

SUMMARY
Cornichon-2 and -3 (CNIH-2/-3) are AMPA receptor (AMPAR) binding proteins that promote receptor trafficking, and markedly slow AMPAR deactivation in heterologous cells, but their role in neurons is unclear. Using CNIH-2 and -3 conditional knock-out mice, we find a profound reduction of AMPAR synaptic transmission in the hippocampus. This deficit is due to the selective loss of surface GluA1-containing AMPARs (GluA1A2 heteromers), leaving a small residual pool of synaptic GluA2A3 heteromers. The kinetics of AMPARs in neurons lacking CNIH2/3 are faster than those in WT neurons due to the fast kinetics of GluA2A3 heteromers. The remarkably selective effect of CNIHs on the GluA1 subunit, is likely mediated by TARP γ-8, which prevents a functional association of CNIHs with non-GluA1 subunits. These results point to a sophisticated interplay between CNIHs and γ-8 that dictates subunit-specific AMPAR trafficking and the strength and kinetics of synaptic AMPAR-mediated transmission.
INTRODUCTION
It is well established that auxiliary proteins play a critical role in the trafficking and function of voltage-gated ion channels. However, until recently it was thought that ionotropic neurotransmitter receptors operated independently of auxiliary subunits. This view changed with the discovery of the tetraspanning membrane protein stargazin, the protein that is mutated in the ataxic mouse stargazer. Cerebellar granule neurons (CGNs), in which stargazin is highly expressed, lack surface AMPA type glutamate receptors (AMPARs) in the stargazer mouse. In addition to controlling AMPAR trafficking, stargazin also controls AMPAR gating, thus establishing it as a bona fide AMPAR auxiliary subunit. Stargazin is a member of a family of proteins termed transmembrane AMPAR regulatory proteins (TARPs), which have both distinct and overlapping properties to stargazin (Coombs and Cull-Candy, 2009; Diaz, 2010; Jackson and Nicoll, 2011; Kato et al., 2010b; Straub and Tomita, 2011). Additional AMPAR auxiliary subunits, unrelated to TARPs, have been identified from a variety of screens (Wang et al., 2008; Zheng et al., 2004). Among these proteins are cornichon-2 and -3 (CNIH-2 and CNIH-3) (Schwenk et al., 2009). In expression systems CNIH-2 markedly slows AMPAR deactivation and desensitization and shares a number of other properties with TARPs (Gill et al., 2011; Gill et al., 2012; Harmel et al., 2012; Kato et al., 2010a; Schwenk et al., 2009; Shi et al., 2010). However, in CGNs and hippocampal neurons no significant effect of CNIH-2 overexpression was observed on AMPAR-mediated synaptic currents (Shi et al., 2010). Thus it was proposed that CNIH-2’s function in neurons was more akin to its yeast and Drosophia homologs, which serve as chaperones in the forward trafficking of EGFR ligands from ER to Golgi (Bokel et al., 2006; Castillon et al., 2009). Additional studies on CNIH-2 supported its role in forward trafficking of neuronal AMPARs (Harmel et al., 2012), but concluded that CNIH-2 remained bound to AMPARs on the surface of neurons (Gill et al., 2011; Harmel et al., 2012; Kato et al., 2010a). Furthermore, it was proposed that CNIH-2 displaced γ-8, the primary TARP expressed in the hippocampus, thus reducing TARP stoichiometry (Gill et al., 2011; Gill et al., 2012; Kato et al., 2010a), which challenged previous work suggesting that all possible γ-8 binding sites on native AMPARs were occupied (Shi et al., 2009).
In the present study we have generated conditional CNIH-2 and CNIH-3 knockout (KO) mice to determine the roles of CNIH-2 and -3 in excitatory synaptic transmission in the hippocampus. We find that CNIHs play a critical role in supporting AMPAR-mediated responses, as AMPAR function is profoundly reduced in neurons lacking both CNIH-2 and -3. However, importantly, CNIH-2/-3 binding to AMPARs is dependent on AMPAR subunit composition and TARPs. Four subunits (GluA1-GluA4) contribute to the formation of tetrameric AMPARs. We have previously shown that ~80% of synaptic AMPARs in CA1 pyramidal neurons are composed of GluA1A2 heteromers, while the rest are GluA2A3 heteromers (Lu et al., 2009). Here our data reveal that CNIH-2/-3 selectively binds to GluA1 in hippocampal neurons, allowing GluA1A2 receptors to reach the surface, and suggest that CNIH-2/-3 interaction with non-GluA1 subunits is prevented by γ-8. Removal of CNIH-2/-3 also speeds up the deactivation kinetics of surface AMPARs, an effect attributable to the loss of GluA1A2 receptors, which deactivate more slowly than GluA2A3 receptors. Thus our data point to a model in which the trafficking and gating of individual AMPARs are determined by the interplay of AMPAR subunits, cornichons and TARPs.
RESULTS
Genetic deletion of CNIH-2 and CNIH-3
Cnih2fl/fl and Cnih3fl/fl mice were generated by standard procedures (Figure S1). Cnih2fl/fl and Cnih3fl/fl mice were first bred as homozygotes and then Cnih2fl/fl mice were bred with Cnih3fl/fl mice and NEX-CRE mice. Importantly, homozygous Cnih2fl/fl and Cnih3fl/fl were indistinguishable from wild-type mice. In addition the NEX-CRE Cnih2fl/fl (NexCnih2−/−) mice, in which CNIH-2 is deleted from all forebrain pyramidal neurons, appeared grossly normal and breeding was Mendelian.
Deletion of CNIH-2 selectively depresses AMPAR synaptic transmission
We used three strategies to study the effects of deleting CNIH-2. Using Cnih2fl/fl mice we: 1) injected AAV-CRE-GFP into the hippocampus of P0–2 mouse pups and then made acute slices three weeks later; 2) made hippocampal slice cultures at P6–9, biolistically transfected neurons with CRE-GFP at DIV4 and recorded 2–3 weeks later (see Experimental Procedures for details); and 3) crossed Cnih2fl/fl mice with the NEX-CRE mouse line. In the first two sets of experiments, simultaneous recordings of AMPAR- and NMDAR-evoked excitatory postsynaptic currents (AMPAR- and NMDAR-eEPSCs, respectively) were made from a green infected/transfected CA1 pyramidal neuron expressing CRE and a neighboring control non-green pyramidal neuron during stimulation of excitatory axons in stratum radiatum. This approach permitted a pair-wise, internally controlled comparison of the consequence of our genetic manipulation. In the third approach using acute slices prepared from NexCnih2−/− mice, the ratio of the AMPAR- and NMDAR-eEPSCs was calculated and compared to wild-type neurons.
CNIH-2 deletion in single neurons by P0–2 injection (red circles) and in slice culture (black circles) caused a 54 ± 6% (n = 19) reduction in AMPAR-eEPSCs (Figure 1A), but no change in NMDAR-eEPSCs (Figure 1B). Because there was no significant difference between the results from acute and cultured slices, the data were combined. CNIH-2 deletion also caused a speeding in the decay of AMPAR-EPSCs in acute slices. This included eEPSCs (Figure 1C) and miniature EPSCs (mEPSCs) (Figure 1E). Furthermore, mEPSC amplitude was reduced (Figure 1D) consistent with a reduction in AMPAR number at individual synapses. The difference in magnitude between the evoked and mEPSCs can be explained by the fact that a threshold is required for detecting mEPSCs and many events fall below this threshold in the absence of CNIH-2. This is reflected in the large decrease in mEPSC frequency (Figure S2A). To quantitatively determine the effects of CNIH-2 on AMPAR kinetics we pulled somatic outside-out patches and used ultra-fast glutamate application to measure AMPAR deactivation (Figure 1F) and desensitization (Figure 1G). Both desensitization and deactivation time constants were faster in the absence of CNIH-2. We also examined AMPAR currents generated from somatic extrasynaptic outside-out patches in the presence of cyclothiazide to block desensitization. Similar to AMPAR-eEPSCs, extrasynaptic currents were reduced by 47 ± 5% in CRE-infected neurons (Figure 1H). Furthermore, if CNIH-2 reduces the stoichiometry of TARP γ-8 binding to AMPARs as previously proposed (Gill et al., 2011; Kato et al., 2010a) then in the absence of CNIH-2 the γ-8/AMPAR stoichiometry should increase and thus, the kainate/glutamate (IKA/IGlu) ratio, a sensitive assay for γ-8/AMPAR stoichiometry (Shi et al., 2009), should also increase. However, no change in IKA/IGlu was seen in neurons lacking CNIH-2 (Figure 1I). We also observed no change in AMPAR-eEPSC rectification in the absence of CNIH-2, indicating no change in GluA2 content (Figure S2B). CNIH-2 deletion also failed to influence paired-pulse ratio indicating an exclusively post-synaptic role for CNIH-2 (Figure S2C).
Figure 1. CNIH-2 deletion selectively reduces synaptic AMPAR-mediated transmission.

