

Abstract
A library of eighty one adenosine antibiotic analogs was prepared under the Pilot Scale Library Program of the NIH Roadmap initiative from 5′-amino-5′-deoxy-2′,3′-O-isopropylidene-adenosine 3. Diverse aldehyde, sulfonyl chloride and carboxylic acid reactant sets were condensed to 3, in solution-phase fashion, leading after acid-mediated hydrolysis to the targeted compounds in good yields and high purity. No marked anti-tuberculosis or anticancer activity was noted on preliminary cellular testing, but these nucleoside analogs should be useful candidates for other types of biological activity.
Keywords: 5′-amino, carboxamide, sulfonyl-adenosine antibiotic analogs
Introduction
Historically, nucleoside chemistry has primarily focused on low throughput production and screening of analogs for anticancer, antifungal and antiviral antimetabolite activities.1–8 Beyond the traditional nucleosides with an available 5′-hydroxyl that can enter nucleoside metabolic pathways, there are numerous examples of relatively simple to complex nucleoside antibiotics that exhibit diverse alternative mechanisms of action.9,10 Natural nucleoside antibiotics, for example, demonstrate potent activities such as protein synthesis inhibition, glycosyltransferase inhibition and methyltransferase inhibition, among others. 9–13 More recently, there has been a trend to develop approaches for the synthesis and screening of this class of compounds, which do not exhibit typical antimetabolite activities based on nucleoside phosphorylation and incorporation into nucleoside metabolic pathways.14–18 In many respects, the nucleoside scaffolds offer an opportunity for numerous diverse and directionally-oriented substitutions in order to probe for activity against diverse protein binding sites. Furthermore, many of these basic scaffolds are not well represented in commercial chemical space, including the Molecular Library Small Molecule Repository (MLSMR) – see PubChem Substance at http://www.ncbi.nlm.nih.gov/pcsubstance. Hence, we have designed and prepared nucleoside antibiotic-like small molecule libraries under the Pilot Scale Library Program of the NIH Roadmap Initiative to probe specific or general biological activities. We report herein the initial phase of this project which is the synthesis of a library of eighty one 5′-amido-, amino- and sulfonylamido-adenosine analogs derived from 5′-amino-5′-deoxy-2′,3′-O-isopropylidene-adenosine 3 (Scheme 1) using parallel solution phase chemistry.
Scheme 1.

Reagents and conditions: i): phthalamide, PPh3, DIAD; ii): H2NNH2, EtOH; iii): R= a part from N-Boc amino acid: HATU, DIEA, CH3CN; R= a part from an aldehyde: MeOH, molecular sieves, 0–40°C, NaBH4; R= a part from sulfonyl chloride: DMF, CsCO3; ; iv) 50% formic acid, 70°C.
Both 3 or its analog 5′-amino-5′-deoxy-adenosine are suitable precursors for the synthesis of a variety of biologically active nucleoside analogs19–36 dating back to the first report describing the synthesis of 5′-amino-5′-deoxy-5′-N-aminoacyl peptide derivatives of guanosine, and adenosine and their effects on cell-free protein synthesis.37 Hence, the 5′-amino group of the nucleoside ribose moiety was chosen as a site of diversification through robust peptidyl chemistry in order to prepare the target library of 5′-amido derivatives 4-34 (Scheme 1). The resulting nucleoside peptide library was expected to show reasonable stability to dissolution, storage and screening as evidenced for similar aminoacyl functions found in various nucleoside antibiotics such as puromycin, gougerotin, amicetin, and blasticidin S, all known inhibitors of protein synthesis. Thus, 3 was synthesized in two steps (Scheme 1) starting from 2′,3′-O-isopropylidene adenosine 1, which was reacted with triphenylphosphine, 1,3-dihydro-1,3-dioxo-2H-isoindole and diisopropyl azodicarboxylate to give 5′-deoxy-5′-(1,3-dihydro-l,3-dioxo-2H-isoindol-2-y1)-2′,3′-O-isopropylidene adenosine 2. Hydrazinolysis of 2 with hydrazine monohydrate in ethanol gave 3 in 78% yield according to the previously reported method.19
In order to achieve our targeted 5′-amido compounds 4-34 (Fig. 1), we explored several methods for peptide coupling of 3 to diverse and readily available carboxylic acid-substituted or N-tert-butyloxycarbonyl (Boc) amino acid derivatives using 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and N,N′-dicyclohexylcarbodiimide (DCC). HATU [(2-(7-Aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate] has been reported to rapidly provide the peptide linkage in high yield and with little or no racemization. In our hands, the best results were obtained with HATU (1 eq.) and N,N-diisoproplyethylamine (DIEA – 1.5 eq.) in acetonitrile for 30–180 minutes. The utility of HATU allowed the facile preparation of the title compounds with a variety substituents and ready adaptation to a parallel format using a Radleys 12-place carousel reaction station on a 0.5 mmol scale. Subsequent acid-mediated deprotection of the acetonide as well as the Boc protecting group using 50% formic acid, again in a parallel format, furnished the desired compounds 4-34 in quantitative yields and high purity.
