

Abstract
Newly activated CD8+ T cells reprogram their metabolism to meet the extraordinary biosynthetic demands of clonal expansion; however, the signals mediating metabolic reprogramming remain poorly defined. Herein, we demonstrate an essential role for sterol regulatory element binding proteins (SREBPs) in the acquisition of effector cell metabolism. Without SREBP signaling, CD8+ T cells are unable to blast, resulting in markedly attenuated clonal expansion during viral infection. Mechanistic studies indicate that SREBPs are essential to meet the heightened lipid requirements of membrane synthesis during blastogenesis. SREBPs are dispensable for homeostatic proliferation, indicating a context-specific requirement for SREBPs in effector responses. These studies provide insights into the molecular signals underlying metabolic reprogramming of CD8+ T cells during the transition from quiescence to activation.
Keywords: SREBP, LCMV, lipids, CD8+ T cell, metabolism, proliferation
A hallmark of adaptive immunity is the ability of responding T lymphocytes to undergo extensive clonal expansion to protect the host from invading pathogens. To fulfill the requirements of clonal expansion, activated T cells must acquire distinct metabolic programs that meet heightened metabolic demands. Quiescent, naive T cells rely on mitochondrial oxidative phosphorylation (OXPHOS) and catabolic metabolism to fulfill the minimal energetic and biosynthetic requirements associated with quiescence and long-term survival. Upon activation, T lymphocytes rapidly switch their metabolic program to rely predominantly on glycolysis in a process highly analogous to the Warburg effect first described for tumor cells 1. Additional changes in intermediary metabolism include increased flux through the pentose phosphate pathway (PPP) and an increased reliance on glutaminolysis. The preferential use of glycolysis, pentose-phosphate and glutaminolytic pathways enables rapidly dividing T cells to satisfy the increased metabolic demands during organelle biogenesis and cellular replication 2, 3. Accordingly, genetic and pharmacological inhibition of glycolytic, glutaminolytic and lipid biosynthetic pathways attenuate T cell survival, cell cycle progression and effector differentiation 4-9. Despite the importance of metabolic reprogramming for effector cell fate and function, the molecular events linking T cell receptor signaling with metabolic reprogramming remain enigmatic.
A key component in the metabolic reprogramming of effector T lymphocytes is the rapid upregulation of lipid biosynthetic pathways 4,8. Early isotopomer enrichment studies demonstrated that activation of lymphocytes results in the rapid increase of de novo cholesterol and fatty acid biosynthesis 4. Critically, addition of specific cholesterol derivatives (e.g., oxysterols) to cultures markedly diminished de novo lipid biosynthesis and inhibited cell cycle progression in G1, suggesting a link between lipid metabolism and cell cycle progression. Subsequent studies using statins, pharmacologic inhibitors of HMG-CoA reductase (the rate-limiting enzyme in de novo cholesterol biosynthesis) also inhibited mitogen-driven lymphocyte expansion 10. More recently, we and others have established that genetic and pharmacologic perturbations in sterol homeostasis, through the action of the Liver X Receptor (LXR) transcriptional axis, also influence T lymphocyte cell cycle progression, survival and effector function 8, 11. Thus, the regulation of intracellular lipid metabolism is critical for proper lymphocyte growth and function. However, the molecular mechanisms linking mitogenic signaling to the lipid anabolic program of activated lymphocytes remain poorly defined.
The sterol regulatory element binding proteins (SREBP1 and 2) are bHLH-zip transcription factors that have a well-defined role in the regulation of cellular lipid homeostasis 12. In mammals there are two SREBP genes that express three SREBP proteins. SREBP1a and SREBP1c are produced via alternative transcriptional start sites on Srebf1, whereas the Srebf2 gene encodes SREBP2. Canonical SREBP1c signaling preferentially drives expression of fatty acid biosynthesis genes whereas SREBP2 predominately transactivates genes involved in cholesterol biosynthesis, intracellular lipid movement and lipoprotein import. The SREBP1a isoform is able to transactivate both SREBP1c and SREBP2 target genes. In addition to their function in regulating lipid biosynthetic and transport gene expression, SREBPs also transactivate key genes involved in the oxidative PPP and the generation of the co-enzyme NADPH 13, ensuring sufficient reducing equivalents to meet anabolic demands.
The influence of SREBP signaling on T cell metabolism and function is not well understood. Herein, we use genetic and pharmacologic models to demonstrate that SREBPs are essential for CD8+ T cells to undergo metabolic reprogramming in response to mitogenic signaling. Loss-of-SREBP function in CD8+ T cells rendered them unable to efficiently blast, resulting in diminished proliferative capacity in vitro, and attenuated clonal expansion during viral infection. SREBP activity was required for the upregulation of glycolytic and oxidative metabolism following activation and to maintain lipid levels sufficient to permit rapid membrane biogenesis and cellular growth. The defect in growth and proliferation was traced to a deficiency in cellular cholesterol during blastogenesis which could be rescued by cholesterol supplementation. However, SREBPs were dispensable for homeostatic proliferation, indicating a specific requirement for SREBP signaling to meet the significant anabolic demands supporting effector responses. This study provides mechanistic insights into the metabolic networks ensuring the growth of effector CD8+ T cells upon activation, and highlights the importance of SREBP proteins in regulating a cholesterol metabolic checkpoint during blastogenesis.
RESULTS
The TCR induces an mTOR-dependent lipid synthetic program
T cells rapidly upregulate a fatty acid and cholesterol lipid biosynthetic program after stimulation with polyclonal mitogens 4. To understand the relationship between mitogenic stimulation and induction of the lipogenic program, we activated purified murine T cells with plate bound anti-CD3 alone or in combination with anti-CD28 for 6 hours. Quantitative RT-PCR revealed that anti-CD3 alone was sufficient for induction of cholesterol (Hmgcr, Hmgcs, Sqle) and fatty acid biosynthesis genes (Acaca, Fasn) (Fig. 1a), and the addition of CD28 costimulation did not provide additional induction of the program. Activation of Pmel TCR transgenic CD8+ T cells with increasing doses of agonist gp100 peptide confirmed a requirement for TCR signaling and showed that induction of the lipogenic program occurs at sub mitogenic doses of antigen (Supplementary Fig. 1a). T cell activation, using the phorbol ester PMA, induced the lipogenic program to levels comparable to that of TCR crosslinking (Fig. 1a). Induction was inhibited by pretreatment of cells with the PKC inhibitor Gö6983 (Supplementary Fig. 1b), suggesting PKC dependence. In contrast, culturing cells with a calcium ionophore (ionomycin) alone did not induce lipogenic genes (Fig. 1a). Taken together, these data indicate that TCR signaling and PKC activation are necessary and sufficient for induction of the lipogenic program in mitogen-stimulated T cells.
Figure 1. The lipid biosynthetic program of activated T cells is SREBP-dependent and sensitive to the PI3K-mTOR pathway.