(A–B) Scatter plots show amplitudes of AMPA and NMDA-eEPSCs for single pairs of neurons from Cnih2fl/fl mice (open circles) and mean ± SEM (filled circles). The scatter plots represent data obtained from acute mouse slices infected with rAAV-CRE-GFP at P0 (red circles) and cultured mouse slices transfected with CRE for 2–3 weeks (black circles). Distributions show a reduction in AMPAR-eEPSC amplitude but no change in NMDAR-eEPSC amplitude. Insets show sample current traces from Control (black) and CRE expressing (green) cells. Bar graphs show mean ± SEM AMPAR and NMDAR-eEPSC amplitudes presented in scatter plots (A, Control [Ctl], 169.2 ± 24.3 pA; CRE, 77.4 ± 10.3 pA; n = 19; *p < 0.001; B, Ctl, 36.5 ± 4.7 pA; CRE, 32.5 ± 5.9 pA; n = 16; p = 0.3). (C) Average AMPAR-eEPSC decay kinetics from pairs of Ctl (black circles) and CRE-infected cells (green circles) (mean Ctl decay ± SEM, 14.4 ± 1.3 ms; mean CRE ± SEM, 11.3 ± 1.4 ms; n = 8; *p < 0.01). Inset shows peak normalized sample traces. (D–E) Bar graphs show mean ± SEM mEPSC amplitude (D, Ctl, 10.0 ± 0.4 pA; n = 8; CRE, 7.6 ± 0.3 pA; n = 8; *p < 0.01) and decay kinetics (E, Ctl, 10.0 ± 0.8 ms; n = 8; CRE, 6.4 ± 0.4 ms; n = 8; *p < 0.01) of Ctl and CRE-infected neurons from Cnih2fl/fl mice. Average traces are shown to the left, and are peak normalized in (E). (F–G) Bar graphs show mean ± SEM AMPAR deactivation (F, Ctl, 3.6 ± 0.2 ms; n = 12; CRE, 2.7 ± 0.2 ms; n = 18; *p < 0.002) and desensitization (G, Ctl, 13.2 ± 0.8 ms; n = 14; CRE, 8.7 ± 0.4 ms; n = 19; *p < 0.0001) from outside-out patches pulled from Ctl and CRE-infected cells and exposed to 1 and 100 ms applications of 1 mM glutamate, respectively. Peak normalized sample traces are shown to the left. (H) Bar graph shows mean ± SEM 1 mM glutamate-induced current amplitudes from outside-out patches pulled from Ctl and CRE-infected cells (Ctl, 870 ± 148 pA; n = 12; CRE, 458 ± 46 pA; n = 17; *p < 0.01). Sample traces are shown to the left. (I) Bar graph shows mean ± SEM IKA/IGlu ratios from outside-out patches pulled from Ctl and CRE-infected cells that were exposed to 1 mM glutamate and 1 mM kainate (Ctl, 0.54 ± 0.03; n = 5; CRE, 0.51 ± 0.03; n = 12; p = 0.56). Sample traces are shown to the left. See also Figure S2.
Deletion of CNIH-3 selectively depresses AMPAR synaptic transmission when combined with CNIH-2 deletion
CNIH-3 is also expressed in hippocampus, although at a lower level than CNIH-2 (Lein et al., 2007). We therefore analyzed Cnih3fl/fl mice (Figures S1B–C). We found that deleting CNIH-3 had no effect on AMPAR- or NMDAR-eEPSCs (Figures 2A–B), suggesting that either CNIH-3 is not expressed in these neurons or that an excess of CNIH-2 compensates for the loss of CNIH-3. To distinguish between these alternatives, we generated Cnih2/3fl/fl mice. Deletion of both CNIH-2 and -3 resulted in a profound and selective reduction in the AMPAR-eEPSC, significantly greater than that seen with CNIH-2 deletion alone (Figures 2C–F). These results suggest that CNIH-2 can compensate for the lack of CNIH-3, that CNIH-2 is the dominant of the two isoforms and that CNIH-2 and -3 are both essential for synaptic AMPAR expression in the hippocampus. Deletion of CNIH-2 and -3 also reduced mEPSC amplitude by ~20% (Figure 2G), similar to that observed with CNIH-2 elimination (Figure 2I), while mEPSC decay was faster than elimination of CNIH-2 alone (Figures 2H and J). In figures 2E,F,I and J our CNIH KO results are summarized and compared to previous results obtained by the conditional KO of GluA1 (Lu et al., 2009). Strikingly, the effects of CNIH-2/-3 elimination on the AMPAR-eEPSC, mEPSC amplitude, and kinetics are indistinguishable from the effects of deleting GluA1. Interestingly, previous studies on the germline GluA1 KO mouse (Andrasfalvy et al., 2003; Zamanillo et al., 1999) did not report a speeding of mEPSCs. We repeated these experiments, however, and observed the same speeding as we found in the conditional GluA1 KO neurons (Figure S3). We have no clear explanation for the difference, although Andrasfalvy et al. (Andrasfalvy et al., 2003) did report faster deactivation in outside out patches from the germline KO mouse. Long-term potentiation (LTP), which is widely held as the cellular basis for learning and memory, is also found to be severely reduced in hippocampal neurons from GluA1 KO mice (Zamanillo et al., 1999). We, therefore, examined LTP in neurons lacking CNIH-2/-3. If GluA1-containing AMPARs are removed from synapses in the absence of CNIH-2/-3, LTP should be compromised. Indeed, when compared to uninfected neurons, LTP was markedly reduced in Cnih2/3fl/fl neurons infected with CRE (Figure 2K). Thus, knocking out CNIH-2/-3 appeared to phenocopy knocking out GluA1 in three key parameters. Previous studies in HEK cells (Kato et al., 2010a) suggested that the absence of CNIH proteins in neurons should result in AMPAR resensitization and alterations in cyclothiazide potentiation of kainate-induced currents. However, neither of these effects were observed (Figure S3C and D).
Figure 2. Deletion of CNIH-2/-3 closely resembles GluA1 elimination.

(A–B) Scatter plots show amplitudes of AMPA and NMDA-eEPSCs of Ctl and CRE-transfected neurons in cultured slices from Cnih3fl/fl mice. Distributions show no change in AMPAR-eEPSCs or NMDAR-eEPSCs. Insets show sample current traces. Bar graphs show mean ± SEM AMPAR and NMDAR eEPSC amplitudes presented in scatter plots (A, Ctl, 77.0 ± 11.1 pA; ΔCNIH-3, 83.6 ± 14.2 pA; n = 10; p = 1; B, Ctl, 42.5 ± 5.6 pA; ΔCNIH-3, 35.7 ± 8.3 pA; n = 10; p = 0.5). (C–D) Scatter plots showing mean amplitudes of AMPA and NMDA-eEPSCs ± SEM of Ctl and CRE-transfected neurons in cultured slices from Cnih2/3fl/fl mice. Distributions show a reduction in AMPAR-eEPSCs (C, Ctl, 242.5 ± 48.4 pA; ΔCNIH-2/-3, 52.6 ± 9.4 pA; n = 10; *p < 0.01) but no reduction of NMDAR-eEPSCs (D, Ctl, 39.8 ± 5.7 pA; ΔCNIH-2/-3, 34.1 ± 4.7 pA; n = 10; p = 0.2). Insets show sample current traces. (E–F) Bar graphs normalized to Ctl ± SEM summarizing eEPSC data from Cnih2fl/fl, Cnih3fl/fl and Cnih2/3fl/fl mice compared to Gria1fl/fl mice. The light brown bars are published data from the Gria1fl/fl mouse (Lu et al., 2009). (G–H) Bar graphs show mean ± SEM mEPSC amplitude (G, Ctl, 10.0 ± 0.4 pA; n = 8; ΔCNIH-2/-3, 8.2 ± 0.3 pA; n = 7; *p < 0.01) and decay kinetics (H, Ctl, 10.0 ± 0.7 ms; n = 8; ΔCNIH-2/-3, 5.0 ± 0.5 ms; n = 7; *p < 0.001) of Ctl and CRE-infected neurons from Cnih2/3fl/fl mice. Average traces are shown to the left and are peak normalized in (H). (I–J) Bar graphs normalized to Ctl ± SEM summarizing mEPSC data from Cnih2fl/fl, Cnih3fl/fl and Cnih2/3fl/fl mice compared to Gria1fl/fl mice (Lu et al., 2009). (K) Mean ± SEM AMPA-eEPSCs in wild-type (black) and ΔCNIH-2/-3 (green) neurons before and after a whole cell LTP pairing protocol (arrow; Vm = 0 mV, 2 Hz Schaffer collateral stimulation for 90 s normalized to average eEPSC amplitude prior to LTP induction. LTP was severely decreased in ΔCNIH-2/-3 neurons (Ctl, n = 6; ΔCNIH-2/-3, n = 8). Sample traces before and 30–45 min after pairing are shown to the right for Ctl (black) and ΔCNIH-2/-3 (green) neurons. See also Figure S3.
