

Abstract
BACKGROUND
Amplification and activating mutations of the epidermal growth factor receptor (EGFR) oncogene are molecular hallmarks of glioblastomas. We hypothesized that deletion of NFKBIA (encoding nuclear factor of κ-light polypeptide gene enhancer in B-cells inhibitor-α), an inhibitor of the EGFR-signaling pathway, promotes tumorigenesis in glioblastomas that do not have alterations of EGFR.
METHODS
We analyzed 790 human glioblastomas for deletions, mutations, or expression of NFKBIA and EGFR. We studied the tumor-suppressor activity of NFKBIA in tumor-cell culture. We compared the molecular results with the outcome of glioblastoma in 570 affected persons.
RESULTS
NFKBIA is often deleted but not mutated in glioblastomas; most deletions occur in nonclassical subtypes of the disease. Deletion of NFKBIA and amplification of EGFR show a pattern of mutual exclusivity. Restoration of the expression of NFKBIA attenuated the malignant phenotype and increased the vulnerability to chemotherapy of cells cultured from tumors with NFKBIA deletion; it also reduced the viability of cells with EGFR amplification but not of cells with normal gene dosages of both NFKBIA and EGFR. Deletion and low expression of NFKBIA were associated with unfavorable outcomes. Patients who had tumors with NFKBIA deletion had outcomes that were similar to those in patients with tumors harboring EGFR amplification. These outcomes were poor as compared with the outcomes in patients with tumors that had normal gene dosages of NFKBIA and EGFR. A two-gene model that was based on expression of NFKBIA and O6-methylguanine DNA methyltransferase was strongly associated with the clinical course of the disease.
CONCLUSIONS
Deletion of NFKBIA has an effect that is similar to the effect of EGFR amplification in the pathogenesis of glioblastoma and is associated with comparatively short survival.
Glioblastoma multiforme is the most common and most deadly primary brain tumor.1 It is a complex disease, in which many signaling pathways are disrupted.2–7 Almost all glioblastomas have excessive activation of the epidermal growth factor receptor (EGFR) pathway,8 often brought about by amplification (see the Glossary for this and other key terms) or activating mutations of the EGFR oncogene.9 Alternative mechanisms of the activation of the EGFR pathway may exist in tumors that do not have alterations of EGFR.
Nuclear factor of κ-light polypeptide gene enhancer in B-cells (NF-κB) is a transcription factor activated by the EGFR pathway.10,11 Aberrant constitutive activation of NF-κB has been observed in glioblastomas.12–15 NF-κB inhibitor-α (NFKBIA) represses NF-κB and, hence, signaling in the NF-κB and EGFR pathways.11,16 The discovery of mutations of NFKBIA, as well as research showing that there is an enrichment of specific single-nucleotide polymorphisms and haplotypes of NFKBIA in Hodgkin’s lymphoma, colorectal cancer, melanoma, hepatocellular carcinoma, breast cancer, and multiple myeloma, suggests that NFKBIA is a tumor suppressor.17–29 This possibility, together with evidence of the activation of NF-κB by EGFR activity in glioblastomas30 and our previous studies showing an association between the down-regulation of NFKBIA in glioblastoma cells and a lack of response to therapy,14 prompted our investigation of deletions, mutations, and expression of NFKBIA in glioblastomas, their associations with EGFR amplification and mutation, and the association between these molecular features and the clinical outcome.
METHODS
TUMOR SAMPLES AND PATIENTS
We used 10 study sets of patients with glioblastoma who were treated between July 26, 1989, and August 12, 2009, and studied the patients and their tumors. The demographic characteristics of the patients, the characteristics of the disease, and the types of data that were used are shown in Table 1.
Table 1.
Patient Characteristics and Types of Data Used in 10 Study Sets.*
| Study Set | No. of Patients | No. with Matched DNA from Non-neoplastic Tissue | Sex | Age(yr) | Vital Status | Median Weeks of Follow-up (Range) | Data Type† | Source |
|---|---|---|---|---|---|---|---|---|
| 1 | 219‡ | 91 | Female: 77 Male: 130 |
55.8±15.1 | Dead: 192 Alive: 15 |
50.6 (1.1–503.4) | G, E, C | Cancer Genome Atlas Project |
| 2 | 182 | 0 | NA | NA | NA | NA | G | REMBRANDT release 1.5.4.1 |
| 3 | 46 | 0 | NA | NA | NA | NA | G | REMBRANDT release 1.5.3 |
| 4 | 36 | 0 | NA | NA | NA | NA | G | Northwestern University |
| 5 | 32 | 0 | NA | NA | NA | NA | S | Stanford University |
| 6 | 15 | 0 | NA | NA | NA | NA | S | Barrow Neurological Institute |
| 7 | 49§ | 0 | Female: 15 Male: 34 |
49.9±12.1 | Dead: 46 Alive: 3 |
70.0 (3.0–313.0) | E, C | M.D. Anderson Cancer Center (GEO accession no., GSE4271) |
| 8 | 47 | 0 | Female: 25 Male: 22 |
50.5±15.6 | Dead: 34 Alive: 13 |
42.0 (1.0–178.0) | E, C | University of California, Los Angeles (GEO accession no., GSE4412) |
| 9 | 191 | 0 | Female: 74 Male: 117 |
53.8±13.6 | Dead: 176 Alive: 15 |
55.6 (1.0–479.0) | E, C | Multi-institutional (GEO accession no., GSE13041) |
| 10 | 76 | 0 | Female: 21 Male: 55 |
51.1±9.1 | Dead: 63 Alive: 13 |
66.9 (6.1–311.9) | E, M, C | Phase 2 trial, EORTC-NCIC phase 3 trial (GEO accession no., GSE7696)¶ |
Plus–minus values are means ± SD. GEO denotes Gene Expression Omnibus, and NA not available.