Figure 1.

Structures of compounds 4-34
Next, we prepared a diverse small library of 5′-N-alkyl-5′-deoxy-adenosine derivatives 35-67 (Fig. 2) based upon the important biological activity and stability exhibited by known natural analogues. Reductive amination is an efficient method that is readily adaptable to parallel format and, hence, we adapted this reaction to couple compound 3 with thirty three commercially available aldehydes. Successful couplings were achieved in methanol in the presence of molecular sieves to efficiently drive intermediate imine formation. It must be noted that the use of molecular sieves was crucial in terms of yield improvement and reaction time. The reaction was also readily adapted to a parallel format on a Radleys 12-place carousel reaction station at room temperature, although occasionally reactions were facilitated with less soluble aldehydes by warming for the first ten minutes at 40 °C. The resulting aldimines were carefully treated in situ with solid sodium borohydride for one half hour and the reaction was then pre-adsorbed and dried on silica gel without further workup followed by flash chromatography purification. Similarly, the hydrolysis of the isopropylidene blocking groups of the resulting intermediates using 50% formic acid was successful leading to the desired targets 35-67 in quantitative yields.
Figure 2.

Structures of compounds 35-67
The synthesis of sulfonylamide adenosine analogs 68-84 (Fig. 3) was achieved by reacting intermediate 3 with seventeen commercially available sulfonyl chlorides. The reactions were carried out in N,N-dimethylformamide (DMF) at room temperature using cesium carbonate as base followed by hydrolysis of the isopropylidene blocking groups as described above.
Figure 3.
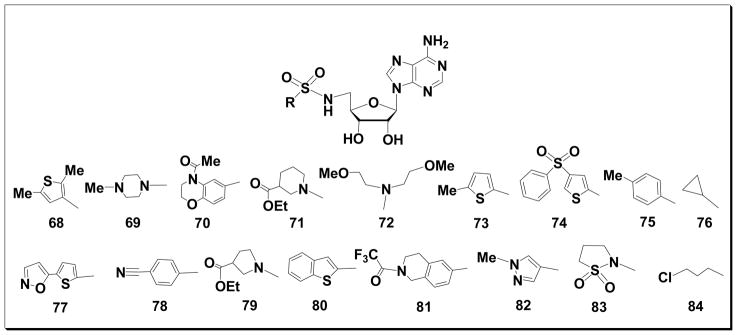
Structures of compounds 68-84
Biological evaluation
The described analogs were tested in vitro for their effectiveness against Mycobacterium tuberculosis (Mtb). None of the compounds showed appreciable activity at compound concentrations lower than 100 μM. Only compound 42 exhibited slight inhibition with an IC50 of 43 μM. Also, they were screened in vitro against three human tumor cell lines (HT29 colon, PC3 prostate and MDA-MB-231 breast). Only compounds 39, 42 and 43 showed toxicity at less than 50 GM in these cells as shown in Table 1 (details of these biological studies are provided in Supporting Information).
Table 1.
Effect of prepared analogs on cell growth
| Compound | Cancer screen | ||
|---|---|---|---|
| HT29 (CC50) | PC3 CC50) | MDA CC50) | |
| 4-38, 40-41 and 44-84 | >50 | >50 | >50 |
| 39 | 14 | 45 | 31 |
| 42 | 9 | 24 | >50 |
| 43 | 12 | >50 | >50 |
CC50 Concentrations of compound required for 50% growth inhibition of cancer cells.