GluA1 is required for the effect of CNIH-2
We next directly tested if the effects of deleting CNIH-2/-3 are specifically related to the regulation of GluA1. To this end we compared the effects of CNIH-2 knock-down on AMPAR-eEPSCs in GluA1 and GluA2 KO mice. The shRNA we generated was highly effective in knocking down CNIH-2 protein levels (Figure S4A) and in wild-type neurons produced a phenotype identical to knocking out CNIH-2 (Figures 1A–B and 3A–B). The knock-down of CNIH-2 in neurons from GluA2 KO mice, which primarily express GluA1 homomers, also resulted in a selective but more pronounced reduction in the AMPAR-eEPSC compared to wild-type mice (Figures 3C–D and G–H). In striking contrast, CNIH-2 knock-down in slices from GluA1 KO mice had no effect on AMPAR-eEPSCs (Figure 3E), AMPAR mEPSC kinetics (Figure S4B) or NMDAR eEPSCs (Figure 3F) demonstrating that CNIH-2 effects on synaptic AMPARs require GluA1. The eEPSC results are summarized in Figures 3G and H. Additionally, residual GluA2A3 receptors in GluA1 KO neurons were found to have a IKA/IGlu ratio of ~0.5 suggesting that all available TARP binding sites on these receptors are occupied (Figure S4C).
Figure 3. GluA1 is required for CNIH-2’s physical and functional interaction with AMPARs.

(A–F) Scatter plots show amplitudes of AMPA and NMDA-eEPSCs of Ctl and CNIH-2 shRNA transfected neurons in cultured slices from wild-type, GluA2 KO and GluA1 KO mice. Distributions show that the CNIH-2 shRNA reduces AMPAR-eEPSCs in wild-type (A, Ctl, 102.5 ± 16.5 pA; CNIH-2 shRNA, 52.0 ± 8.6 pA; n = 11; *p < 0.05) and GluA2 KO mice (C, Ctl, 128.9 ± 18.2 pA; CNIH-2 shRNA, 40.2 ± 5.1 pA; n = 10; *p < 0.05) but not GluA1 KO mice (E, Ctl, 54.8 ± 13.1 pA; CNIH-2 shRNA, 58.1 ± 12.3 pA; n = 9; p = 0.4). No effects were seen on NMDAR-eEPSCs (B, Ctl, 44.3 ± 7.0 pA; CNIH-2 shRNA, 42.0 ± 4.5 pA; n=10; p = 1; D, Ctl, 39.4 ± 5.1 pA; CNIH-2 shRNA, 34.9 ± 7.4 pA; n = 9; p = 0.4; F, Ctl, 84.9 ± 18.1 pA; CNIH-2 shRNA, 79.5 ± 23.3 pA; n = 8; p = 0.8). Insets show sample current traces. Bar graphs to the right show mean ± SEM AMPAR and NMDAR-eEPSC amplitudes presented in scatter plots. (G–H) Bar graphs normalized to Ctl ± SEM summarizing AMPAR and NMDAR-eEPSC data from CNIH-2 shRNA transfection of wild-type, GluA2 KO and GluA1 KO mice. (I) Immunoprecipitation of GluA2, GluA1 and CNIH-2 from hippocampal lysates of one wild-type mouse and two GluA1 KO mice using antibodies against GluA2 and GluA2/3. See also Figure S4.
Although it is well established that CNIH-2 binds to AMPARs (Kato et al., 2010a; Schwenk et al., 2009; Shi et al., 2010), the relative binding to GluA subunits has not been reported. Because CNIH-2 knock-down has a profound and selective effect on GluA1-containing AMPARs, we compared GluA1 and GluA2 binding to CNIH-2. We first immunoprecipitated GluA2 from wild-type hippocampal lysates using two different antibodies (anti-GluA2 or anti-GluA2/3). We found that CNIH-2 coimmunoprecipitated with GluA2 from wild-type hippocampal lysates, as expected (Figure 3I). In sharp contrast, we observed no coimmunoprecipitation of CNIH-2 with GluA2 when using GluA1 KO lysates. However, CNIH-2 coimmunoprecipitated with GluA1 from GluA2 KO lysates (Figure S8B), and γ-8 was coimmunoprecipitated with GluA2 from both wild-type and GluA1 KO lysates (Figure S4D). These biochemical studies demonstrate a striking specificity of CNIH-2 binding to GluA1 subunits in hippocampal lysates. Together these data indicate that both the physical and functional interaction of CNIH-2 with native AMPARs requires the GluA1 subunit.
To evaluate the surface expression of GluA1 using immunofluorescence microscopy we cultured dissociated rat hippocampal neurons transfected with CNIH-2 shRNA and visualized somatic and dendritic surface GluA1 immunoreactivity ~20 days later. CNIH-2 shRNA transfected neurons were compared to adjacent untransfected neurons. CNIH-2 knock-down dramatically reduced surface GluA1 (Figures 4A and S5A), consistent with our findings showing reduction of synaptic currents. Transfection of a scrambled shRNA or GFP alone had no effect on surface GluA1 staining (Figures 4B and S5B–C).
Figure 4. Residual GluA2A3 heteromers can account for the effects of CNIH elimination on AMPAR kinetics.
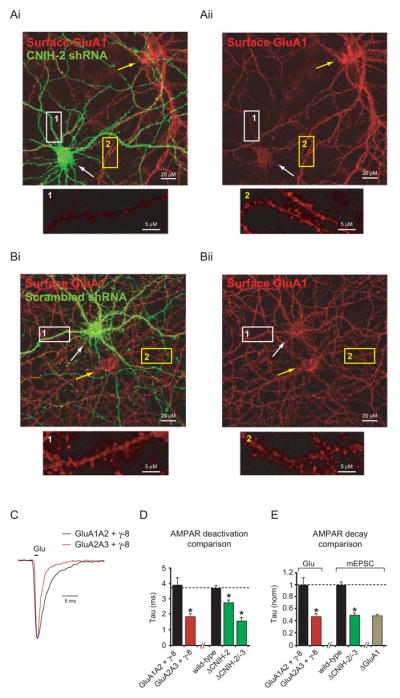
(A–B) Immunolabeling of surface GluA1 in untransfected dissociated rat hippocampal neurons (yellow arrows) compared to neurons transfected with either CNIH-2 shRNA (A) or a scrambled shRNA (B) (white arrows). Somatic dark regions are by-products of the confocal image thickness. Dendritic regions of transfected (1) and untransfected (2) neurons are shown at a higher magnification below. (C) Peak normalized sample traces showing AMPAR deactivation in outside-out patches from HEK cells transfected with GluA1A2 and γ-8 or GluA2A3 and γ-8. (D) Bar graph showing mean ± SEM deactivation of GluA1A2γ-8 and GluA2A3γ-8 complexes and the change in AMPAR deactivation kinetics in outside-out patches from ΔCNIH-2 and ΔCNIH-2/-3 CA1 pyramidal neurons (GluA1A2 + γ-8, 3.9 ± 0.4 ms; n = 10; GluA2A3 + γ-8, 1.8 ± 0.2 ms; n = 10; p < 0.001; wild-type. 3.6 ± 0.2 ms; n = 12; ΔCNIH-2, 2.7 ± 0.2 ms; n = 18; *p < 0.002; ΔCNIH-2/-3, 1.6 ± 0.2 ms; n = 6; *p < 0.0001). (E) Bar graph showing mean ± SEM deactivation of GluA2A3γ-8 complexes normalized to GluA1A2γ-8 complexes in outside-out patches from HEK cells (Glu) compared to the percent change in mEPSC decay (mEPSC) in ΔCNIH-2/-3 and ΔGluA1 CA1 pyramidal neurons. See also Figure S5.
Our data, thus far, demonstrate that synaptic expression of GluA1A2 AMPARs is eliminated in the absence of CNIH-2/-3. What then accounts for the fast kinetics of the remaining AMPARs observed after deleting CNIH-2/-3? Importantly, deletion of GluA1 results in the same fast kinetics, suggesting that the kinetics are a direct result of the specific molecular composition of the remaining receptors, which are primarily GluA2A3γ-8 complexes (Lu et al., 2009). Therefore, we next used heterologous cells to evaluate if CNIH-2 affects AMPAR kinetics by specifically regulating GluA1A2 trafficking. We co-expressed GluA2, GluA3 and γ-8 in HEK cells and measured the deactivation of this receptor complex (Figures 4C–E). For all experiments, flip-type AMPAR subunits were evaluated (see Supplemental Experimental Procedures). GluA2A3γ-8 complex deactivation is twice as fast as GluA1A2γ-8, with GluA2A3γ-8 deactivation being virtually identical to the deactivation of AMPARs in CRE-expressing Cnih2/3fl/fl neurons (Figure 4D). Furthermore, the difference in deactivation between GluA1A2γ-8 and GluA2A3γ-8 complexes is virtually identical to the magnitude of change in mEPSC decay in both CRE-expressing conditional GluA1 and CNIH-2/-3 (Gria1fl/fl and Cnih2/3fl/fl) KO neurons (Figure 4E). Thus, these findings indicate that the kinetic changes caused by the deletion of CNIH-2/-3 in neurons can be fully explained by the selective removal of the GluA1 subunit, leaving GluA2A3γ-8 complexes with faster kinetics.