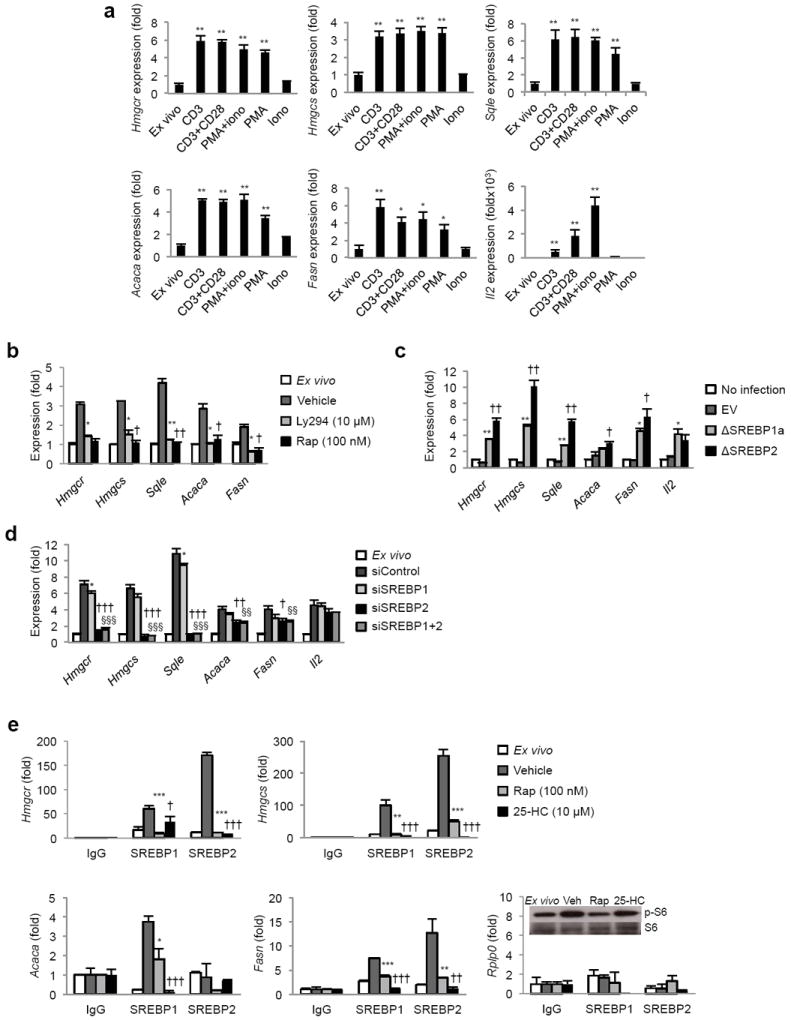
(a) Real-time PCR analysis of lipogenic genes from murine spleen and LN T cells activated for 6 h with plate-bound CD3+/-CD28, PMA and ionomycin as indicated. (b) Real-time PCR analysis of quiescent or 6 h activated T cells pretreated for 30 min with PI3K inhibitor LY294002 (10 μM) or mTOR inhibitor rapamycin (100 nM) before activation with CD3-28. (c) Real-time PCR analysis of lipogenic genes in activated T cells transduced with active SREBP (ΔSREBP1a or ΔSREBP2) or empty vector (EV) for 24 h. * denotes comparisons between empty vector and ΔSREBP1a, † denotes comparisons between empty vector and ΔSREBP2. Number of symbols indicates the p value. (d) Real-time PCR analysis of quiescent or T cells transfected with siSREBP1 alone, siSREBP2 alone, siSREBP1 and siSREBP2 or non-target control (siControl) and then activated with PMA-iono for 5 h. * denotes comparisons between siControl and siSREBP1, † denotes comparisons between siControl and siSREBP2, § denotes comparisons between siControl and siSREBP1+2. (e) Chromatin Immunoprecipitation analysis of SREBP1 and SREBP2 at the promoters of indicated genes in splenocytes ex vivo or 4 hours after activation with PMA. In addition, some cultures were pretreated for 30 minutes with rapamycin (100 nM) or 25-hydroxycholesterol (25-HC, 10 μM) before activation. Data is normalized to input and expressed relative to IgG control. Rplp0: non-SREBP target gene control. * Denotes comparisons between vehicle and rapamycin treated samples, † denotes comparisons between vehicle and 25-HC treated samples. Inset: Immunoblot of phospho and total S6 from whole cell lysates to confirm rapamycin function. For all experiments, data are presented as a mean of triplicates with standard deviation, and representative of at least three experiments. *p<0.05, **p<0.01, ***p<0.001 (statistical difference between other experimental groups is also noted by the number of other symbols, two-tailed unpaired Student’s t-test).
Recent studies indicate that loss of Tsc1, a negative regulator of mTOR kinase signaling, resulted in the upregulation of sterol biosynthesis and propanoate metabolism genes in naive CD4+ T cells14. Thus, we asked if induction of lipid biosynthetic programs by mitogens was dependent on mTOR signaling. To address this, purified splenic T cells were pretreated for 30 minutes with 100 nM rapamycin or vehicle and then stimulated up to 6 hours. As expected, activation of T cells robustly induced fatty acid and cholesterol biosynthetic genes, whereas 30 minutes pretreatment of cultures with rapamycin completely inhibited the induction of the lipogenic program (Fig. 1b). Immunoblots on whole cell and nuclear extracts revealed that rapamycin prevented processing of full length SREBP resulting in a failure to accumulate mature SREBP in the nucleus (Supplementary Fig. 1c). Inactivation of the PI3K pathway with LY294002 (Ly294) treatment similarly repressed lipogenic gene expression (Fig. 1b). These data indicate that signaling through the PI3K-mTOR pathway is necessary for the induction of the lipid anabolic gene program of activated lymphocytes.
SREBPs mediate the lipid anabolic program of T cells
To directly address the function of SREBPs in the induction of the lipogenic program by mitogenic signals, purified splenic and lymph node (LN) T cells were retrovirally transduced with a truncated form of SREBP1a or SREBP2 that is constitutively active (designated ΔSREBP1a or ΔSREBP2 herein, Supplementary Fig. 1d,e). mRNA was collected 24 h post infection and expression of the lipid biosynthetic genes were determined by RT-PCR. Both ΔSREBP1a and ΔSREBP2 markedly increased the expression of cholesterol and fatty acid biosynthesis genes when compared to empty vector controls (Fig. 1c), indicating that the lipogenic program of T cells is responsive to SREBP signaling.
In complementary experiments, we asked if SREBPs were required for induction of the lipogenic program by mitogenic signaling. To directly address this, purified mouse spleen and LN T cells were transiently transfected with siRNAs targeting SREBP1 and SREBP2 before stimulation with PMA-ionomycin. After 5 h of stimulation, mRNA was collected and expression of lipid biosynthetic genes assessed. Activation of cells transfected with control siRNA increased expression of genes involved in de novo lipid biosynthesis (Fig. 1d). In contrast, siSREBP1 and siSREBP2 transfected cells were unable to upregulate cholesterol synthetic genes (Fig. 1d, Supplementary Fig. 1f). Upregulation of fatty acid biosynthetic genes was inhibited, albeit to a lesser extent. Knockdown of SREBP2 alone was sufficient to inhibit the induction of both cholesterol and fatty acid synthetic genes (Fig. 1d). We were only able to achieve a partial knockdown of SREBP1 (Supplementary Fig. 1f) and correspondingly, we observed a small, but statistically significant effect on fatty acid synthetic genes (Fig. 1d). However, we were unable to inhibit sterol synthetic genes with this knockdown.
The observation that over-expression of ΔSREBP1a or ΔSREBP2 upregulates both fatty acid and cholesterol biosynthetic genes in activated T cells lead us to hypothesize that SREBP1 and SREBP2 might cooperate, or share occupancy, at the promoters of lipogenic genes. Thus, we performed chromatin immunoprecipitations (ChIP) on SREBP1 and 2 from quiescent and activated T cell lysates. In quiescent cells, SREBP2 was readily detectable at the promoters of Hmgcr and Hmgcs (Fig. 1e). Activation of T cells resulted in a 10-fold or greater enrichment of SREBP2 at the promoters of Hmgcr and Hmgcs (Fig. 1e). Clear enrichment of SREBP1 was also detectable at the promoters of Hmgcr and Hmgcs, canonical SREBP2 target genes. Likewise, we could find both SREBP1 and 2 at the promoters of fatty acid synthetic genes (Fig. 1e). These data suggest that both SREBP1 and 2 may cooperate in driving the lipogenic program of activated T cells. Pre-treatment of cultures with 25-hydroxycholesterol (25-HC, 10 μM), a well-described sterol inhibitor of SREBP processing markedly reduced enrichment of SREBP1 and 2 at the promoters of lipogenic genes (Fig. 1e), and consequently induction of the lipid synthetic program (Supplementary Fig. 1g). Lastly, pretreating cultures with 100 nM rapamycin also reduced SREBP1 and SREBP2 enrichment to background (Fig. 1e); further indicating that mTOR is required to link TCR signaling with induction of the lipid synthetic program via SREBP.
Deletion of the SCAP inhibits SREBP activity in T cells
Previous studies on SREBP proteins indicate that SREBP1 and 2 can compensate for each other when one protein is deleted 15, 16. Moreover, our overexpression and ChIP studies suggested that both SREBP1 and SREBP2 could potentially influence the lipid biosynthetic program in mitogen-stimulated T cells. Thus, we reasoned that loss of both SREBP1 and SREBP2 would be required to fully ablate the lipogenic program of activated T cells. To achieve this, we crossed mice containing floxed alleles of the SREBP chaperone protein SCAP (SREBP Cleavage Activating Protein) with Cd4-Cre mice (hereafter referred to as Scapfl/fl). SCAP protein is associated with SREBPs in the ER membrane and regulates the processing and subsequent transcriptional activity of both SREBP1 and SREBP2 17. Examination of 5-6 week-old Scapfl/fl and littermate control mice revealed no significant differences in the cellularity of thymus, spleen and LN (Fig. 2a). Phenotypic analysis of thymic and peripheral T cell subsets from Scapfl/fl and control mice demonstrated no demonstrable differences in the frequency of CD4+ and CD8+ T cells (Fig. 2b). Nor were there differences in the expression pattern of the activation markers (e.g., CD25 and CD44, Supplementary Fig. 2a), or total numbers of CD4+CD25+ and CD8+CD44+ cells observed (Supplementary Fig. 2b). Finally, BrdU pulse chase experiments to follow homeostatic maintenance of peripheral T cells revealed no significant difference in the labeling or decay between control and Scapfl/fl mice over a 42 day period (Supplementary Fig. 2c).
Figure 2. Deletion of Scap inhibits SREBP activity but does not affect T cell homeostasis.
