: 10.1016/j.molmed.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 30.Dambacher J, Beigel F, Zitzmann K, de Toni E, Goke B, Diepolder HM, Auernhammer CJ, Brand S. The role of the novel Th17 cytokine IL-26 in intestinal inflammation. Gut. 2008;15:15. doi: 10.1136/gut.2007.130112. [DOI] [PubMed] [Google Scholar]
- 31.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 32.Laprise P, Langlois MJ, Boucher MJ, Jobin C, Rivard N. Down-regulation of MEK/ERK signaling by E-cadherin-dependent PI3K/Akt pathway in differentiating intestinal epithelial cells. J Cell Physiol. 2004;199:32–39. doi: 10.1002/jcp.10432. [DOI] [PubMed] [Google Scholar]
- 33.Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000;68:7010–7017. doi: 10.1128/iai.68.12.7010-7017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, Hsu D, Xu R, Harpaz N, Dannenberg AJ, Subbaramaiah K, Cooper HS, Itzkowitz SH, Abreu MT. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology. 2007;133:1869–1881. doi: 10.1053/j.gastro.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cario E, Gerken G, Podolsky DK. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology. 2007;132:1359–1374. doi: 10.1053/j.gastro.2007.02.056. [DOI] [PubMed] [Google Scholar]
- 36.Vijay-Kumar M, Gewirtz AT. Guardians of the gut: newly appreciated role of epithelial toll-like receptors in protecting the intestine. Gastroenterology. 2008;135:351–354. doi: 10.1053/j.gastro.2008.06.064. [DOI] [PubMed] [Google Scholar]