Effect of CNIH-2 deletion on synaptic proteins
The lack of synaptic GluA1-containing AMPARs in the absence of CNIH-2/-3 expression may be explained by either a selective loss in total GluA1 protein expression or a specific involvement of CNIH proteins in the forward trafficking of GluA1-containing AMPARs to synapses. To examine potential effects of CNIH-2 on synaptic protein expression, Cnih2fl/fl mice were crossed to the Nex-CRE mouse line to create NexCnih2−/− mice. CRE expression in these mice includes pyramidal neurons of the neocortex and hippocampus as well as mossy and granule cells in the dentate gyrus (Goebbels et al., 2006).
We first used AMPA/NMDA ratios to ensure that similar synaptic defects were present in the hippocampus of NexCnih2−/− mice. Because CNIH-2 has no effect on NMDAR-eEPSCs, a change in this ratio should be an accurate reflection of synaptic AMPAR content. AMPA/NMDA ratios were reduced by half in CA1 pyramidal neurons lacking CNIH-2 (Figure 5A). We also observed similar reductions in dentate granule neurons and layer 2/3 pyramidal neurons in barrel cortex (Figure 5A). Interestingly, no change in the ratio was found in the heterozygous (NexCnih2+/−) mice (Figure 5A) despite a 30–50% reduction in total CNIH-2 expression (Figure S6A), thus providing further evidence that CNIH-2 is expressed in excess in CA1 pyramidal neurons and that all available CNIH-2 binding sites on AMPARs are occupied or “saturated”. In paired recordings from slice cultures from NexCnih2−/− mice, transfection of CA1 pyramidal neurons with CNIH-2 fully rescued AMPAR-eEPSCs (Figure 5B). No change in the NMDAR-eEPSC was observed (Figure 5C). As previously shown, CNIH-2 overexpression in wild-type neurons has no effect on AMPAR- or NMDAR-eEPSCs (Figures S6B–C) (Shi et al., 2010) again indicating saturation of CNIH binding sites on native AMPARs.
Figure 5. CNIH-2 deletion impedes AMPAR trafficking with little effect on other synaptic proteins.

(A) Bar graphs show mean ± SEM AMPA/NMDA ratios of primary neurons in CA1, dentate gryrus and barrel cortex from wild-type, NexCnih2+/− and NexCnih2−/− mice. (CA1, Ctl, 3.6 ± 0.5; n = 8; NexCnih2+/−, 3.7 ± 0.5; n = 5; NexCnih2−/−, 1.8 ± 0.2; n = 8, *p < 0.001), (DG, Ctl, 3.6 ± 0.3; n = 8; NexCnih2−/−, 1.7 ± 0.2; n = 8) and (BC, Ctl, 2.9 ± 0.5; n = 5; NexCnih2−/−, 1.6 ± 0.2; n = 6; *p < 0.05). AMPA and NMDA sample current traces from CA1 of wild-type and NexCnih2−/− mice normalized to NMDAR current at 150 ms are shown to the left. (B–C) Scatter plots showing that transfection of NexCnih2−/− neurons with CNIH-2 restores the AMPAR-eEPSC amplitude to wild-type levels. Bar graphs to the right of scatter plots show corresponding mean ± SEM eEPSC amplitudes (B, NexCnih2−/−, 53.3 ± 16.9 pA; NexCnih2−/− + CNIH-2, 109.1 ± 29.6 pA; n = 7; *p < 0.05; C, NexCnih2−/−, 60.2 ± 8.7 pA; NexCnih2−/− + CNIH-2, 55.5 ± 7.7 pA; n = 7; p = 0.8). Insets show corresponding sample traces. (D) Immunoblots from hippocampal lysates of wild-type and NexCnih2−/− mice comparing expression levels of synaptic proteins. Bar graph to the right shows average synaptic protein levels in NexCnih2−/− mice normalized to wild-type mice ± SEM (CNIH-2, 0.04 ± 0.008; GluA1, 0.84 ± 0.033; GluA2, 0.82 ± 0.057; γ-8, 0.97 ± 0.062; PSD-95, 0.97 ± 0.039; NR2A, 1.01 ± 0.081; n = 3–5; *p < 0.05). (E) Immunoblots from hippocampal lysates of wild-type NexCnih2−/− and γ-8 KO mice comparing total GluA1, GluA2, γ-8 and CNIH-2 expression levels. Bar graph to the right shows average GluA1, GluA2, γ-8 and CNIH-2 expression levels in NexCnih2−/− and γ-8 KO mice normalized to wild-type mice ± SEM (NexCnih2−/− mice: GluA1, 0.83 ± 0.03; GluA2, 0.89 ± 0.02; γ-8, 0.99 ± 0.05; CNIH-2, 0.05 ± 0.02; n = 3; γ-8 KO mice: GluA1, 0.49 ± 0.05; GluA2, 0.50 ± 0.04; γ-8, 0.03 ± 0.01; CNIH-2, 0.28 ± 0.02; n = 3; *p < 0.05). (F) Glycosylation analysis of GluA1 and GluA2 in wild-type and NexCnih2−/− mice. The representative blot to the left shows the relative amount of mature GluA1 receptor subunits (blue arrows) to immature GluA1 subunits (red arrows) in hippocampal lysates from wild-type and NexCnih2−/− mice. Bar graph to the right shows the average ratio of immature to mature GluA1 and GluA2 subunits in NexCnih2−/− mice normalized to wild-type mice ± SEM (GluA1, 1.99 ± 0.28; GluA2, 1.70 ± 0.13; n = 3–5; *p < 0.05). (G) Biotinylation analysis of GluA1, GluA2, γ-8 and CNIH-2 in dissociated hippocampal neurons. See also Figure S6.
We next examined the total expression level of a number of synaptic proteins in NexCnih2−/− mice. Importantly, no CNIH-2 protein was detected in hippocampal lysates confirming that CNIIH-2 is absent in the hippocampus of these mice (Figures 5D–E). We found that GluA1 and GluA2 were reduced by about 15%, but no change was observed for γ-8, PSD-95 or the NMDAR subunit GluN2A (Figure 5D). Infection of dissociated hippocampal neurons with the CNIH-2 shRNA also produced little effect on total GluA1/A2 expression levels (Figure S4A). We then compared the consequences of deleting CNIH-2 to γ-8 deletion (Figure 5E). Total expression of GluA1 and GluA2 are more severely reduced in γ-8 KO mice than in NexCnih2−/− mice and, unlike the lack of change in γ-8 levels in NexCnih2−/− mice, total CNIH-2 expression is markedly reduced in γ-8 KO mice, as reported previously (Kato et al., 2010a).
As the modest loss of AMPAR protein in the absence of CNIH-2 expression is unlikely to explain the profound effects on physiology, we next examined the effect of deleting CNIH-2 on AMPAR trafficking to the cell surface. AMPARs are glycoproteins, which traffic through the biosynthetic pathway. To determine if CNIH-2 affects AMPAR maturation, we examined receptor glycosylation using endoglycosidase H (Endo H), which digests immature high mannose sugars and PNGase F, which removes all N-linked carbohydrates. Relative to wild-type brains both GluA1 and GluA2 showed increased sensitivity to Endo H in NexCnih2−/− brains (Figure 5F and S6D), as demonstrated by stronger Endo H-sensitive immature bands (red arrows) compared to Endo H-resistant mature bands (blue arrows). These data suggest that a large pool of immature receptors are retained in the ER or cis-Golgi in the absence of CNIH-2. The Endo H-sensitive band co-migrates with completely deglycosylated receptors following treatment with PNGase F.
We also re-examined the distribution of CNIH-2 protein in the hippocampus, using an antibody we recently generated using the same epitope as Kato et al. (Kato et al., 2010a). As in our previous study (Shi et al., 2010), the large majority of CNIH-2 was intracellular. However, with the new antibody, CNIH-2 could also be detected on the cell surface (Figure 5G).
TARP γ γγ-8 reverses the CNIH-2 induced slowing of GluA2-containing AMPARs but not homomeric GluA1 receptors
In heterologous cells CNIH-2 has marked effects on GluA1-containing and lacking AMPARs (Schwenk et al., 2009). What then accounts for the selective effects of CNIH-2 deletion on native GluA1-containing receptors? Furthermore, how can one reconcile the fact that all CNIH binding sites appear to be occupied in CA1 neurons and yet endogenous AMPAR kinetics are considerably faster than the kinetics of AMPARs co-expressed with CNIH-2 in expression systems? To better understand the AMPAR kinetics in expression systems, we examined a variety of conditions. Initially, we measured the effects of CNIH-2 and γ-8, the primary TARP in the hippocampus (Rouach et al., 2005), on receptors of defined subunit composition in HEK cells. As seen previously, CNIH-2 significantly slowed deactivation of GluA1 homomeric receptors and to a greater extent than γ-8 (Figure 6Ai). Expression of both CNIH-2 and γ-8 did not significantly change the slowing seen with CNIH-2 alone (Figure 6Ai).
Figure 6. γ γγ-8 blocks CNIH-2’s functional interaction with GluA2 but not GluA1.