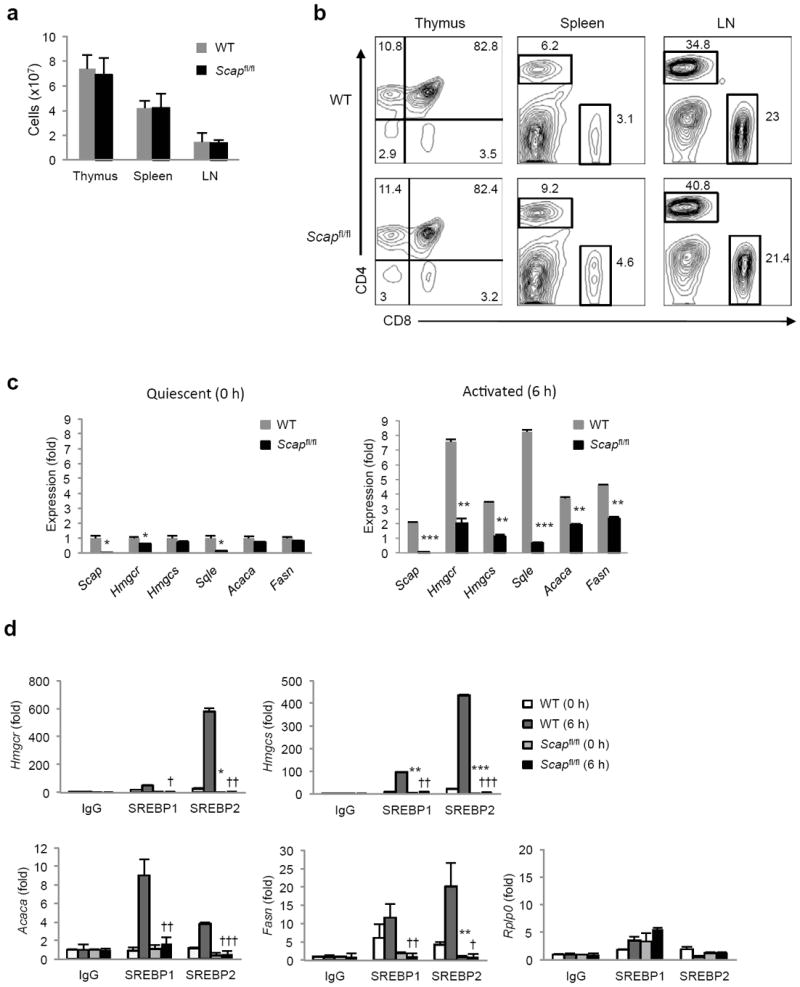
(a) Total cell number in thymus, spleen and lymph nodes (LN) from Cd4-Cre-Scapfl/fl mice (designated Scapfl/fl herein) and littermate controls (WT). Data is expressed as mean with standard deviation. (b) Flow cytometric analysis of CD4+ and CD8+ T cells in thymus, spleen and LN from Scapfl/fl and littermate control mice. Relative frequency indicated in plots. (c) Real-time PCR analysis of control and Scapfl/fl CD8+ T cells ex vivo or activated for 6 h with anti-CD3-28. (d) Chromatin immunoprecipitation analysis of SREBP1 and SREBP2 at the promoters of indicated genes in T cells ex vivo or 6 hours after activation (anti-CD3-28). Data are normalized to input and plotted relative to IgG control. Rplp0: non-SREBP target gene control. * denotes comparisons between WT T=0 and Scapfl/fl T=0, † denotes comparisons between WT T=6 and Scapfl/fl T=6. Data are mean of triplicates with standard deviation (c,d). *p<0.05, **p<0.01, ***p<0.001 (statistical difference between other experimental groups is also noted by the number of other symbols, two-tailed unpaired Student’s t-test). Data are representative of four independent experiments with three mice per group (a,b), or three independent experiments (c,d).
Gene expression studies confirmed a near complete deletion of Scap in quiescent peripheral Scapfl/fl T cells, and we observed a modest change in the expression of lipogenic genes (Fig. 2c). ChIP studies demonstrated that T cell specific deletion of Scap modestly reduced the amount of detectable SREBP protein at target gene promoters in quiescent cells (Fig. 2d). As expected, control cells rapidly upregulated the lipid biosynthetic program (Fig. 2c) and led to increased SREBP1 and SREBP2 at target gene promoters (Fig. 2d). In contrast, the induction of lipogenic genes was markedly attenuated in Scap-deficient CD8+ T cells (Fig. 2c). Correspondingly, we observed a significant reduction in the amount of both SREBP1 and SREBP2 protein occupying promoters of lipid biosynthesis genes (Fig. 2d). Taken together, our data indicate that the SREBP-dependent lipid biosynthetic program is largely dispensable for quiescent peripheral T cells; however, genetic deletion of Scap attenuates the upregulation of the SREBP transcriptome of mitogen-stimulated T cells.
SREBPs influence CD8+ T cell growth and proliferation
To determine if loss of SREBP activity would influence T cell blastogenesis and proliferation in vitro, purified control and Scapfl/fl CD8+ T cells were stimulated with anti-CD3 with or without anti-CD28 or IL-2 for 72 h. Control T cells enlarged by 24 h (Fig. 3a and Supplementary Fig. 3a) and diluted CFSE by 72 h after activation (Fig. 3b). In contrast, Scapfl/fl CD8+ T cells did not undergo significant enlargement (Fig. 3a and Supplementary Fig. 3a), and only modestly proliferated in response to anti-CD3 (Fig. 3b). The addition of exogenous IL-2 and/or anti-CD28, or PMA-ionomycin to cultures provided only a slight or no increase in the proliferation of Scapfl/fl T cells (Fig. 3b). Cell cycle analysis indicated that activated Scapfl/fl CD8+ T cells remained in G0-G1 of cell cycle (Fig. 3c). The addition of CD28 and/or IL-2 to cultures very modestly increased the proportion of cells in S or G2-M phase (Fig. 3c). These data clearly indicate that SREBP signaling is essential for mitogen-driven CD8+ T cell blastogenesis, proliferation and survival.
Figure 3. SREBP activity influences CD8+ T cell growth and proliferation.
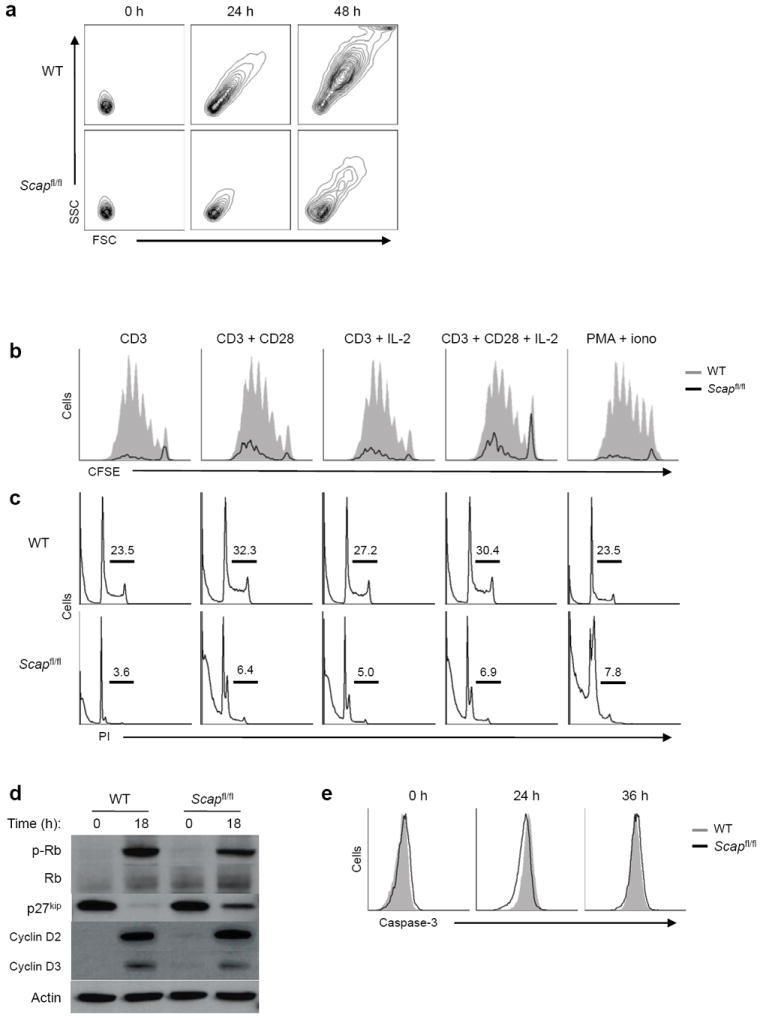
(a) Forward and side scatter plots of DAPI-negative (live) WT and Scapfl/fl CD8+ T cells activated with anti-CD3-28 for indicated time. (b) CFSE dilution of DAPI negative (live) WT and Scapfl/fl CD8+ T cells activated for 72 hours with indicated mitogens. (c) Flow cytometric analysis of DNA content from WT and Scapfl/fl CD8+ T cells activated for 48 hours with indicated stimuli. Cells were stained for DNA content with propidium iodide. Frequency of S and G2/M phase indicated in plots. (d) Immunoblots of G1 associated cell cycle proteins from whole cell lysates of quiescent or 18 h activated WT and Scapfl/fl CD8+ T cells with anti-CD3-28. (e) Flow cytometric analysis of intracellular cleaved caspase-3 in WT and Scapfl/fl CD8+ T cells activated with anti-CD3-28 for indicated time. Data are representative of three (a), or two (b,c,d,e) independent experiments.