All the prepared analogs have been submitted (20 mg) in the Molecular Libraries Small Molecule Repository (MLSMR) to be screened against a wide range of biological assays (see: www.ncbi.nlm.nih.gov/pcsubstance search term Robert Reynolds). Certain analogs (Fig. 4) exhibited interesting activities in these primary screens. For example, adenosine peptides 9 and 21 were found to be malaria HSP40-mediated yeast toxicity inhibitors; 16 was identified as an inhibitor of G-Protein coupled Receptor Kinase-2 (GRK2) (protein target: beta-adrenergic receptor kinase 1); 39 was identified as an activator of methionine sulfoxide reductase A (MsrA); compound 48 was found to be inhibitor of prion protein 5′ UTR mRNA; 58 and 63 were identified as inhibitors of the histone demethylase GASC-1 (gene amplified in squamous cell carcinoma-1) and vitamin D receptor (VDR) respectively; sulfonylamide adenosine compound 71 inhibited the interaction of LANA 1–23 peptide with purified nucleosomes (containing H2A/H2B histone dimers) while the derivative 74 was found to inhibit scavenger receptor class B using DiI-HDL.
Figure 4.

Some active adenosine analogues
Conclusion
In conclusion, a small library of eighty one adenosine antibiotic-like analogs derived from 5′-amino-5′deoxy-2′,3′-O-isopropylideneadenosine was prepared in good yields and high purity using a parallel solution phase format. No marked anti-tuberculosis nor anticancer activity was witnessed and all the prepared analogs have been submitted for screening in the Molecular Libraries Probe Production Centers Network (MLPCN), preliminary screening via the MLPCN has indicated a variety of interesting activities although full evaluation of the libraries is still under way (see http://www.ncbi.nlm.nih.gov/pcsubstance search term Robert Reynolds).
Supplementary Material
Acknowledgments
This investigation was supported by NIH grant No. 1P41 GM086163-01 (entitled: Pilot-Scale Libraries Based on Nucleoside Templates for the ML Initiative, Robert C. Reynolds, PI.). We thank James M. Riordan, Jackie Truss, Mark Richardson and David Poon of the Molecular and Spectroscopy Section of Southern Research Institute for analytical and spectral data.
References
- 1.Robins RK, Revankar GR. Purine analogs and related nucleosides and nucleotides as antitumor agents. Med Res Rev. 1985;5:273–296. doi: 10.1002/med.2610050302. [DOI] [PubMed] [Google Scholar]
- 2.Plunkett W, Saunders PP. Metabolism and action of purine nucleoside analogs. Pharmacol Ther. 1991;49:239–268. doi: 10.1016/0163-7258(91)90057-s. [DOI] [PubMed] [Google Scholar]
- 3.Huryn DM, Okabe M. AIDS-Driven Nucleoside Chemistry. Chem Rev. 1992;92:1745–1768. [Google Scholar]
- 4.Kolesar JM, Morris AK, Kuhn JG. Purine nucleoside analogues: fludarabine, pentostatin, and cladribine: Part 2: Pentostatin. J Oncol Pharm Pract. 1996;2:211–224. [Google Scholar]
- 5.Mansour TS, Storer R. Antiviral nucleosides. Curr Pharm Des. 1997;3:227–264. [Google Scholar]
- 6.Tan X, Chu CK, Boudinot FD. Development and optimization of anti-HIV nucleoside analogs and prodrugs: A review of their cellular pharmacology, structure-activity relationships and pharmacokinetics. Adv Drug Deliv Rev. 1999;39:117–151. doi: 10.1016/s0169-409x(99)00023-x. [DOI] [PubMed] [Google Scholar]
- 7.Cheson BD, Keating MJ, Plunkett W. Nucleoside Analogues in Cancer Therapy. Vol. 12 Marcel Dekker; New York: 1997. [Google Scholar]
- 8.Shuter J. Antifungal and antiviral agents: a review. Cancer Invest. 1999;17:145–152. [PubMed] [Google Scholar]
- 9.Isono K. Current progress on nucleoside antibiotics. Pharmacol Ther. 1991;52:269–286. doi: 10.1016/0163-7258(91)90028-k. [DOI] [PubMed] [Google Scholar]