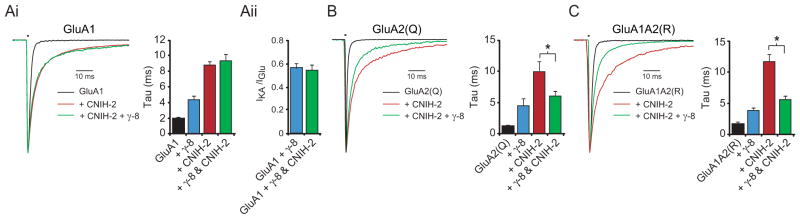
(A) Bar graph shows mean ± SEM deactivation kinetics of GluA1 homomers expressed in HEK cells alone, with γ-8, with CNIH-2 and with γ-8 and CNIH-2 (Ai, GluA1, 1.8 ± 0.2 ms, n = 10; GluA1 + γ-8, 4.9 ± 0.3 ms, n = 8; GluA1 + CNIH-2, 8.7 ± 0.6 ms, n = 11; GluA1 + γ-8 + CNIH-2, 9.4 ± 0.7 ms, n = 12). Mean ± SEM IKA/IGlu ratios for GluA1 + γ-8 and GluA1 + γ-8 + CNIH-2 were also compared (Aii, GluA1 + γ-8, 0.57 ± 0.03, n = 8; GluA1 + γ-8 + CNIH-2, 0.54 ± 0.05, n = 5). (B–C) Bar graphs show mean ± SEM deactivation kinetics of GluA2(Q) homomers and GluA1A2(R) heteromers expressed in HEK cells alone, with γ-8, with CNIH-2 and with γ-8 and CNIH-2 (B, GluA2(Q), 1.2 ± 0.2 ms, n = 8; GluA2(Q) + γ-8, 4.5 ± 1.0 ms, n = 4; GluA2(Q) + CNIH-2, 10.0 ± 1.5 ms, n = 7; GluA2(Q) + γ-8 + CNIH-2, 6.0 ± 0.7 ms, n = 6) (C, GluA1A2(R), 1.7 ± 0.4 ms, n = 6; GluA1A2(R) + γ-8, 3.9 ± 0.4 ms, n = 10; GluA1A2(R) + CNIH-2, 11.7 ± 1.0 ms, n = 6; GluA1A2(R) + γ-8 + CNIH-2, 5.7 ± 0.5 ms, n = 9). Corresponding peak normalized sample traces are shown to the left of bar graphs. See also Figure S7.
These findings could be explained by CNIH-2 and γ-8 binding to the same site on GluA1 subunits with CNIH-2 displacing γ-8, or the two proteins binding to separate sites. The fact that the slowing of kinetics seen with CNIH-2 is the same in GluA1-containing AMPARs with covalently attached γ-8 (Shi et al., 2010), suggests that CNIH-2 is not displacing γ-8. Furthermore, the fact that the IKA/IGlu ratio, a sensitive measure of γ-8/AMPAR stoichiometry, is unchanged (Figure 6Aii) also strongly argues that CNIH-2 is not displacing γ-8 and that γ-8 and CNIH-2 are able to co-occupy GluA1 subunits. These results, however, do not explain why CNIH-2 appears to occupy all available binding sites on neuronal AMPARs and yet native neuronal AMPAR kinetics are substantially faster than what is observed when CNIH-2 and γ-8 are expressed with homomeric GluA1. Might GluA2 behave differently from GluA1, in that essentially all native AMPARs in CA1 pyramidal neurons contain the GluA2 subunit (Lu et al., 2009)? We therefore examined the effect of CNIH-2 on GluA2 homomers in HEK cells. Unedited GluA2(Q) was used owing to its ability to form functional channels at higher levels than GluA2(R). Like GluA1 we found that CNIH-2 slowed deactivation of GluA2 homomers (Figures 6B). However, in sharp contrast to GluA1, the co-expression of γ-8 reversed the slowing of homomeric GluA2 kinetics caused by CNIH-2 (Figure 6B). The IKA/IGlu ratio of GluA2(Q) in the presence of both γ-8 and CNIH-2 was 0.48 ± 0.04 (n = 6), indicating a 4 γ-8 receptor (Figure S7). We repeated the experiments with GluA1A2(R) heteromers, the subunit composition that accounts for the majority of endogenous AMPARs in CA1 neurons (Lu et al., 2009). When GluA1A2 heteromers were co-expressed with either γ-8 or CNIH-2, CNIH-2 produced a much stronger slowing of deactivation compared to γ-8, as expected. Remarkably, however, co-expression of γ-8 and CNIH-2 with GluA1A2 heteromers reversed CNIH-2-induced slowing (Figure 6C). Together these findings are of considerable interest for two main reasons. One, such data are consistent with a model in which γ-8 prevents the physical interaction of CNIH with non-GluA1 subunits, thus explaining the observed CNIH subunit specificity. And two, when CNIH-2 is bound to GluA1 but prevented from functionally interacting with GluA2 by γ-8, as would be expected in neurons, CNIH-2 has little influence on the kinetics of GluA1A2 heteromers. It is important to note that previous efforts to understand CNIH function have focused heavily on whether or not CNIH proteins are associated with synaptic AMPARs or sequestered in the ER. The present data appear to diminish the relevance of this issue owing to the fact that all of the physiological consequences of deleting CNIH proteins can be explained by the selective loss of synaptic GluA1A2 heteromers.
Based on the results in Figure 6Ai, one might expect the kinetics of the AMPAR EPSC to be slow in pyramidal neurons from GluA2 KO mice, because most receptors are composed of GluA1 homomers (Lu et al., 2009), presumably bound to CNIH-2/-3. This, however, is not the case (Lu et al., 2009). Surprisingly, we find a marked enhancement in the total expression and association of γ-2 with GluA1-containing receptors when GluA2 expression is reduced (Figure S8A–C). γ-2 has been shown to reverse the kinetic effects of CNIH-2/-3 on GluA1 homomers (Gill et al., 2012 and Figure S8C). Indeed, in neurons from stargazer mice (a γ-2 deficient mouse line), GluA2 KD leads to slowing of AMPA mEPSC decay kinetics as expected (Figure S8D–E). See Figure S8 for more details.
CNIH-2 enhances AMPAR-mediated responses in the γ γγ-8 KO
The above results provide an explanation for the paradox that, while all CNIH-2 binding sites of native AMPARs seem to be occupied, the kinetics of neuronal AMPARs are fast. That is, under normal conditions γ-8 prevents a functional association of CNIH-2/-3 to GluA2 and thus prevents the expected slowing of GluA1A2 heteromers. If this model is correct and CNIH proteins are able to associate with AMPARs on the surface then deleting γ-8 should cause a marked slowing in mEPSCs. However, while we confirmed a reduction in mEPSC amplitude in γ-8 KO mice (Figure 7A), no effect on mEPSC decay was observed (Figure 7B) (Rouach et al., 2005). One explanation for this might be that CNIH-2 is expressed at severely reduced levels in γ-8 KO mice (Figure 5E) (Kato et al., 2010a). Therefore, we expressed CNIH-2 in slice cultures made from γ-8 KO mice and found that CNIH-2 not only rescued the amplitude of the AMPAR-mEPSCs (Figure 7A), but also markedly slowed mEPSC responses, such that the kinetics were considerably slower than what is seen in wild-type neurons or when CNIH-2 is overexpressed in wild-type neurons (Figure 7B). These data are compelling for several reasons. One, they show that CNIH-2 effects on AMPAR kinetics are similar in HEK cells and in neurons lacking γ-8. Two, they emphasize the critical role that γ-8 has in determining the effects of CNIH-2/-3 on AMPAR kinetics. And three, they demonstrate that CNIH proteins are able to associate with synaptic AMPARs. While we maintain that the primary role for CNIH proteins is in the selective trafficking of GluA1A2 heteromers to synapses, the presence of CNIH protein on the surface of neurons (Figure 5G) and the ability CNIH-2 to influence gating properties of synaptic AMPARs in the absence of γ-8 (Figure 7B) are consistent with a selective and likely inert association of CNIH protein with GluA1 subunits of native synaptic GluA1A2 heteromers in the presence of γ-8.
Figure 7. CNIH-2 slows synaptic AMPAR currents in the absence of γγ γ-8.

(A–B) Bar graphs show mean ± SEM mEPSC amplitude (A) and decay (B) of wild-type, NexCnih2−/−, CNIH-2 overexpressing (OE), γ-8 KO and γ-8 KO + CNIH-2 CA1 neurons in slice culture (A, wild-type, 17.4 ± 1.6 pA; n = 8; NexCnih2−/−, 9.5 ± 0.5 pA; n = 9; CNIH-2 OE, 17.3 ± 1.6 pA; n = 5; γ-8 KO, 10.7 ± 0.9 pA; n = 9; γ-8 KO + CNIH-2, 19.1 ± 3.5 pA; n = 7; *p < 0.05) (B, wild-type, 6.3 ± 0.4 ms; n = 8; NexCnih2−/−, 4.4 ± 0.3 ms; n = 9; CNIH-2 OE, 6.4 ± 0.4 ms; n = 5; γ-8 KO, 7.8 ± 0.6 ms; n = 9; γ-8 KO + CNIH-2, 14.2 ± 0.63 ms; n = 7; *p < 0.05). Select color-matched sample traces are shown above bar graphs. Sample traces in (B) are peak normalized. Note that compared to acute slices baseline mEPSC amplitude is larger and mEPSC kinetics are faster in slice culture (see Supplemental Experimental Procedures).