Our cell cycle data indicates that Scap deficient T cells remain in G0-G1 phase. To better define whether Scap-deficient T cells were capable of moving out of G0 into G1 of cell cycle, we examined the expression of the G1 associated cyclins and restriction point proteins. Immunoblots from control and Scapfl/fl CD8+ T cells activated for 18h indicated Scap-deficient T cells upregulated cyclins D2 and D3 (Fig. 3d). We also observed decreased phosphorylation of the cell cycle checkpoint protein Rb and maintenance of p27kip (Fig. 3d), suggesting that Scap-deficient cells were moving into G1 of cell cycle, but were not entering S phase. We also examined if the loss of SREBP activity was resulting in apoptosis. However, kinetic studies indicated no difference in the frequency of cleaved caspase-3-positive cells up to 36 h post-activation (Fig. 3e). Similarly, we observed no difference in the frequency of annexin V-positive Scapfl/fl CD8+ T cells (data not shown). However, we did observe a significant decrease in the frequency of DAPI-negative cells by 48 h (Supplementary Fig. 3b), suggesting that loss of SREBP activity results in non-apoptotic death late in cultures. Taken together these data indicate that Scap-deficient T cells move efficiently into G1 of cell cycle, but are unable to exit before undergoing a caspase-independent cell death.
Loss of SREBP impacts lipid homeostasis of T cells
Next, we asked if genetic ablation of SREBP activity grossly perturbed lipid homeostasis in T cells. To address this, splenic T cells were purified from 5-8 week old Cd4-Cre × Scapfl/fl or littermate control mice. Total cholesterol and fatty acid (myristate, palmitate and stearate) content were measured by Gas chromatography-Mass spectroscopy (GC-MS) directly ex vivo. Scapfl/fl T cells contained an approximate 2-fold decrease in cellular cholesterol compared to littermate control T cells (Fig. 4a). Palmitate was also modestly decreased in quiescent Scap-deficient T cells (Fig. 4b), however no significant difference in myristate or stearate was observed in resting cells. Activation of control T cells with anti-CD3-28 for 24 h increased cholesterol, myristate, palmitate and stearate (Fig. 4a,b). In contrast, Scapfl/fl T cells were unable to increase their cholesterol, myristate, palmitate and stearate content in response to activation (Fig. 4a,b). Taken together, these data indicate that SREBP signaling is required for defining a lipid homeostatic set point in quiescent T cells, and plays an essential role in meeting the increased lipid biosynthetic demands during blastogenesis.
Figure 4. Loss of SREBP signaling impacts lipid homeostasis but does not perturb proximal TCR signaling.
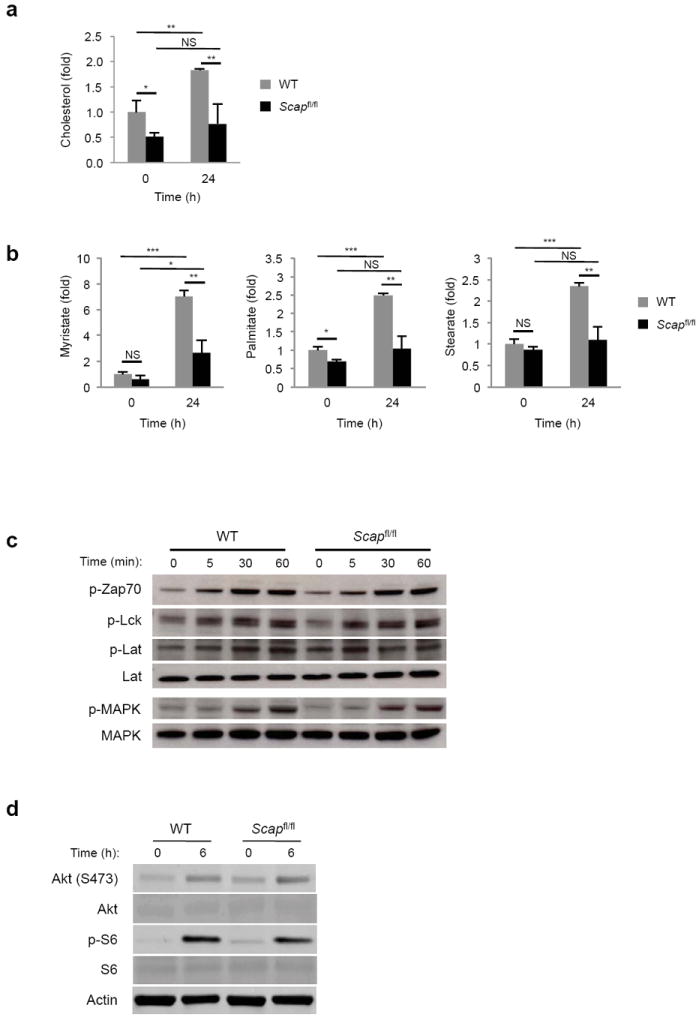
(a,b) GC-MS determination of cholesterol (a) or indicated long-chain fatty acid (b) content of control and Scapfl/fl T cells ex vivo or 24 h after activation with anti-CD3-28. Data are normalized to cell numbers and plotted relative to control ex vivo (WT 0 h). Data is expressed as mean of triplicates with standard deviations. (c) Immunoblot analysis of WT and Scapfl/fl lymphocytes ex vivo or after stimulation with 1 μg/ml of soluble anti-CD3e for the indicated time. Whole cell lysates were blotted for indicated total and phosphorylated proteins. (d) Immunoblot analysis of the Akt pathway from WT and Scapfl/fl CD8+ T cells ex vivo or after 6 h of activation with anti-CD3-28. NS: not significant, *p<0.05, **p<0.01, ***p<0.001 (two-tailed unpaired Student’s t-test).
Loss of SREBP does not perturb proximal TCR signals
Given the importance of cholesterol in plasma membrane fluidity18, lipid raft formation and TCR signaling 19, we considered the possibility that the diminished proliferative capacity and survival of Scapfl/fl CD8+ T cells was a function of altered proximal signaling. To address this, T cells were stimulated with anti-CD3 for 60 min and whole cell lysates were probed for phosphorylation of the upstream signaling molecules Zap-70, Lck, Lat and p44/42 MAPK. Regardless of genotype, we observed normal induction of phosphorylation as early as 5 minutes after stimulation, which was maintained out to 60 min (Fig. 4c). We also considered the possibility that we would observe alterations in the PI3K-AKT pathway given the importance of lipids (e.g., myristate) in proper activation of this pathway. However, we did not observe any difference in phosphorylation of AKT and the downstream target S6 in response to activation (Fig. 4d). Moreover, activation with PMA and ionomycin, which obviate the requirement for TCR and co-stimulation, did not provide a significant rescue in the cell cycle progression, viability and proliferative capacity of Scap-deficient T cells (Fig. 3b,c). These data suggest that it is unlikely that the inability of Scap-deficient T cells to grow is a function of changes in TCR and co-receptor signaling.
SREBPs regulate a gene program required for T cell growth
To better understand how SREBP was influencing CD8+ T cell growth and survival, we performed global gene expression studies on quiescent and 6 h stimulated CD8+ T cells. A number of genes were either up or downregulated with anti-CD3-CD28 stimulation in control CD8+ T cells. Importantly, we observed a very similar pattern of gene expression in activated Scap-deficient cells indicating that loss of SREBP activity does not globally perturb T cell activation. Pathway analysis of genes differentially expressed between quiescent control and Scap-deficient CD8+ T cells revealed a small set of genes involved in cellular survival was downregulated in the absence of SREBP activity (Supplementary Table 1).
Analysis on genes significantly (P<0.001) less-induced in Scap-deficient CD8+ T cells after activation using DAVID software analysis20, 21 revealed significant enrichment in processes associated with sterol biosynthesis, confirming the effect of Scap-deficiency in reducing lipid anabolism in the context of T-cell activation (Fig. 5, Table 1, and Supplementary Table 1). Other gene ontology clusters significantly enriched were associated with RNA homeostasis (e.g., RNA processing, ribosome biogenesis and rRNA metabolic processes), nuclear and organelle lumen and nucleolus biogenesis (Table 1). Analysis of predicted transcription factors mediating differential gene expression between activated control and Scapfl/fl CD8+ T lymphocytes revealed significant enrichment in SREBP1 and SREBP2, further validating that loss of SCAP protein influences SREBP-mediated lipid anabolism following T cell activation (Table 2). Loss of the transcriptional regulators CREBBP, NFY, and YY1 was also predicted, and since these have well-positioned roles in lipid metabolism, this likely reflect loss of the lipid anabolic program in Scap-deficient T cells. Taken together, these data indicate that SREBP signaling specifically regulates a gene program necessary for lipid anabolism and growth of T cells, but does not grossly influence T cell activation.
Figure 5. SREBP activity influences a transcriptional program related to lipid and RNA metabolism.