- 10.Isono K. Nucleoside antibiotics: structure, biological activity and biosynthesis. J Antibiot. 1988;41:1711–1739. doi: 10.7164/antibiotics.41.1711. [DOI] [PubMed] [Google Scholar]
- 11.Knapp S. Synthesis of Complex Nucleoside Antibiotics. Chem Rev. 1995;95:1859–1876. [Google Scholar]
- 12.Rosemeyer H. The chemodiversity of purine as a constituent of natural products. Chem Biodivers. 2004;1:361–401. doi: 10.1002/cbdv.200490033. [DOI] [PubMed] [Google Scholar]
- 13.Lagoja IM. Pyrimidines as constituent of natural biologically active compounds. Chem Biodivers. 2005;2:1–50. doi: 10.1002/cbdv.200490173. [DOI] [PubMed] [Google Scholar]
- 14.Herforth C, Wiesner J, Franke S, Golisade A, Jomaa A, Link A. Antimalarial Activity of N6-Substituted Adenosine Derivatives. J Comb Chem. 2002;4:302–314. doi: 10.1021/cc0100823. [DOI] [PubMed] [Google Scholar]
- 15.Winans KA, Bertozzi CR. An inhibitor of the human UDP-GlcNAc 4-epimerase identified from a uridine-based library: A strategy to inhibit O-linked glycosylation. Chem Biol. 2002;9:113–129. doi: 10.1016/s1074-5521(02)00093-5. [DOI] [PubMed] [Google Scholar]
- 16.Epple R, Kudirka R, Greenberg WA. Solid-Phase Synthesis of Nucleoside Analogues. J Comb Chem. 2003;5:292–310. doi: 10.1021/cc020087f. [DOI] [PubMed] [Google Scholar]
- 17.Valade A, Urban D, Beau JM. Target-Assisted Selection of galactosyltransferase binders from dynamic combinatorial libraries. An unexpected solution with restricted amounts of the enzyme. ChemBioChem. 2006;7:1023– 1027. doi: 10.1002/cbic.200600022. [DOI] [PubMed] [Google Scholar]
- 18.Townsend AP, Roth S, Williams HEL, Stylianou E, Thomas NR. New S-adenosyl-L-methionine analogues: synthesis and reactivity studies. Org Lett. 2009;11:2976–2979. doi: 10.1021/ol9009859. [DOI] [PubMed] [Google Scholar]
- 19.Kolb M, Danzin C, Barth J, Claverie N. Synthesis and biochemical and properties of chemically stable product analogues of the reaction catalyzed by S-adenosyl l-methionine decarboxylase. J Med Chem. 1982;25:550–556. doi: 10.1021/jm00347a014. [DOI] [PubMed] [Google Scholar]
- 20.Kolb M, Barth J. Synthesis of 5′-[(3-aminooxypropyl)amino]-5′-deoxyadenosine. Lieb Annal der Chem. 1985;5:1036–1040. [Google Scholar]
- 21.Lever OW, Jr, Vestal BR. Bridged isocytosine-adenosine compounds: synthesis and antibacterial evaluation. J Heter Chem. 1986;23:901–903. [Google Scholar]
- 22.Minnick AA, Kenyon GL. General synthetic approach to stable nitrogen analogs of S-adenosylmethionine. J Org Chem. 1988;53:4952–61. [Google Scholar]
- 23.Kvasyuk EI, Kulak T, Mikhailopulo IA, Charubala R, Pfleiderer W. Nucleotides. Part XLV. Synthesis of new (2′-5′) adenylate trimers, containing 5′-amino-5′-deoxyadenosine residues at the 5′-end of the oligoadenylate chain, and of its analogs, carrying a 9-[(2-hydroxyethoxy)methyl]adenine residue at the 2′-terminus. Helv Chim Acta. 1995;78:1777–84. [Google Scholar]
- 24.Gareava LD, Goryunova OV, Yartseva IV, Mashalova NA, Ya Mel’nik S. Synthesis of modified purine ribonucleosides from 3′ (5′)-O-succinyladenosine and 5′-amino-5′-deoxyadenosine. Bioorg Khim. 1995;21:717–723. [Google Scholar]
- 25.Ceulemans G, Vandendriessche F, Rozenski J, Herdewijn P. Synthesis of an uncharged cAMP analog. Nucl Nucl. 1995;14:117–127. [Google Scholar]
- 26.Comstock LR, Rajski SR. Expeditious synthesis of aziridine-based cofactor mimics. Tetrahedron. 2002;58:6019–6026. [Google Scholar]
- 27.Ciuffreda P, Loseto A, Santaniello E. Deamination of 5′-substituted-2′,3′-isopropylidene adenosine derivatives catalyzed by adenosine deaminase (ADA, EC 3.5.4.4) and complementary enzymatic biotransformations catalyzed by adenylate deaminase (AMPDA, EC 3.5.4. 6): a viable route for the preparation of 5′-substituted inosine derivatives. Tetrahedron. 2002;58:5767–5771. [Google Scholar]