The types of data include clinical-outcome data (C), gene-expression data (E), gene copy number data (G), methylation status of the MGMT promoter (M), and sequencing data (S).
There were 219 patients in study set 1; clinical data were available for 207 of those patients.
Twenty-two patients had matched tumor pairs from the initial diagnosis and recurrent disease; therefore, 71 tumors were assessed in study set 7.
Study set 10 included 76 patients with glioblastoma who were treated as part of a phase 2 trial or a European Organization for Research and Treatment of Cancer (EORTC)/National Cancer Institute of Canada (NCIC) randomized phase 3 trial (22981-26981/CE.38), both of which evaluated the addition of concomitant and adjuvant temozolomide to radiotherapy.31,32
CELL LINES AND PREPARATION OF GENOMIC DNA
We obtained glioblastoma cell lines LN229, U87, and U118 from the American Type Culture Collection. PT67 retroviral packaging cells were grown according to the instructions of the manufacturer (Clontech). Primary tumor-cell cultures were generated from malignant glioma specimens from patients enrolled in a study that was conducted at Northwestern University with approval from the institutional review board. Primary cancer stem-like cell cultures were generated from nine glioblastomas in Study Set 4. Genomic DNA from tumor samples and cell lines was isolated with the use of DNeasy kits (Qiagen) and was quantified with the use of spectrophotometry. Detailed descriptions of cell biologic and molecular biologic analyses and experimental design are provided in the Supplementary Appendix, available with the full text of this article at NEJM.org.
COPY-NUMBER VARIATION AND MUTATIONAL ANALYSES
Details of the tissue collection, methods of generation and preprocessing of multidimensional genomic data, analysis of copy-number variation, and sequence analysis are provided in the Supplementary Appendix. We sequenced the NFKBIA coding region in 32 glioblastomas in study set 5 and, along with the promoter region, in 15 cell lines in study set 6. We analyzed activating EGFR mutations in 91 patients with glioblastoma in study set 1 and DNA samples from non-neoplastic tissue from those patients7 and tested for an association between the presence of activating EGFR mutations and the presence of a deletion affecting NFKBIA.
STATISTICAL ANALYSIS
Survival curves were estimated with the use of the Kaplan–Meier product-limit method, and survival distributions were compared across groups with the use of the log-rank test. We performed univariate and multivariate Cox proportional-hazards regression analyses, with overall survival as the dependent variable and NFKBIA and EGFR dosage or NFKBIA and O6-methylguanine DNA methyl-transferase (MGMT) expression as the primary predictor. In interpreting hazard ratios, we dichotomized NFKBIA expression (in all models) at the median, and in the NFKBIA–MGMT combined risk-group model, we dichotomized MGMT expression at the 60th percentile (i.e., 60% of tumors with comparatively high MGMT expression vs. 40% of tumors with comparatively low MGMT expression). The 60th percentile of MGMT expression was prespecified to define MGMT “high-risk” tumors (i.e., the 60% of tumors that showed the highest expression of MGMT) on the basis of previously noted frequencies of MGMT promoter methylation in glioblastoma.33–35 We also used alterations of gene dosage (wild-type vs. deleted in the case of NFKBIA and wild-type vs. amplified in the case of EGFR) as binary predictors and survival as the outcome.
We used, where appropriate, Wilcoxon rank-sum and signed-rank tests, an unpaired t-test, and a two-way contingency table analysis that was based on Pearson’s chi-square test and Fisher’s exact test. We used linear regression analysis to assess the relationship between NFKBIA and EGFR expression. We computed odds ratios in the two-way contingency-table analysis using Woolf’s method for variance estimation.36
RESULTS
DELETIONS OF NFKBIA
We observed a common heterozygous deletion encompassing NFKBIA in 53 of the 219 glioblastomas (24.2%) in study set 1 (Fig. 1A). An analysis of NFKBIA copy number in the glioblastomas in study set 2 revealed fewer than 1.5 copies of NFKBIA in 37 of the 182 tumors (20.3%). There were heterozygous deletions of NFKBIA in 13 of the 46 glioblastomas (28.3%) in study set 3 and in 6 of 27 glioblastomas (22.2%) and 2 of 9 glioblastoma-derived cancer stemlike cell populations (22.2%) in study set 4.
Figure 1. NFKBIA Deletions in Glioblastomas.

The analysis of copy-number variation for chromosome 14 (Panel A) was based on circular binary segmentation in 219 glioblastomas in study set 1. Gene dosages are mapped according to gene order on chromosome 14. NFKBIA is deleted (del) in 24.2% of tumors (yellow line, NFKBIA locus on 14q13). The bar diagram at the bottom of the panel shows the gene-dosage profiles for NFKBIA. Gene-dosage values indicate the log2 ratio of red (R, Cy5) to green (G, Cy3) intensity of the fluorescence dye (or log2R/G), as estimated with the use of the circular binary segmentation algorithm. The deletion of NFKBIA is associated with significant loss of NFKBIA expression in the 175 glioblastomas in study set 1 that had combined gene and transcript data (Panel B). Values for gene dosage and gene expression are presented as log2R/G ratios, as estimated by the circular binary segmentation and robust multigene average preprocessing algorithms, respectively. The box plots show the smallest and largest observations (upper and lower whiskers, respectively), the interquartile range (box), and the median (red line). Data points that are more than 1.5 times the interquartile range lower than the first quartile or 1.5 times the interquartile range higher than the third quartile were considered to be outliers. The P value was calculated with the use of the Wilcoxon rank-sum test. Gene-dosage profiles for NFKBIA and EGFR across 188 glioblastomas in study set 1 are shown (Panel C), along with their relationship to four molecular subtypes of glioblastoma (classical, mesenchymal, neural, and proneural). A corresponding two-way contingency-table analysis reveals a significant association of NFKBIA deletion with the nonclassical subtypes. CI denotes confidence interval.