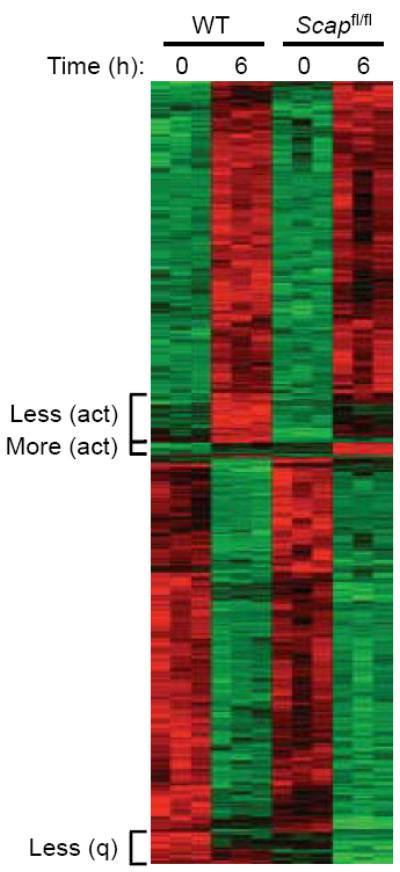
Heat map of genes expressed in purified quiescent or 6 h stimulated (anti-CD3-28) WT and Scapfl/fl CD8+ T cells (all changes in gene expression met criteria of p<0.001 to be considered as significant). Less (act): less induced in activated Scapfl/fl CD8+ T cells. More (act): more induced in activated Scapfl/fl CD8+ T cells. Less (q): less induced in quiescent Scapfl/fl CD8+ T cells.
Table 1. Pathway enrichment analysis of genes less induced in 6 h activated Scapfl/fl versus WT CD8+ T cells using DAVID (NIAID).
The number of genes enriched in each pathway (designated “count”) and associated p values are indicated.
| Enrichment cluster 1: Score 7.09 | Count | P Value |
|---|---|---|
| sterol biosynthetic process | 10 | 6.20 × 10-14 |
| sterol metabolic process | 12 | 6.10 × 10-13 |
| steroid biosynthetic process | 11 | 8.50 × 10-12 |
| cholesterol biosynthetic process | 8 | 3.90 × 10-11 |
| steroid metabolic process | 13 | 1.30 × 10-10 |
| cholesterol metabolic process | 10 | 2.30 × 10-10 |
| lipid biosynthetic process | 14 | 8.90 × 10-9 |
| isoprenoid biosynthetic process | 4 | 2.70 × 10-4 |
| isoprenoid metabolic process | 4 | 2.90 × 10-3 |
| oxidation reduction | 11 | 5.40 × 10-3 |
|
| ||
| Enrichment cluster 2: Score 4.33 | Count | P Value |
|
| ||
| nuclear lumen | 20 | 2.90 × 10-7 |
| intracellular organelle lumen | 22 | 7.00 × 10-7 |
| organelle lumen | 22 | 7.30 × 10-7 |
| nucleolus | 12 | 1.20 × 10-6 |
| membrane-enclosed lumen | 22 | 1.20 × 10-6 |
| nucleoplasm | 10 | 6.60 × 10-3 |
| intracellular non-membrane-bounded organelle | 16 | 1.20 × 10-1 |
| non-membrane-bounded organelle | 16 | 1.20 × 10-1 |
|
| ||
| Enrichment cluster 3: Score 2.75 | Count | P Value |
|
| ||
| ribonucleoprotein complex biogenesis | 8 | 1.40 × 10-5 |
| ribosome biogenesis | 7 | 4.70 × 10-5 |
| rRNA processing | 5 | 8.90 × 10-4 |
| rRNA metabolic process | 5 | 9.40 × 10-4 |
| RNA processing | 9 | 3.80 × 10-3 |
| ribonucleoprotein complex | 9 | 4.50 × 10-3 |
| ncRNA processing | 5 | 1.30 × 10-2 |
| ncRNA metabolic process | 5 | 3.00 × 10-2 |
|
| ||
| Enrichment cluster 4: Score 2.72 | Count | P Value |
|
| ||
| RNA binding | 16 | 6.90 × 10-6 |
| RNA recognition motif, RNP-1 | 7 | 7.10 × 10-4 |
| Nucleotide-binding, alpha-beta plait | 7 | 1.10 × 10-3 |
| mRNA metabolic process | 6 | 3.10 × 10-2 |
| mrna splicing | 4 | 8.40 × 10-2 |
| RNA splicing | 4 | 1.10 ×10-1 |
| mrna processing | 4 | 1.40 × 10-1 |
| mRNA processing | 4 | 2.00 × 10-1 |
Table 2.
Predicted transcription factors for the genes less induced in 6 h activated Scapfl/fl versus WT CD8+ T cells.
| Transcription Factor | P value | FDR |
|---|---|---|
| CREBBP | 2.47 × 10-9 | 4.55 × 10-3 |
| SREBF2 | 2.48 × 10-9 | 6.82 × 10-3 |
| SREBF1 | 2.65 × 10-8 | 9.09 × 10-3 |
| NFYA | 3.86 × 10-5 | 1.14 × 10-2 |
| YY1 | 6.69 × 10-4 | 1.36 × 10-2 |
SREBP is required for metabolic reprogramming
To further interrogate the metabolic state of Scap-deficient T cells, we performed cellular bioenergetics analysis on quiescent and mitogen stimulated T cells. No difference in the basal extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) was observed between quiescent control and Scapfl/fl CD8+ T cells (Fig. 6a). Stimulation of control CD8+ T cells with anti-CD3-28 for 24 h resulted in a 10-fold increase in ECAR and a 4-fold increase in OCR. In contrast, Scapfl/fl CD8+ T cells only modestly increased OCR and ECAR, but were unable achieve the levels of control cells (Fig. 6a).
Figure 6. SREBP signaling is required for metabolic reprograming of activated CD8+ T cells.
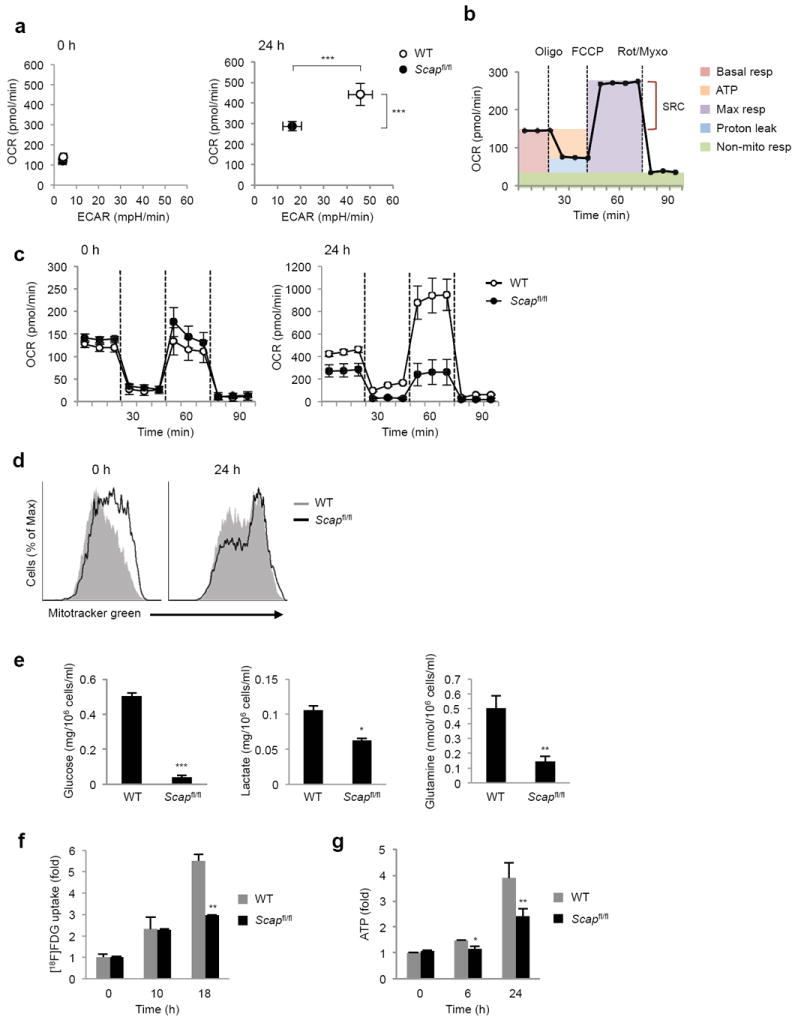
(a) Basal oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) of quiescent and 24 h stimulated WT and Scapfl/fl CD8+ T cells. (b) A schematic of OCR in basal condition and in response to sequential treatment with oligomycin (oligo: ATPase inhibitor), FCCP (uncoupling agent) and rotenone-myxothiazol (Rot-Myxo: electron transport chain inhibitor). Basal resp: basal respiration, ATP: ATP production, Max resp: maximal respiration, Non-mito resp: non-mitochondrial respiration, SRC: spare respiratory capacity (c) OCR of 24 h activated WT and Scapfl/fl CD8+ T cells in basal state and in response to sequential treatment with oligomycin, FCCP and rotenone-myxothiazol. (d) Mitotracker staining of WT and Scapfl/fl CD8+ T cells ex vivo and after 24h of activation with anti-CD3-28. (e) Glucose and glutamine consumption, and lactate production measurement in spent media from activated WT and Scapfl/fl CD8+ T cell activated for 24 h with anti-CD3-28. (f) 2-deoxy-2-(18F)fluoro-D-glucose (FDG) uptake assay. CD8+ T cells were activated with anti-CD3-28 for indicated time. Cultures were pulsed with [18F]FDG for 60 min before analysis. (g) Cellular ATP levels in quiescent, 6 or 24 h activated WT and Scapfl/fl CD8+ T cells (anti-CD3-28). Data are mean of triplicates with standard deviation and representative of two (c) or three (d) independent experiments. *p<0.05, **p<0.01, ***p<0.001 (two-tailed unpaired Student’s t-test).