- 28.Manfredini S, Solaroli N, Angusti A, Nalin F, Durini E, Vertuani S, Pricl S, Ferrone M, Spadari S, Focher F, Verri A, De Clercq E, Balzarini J. Design and synthesis of phosphonoacetic acid (PPA) ester and amide bioisosters of ribofuranosylnucleoside diphosphates as potential ribonucleotide reductase inhibitors and evaluation of their enzyme inhibitory, cytostatic, and antiviral activity. Antiviral Chem Chemo. 2003;4:183–194. doi: 10.1177/095632020301400403. [DOI] [PubMed] [Google Scholar]
- 29.Jagtap PG, Southan GJ, Baloglu E, Ram S, Mabley JG, Marton A, Salzman A, Szabo C. The discovery and synthesis of novel adenosine substituted 2:3-dihydro-1H-isoindol-1-ones: potent inhibitors of poly(ADP-ribose) polymerase-1 (PARP-1) Bioorg Med Chem Lett. 2004;14:81–85. doi: 10.1016/j.bmcl.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 30.Enkvis E, Lavogina D, Raidaru G, Vaasa A, Viil I, Lust M, Viht K, Uri A. Conjugation of Adenosine and Hexa-(D-arginine) Leads to a Nanomolar Bisubstrate-Analog Inhibitor of Basophilic Protein Kinases. J Med Chem. 2006;49:7150–7159. doi: 10.1021/jm0605942. [DOI] [PubMed] [Google Scholar]
- 31.Bisseret P, Thielges S, Bourg S, Miethke M, Marahiel MA, Eustache J. Synthesis of a 2-indolylphosphonamide derivative with inhibitory activity against yersiniabactin biosynthesis. Tetrahedron Lett. 2007;48:6080–6083. [Google Scholar]
- 32.Dowden J, Hong W, Parry RV, Pike RA, Ward SG. Toward the development of potent and selective bisubstrate inhibitors of protein arginine methyltransferases. Bioorg Med Chem Lett. 2010;20:2103–2105. doi: 10.1016/j.bmcl.2010.02.069. [DOI] [PubMed] [Google Scholar]
- 33.Mori S, Iwase K, Iwanami N, Tanaka Y, Kagechika H, Hirano T. Development of novel bisubstrate-type inhibitors of histone methyltransferase SET7/9. Bioorg Med Chem. 2010;18:8158–8166. doi: 10.1016/j.bmc.2010.10.022. [DOI] [PubMed] [Google Scholar]
- 34.Dowden J, Pike RA, Parry RV, Hong W, Muhsen UA, Ward SG. Small molecule inhibitors that discriminate between protein arginine N-methyltransferases PRMT1 and CARM1. Org. Biom. Chem. 2011; 9: 7814–7821. Hong, W. Dowden J. Facile synthesis of N-6 adenosine modified analogue toward S-adenosyl methionine derived probe for protein arginine methyltransferases. Chinese Chem Lett. 2011;22:1439–1442. doi: 10.1039/c1ob06100c. [DOI] [PubMed] [Google Scholar]
- 35.Yao Y, Chen P, Diao J, Cheng G, Deng L, Anglin JL, Prasad BVV, Song Y. Selective Inhibitors of Histone Methyltransferase DOT1L: Design, Synthesis, and Crystallographic Studies. J Amer Chem Soc. 2011;133:16746–16749. doi: 10.1021/ja206312b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Felczak K, Chen L, Wilson D, Williams J, Vince R, Petrelli R, Jayaram HN, Kusumanchi P, Kumar M, Pankiewicz KW. Cofactor-type inhibitors of inosine monophosphate dehydrogenase via modular approach: targeting the pyrophosphate binding sub-domain. Bioorg Med Chem. 2011;19:1594–1605. doi: 10.1016/j.bmc.2011.01.042. [DOI] [PubMed] [Google Scholar]
- 37.Robins MJ, Simon LN, Stout MG, Ivanovics GA, Schweizer MP, Rousseau RJ, Robins RK. Nucleoside peptides. I. Synthesis of 5′-deoxy-5′-amino-5′-N-aminoacyl peptide derivatives of guanosine, adenosine, and 2′-deoxyadenosine and their effect of cell-free protein synthesis. J Amer Chem Soc. 1971;98:1474–1480. doi: 10.1021/ja00735a025. [DOI] [PubMed] [Google Scholar]
- 38.Ananthan S, Faaleolea ER, Goldman RC, Hobrath JV, Kwong CD, Laughon BE, Maddry JA, Mehta A, Rasmussen L, Reynolds RC, Secrist JA. 3rd; Shindo, N., Showe, D. N., Sosa, M. I., Suling, W. J., White, E. L. High-throughput screening for inhibitors of Mycobacterium tuberculosis H37Rv. Tuberculosis. 2009;89:334–353. doi: 10.1016/j.tube.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.