In the 175 tumors in study set 1 with data on NFKBIA dosage and expression, we found significantly lower NFKBIA mRNA expression in tumors in which NFKBIA was deleted than in those with two intact copies of NFKBIA (P = 8×10−9 by the Wilcoxon rank-sum test) (Fig. 1B).
We sequenced the coding region of NFKBIA in the 32 glioblastomas in study set 5 and both promoter and coding regions of NFKBIA in the 15 cell lines in study set 6. We found no mutations in either coding or promoter sequences, suggesting that inactivation of NFKBIA in glioblastoma cells occurs primarily through the loss of gene copy number (i.e., a reduction of gene dosage).
NFKBIA DELETION AND EGFR ALTERATION
Recent studies have distinguished between classical and nonclassical (i.e., mesenchymal, neural, and proneural) subtypes of glioblastoma.9 EGFR amplifications are common (80.0%) in the classical subtype (Fig. 1C). Among the 188 glioblastomas in study set 1 with data on gene dosage and subtype, we found that NFKBIA deletions are rare (5.9%) in classical glioblastomas and more common (32.1%) in nonclassical glioblastomas (P = 5×10−4 by Pearson’s chi-square test; odds ratio for deletions in classical glioblastomas, 0.13; 95% confidence interval [CI], 0.04 to 0.42) (Fig. 1C). Irrespective of subtype, we observed a pattern suggesting a degree of mutual exclusivity between NFKBIA deletion and EGFR amplification (Fig. 2). In study set 1, we observed NFKBIA deletion or EGFR amplification, but not both, in 115 of 219 tumors (52.5%); only 11 tumors (5.0%) harbored concomitant NFKBIA deletion and EGFR amplification (P = 2×10−3 by Pearson’s chi-square test; odds ratio for concomitant deletion and amplification, 0.33; 95% CI, 0.16 to 0.69) (Fig. 2A). We observed a similar pattern in 46 glioblastomas in study set 3 (P = 0.01 by Fisher’s exact test; odds ratio, 0.00; 95% CI, 0.00 to 0.56): no tumor harbored both alterations (Fig. 2B).
Figure 2. Pattern of NFKBIA Deletion and EGFR Amplification in Glioblastomas.
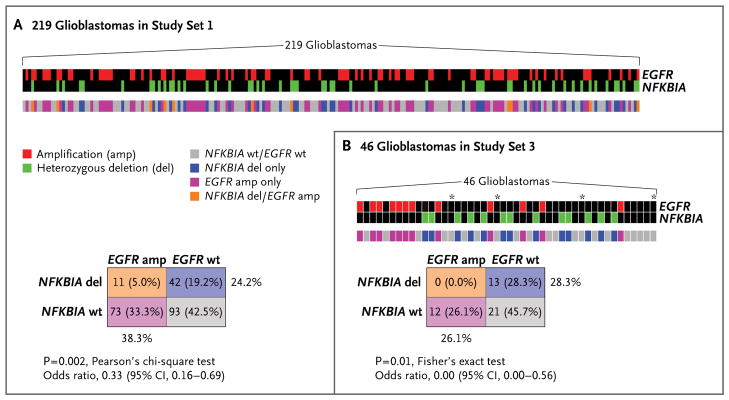
Panel A shows gene-dosage profiles for NFKBIA and EGFR across 219 glioblastomas in study set 1 and their relationship to each other; the color key indicates the status of both genes in individual tumors. A corresponding two-way contingency table shows that NFKBIA deletion and EGFR amplification occur mutually exclusively in glioblastomas. Panel B shows the relationship between NFKBIA deletion and EGFR amplification in 46 glioblastomas in study set 3 and a corresponding two-way contingency table. Asterisks indicate samples in which there was potential contamination by nontumor tissue.
In the 83 tumors in study set 1 for which data on gene dosage and somatic mutation for EGFR were available, NFKBIA deletions and EGFR alteration (amplification, activating mutation, or both) were unlikely to occur in the same tumor, although the relative mutual exclusivity of these events reached only marginal significance (P = 0.05 by Fisher’s exact test; odds ratio, 0.35; 95% CI, 0.13 to 1.00). The pattern of relative mutual exclusivity between alterations of NFKBIA and EGFR extended to gene expression; tumors with diminished NFKBIA expression from gene deletion had comparatively low EGFR expression, and tumors with elevated expression of EGFR from gene amplification expressed NFKBIA (P = 9×10−3 by linear regression) (Fig. 1 in the Supplementary Appendix).
TUMOR SUPPRESSION IN CELL CULTURE
Retrovirally mediated reexpression of NFKBIA in established glioblastoma cell lines with heterozygous NFKBIA deletions (Fig. 2 in the Supplementary Appendix) inhibited malignant cell behaviors, including cell-cycle transition, growth, migration, and colony formation; it also reduced cell viability and induced cellular senescence (Fig. 3 in the Supplementary Appendix). Furthermore, tumor cells became more sensitive to temozolomide — the preferred chemotherapy for glioblastoma32 — and its induction of programmed cell death (Fig. 4 in the Supplementary Appendix). These data establish the tumor-suppressor activity of NFKBIA in glioblastoma cells.