Next, we directly assessed mitochondrial function in Scap-deficient CD8+ T cells (ATP production and spare respiratory capacity) by monitoring the OCR in response to sequential treatment with oligomycin (ATPase inhibitor), FCCP (uncoupling agent) and the electron transport chain inhibitors rotenone and myxothiazol (Fig. 6b). Treatment of freshly isolated CD8+ T cells with oligomycin, FCCP and rotenone-myxothiazol revealed no difference in basal respiration, ATP production or spare respiratory capacity of quiescent control and Scapfl/fl CD8+ T cells (Fig. 6c). Activated control CD8+ T cells increased basal OCR, mitochondrial ATP production and spare respiratory capacity (Fig. 6c), consistent with significantly heightened mitochondrial function in blasting CD8+ T cells. In contrast, activated Scapfl/fl CD8+ T cells had significant deficiencies in basal respiration, ATP production and spare respiratory capacity (Fig. 6c), suggesting profound respiratory defects. We considered the possibility that the decrease in respiratory capacity was a function of perturbations in mitochondrial mass, however no difference in mitotracker staining was observed between control and Scap-deficient CD8+ T cells (Fig. 6d). Taken together, these data suggest that SREBP signaling is required for the acquisition of glycolytic metabolism and heightened mitochondrial respiration during CD8+ blastogenesis.
Glucose and glutamine are important carbon sources for energetics, anaplerosis (replenishment of TCA cycle intermediates) and macromolecular biosynthesis in activated lymphocytes7. Thus, we asked if SREBP signaling was influencing glucose and glutamine utilization in blasting T cells by directly measuring glucose, glutamine and lactate concentrations in culture media 24 hours after activation. Consistent with our ECAR data, we observed a marked decrease in lactate production by activated Scapfl/fl CD8+ T cells (Fig. 6e). We also observed a corresponding decrease in both glucose and glutamine consumption in activated Scapfl/fl CD8+ T cells (Fig. 6e). Kinetic analysis of [18F]-FDG (an 18F-labeled glucose derivative) uptake by CD8+ T cells revealed that both control and Scap-deficient CD8+ T cells initially increase their glucose uptake in response to activation (Fig. 6f). However, Scap-deficient CD8+ T cells were unable to increase glucose uptake, resulting in an approximate 50% reduction in [18F]-FDG signals at 18 hours after activation.
RT-PCR analysis revealed that Scapfl/fl CD8+ T cells have significantly lower expression of glycolysis genes (Supplementary Fig. 4a). Both Myc and HIF1α have important roles in the upregulation of glycolytic program 22, thus we considered the possibility that Scap-deficient T cells might have perturbations in these pathways. However, immunoblots indicated normal upregulation of Myc, and increased stabilization of HIF1α in activated Scap-deficient CD8+ T cells (Supplementary Fig. 4b). Thus, the inability of Scapfl/fl CD8+ T cells to acquire a glycolytic program during activation is likely not a direct function of altered Myc or HIF1α signaling.
A logical prediction of our metabolic studies is that blasting Scap-deficient T cells would have altered bioenergetics. To directly address this, we determined intracellular ATP concentrations in quiescent and anti-CD3-28 activated CD8+ T cells. No difference in ATP concentration was observed between control and Scapfl/fl CD8+ T cells directly ex vivo (Fig. 6g). Activation of control CD8+ T cells modestly increased ATP levels by 6 h, and resulted in a 4-fold increase in cellular ATP by 24 h (Fig. 6g). In contrast, Scapfl/fl CD8+ T cells did not increase their cellular ATP concentration at 6h and increased ATP by 2-fold at 24 h post stimulation (Fig. 6g). We considered the possibility that perturbations in ATP levels would activate the AMP-activated protein kinase (AMPK) pathway in Scap-deficient CD8+ T cells. Consistent with earlier studies23, activation of control T cells resulted in phosphorylation of AMPK and the AMPK target acetyl-CoA carboxylase (ACC). No difference in p-AMPK and p-ACC from Scapfl/fl T cells was noted (Supplementary Fig. 4c), suggesting that the difference in glycolysis and cellular growth are not a function of exaggerated AMPK signaling. Taken together, these data support the notion that SREBP signaling influences cellular bioenergetics during blastogenesis independent of the Myc, HIF1α and AMPK pathways.
Cholesterol restores growth of Scap-deficient T cell
Our gene expression data indicates that loss of Scap primarily influences lipid homeostasis, in particular flux through the mevalonate pathway. In addition to cholesterol synthesis, the mevalonate pathway is responsible for the generation of non-steroidal lipid modification to proteins, such as prenylation (e.g., Ras) 8, 24. Thus, we asked if addition of excess mevalonic acid to cultures would increase flux into isoprenoid synthesis, and rescue cellular growth to Scap-deficient T cells. However, the addition of mevalonic acid (50-100μM) did not impact survival or blastogenesis (Supplementary Fig. 5a), suggesting that a defect in the prenylation of proteins could not mechanistically explain the phenotype of Scapfl/fl CD8+ T cells.
These data led us instead to posit that cholesterol was limiting in Scapfl/fl CD8+ T cells. To directly test this predication, control and Scap-deficient T cells were activated in complete media supplemented with 5μg/ml of cholesterol conjugated to Methyl-β-cyclodextrin (MBCD-Chol). Analysis of cultures up to 48 h revealed that addition of cholesterol to control CD8+ cells provided a modest increase in cellular survival, with no influence on size (Fig. 7a, Supplementary Fig. 5b). In contrast, Scap-deficient T cells doubled their survival at 48 h from 21 % to 45%, approaching that of control T cells without supplemental cholesterol (54%). Furthermore, supplementation with cholesterol restored the cellular enlargement of Scapfl/fl CD8+ T cells following activation with anti-CD3-28 (Fig. 7a,b). Analysis of cell cycle 48 h after stimulation indicated that addition of cholesterol markedly increased the frequency of Scapfl/fl CD8+ T cells in S and G2-M (Fig. 7c). Likewise, CFSE analysis indicated that Scap-deficient T cells were now able to undergo robust proliferation in response to mitogenic stimulation (Fig. 7d).
Figure 7. Addition of cholesterol rescues growth and proliferation impairment of Scap-deficient CD8+ T cells.
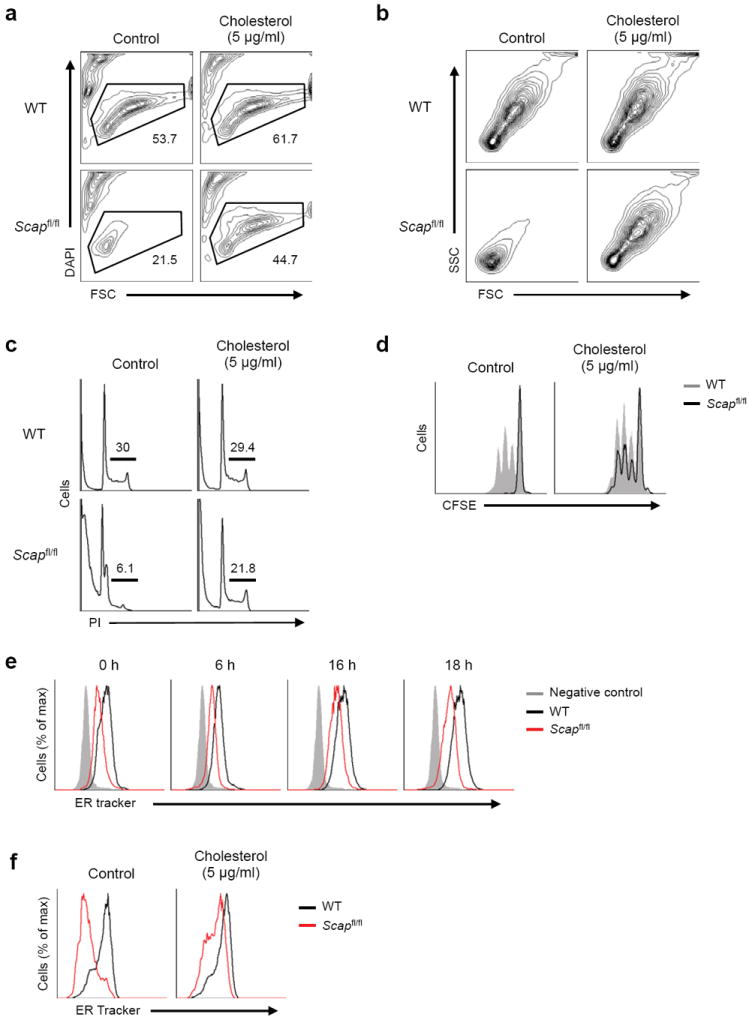
(a) Viability of WT and Scapfl/fl CD8+ T cell activated with anti-CD3-28 for 48 h with or without 5 μg/ml of MβCD conjugated cholesterol in 10% FBS supplemented media. DAPI-negative (live) cell populations are gated and the frequencies are indicated in plots. (b) Forward and side scatter plots of DAPI-negative (live) WT and Scapfl/fl CD8+ T cell activated with anti-CD3-28 for 48 h with or without 5 μg/ml of MβCD-cholesterol in 10% FBS supplemented media. (c) Flow cytometric analysis of DNA content from WT and Scapfl/fl CD8+ T cells activated for 48 hours with or without 5 μg/ml of MβCD-cholesterol in 10% FBS supplemented media. Cells were stained for DNA content with propidium iodide. Frequency of S and G2/M phase indicated in plots. (d) CFSE dilution of DAPI negative (live) WT and Scapfl/fl CD8+ T cells activated for 48 hours with or without 5 μg/ml of MβCD-cholesterol in 10% FBS supplemented media. (e) ER-tracker staining of WT and Scapfl/fl CD8+ T cells ex vivo or after activation with anti-CD3-28 for indicated time. (f) ER-tracker staining of WT and Scapfl/fl CD8+ T cells activated for 48 hours with or without 5 μg/ml of MβCD-cholesterol in 10% FBS supplemented media.