DISCUSSION
In this study, we used a variety of approaches, including the generation of conditional KO mice for CNIH-2 and CNIH-3, to determine the role of cornichon proteins in the regulation of neuronal AMPARs. By deleting CNIHs from neurons, we reveal a critical role for these proteins in regulating AMPAR-mediated synaptic transmission, as there is a profound loss of AMPAR currents in KO neurons. We have demonstrated that under native conditions CNIH is saturating and the knock-down or KO of CNIHs is essential for studying their roles in neurons. Furthermore, we find an unanticipated subunit specificity, in that CNIH-2/-3 preferentially interact with, and functionally regulate GluA1-containing AMPARs. Strikingly, CNIH-2/-3 KO neurons phenocopy GluA1 KO neurons with respect to their current amplitudes, kinetics and synaptic plasticity. All of our findings are most consistent with a model in which the primary role of CNIH-2/-3 in CA1 pyramidal neurons is the selective trafficking of GluA1-containing receptors to synapses.
A model for the interaction between γ-8 and CNIH with AMPAR subunits
Figure 8 summarizes the proposed interactions between γ-8 and CNIH with surface AMPAR subunits. This model is based primarily on data in which γ-8 and CNIH are expressed with the various AMPAR subunits in HEK cells, but, as we discuss below, is strongly supported by our data from CA1 pyramidal neurons. We propose based on the IKA/IGlu ratio, a sensitive assay for TARP stoichiometry (Shi et al., 2009), that all AMPAR subunit combinations presented in Figure 8 contain 4 γ-8 as shown in HEK cells for AMPAR homomers (Figures 6Aii and S7) and in neurons for AMPAR heteromers (Figures 1I and S4C). The rest of this discussion concerns the number of CNIH proteins associated with the various AMPAR subunit combinations.
Figure 8. Model of CNIH and γ γγ-8 interactions with AMPARs.
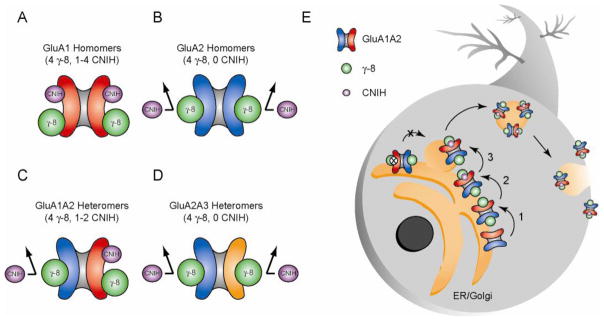
(A) GluA1 AMPAR subunits simultaneously associate with CNIH proteins and TARP γ-8. Therefore, we propose surface tetrameric GluA1 homomers associate with 4 γ-8 molecules and 1–4 CNIH molecules. (B) CNIH protein association with GluA2 AMPAR subunits appears to be prevented by γ-8. Therefore, in the presence of γ-8, we propose surface GluA2 homomers associate with 4 γ-8 molecules and 0 CNIH molecules. (C) Because of GluA1 and GluA2’s respective relationships with γ-8 and CNIH proteins, we propose surface GluA1A2 heteromers associate with 4 γ-8 molecules and 1–2 CNIH molecules. (D) Because GluA1 is required for the physical association of CNIH proteins but not γ-8 with AMPARs in neurons, we propose surface GluA2/3 heteromers associate with 4 γ-8 molecules and 0 CNIH molecules. (E) In neurons CNIH proteins selectively promote the trafficking of GluA1A2 heteromers but not GluA2A3 heteromers to the neuronal surface. γ-8 prevents CNIH interaction with non-GluA1 subunits and provides a mechanism for the subunit specific action of CNIH on GluA1A2 receptor trafficking. Overexpression of CNIH in wild-type neurons does not slow AMPAR gating kinetics indicating CNIH cannot displace γ-8 on non-GluA1 subunits. Together these data suggest a model whereby in the ER/Golgi γ-8 associates with AMPARs prior to CNIH (1) thus limiting subsequent CNIH interactions to only GluA1 subunits, which uniquely associate with both γ-8 and CNIH (2). CNIH proteins would then selectively enable the forward trafficking of GluA1A2 heteromers to the neuronal surface (3). CNIH deletion prevents GluA1A2 receptors from leaving the ER/Golgi.
GluA1 homomers
The deactivation of GluA1 homomers in the presence of both γ-8 and CNIH in HEK cells is at least as slow as that observed with CNIH alone (Figure 6Ai). Furthermore, the IKA/IGlu ratio of GluA1 homomers is the same when expressed in HEK cells with only γ-8 and when CNIH-2 and γ-8 are co-expressed (Figure 6Aii). Therefore, these receptors must be associated with at least 1 and possibly 4 CNIH molecules, in addition to the 4 γ-8 (Figure 8A). We cannot be more precise on the CNIH stoichiometry but γ-8 and CNIH-2 are capable of co-occupying GluA1 subunits.
GluA2 homomers
In HEK cells, based on the same fast kinetics of GluA2 homomers in the presence of both γ-8 and CNIH or with γ-8 alone (Figure 6B), we propose that γ-8 prevents GluA2 subunits from associating with CNIH, with GluA2 homomers containing 4 γ-8 and 0 CNIH (Figure 8B). This model is supported by the ability of GluA2A3 receptors to coimmunoprecipitate γ-8 but not CNIH-2 from hippocampal lysates (Figures 3I and S4D).
GluA1A2 heteromers
The fast kinetics seen with this heteromer in the presence of γ-8 and CNIH in HEK cells indicates that CNIH has little effect suggesting the absence of CNIH on this heteromer on the surface (Figure 6C). Alternatively, because CNIH does interact with surface GluA1 homomers in the presence of γ-8 (Figure 6A), CNIH could be associated with GluA1 subunits of surface GluA1A2 heteromers but not affect the kinetics of these heteromers. Wild-type AMPARs in CA1 pyramidal neurons are primarily GluA1A2 (Lu et al., 2009) and exhibit deactivation kinetics characteristic of limited CNIH influence on GluA1A2 gating kinetics (Figure 4D and 6C). In the hippocampus our biochemical data show that CNIH-2 associates exclusively with GluA1A2 receptors through the GluA1 subunit (Figure 3I and S8B), and we do observe CNIH-2 on the surface of hippocampal neurons (Figure 5G). Because of such data we would argue that native surface GluA1A2 receptors could have up to 2 CNIH associated with the GluA1 subunits, but that, if present, they exert no effect on gating kinetics due to γ-8’s prevention of a functional CNIH association with the GluA2 subunit (Figure 8C). If this is the case, CNIH expression in the absence of γ-8 should slow the gating kinetics of surface AMPARs in neurons. Indeed, when CNIH-2 is expressed in pyramidal neurons from γ-8 KO mice the gating kinetics of surface AMPARs at synapses are markedly slowed (Figure 7B).
GluA2A3 heteromers
In GluA1 KO mice the remaining GluA2A3 receptors bind to γ-8 and have a high IKA/IGlu ratio indicating that they also contain 4 γ-8 (Figure S4C and D). The fast kinetics of native neuronal GluA2A3 receptors in GluA1 conditional KO mice (Figure 4E), the inability of CNIH-2 knock-down to influence AMPA EPSCs of neurons from GluA1 KO mice (Figure 3E and S4B) and the failure of neuronal GluA2A3 receptors to coimmunoprecipitate CNIH-2 (Figure 3I) argue that CNIH is prevented from associating with these receptors. Thus we assert that GluA2A3 receptors contain 4 γ-8 and 0 CNIH molecules (Figure 8D). While speculative, given the likelihood that γ-8 inhibits the interaction of CNIH on GluA2 subunits, we believe γ-8 may similarly inhibit CNIH interaction with GluA3.