In primary tumor cultures from three human glioblastomas with different NFKBIA and EGFR status — deleted NFKBIA and wild-type (i.e., normal-gene-dosage) EGFR, wild-type NFKBIA and amplified EGFR, and wild-type NFKBIA and EGFR (i.e., both genes present in two copies) — retroviral expression of NFKBIA substantially reduced cell viability in the NFKBIA-deleted tumor and in the EGFR-amplified tumor (P = 2×10−4 and P = 0.02, respectively, by unpaired t-test) but not in the tumor with normal dosages of each gene (P = 0.21) (Fig. 3). These data support our conclusion that NFKBIA suppresses the growth of glioblastomas in which EGFR signaling pathway dependence is brought about by deletion of NFKBIA or amplification of EGFR.
Figure 3. Effect of NFKBIA Expression in Patient-Derived Glioblastoma Cultures.

Primary cultures derived from three patients with glioblastoma are shown, each with a distinct NFKBIA and EGFR status (Patient 1: NFKBIA deleted [del, green font] and EGFR wild type [wt]; Patient 2: NFKBIA wt and EGFR amplified [amp, red font]; Patient 3: NFKBIA wt and EGFR wt). Cultures were infected with a retroviral vector expressing Flag-tagged NFKBIA (NFKBIA+). Protein lysates were subjected to immunoblotting for the detection of Flag-NFKBIA protein expression in relation to α-tubulin loading control. Mean cell viability, measured spectrophotometrically through bio-reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide dye by dehydrogenase enzymes of metabolically active cells, reveals a significant reduction in cell viability after expression of NFKBIA in NFKBIA-deleted tumor cells or in EGFR-amplified tumor cells, but no effect in tumor cells with wild-type status for both genes. P values were calculated with the use of an unpaired t-test. T bars indicate standard errors.
NFKBIA AND OUTCOMES IN PATIENTS WITH GLIOBLASTOMA
Cox proportional-hazards regression analysis of the 188 glioblastomas in study set 1 for which data on gene copy number and survival were available showed that patients with two copies of NFKBIA survived significantly longer than did patients with tumors harboring a deletion of NFKBIA (hazard ratio for death with two copies vs. deletion of NFKBIA, 0.45; 95% CI, 0.23 to 0.89; P = 0.02). A multivariate Cox model suggested that this association is independent of the prognostic covariate, the age of the patient (hazard ratio, 0.40; 95% CI, 0.21 to 0.79; P = 8×10−3). Similarly, models incorporating the patient’s age and either EGFR dosage or clinically relevant molecular subtypes of glioblastoma9 confirmed an independent association between survival and normal dosage of NFKBIA (hazard ratio for death, 0.40; 95% CI, 0.20 to 0.78; P = 7×10−3; and hazard ratio, 0.39; 95% CI, 0.20 to 0.77; P = 7×10−3, respectively).
Among the 171 patients in study set 1 with newly diagnosed glioblastoma for whom data on both gene dosage and survival were available, we found no difference in time to death between the patients with an isolated NFKBIA deletion (i.e, NFKBIA deletion without EGFR amplification) and those with isolated EGFR amplification (i.e., EGFR amplification without NFKBIA deletion) (hazard ratio for death with isolated NFKBIA deletion vs. isolated EGFR amplification, 1.13; 95% CI, 0.72 to 1.79; P = 0.57 by the Cox model) (Fig. 4A). In contrast, patients with tumors that had either an NFKBIA deletion or EGFR amplification had shorter survival, as measured from the time of diagnosis, than did those with normal dosages of both NFKBIA and EGFR (hazard ratio for death with NFKBIA deletion, 1.69; 95% CI, 1.09 to 2.63; P = 0.02 by the Cox model; and hazard ratio for death with EGFR amplification, 1.48; 95% CI, 1.02 to 2.13; P = 0.04 by the Cox model) (Fig. 4A). The estimated median survival times were 46 weeks for patients whose tumors harbored an isolated NFKBIA deletion, 53 weeks for those whose tumors had isolated EGFR amplification, and 67 weeks for those whose tumors had normal dosages of both NFKBIA and EGFR.
Figure 4. NFKBIA and Survivalin Patients with Glioblastomas.

Kaplan–Meier estimates of overall survival are shown for 171 patients in study set 1 with newly diagnosed glioblastomas (Panel A), with patients stratified into three subgroups according to the presence of tumors with NFKBIA and EGFR wild-type (wt) status, NFKBIA deletion (del) without EGFR amplification (amp), or EGFR amplification without NFKBIA deletion (9 patients with tumors that had alteration of both NFKBIA and EGFR were omitted owing to the small sample size). Kaplan–Meier estimates of survival are shown for the 49 patients in study set 7 (Panel B), with patients stratified according to median NFKBIA expression. The combined NFKBIA and O6-methylguanine DNA methyltransferase (MGMT) risk-group models are shown for 191 patients with glioblastomas in study set 9 (Panel C) and for 42 patients with newly diagnosed glioblastomas in study set 10 who were treated with radiotherapy plus concomitant and adjuvant temozolomide (Panel D). Assignment of patients to low-, intermediate-, or high-risk groups was based on NFKBIA expression (dichotomized at the median) and MGMT status (MGMT expression dichotomized at the 60th percentile or based on MGMT promoter methylation status). In Panel C, NFKBIA expression higher than the median combined with MGMT expression lower than the 40th percentile denotes a low-risk group, and NFKBIA expression lower than the median combined with MGMT expression higher than the 60th percentile denotes a high-risk group. In Panel D, NFKBIA expression higher than the median combined with methylated MGMT promoter status denotes a low-risk group, and NFKBIA expression lower than the median combined with unmethylated MGMT promoter status denotes a high-risk group; all other cases were assigned to an intermediate-risk group. Small vertical lines indicate patients who were alive at the last follow-up assessment. P values were calculated with the use of the log-rank test.