Cellular cholesterol levels have been implicated in regulating phospholipid synthesis. The observation that replenishing cholesterol was sufficient to significantly restore the growth of Scap-deficient T cells led us to ask if membrane biogenesis was diminished in the absence of SREBP activity. Membrane biosynthesis occurs in the endoplasmic reticulum (ER) before shuttling to other intracellular organelles or the plasma membrane. Thus, we assessed total ER content by flow cytometry in quiescent and newly activated cells using ER tracker. Quiescent Scapfl/fl CD8+ T cells had decreased ER content when compared to their control counterparts (Fig. 7e). Activation of control cells led to the addition of ER (Fig. 7e). In contrast, Scapfl/fl CD8+ T cells were unable to expand the ER during blastogenesis (Fig. 7e). However, the addition of exogenous cholesterol restored ER growth (Fig. 7f). These data are consistent with previous work implicating SREBPs in membrane biogenesis 25, and support the notion that intracellular cholesterol serves as a metabolic checkpoint in cell cycle progression via regulation of ER membrane biogenesis.
SREBPs intrinsically regulate virus-specific immunity
To determine if SREBP signaling was also required for efficient T cell clonal expansion in vivo, Scapfl/fl and control mice were challenged with LCMV Armstrong (Arm). LCMV-Arm infection of wild type mice generates a robust virus-specific T cell response leading to rapid control of virus infection within 10-12 days 26. On day 8 following infection, splenocytes were counted and stained with LCMV-specific DbGP33 tetramers to determine the level of virus-specific CD8+ T cell. Scapfl/fl mice exhibited a significant decrease in the frequency and total number of DbGP33-specific CD8+ T cells (Fig. 8a-c). A similar decrease was observed in the DbNP396-specific CD8+ T cell, indicating a globally diminished antiviral T cell response (Fig. 8c). Intracellular interferon-γ (IFN-γ) and tumor necrosis factor (TNF) staining in response to GP33 peptide challenge ex vivo confirmed a decrease in the frequency of responding CD8+ T cells (Fig. 8d,e). The few LCMV-specific CD8+ T cells that were generated in Scap-deficient mice, displayed decreased expression of IFN-γ and TNF (Fig. 8d), suggesting a potential functional defect in the absence of SREBP activity.
Figure 8. Loss of SREBP activity impairs clonal expansion of antigen specific effector CD8+ T cells, but does not perturb homeostatic proliferation.
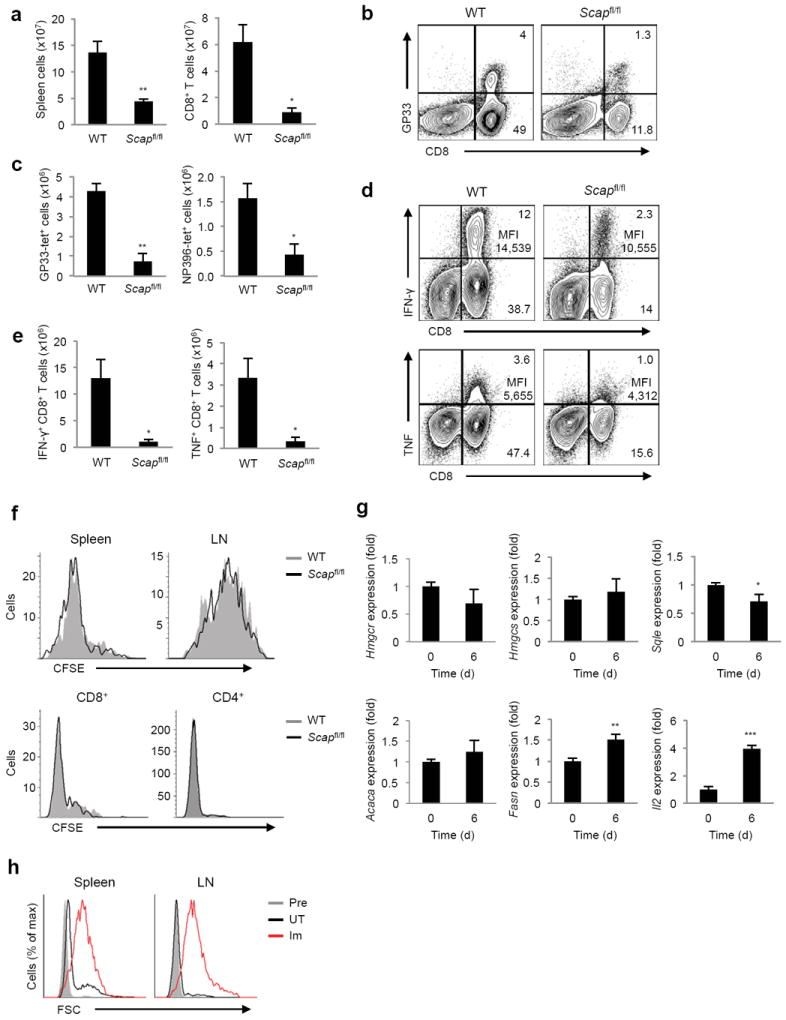
(a) Absolute numbers of splenocytes and CD8+ T cells from spleen harvested on day 8 of LCMV Armstrong (Arm) infection. (b) Frequency of DbGP33-tetramer specific CD8+ T cells from spleen harvested on day 8 of LCMV-Arm infection. Frequency of cells marked in quadrants of FACS plots. (c) Absolute numbers of CD8+DbGP33-tetramer+ cells and CD8+NP396-tetramer+ cells from spleen harvested on day 8 of LCMV-Arm infection. (d,e) Frequency (d) and absolute number (e) of IFN-γ or TNF producing WT and Scapfl/fl CD8+ T cells harvested on day 8 of LCMV-Arm infection and stimulated with GP33-peptide for 5h in vitro. Frequency and mean fluorescent intensity (MFI) of cytokine producing CD8+ T cells indicated in right upper quadrant of FACS plots. (f) CFSE dilution gated on Thy1.2+ CD8+(top), and CD3+CD4+or CD3+CD8+ (bottom) cells on day 6 after adoptive transfer into RAG-deficient Thy1.1+ mice (top) or irradiated littermate WT mice (bottom). (g) Real-time PCR analysis of Thy1.1+ T cells adoptively transferred into RAG-deficient Thy1.2+ mice and sorted from spleens and LNs after 6 days. (h) Forward scatter plots of OT-I cells. Cells were adoptively transferred into RAG-deficient mice and either left untreated (UT) or immunized (Im) with OVA/IFA subcutaneously. Twenty-four hours later, cells from spleens and LNs were stained as described in Methods and analyzed on flow cytometry. Pre: pre-transfer. *p<0.05, **p<0.01 (two-tailed unpaired Student’s t-test).