CNIH-2/-3 selectively interact with GluA1 subunits and are required for synaptic expression of GluA1-containing AMPARs in CA1 pyramidal neurons
Previous studies, including our own, report little effect of CNIH overexpression on endogenous AMPARs. However, CNIHs clearly interact with AMPARs in heterologous cells and in neurons (Harmel et al., 2012; Shi et al., 2009; Schwenk et al., 2009; Kato et al., 2010; Gill et al., 2011; Gill et al., 2012). To test if CNIHs have an important role in neurons but are expressed at saturating levels, we performed extensive analyses using genetic deletion and knock-down of CNIHs. Indeed, we found that CNIH-2/-3 deletion causes a profound and selective reduction in AMPAR-eEPSC amplitude. This is accompanied by faster decay of mEPSCs, faster deactivation and desensitization of glutamate-evoked currents from somatic patches and compromised LTP induction. These results demonstrate a critical role for CNIHs in neuronal AMPAR regulation and are particularly fascinating given that the profound synaptic changes seen with the deletion of CNIH-2/-3 match those seen with the selective deletion of GluA1 (Lu et al., 2009). Because neurons lacking CNIH proteins look physiologically similar to neurons lacking GluA1, we hypothesized that removal of CNIH-2/-3 might have different effects in various AMPAR KO mice and therefore used these tools to probe CNIH-2 function. Knocking down CNIH-2 in hippocampal slices from GluA2 KO mice causes a profound reduction of AMPAR-eEPSCs, whereas knocking down CNIH-2 in slices from GluA1 KO mice has no effect, either on the amplitude or kinetics of AMPAR EPSCs. These physiological results support a selective action of CNIH-2/-3 on GluA1-containing receptors. We also found that CNIH-2 and GluA1 co-IP with GluA2 when using wild-type hippocampal homogenates. However, in striking contrast, when using homogenates from GluA1 KO mice, CNIH-2 does not co-IP with GluA2. Furthermore, GluA2A3/γ-8 receptors, the most likely composition of the receptors remaining in neurons lacking GluA1 or CNIH-2/-3, are twice as fast as GluA1A2/γ-8 receptors. Thus, the 50% reduction in mEPSC decay observed in neurons lacking GluA1 and CNIH-2/-3 can be explained by the selective loss of synaptic GluA1-containing AMPARs.
γ-8 prevents the action of CNIH-2/-3 on non-GluA1 subunits
Why is the action of CNIH-2/-3 confined to the GluA1 subunit? Previous studies in heterologous systems have shown that CNIH-2 has significant effects on AMPARs containing and lacking GluA1 subunits (Schwenk et al., 2009). To address this seeming contradiction, we examined the interactions between CNIH-2 and γ-8, the most prevalent TARP in the hippocampus (Rouach et al., 2005), on the kinetics of AMPARs of defined subunit composition. Remarkably, while γ-8 was incapable of reversing the slowing caused by CNIH-2 on homomeric GluA1 receptors, it fully reversed the slowing caused by CNIH-2 on homomeric GluA2 receptors, revealing exquisite selectivity of CNIH-2 for GluA1 over GluA2 due to γ-8 preventing a functional association of CNIH-2 with GluA2.
The marked slowing of deactivation is one of the most prominent effects of CNIH-2 on heterologously expressed AMPARs. Does CNIH-2 make any contribution to the kinetics of AMPARs in CA1 pyramidal neurons? As discussed above, the speeding of AMPAR kinetics in neurons lacking CNIH-2/-3 can be fully accounted for by the selective loss of GluA1-containing receptors without any need for a direct action of CNIH-2 on the gating of surface/synaptic AMPARs, raising the question as to whether CNIH-2 is, in fact, associated with surface/synaptic AMPARs. Results from other groups (Gill et al., 2011; Kato et al., 2010a), based largely on data from heterologous cells, found that CNIH proteins prevent AMPAR resensitization, suggesting that the lack of resensitization in neurons is due to the presence of CNIH proteins. However, we failed to see resensitization in neurons lacking CNIH proteins (Figure S3C). We also found that γ-8 reverses the effects of CNIH-2 on the deactivation of GluA1A2 heteromers. Taken together these findings may leave very little room for a physiologically relevant role for CNIH proteins on synaptic AMPAR gating in neurons and perhaps diminish the relevance of arguments concerning the presence of CNIH proteins on surface AMPARs. However, we do detect the expression of endogenous CNIH on the surface of neurons and are able to observe effects of CNIH-2 on synaptic AMPAR gating in the absence of γ-8. Therefore it is possible for CNIH proteins to associate with synaptic AMPARs. As stated above, such data point to a selective and potentially inert association of CNIH proteins with GluA1 subunits of synaptic GluA1A2 heteromers, with γ-8 bound to all four subunits, as previously proposed (Shi et al., 2009).
CNIH-2/-3 promotes forward trafficking of GluA1-containing AMPARs in the ER/Golgi
How does CNIH-2/-3 control the level of AMPARs on the surface of hippocampal pyramidal neurons? One possibility is that in the absence of CNIH-2/-3 AMPAR protein is lost, similar to what is seen in γ-8 KO mice (Rouach et al., 2005). However, the modest loss of AMPAR protein seen in the NexCnih2−/− mice cannot explain the profound loss of surface AMPARs. Rather our data suggest that the maturation of AMPARs is impaired and that the immature receptors are retained in the ER/cis-Golgi. As pointed out previously (Shi et al, 2009), such a role is remarkably similar to the established role of the yeast (Erv14p) and Drosophila (Cni) CNIH homologues, in which these proteins serve as chaperones that aid in the forward trafficking of EGFR ligands from the ER to Golgi (Bokel et al., 2006; Castillon et al., 2009; Roth et al., 1995). However, unlike the yeast and Drosophia homologues, but analogous to its effects in HEK cells, CNIH-2 can remain associated with neuronal AMPARs, at least in the absence of γ-8 protein.
More specifically our results indicate that CNIH is essential for the functional expression of GluA1-containing receptors on the surface. Although, at present, we cannot say at what forward trafficking step of these receptors requires CNIH proteins. It is possible that CNIH proteins are required for the transport of GluA1-containing AMPARs from the ER to the Golgi, from the Golgi to the neuronal surface or both. Future study will undoubtedly be necessary to answer these questions. However, our data would suggest that γ-8 proteins associate with AMPARs prior to CNIH proteins as AMPARs progress through the secretory pathway due to γ-8 seemingly being required for the subunit-specific actions of CNIH proteins on the surface trafficking of GluA1A2 heteromers (Figure 8E).
Our results raise two related issues. First, the delivery of the GluA1 subunit to the surface of CA1 pyramidal neurons requires CNIHs. Yet this is clearly not the case in heterologous expression systems. What accounts for the difference? The situation may be analogous to TARP γ-2, which is essential for the surface delivery of AMPARs in cerebellar granule neurons and greatly facilitates surface delivery of AMPARs in heterologous cells, but is not essential for their delivery. Second, can the results obtained in CA1 pyramidal neurons be applied to other neurons? Our results suggest that CNIH-2 plays a similar role in AMPAR trafficking in both dentate granule neurons and layer 2/3 neocortical neurons. However, these neurons are likely to be similar to CA1 neurons in their expression of GluA1A2 heteromers and TARP γ-8. Is there an example of a neuron that expresses GluA1 subunits but not CNIH-2? Our results would suggest not, since the surface expression of GluA1 in neurons requires CNIH-2. Also of interest are Purkinje neurons, which express high levels of CNIH-2, but only transiently express GluA1 (Douyard et al., 2007). It is also worth noting that additional AMPAR auxiliary proteins have been identified, such as CKamp44, which is expressed in DG but not CA1 pyramidal neurons (von Engelhardt et al., 2010). Whether a functional relationship between CKamp44 and CNIH proteins exists in DG remains to be determined. Another interesting question is whether the ability of CNIH proteins to influence AMPAR gating is utilized in other types of neurons.
Conclusion
Our results reveal an intricate interplay between CNIHs and γ-8 that allow for trafficking of GluA1-containing AMPARs to synapses. Because of the selective interaction of CNIHs with GluA1, GluA1A2 heteromers are allowed to dominate the population of neuronal AMPARs in CA1 pyramidal neurons. GluA1A2 heteromers are required for LTP and display slower deactivation kinetics than GluA2A3 heteromers likely allowing for greater dendritic signal integration. Furthermore, GluA1 subunits possess an intracellular loop and long C-tails that are subject to posttranslational modification and protein interactions that have been shown to play roles in activity-dependent synaptic plasticity. It will be of interest to know if CNIHs themselves are also subject to such forms of regulation and thus contribute to activity-dependent trafficking and function of synaptic GluA1-containing AMPARs. Finally, what is the structural basis that allows CNIH and γ-8 to associate with GluA1 whereas for GluA2, γ-8 prevents a functional CNIH association? Future work toward a more complete understanding of the uniqueness of GluA1-containing AMPARs and the mechanisms that regulate their function will be invaluable to our understanding of how primary neurons of numerous brain structures communicate with one another.
EXPERIMENTAL PROCEDURES
Generation of Cnih2fl/fl and Cnih3fl/fl mice
Cnih2fl/fl and Cnih3fl/fl mice were generated using standard procedures by inGenious Targeting Laboratory (Ronkonkoma, NY). For Cnih2fl/fl and Cnih3fl/f mice homologous recombination introduced loxP sites allowing for the excision of exons 2–5 and exon 4, respectively. Both lines were crossed to a FLP deleter line to remove the neomycin resistance cassette.
Electrophysiology
Acute transverse 300 μm hippocampal slices were prepared from P17–P21 mice. Cultured hippocampal slices were prepared from P6–P9 mice as previously described (Schnell et al., 2002). Paired recordings of eEPSC’s involved simultaneous whole-cell recordings at room temperature from one infected/transfected GFP-positive neuron and a neighboring GFP-negative neuron while stimulating Schaffer collaterals. Series resistance was monitored and not compensated, and cells in which series resistance was above 30 MΩ or varied by 25% during a recording session were discarded. mEPSCs were recorded in the presence of 0.5 μM TTX. mEPSCs with an amplitude of ≥ 5 pA and a rate of rise of ≥ 4 pA/ms were automatically detected and analyzed offline with customized software in (IGOR). Fast application of 1 mM glutamate to somatic and HEK cell outside-out patches for 1 and 100 ms by a piezoelectric controller (Siskiyou) was used to determine AMPAR deactivation and desensitization kinetics, respectively. Our open-tip response experiments show the 20%–80% exchange times to be less than 200 μs.