A correlation between NFKBIA expression, as assessed by microarray analysis, and survival was established in three different groups. In a Cox proportional-hazards regression analysis of the 49 glioblastomas in study set 7, greater NFKBIA expression was associated with longer survival (hazard ratio for death, 0.51; 95% CI, 0.35 to 0.75; P = 6×10−4). A multivariate Cox model incorporating the patient’s age, molecular subtype,4,37 and MGMT expression — currently the most potent predictor of response to temozolomide therapy33 — yielded an independent association of NFKBIA with survival (hazard ratio, 0.44; 95% CI, 0.29 to 0.66; P = 7×10−5). A two-class model in which patients were stratified according to median NFKBIA expression also showed an association between NFKBIA expression and longer survival (hazard ratio with high vs. low expression of NFKBIA, calculated with the use of a Cox model, 0.31; 95% CI, 0.16 to 0.58; P = 2×10−5 by the log-rank test); the estimated median survival for patients with tumors that had high NFKBIA expression was 131 weeks, as compared with 57 weeks for patients with low NFKBIA expression (Fig. 4B). This relationship was also present in the 47 glioblastomas in study set 8 and the 191 glioblastomas in study set 9, both in multivariate models (hazard ratio, 0.57; 95% CI, 0.33 to 0.98; P = 0.04; and hazard ratio, 0.73; 95% CI, 0.60 to 0.90; P = 3×10−3, respectively) and in two-class models (hazard ratio for high vs. low NFKBIA expression, calculated with the use of a Cox model, 0.43; 95% CI, 0.21 to 0.86; P = 0.02 by the log-rank test; and hazard ratio, 0.61; 95% CI, 0.45 to 0.82; P = 1×10−3, respectively) (Fig. 5 in the Supplementary Appendix).
An analysis of tumor recurrence in 22 patients in study set 7 showed that tumors with high expression of NFKBIA in the primary tumor had significantly lower levels of NFKBIA in the recurrent tumor (P = 0.02 by Wilcoxon signed-rank test), and expression levels in recurrent tumors were similar to those in tumors with low expression initially (Fig. 6 in the Supplementary Appendix). Nonetheless — and consistent with data obtained from tumors in study sets 7, 8, and 9 — NFKBIA expression in the primary tumor was associated with comparatively long survival (hazard ratio for death with high vs. low NFKBIA expression, calculated with the use of a Cox model, 0.32; 95% CI, 0.12 to 0.88; P=0.02 by the log-rank test) (Fig. 6 in the Supplementary Appendix).
The association between NFKBIA expression and longer survival was also present in the case of tumors with high-risk MGMT status. One third to 45% of glioblastomas have a comparatively methylated (commonly referred to as hypermethylated) MGMT promoter, which silences expression of the corresponding MGMT mRNA transcript.33–35 These low-risk tumors have a favorable response to temozolomide and radiation therapy.33 The remaining 55 to 67% of tumors (with hypomethylated MGMT promoter) are at high risk for rapid treatment failure and disease progression since they have at least one copy of MGMT with an unmethylated promoter.
Since there is a strong inverse correlation between MGMT promoter methylation and MGMT gene expression in glioblastoma,34 we considered the 60% of tumors with the highest MGMT expression (and thus putatively hypomethylated promoter) in microarray-based gene-expression analysis to represent MGMT high-risk tumors. Among 29 patients in study set 7 with MGMT high-risk tumors, those with NFKBIA expression above the median lived longer than did those with lower NFKBIA expression (estimated median survival, 118 weeks vs. 53 weeks; hazard ratio for death with high vs. low NFKBIA expression, 0.14; 95% CI, 0.05 to 0.40; P = 3×10−5 by the log-rank test) (Fig. 7A in the Supplementary Appendix). Similar analyses of data from 28 patients in study set 8 and 114 patients in study set 9 yielded similar findings (Fig. 7B and 7C in the Supplementary Appendix). A total of 21 of the 76 patients in study set 10 had glioblastomas with an unmethylated MGMT promoter and were treated with radiotherapy and temozolomide. In these 21 patients, high NFKBIA expression (defined as expression above the median), as compared with low NFKBIA expression, was associated with longer survival (estimated median survival of 64 weeks vs. 40 weeks; hazard ratio for death with high vs. low NFKBIA expression, 0.27; 95% CI, 0.09 to 0.79; P = 0.01 by the log-rank test) (Fig. 7D in the Supplementary Appendix).
NFKBIA, MGMT, AND THE COURSE OF DISEASE
To determine the usefulness of a two-gene outcome-predictor model that is based on the status of both NFKBIA and MGMT, we divided the 191 glioblastomas in study set 9 into groups that were defined according to the prespecified cutoff points for expression of NFKBIA (median) and MGMT (60% of tumors with comparatively high MGMT expression vs. 40% of tumors with comparatively low MGMT expression): one high-risk group with low NFKBIA and high MGMT expression, one low-risk group with high NFKBIA and low MGMT expression, and one intermediate-risk group with either low NFKBIA and low MGMT expression or high NFKBIA and high MGMT expression. This model yielded a strong association between risk status and survival (P = 2×10−4 by the log-rank test) (Fig. 4C). The estimated median survival in the low-risk, intermediate-risk, and high-risk groups was 92 weeks, 59 weeks, and 44 weeks, respectively. A model with groups defined according to the median expression of both NFKBIA and MGMT produced similar results; the estimated median survival was 89 weeks in the low-risk group, 57 weeks in the intermediate-risk group, and 45 weeks in the high-risk group (P = 6×10−4 by the log-rank test) (Fig. 8A in the Supplementary Appendix).
When the 74 patients in study set 10 with known MGMT promoter status (methylated vs. un-methylated) were similarly stratified into three risk groups, we found significant differences in estimated median survival: 91 weeks in the low-risk group, 63 weeks in the intermediate-risk group, and 45 weeks in the high-risk group (P = 7×10−4 by the log-rank test) (Fig. 8B in the Supplementary Appendix). The association between risk and survival was even more pronounced in the case of patients with newly diagnosed tumors who were treated with radiotherapy and temozolomide (P = 3×10−6 by the log-rank test) (Fig. 4D): the estimated median survival in the low-risk, intermediate-risk, and high-risk groups was 122 weeks, 71 weeks, and 35 weeks, respectively.
DISCUSSION
Our data support a role for NFKBIA in the suppression of glioblastoma tumors. The presence of NFKBIA deletions in some glioblastoma cancer stem cells suggests that such deletions can emerge early in the pathogenesis of glioblastoma. Our data show that loss of NFKBIA can also be associated with disease progression and tumor recurrence.
The general, albeit not absolute, mutual exclusivity of NFKBIA deletion and EGFR amplification has been reported in the case of other gene pairs in signaling pathways pertinent to the biologic nature of glioblastomas. For example, a decrease in retinoblastoma pathway signaling is achieved through a mutually exclusive mutation of the tumor-suppressor gene RB1 or deletion of the tumor-suppressor genes CDKN2A and CDKN2B.7 Similarly, mutations in the tumor-suppressor gene TP53 and deletions affecting CDKN2A, both of which reduce TP53 pathway signaling, appear to be mutually exclusive in glioblastomas.38
The fact that tumors with deletion of NFKBIA and those with EGFR amplification have similarly poor outcomes suggests that NFKBIA deletion can substitute for EGFR amplification in the pathogenesis of glioblastoma. This finding is consistent with our observation that deletion of NFKBIA occurs more commonly in nonclassical glioblastomas than in classical glioblastomas, which have EGFR amplification more often than do nonclassical glioblastomas. Which aberration occurs may depend on the tumor’s cell of origin and its pattern of accumulation of the other genetic lesions that define glioblastoma subtypes.9
We have observed, in a previous study, that glioblastoma cells that do not respond to temozolomide chemotherapy have comparatively low expression of NFKBIA14 and, in this study, that increasing NFKBIA expression in these cells sensitizes them to temozolomide. Our findings, together with research showing the role of NFKBIA as a gatekeeper for EGFR signaling11 and the involvement of EGFR activation in the lack of response of glioblastoma cells to chemotherapy and radiotherapy,39 collectively suggest that NFKBIA-mediated sensitization of glioblastoma cells to temozolomide reflects NFKBIA abrogation of EGFR signaling.
Our observation that NFKBIA status is independently associated with survival in several patient groups supports the importance of NFKBIA as a determinant of glioblastoma behavior, including the response to temozolomide, and suggests that it would be useful to include the gene dosage or expression of NFKBIA in models predicting survival. Our data show that a risk model combining NFKBIA status and MGMT status (currently the best single predictor of response to temozolomide therapy for glioblastomas33) was strongly associated with the clinical course of the disease. This makes sense mechanistically: concomitant down-regulation of NFKBIA (enhancing the pro-survival effect of NF-κB) and up-regulation of MGMT (enhancing the repair of DNA damage) could have a synergistic, positive effect on resistance to therapeutic response and cell death.
Our finding that increased expression of NFKBIA inhibited the malignant behavior of tumors that had amplified EGFR and normal dosage of NFKBIA (in addition to tumors with deletions of NFKBIA) suggests that NFKBIA-stabilizing therapies may be effective against glioblastomas that have alterations of EGFR. The limited efficacy of molecular therapies targeting EGFR in glioblastomas suggests that the therapeutic effect of EGFR inhibition can be circumvented through cross-coupled signaling from other growth factor receptors that are mutated, amplified, or overexpressed in these tumors, such as PDGFRA, ERBB2, or MET.7 Because NFKBIA is a major node downstream of such cross-coupled signaling, therapies that stabilize NFKBIA might more effectively restrain oncogenic signaling.
Supplementary Material
Acknowledgments
Supported by a Project Award of Accelerate Brain Cancer Cure, a Mike Gardner/American Brain Tumor Association Grant Award, a German Cancer Aid Grant Award (107714), a State of Illinois Excellence in Academic Medicine Program Award (211), State of Alabama Investment Pool for Action (IMPACT) funds, grants from the National Cancer Institute (RO1CA108633, RC2CA148190, P50CA127001, RTOG U10CA21661, and CCOP U10CA37422), a Goldhirsh Brain Tumor Research Award, a Leach Foundation Research Award, the Brain Tumor Funders’ Collaborative, the Lou Malnati’s Cancer Benefit Committee, and the Mazza Foundation.