Western blotting, immunoprecipitation and glycosidase treatment
Adult mouse hippocampi were homogenized and the nuclear pellet was removed by centrifugation and resuspended in 1% Triton X-100. Pre-cleared lysates were incubated with antibody-bound Sepharose beads (Sigma). Beads were washed with lysis buffer and analyzed by immunoblotting with the relevant antibodies as indicated. For glycosylation analysis, the precleared lysate was immunoprecipitated with GluA1 or GluA2 antibody, and treated with endoglycosidase Hf (Endo H) or PNGase F overnight at 37°C, resolved by SDS-PAGE and analyzed by immunoblotting with indicated antibodies.
Surface immunolabeling/imaging
Hippocampal neurons were cultured on coverslips from E18 rat hippocampus as previously described (Roche and Huganir, 1995). The neurons were transfected at 7 days in vitro (DIV). ~20 days after transfection, neurons were incubated with GluA1 antibody and then fixed. After blocking the neurons were incubated with the Alexa Fluor 555–conjugated secondary antibody. The neurons were mounted and imaged under a Zeiss LSM 710 confocal microscope.
Supplementary Material
HIGHLIGHTS.
Cornichons are required for synaptic expression of GluA1-containing AMPA receptors.
Cornichons are required for GluA1-containing AMPA receptor trafficking.
TARP γ-8 prevents a functional association of cornichons with non-GluA1 subunits.
Acknowledgments
This work was funded by grants from the US NIMH to R.A.N., a grant from the Brain and Behavior Research Fund to B.E.H.. C.Z. and K.W.R. were supported by the Intramural Research Program of NINDS. Y.H.S. was supported by the Basic Science Research Program (2011-0011694) and MRC (2012048183) through the National Research Foundation of Korea.
Footnotes
Supplemental information includes Extended Experimental Procedures and eight figures and can be found with this article online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrasfalvy BK, Smith MA, Borchardt T, Sprengel R, Magee JC. Impaired regulation of synaptic strength in hippocampal neurons from GluR1-deficient mice. J Physiol. 2003;552:35–45. doi: 10.1113/jphysiol.2003.045575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokel C, Dass S, Wilsch-Brauninger M, Roth S. Drosophila Cornichon acts as cargo receptor for ER export of the TGFalpha-like growth factor Gurken. Development. 2006;133:459–470. doi: 10.1242/dev.02219. [DOI] [PubMed] [Google Scholar]
- Castillon GA, Watanabe R, Taylor M, Schwabe TM, Riezman H. Concentration of GPI-anchored proteins upon ER exit in yeast. Traffic. 2009;10:186–200. doi: 10.1111/j.1600-0854.2008.00857.x. [DOI] [PubMed] [Google Scholar]
- Coombs ID, Cull-Candy SG. Transmembrane AMPA receptor regulatory proteins and AMPA receptor function in the cerebellum. Neuroscience. 2009;162:656–665. doi: 10.1016/j.neuroscience.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz E. Regulation of AMPA receptors by transmembrane accessory proteins. Eur J Neurosci. 2010;32:261–268. doi: 10.1111/j.1460-9568.2010.07357.x. [DOI] [PubMed] [Google Scholar]
- Douyard J, Shen L, Huganir RL, Rubio ME. Differential neuronal and glial expression of GluR1 AMPA receptor subunit and the scaffolding proteins SAP97 and 4.1N during rat cerebellar development. J Comp Neurol. 2007;502:141–156. doi: 10.1002/cne.21294. [DOI] [PubMed] [Google Scholar]
- Gill MB, Kato AS, Roberts MF, Yu H, Wang H, Tomita S, Bredt DS. Cornichon-2 modulates AMPA receptor-transmembrane AMPA receptor regulatory protein assembly to dictate gating and pharmacology. J Neurosci. 2011;31:6928–6938. doi: 10.1523/JNEUROSCI.6271-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill MB, Kato AS, Wang H, Bredt DS. AMPA receptor modulation by cornichon-2 dictated by transmembrane AMPA receptor regulatory protein isoform. Eur J Neurosci. 2012;35:182–194. doi: 10.1111/j.1460-9568.2011.07948.x. [DOI] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, Nave KA. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis. 2006;44:611–621. doi: 10.1002/dvg.20256. [DOI] [PubMed] [Google Scholar]
- Harmel N, Cokic B, Zolles G, Berkefeld H, Mauric V, Fakler B, Stein V, Klocker N. AMPA receptors commandeer an ancient cargo exporter for use as an auxiliary subunit for signaling. PLoS ONE. 2012;7:e30681. doi: 10.1371/journal.pone.0030681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AC, Nicoll RA. The expanding social network of ionotropic glutamate receptors: TARPs and other transmembrane auxiliary subunits. Neuron. 2011;70:178–199. doi: 10.1016/j.neuron.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato AS, Gill MB, Ho MT, Yu H, Tu Y, Siuda ER, Wang H, Qian YW, Nisenbaum ES, Tomita S, Bredt DS. Hippocampal AMPA receptor gating controlled by both TARP and cornichon proteins. Neuron. 2010a;68:1082–1096. doi: 10.1016/j.neuron.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato AS, Gill MB, Yu H, Nisenbaum ES, Bredt DS. TARPs differentially decorate AMPA receptors to specify neuropharmacology. Trends Neurosci. 2010b;33:241–248. doi: 10.1016/j.tins.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron. 2009;62:254–268. doi: 10.1016/j.neuron.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, Huganir RL. Synaptic expression of the high-affinity kainate receptor subunit KA2 in hippocampal cultures. Neuroscience. 1995;69:383–393. doi: 10.1016/0306-4522(95)00253-f. [DOI] [PubMed] [Google Scholar]
- Roth S, Neuman-Silberberg FS, Barcelo G, Schupbach T. Cornichon and the EGF receptor signaling process are necessary for both anterior-posterior and dorsal-ventral pattern formation in Drosophila. Cell. 1995;81:967–978. doi: 10.1016/0092-8674(95)90016-0. [DOI] [PubMed] [Google Scholar]
- Rouach N, Byrd K, Petralia RS, Elias GM, Adesnik H, Tomita S, Karimzadegan S, Kealey C, Bredt DS, Nicoll RA. TARP gamma-8 controls hippocampal AMPA receptor number, distribution and synaptic plasticity. Nat Neurosci. 2005;8:1525–1533. doi: 10.1038/nn1551. [DOI] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci U S A. 2002;99:13902–13907. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk J, Harmel N, Zolles G, Bildl W, Kulik A, Heimrich B, Chisaka O, Jonas P, Schulte U, Fakler B, Klocker N. Functional proteomics identify cornichon proteins as auxiliary subunits of AMPA receptors. Science. 2009;323:1313–1319. doi: 10.1126/science.1167852. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lu W, Milstein AD, Nicoll RA. The stoichiometry of AMPA receptors and TARPs varies by neuronal cell type. Neuron. 2009;62:633–640. doi: 10.1016/j.neuron.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Suh YH, Milstein AD, Isozaki K, Schmid SM, Roche KW, Nicoll RA. Functional comparison of the effects of TARPs and cornichons on AMPA receptor trafficking and gating. Proc Natl Acad Sci U S A. 2010;107:16315–16319. doi: 10.1073/pnas.1011706107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub C, Tomita S. The regulation of glutamate receptor trafficking and function by TARPs and other transmembrane auxiliary subunits. Curr Opin Neurobiol. 2011;22:488–495. doi: 10.1016/j.conb.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Engelhardt J, Mack V, Sprengel R, Kavenstock N, Li KW, Stern-Bach Y, Smit AB, Seeburg PH, Monyer H. CKAMP44: a brain-specific protein attenuating short-term synaptic plasticity in the dentate gyrus. Science. 2010;327:1518–1522. doi: 10.1126/science.1184178. [DOI] [PubMed] [Google Scholar]
- Wang R, Walker CS, Brockie PJ, Francis MM, Mellem JE, Madsen DM, Maricq AV. Evolutionary conserved role for TARPs in the gating of glutamate receptors and tuning of synaptic function. Neuron. 2008;59:997–1008. doi: 10.1016/j.neuron.2008.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanillo D, Sprengel R, Hvalby O, Jensen V, Burnashev N, Rozov A, Kaiser KM, Koster HJ, Borchardt T, Worley P, et al. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science. 1999;284:1805–1811. doi: 10.1126/science.284.5421.1805. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Mellem JE, Brockie PJ, Madsen DM, Maricq AV. SOL-1 is a CUB-domain protein required for GLR-1 glutamate receptor function in C. elegans. Nature. 2004;427:451–457. doi: 10.1038/nature02244. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.