Glossary
- Amplification
An increase in the copy number of a particular gene, which can be either inherited or somatic. Amplification of oncogenes is a preeminent event in the pathogenesis of many types of human cancer
- Cancer stemlike cells
Cancer cells found within tumors or hematologic cancers that possess characteristics associated with normal stem cells. Cancer stemlike cells are probably tumorigenic (tumor-forming) through the stem-cell processes of self-renewal and differentiation into multiple cell types. Such cells are proposed to persist in tumors and cause relapse and metastasis by giving rise to new tumors
- Codingregion
The portion of a gene’s DNA or RNA that codes for its corresponding gene product — the protein
- Colony formation or colony-forming activity
A phenotypically recognizable characteristic of cell transformation and a measure of malignant tumor-cell behavior. It indicates that individual cells develop into cell clones that are identified as single colonies
- Copy-number variation
A segment of DNA in which differences in copy number have been found by means of a comparison of two or more genomes (e.g., a tumor genome and a normal human genome). Cancer cells typically show complex patterns of increased copy numbers (or dosage) of oncogenes and reduced copy numbers of tumor suppressor genes
- Deletion
The absence of one (heterozygous deletion) or both (homozygous deletion) copies of a gene in a diploid cell. Heterozygous deletions may or may not disrupt gene or protein function and cell function as a result
- Genedosage
The copy number for a specific gene as determined in analytic approaches that do not assess single cells but describe the average copy-number profile of a complex tumor in which some cell populations may harbor copy-number alterations of the gene and some may not
- Haplotype
A set of single-nucleotide polymorphisms on an allele that are statistically associated and might provide valuable insights into the genetic variables associated with common diseases
- Promoter methylation
An epigenetic mechanism to regulate the expression of a gene. Hypermethylation is associated with a silencing of the promoter and thus reduced gene expression; hypomethylation leads to increased gene transcription
- Senescence
The phenomenon by which normal diploid cells lose the ability to divide
- Single-nucleotide polymorphism
A variation of a single nucleotide at a specific location of the genome that is due to a single-base substitution and that is present at an appreciable frequency between individuals of a single interbreeding population
- Transcription factor
A protein that binds to specific DNA sequences and thereby controls the transfer (or transcription) of genetic information from DNA to mRNA
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [Erratum, N Engl J Med 2008;359:877.] [DOI] [PubMed] [Google Scholar]
- 2.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–73. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–73. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 5.Bredel M, Scholtens DM, Harsh GR, et al. A network model of a cooperative genetic landscape in brain tumors. JAMA. 2009;302:261–75. doi: 10.1001/jama.2009.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yadav AK, Renfrow JJ, Scholtens DM, et al. Monosomy of chromosome 10 associated with dysregulation of epidermal growth factor signaling in glioblastomas. JAMA. 2009;302:276–89. doi: 10.1001/jama.2009.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watanabe K, Tachibana O, Sata K, Yonekawa Y, Kleihues P, Ohgaki H. Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol. 1996;6:217–23. doi: 10.1111/j.1750-3639.1996.tb00848.x. [DOI] [PubMed] [Google Scholar]
- 9.Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Habib AA, Chatterjee S, Park SK, Ratan RR, Lefebvre S, Vartanian T. The epidermal growth factor receptor engages receptor interacting protein and nuclear factor-kappa B (NF-kappa B)-inducing kinase to activate NF-kappa B: identification of a novel receptor-tyrosine kinase signalosome. J Biol Chem. 2001;276:8865–74. doi: 10.1074/jbc.M008458200. [DOI] [PubMed] [Google Scholar]
- 11.Sethi G, Ahn KS, Chaturvedi MM, Aggarwal BB. Epidermal growth factor (EGF) activates nuclear factor-kappaB through IkappaBalpha kinase-independent but EGF receptor-kinase dependent tyrosine 42 phosphorylation of IkappaBalpha. Oncogene. 2007;26:7324–32. doi: 10.1038/sj.onc.1210544. [DOI] [PubMed] [Google Scholar]
- 12.Raychaudhuri B, Han Y, Lu T, Vogelbaum MA. Aberrant constitutive activation of nuclear factor kappaB in glioblastoma multiforme drives invasive phenotype. J Neurooncol. 2007;85:39–47. doi: 10.1007/s11060-007-9390-7. [DOI] [PubMed] [Google Scholar]
- 13.Nagai S, Washiyama K, Kurimoto M, Takaku A, Endo S, Kumanishi T. Aberrant nuclear factor-kappaB activity and its participation in the growth of human malignant astrocytoma. J Neurosurg. 2002;96:909–17. doi: 10.3171/jns.2002.96.5.0909. [DOI] [PubMed] [Google Scholar]
- 14.Bredel M, Bredel C, Juric D, et al. Tumor necrosis factor-alpha-induced protein 3 as a putative regulator of nuclear factor-kappaB-mediated resistance to O6-alkylating agents in human glioblastomas. J Clin Oncol. 2006;24:274–87. doi: 10.1200/JCO.2005.02.9405. [DOI] [PubMed] [Google Scholar]
- 15.Tsunoda K, Kitange G, Anda T, et al. Expression of the constitutively activated RelA/NF-kappaB in human astrocytic tumors and the in vitro implication in the regulation of urokinase-type plasminogen activator, migration, and invasion. Brain Tumor Pathol. 2005;22:79–87. doi: 10.1007/s10014-005-0186-1. [DOI] [PubMed] [Google Scholar]
- 16.Bassères DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817–30. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 17.Spink CF, Gray LC, Davies FE, Morgan GJ, Bidwell JL. Haplotypic structure across the I kappa B alpha gene (NFKBIA) and association with multiple myeloma. Cancer Lett. 2007;246:92–9. doi: 10.1016/j.canlet.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 18.Krappmann D, Emmerich F, Kordes U, Scharschmidt E, Dörken B, Scheidereit C. Molecular mechanisms of constitutive NF-kappaB/Rel activation in Hodgkin/Reed-Sternberg cells. Oncogene. 1999;18:943–53. doi: 10.1038/sj.onc.1202351. [DOI] [PubMed] [Google Scholar]
- 19.Cabannes E, Khan G, Aillet F, Jarrett RF, Hay RT. Mutations in the IkBa gene in Hodgkin’s disease suggest a tumour suppressor role for IkappaBalpha. Oncogene. 1999;18:3063–70. doi: 10.1038/sj.onc.1202893. [DOI] [PubMed] [Google Scholar]
- 20.Emmerich F, Meiser M, Hummel M, et al. Overexpression of I kappa B alpha without inhibition of NF-kappaB activity and mutations in the I kappa B alpha gene in Reed-Sternberg cells. Blood. 1999;94:3129–34. [PubMed] [Google Scholar]
- 21.Jungnickel B, Staratschek-Jox A, Bräuninger A, et al. Clonal deleterious mutations in the IkappaBalpha gene in the malignant cells in Hodgkin’s lymphoma. J Exp Med. 2000;191:395–402. doi: 10.1084/jem.191.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lake A, Shield LA, Cordano P, et al. Mutations of NFKBIA, encoding IkappaBalpha, are a recurrent finding in classical Hodgkin lymphoma but are not a unifying feature of non-EBV-associated cases. Int J Cancer. 2009;125:1334–42. doi: 10.1002/ijc.24502. [DOI] [PubMed] [Google Scholar]
- 23.Sjöblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 24.Gao J, Pfeifer D, He LJ, et al. Association of NFKBIA polymorphism with colorectal cancer risk and prognosis in Swedish and Chinese populations. Scand J Gastroenterol. 2007;42:345–50. doi: 10.1080/00365520600880856. [DOI] [PubMed] [Google Scholar]
- 25.Osborne J, Lake A, Alexander FE, Taylor GM, Jarrett RF. Germline mutations and polymorphisms in the NFKBIA gene in Hodgkin lymphoma. Int J Cancer. 2005;116:646–51. doi: 10.1002/ijc.21036. [DOI] [PubMed] [Google Scholar]
- 26.He Y, Zhang H, Yin J, et al. IkappaBalpha gene promoter polymorphisms are associated with hepatocarcinogenesis in patients infected with hepatitis B virus genotype C. Carcinogenesis. 2009;30:1916–22. doi: 10.1093/carcin/bgp226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bu H, Rosdahl I, Sun XF, Zhang H. Importance of polymorphisms in NF-kappaB1 and NF-kappaBIalpha genes for melanoma risk, clinicopathological features and tumor progression in Swedish melanoma patients. J Cancer Res Clin Oncol. 2007;133:859–66. doi: 10.1007/s00432-007-0228-7. [DOI] [PubMed] [Google Scholar]
- 28.Liu X, Yu H, Yang W, Zhou X, Lu H, Shi D. Mutations of NFKBIA in biopsy specimens from Hodgkin lymphoma. Cancer Genet Cytogenet. 2010;197:152–7. doi: 10.1016/j.cancergencyto.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 29.Parker KM, Ma MH, Manyak S, et al. Identification of polymorphisms of the IkappaBalpha gene associated with an increased risk of multiple myeloma. Cancer Genet Cytogenet. 2002;137:43–8. doi: 10.1016/s0165-4608(02)00541-1. [DOI] [PubMed] [Google Scholar]
- 30.Kapoor GS, Zhan Y, Johnson GR, O’Rourke DM. Distinct domains in the SHP-2 phosphatase differentially regulate epidermal growth factor receptor/NF-kappaB activation through Gab1 in glioblastoma cells. Mol Cell Biol. 2004;24:823–36. doi: 10.1128/MCB.24.2.823-836.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stupp R, Dietrich PY, Ostermann Kraljevic S, et al. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J Clin Oncol. 2002;20:1375–82. doi: 10.1200/JCO.2002.20.5.1375. [DOI] [PubMed] [Google Scholar]
- 32.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 33.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 34.Everhard S, Tost J, El Abdalaoui H, et al. Identification of regions correlating MGMT promoter methylation and gene expression in glioblastomas. Neuro Oncol. 2009;11:348–56. doi: 10.1215/15228517-2009-001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mikeska T, Bock C, El-Maarri O, et al. Optimization of quantitative MGMT promoter methylation analysis using pyrosequencing and combined bisulfite restriction analysis. J Mol Diagn. 2007;9:368–81. doi: 10.2353/jmoldx.2007.060167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woolf B. On estimating the relation between blood group and disease. Ann Hum Genet. 1955;19:251–3. doi: 10.1111/j.1469-1809.1955.tb01348.x. [DOI] [PubMed] [Google Scholar]
- 37.Lee Y, Scheck AC, Cloughesy TF, et al. Gene expression analysis of glioblastomas identifies the major molecular basis for the prognostic benefit of younger age. BMC Med Genomics. 2008;1:52. doi: 10.1186/1755-8794-1-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fulci G, Labuhn M, Maier D, et al. p53 Gene mutation and ink4a-arf deletion appear to be two mutually exclusive events in human glioblastoma. Oncogene. 2000;19:3816–22. doi: 10.1038/sj.onc.1203700. [DOI] [PubMed] [Google Scholar]
- 39.Chakravarti A, Chakladar A, Delaney MA, Latham DE, Loeffler JS. The epidermal growth factor receptor pathway mediates resistance to sequential administration of radiation and chemotherapy in primary human glioblastoma cells in a RAS-dependent manner. Cancer Res. 2002;62:4307–15. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.