Finally, our data indicates an essential requirement for SREBPs in mitogen-driven proliferation in vitro and clonal expansion of viral-specific CD8+ effector cells in vivo. However, we did not observe significant perturbations in the frequency or absolute numbers of peripheral T cells from naive Cd4-Cre-Scapfl/fl mice (Fig. 2). These data led us to posit that SREBP activity might not be required under homeostatic conditions. To initially address this, we examined the homeostatic proliferation of purified, CFSE-labeled CD8+ T cells adoptively transferred in to Thy1.1+RAG2-deficeint hosts. On day 6 following adoptive transfer, CFSE dilution of Thy1.2+ T cells from LN and spleen was assessed. Consistent with our hypothesis, we observed no difference in the proliferative patterns of Scap-deficient and control T cells (Fig. 8f). Similar proliferative results were seen in a sub-lethal irradiation model of homeostatic proliferation 27 (Fig. 8f). RT-PCR revealed that homeostatically proliferating cells did not upregulate the SREBP driven cholesterol synthetic genes (Fig. 8g), although we did observe a very modest upregulation of Fasn. Thus we posited that a major difference between T cells undergoing homeostatic proliferation versus conventional antigen-driven proliferation might be cellular growth. Indeed, analysis of cell size indicated that homeostatically proliferating OT-I cells remained small despite undergoing significant proliferation whereas OVA immunized OT-I cells underwent conventional blastogenesis (Fig. 8h). Taken together, these data reveal a context specific requirement for SREBP signaling during conventional antigen-driven responses to support cellular enlargement and effector function.
DISCUSSION
The molecular events guiding the metabolic reprogramming of T cells during activation remain poorly understood. Our studies convincingly demonstrate that SREBPs are essential to coordinate TCR signaling with a lipid anabolic program requisite for rapid membrane biosynthesis and cellular growth. In the absence of SREBP signaling, newly activated CD8+ T cells move into G1 of the cell cycle but are unable to grow, resulting in decreased proliferative capacity and markedly attenuated anti-viral immune responses. Strikingly, this defect in growth and proliferation was traced to an insufficiency in cholesterol. These data support a model where an intracellular cholesterol pool, likely in the endoplasmic reticulum – the site of lipid biosynthesis – serves as a metabolic signal conveying the fitness of a blasting T cells. Consequently, the SREBP gene program facilitates the transition of mitogen-stimulated T cells through a critical metabolic checkpoint during blastogenesis. These signals ensure coordination of cellular metabolism with cell cycle. Simply put, without sufficient cholesterol, effector T cells are unable to grow in response to conventional antigen stimulation.
One quite surprising aspect of our study is that SREBP activity is dispensable for homeostatic proliferation of CD8+ T cells. SREBPs have importance in regulating the proliferative capacity and survival of cancer cells 28, 29, albeit through poorly described and disparate mechanisms. The influence of SREBPs on primary cell proliferation is even less well understood. Our studies on conventional antigen-driven proliferation and anti-viral immunity clearly support the concept that SREBPs influence rapid cellular growth. Nevertheless, the observation that Scap-deficient T cells have no observable defects in homeostatic proliferation is the first clear evidence that there are differential requirements for SREBP signaling during proliferation. As such, the emerging notion that engagement of the SREBP pathway is universally required for cellular proliferation and survival is likely incorrect. Alternatively, we would propose that SREBP signaling is necessary under circumstances where cells require the rapid addition of biomass, rather than proliferation per se.
An interrelated and important concept centers on the observations that effector and memory CD8+ T cells employ distinct metabolic programs. Generation of effector CD8+ T cells appears to be favored by glycolytic and anabolic programs, whereas memory T cell generation is enhanced under heightened OXPHOS and catabolism 2, 30. Our metabolic studies indicate that SREBP signaling is required for a CD8+ T cell to fully acquire a glycolytic phenotype. One intriguing possibility is that inhibition of SREBP in responding T cells could preferentially enrich for memory at the expense of effector cell generation. Recent studies have indicated that moderate inhibition of mTOR signaling with low dose rapamycin favors the generation of memory CD8+ T cells 31, 32. Other studies indicate that mTOR signaling controls cellular metabolism through the HIF and SREBP transcription factors 33, and the data presented here clearly indicate that SREBP in CD8+ T cells is rapamycin-sensitive during T cell activation. A logical prediction of these studies is that low dose rapamycin is favoring the development of CD8+ memory cells through its effects on SREBP activity. Future studies will be required to rigorously test this prediction. Nevertheless, our data raise the possibility that generation of memory and effector cells could be uncoupled via manipulation of sterol metabolism.
In conclusion, these studies delineate a role for SREBP in controlling conventional antigen-driven clonal expansion, but not homeostatic proliferation. These observations could have significant implications for developing novel therapeutic strategies targeting this pathway to control unwanted effector T cell proliferation, without perturbing homeostasis or development.
Methods
Mice
Scapflox/flox 17 and C57/BL6 mice were purchased from the Jackson Laboratory. Cd4-Cre mice were purchased from Taconic. Scapflox/flox mice were crossed with Cd4-Cre mice to generate Cd4-Cre/Scapflox/flox mice. Cd4-Cre/Scap+/+ mice were used as littermate control for all experiments using Cd4-Cre/Scapflox/flox mice. All mice were maintained in pathogen free facilities of the University of California, Los Angeles. All experiments on mice and tissues collected from mice were performed in strict accordance with University of California, Los Angeles policy on the humane and ethical treatment of animals.
Cell culture and reagents
Cells were cultured in IMDM supplemented with 10% heat inactivated FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 uM 2-ME. Total T cells or CD8+ T cells were purified using negative enrichment kit (STEMCELL Technologies). T cells were cultured in 96-well plates coated with 5 μg/ml of anti-CD3 (2C11;Bio X Cell) alone or in combination with 2 μg/ml of soluble anti-CD28 (37.51;Bio X Cell) or 100 IU/ml of human IL-2. Alternatively, cells were cultured with 50 ng/ml of PMA (EMD Chemicals) alone or in combination with 500 ng/ml of ionomycin (Sigma-Aldrich). For proximal TCR signaling analysis, splenocytes were cultured with 1 μg/ml of soluble anti-CD3. In some experiments, cells were cultured with following inhibitors for 30 min prior to activation: Gö6983 (Sigma-Aldrich), Ly294002 (EMD Chemicals), Rapamycin (EMD Chemicals), 25-hydroxycholesterol (Sigma-Aldrich).
Virus transduction
Truncated forms of SREBP1a or SREBP2 (Supplementary Fig. 1c) were cloned into the retrovirus vector MIGR1 (a gift from Dr. Yukiko Tone and Dr. Masahide Tone, Cedars-Sinai, Los Angeles, USA). Virions were produced using the 293T cell line by transfection using Lipofectamine 2000 (Invitrogen). Purified naiveT cells were stimulated in 6-well plates pre-coated with 5 μg/ml of anti-CD3 supplemented with 2 μg/ml of anti-CD28 at 2 × 106 cells/well. At 24 hours, cells were transduced by centrifugation in viral medium plus 8 μg/ml of polybrene and 100 IU/ml of IL-2 at 1200 rpm for 90 min at 37 °C. Medium was then replaced with complete IMDM supplemented with 100 IU/ml of IL-2 and cells were incubated for additional 24 hours. Control cells were subjected to the same stimulation and centrifugation as transduced cells but without virus. After a total of 48 hours of stimulation, cells were harvested and transduction efficiencies were evaluated by GFP expression level on flow cytometry.
RNA interference
Purified T cells were transfected with non-target siRNA, SREBP1 siRNA, or SREBP2 siRNA (Dharmacon) using Nucleofector according to manufacture’s protocol (Lonza). Transfected cells were rested for 18 hours at 37 °C and then stimulated with PMA and ionomycin for 5 hours.
Quantitative real-time PCR analysis
RNA was isolated using Trizol (Invitrogen). cDNA was synthesized using iScript cDNA Synthesis Kit (BIO-RAD). qRT-PCR was performed using SYBR Green I Master (Roche) on LightCycler 480 (Roche). Primer sequences are available upon request.
Flow cytometry analysis
7-ADD, anti-mouse CD4 (RM4-5), CD8 (53-6.7), CD25 (PC61.5), CD44 (IM7), Thy1.2 (53-2.1), IL-7R (CD127), KLRG1 (2F1), INFγ (XMG1.2), TNFα (MP6-XT22), and IL-2 (JES6-5H4) were purchased from eBioscience, BioLegend, or BD Biosciences. Active caspase3 and annexin V staining kits were purchased from BD Bioscience. CFDA SE, ER-Tracker Blue-White DPX, and DAPI were purchased from molecular probe. Propidium Iodide was purchased from Calbiochem. Cells were analyzed on LSRII or FACSVersa with FlowJo software (Treestar).
Immunoblot analysis
Whole cell extracts were produced from equivalent number of cells with RIPA buffer (50mM Tris-HCl [pH 8.0], 150mM NaCl, 1% NP40, 0.5% Deoxycholate, 0.1% SDS) supplemented with Calyculin A (Cell Signaling) and protease inhibitor cocktail (Sigma-Aldrich). Samples were separated on a 4-12% Bis-Tris gel and transferred to nitrocellulose. Antibodies for phospho-Akt (S473), Akt1 (5C10), Actin (I-19) were purchased from Santa Cruz Biotechnology. Antibody for HIF1α was from Cayman Chemical. Antibodies for S6 ribosomal protein (54D2), phospho-S6 (S235/236)(D57.2.2E), Rb (D20), phospho-Rb (Ser807/811), p27Kip1, Cyclin D2 (D52F9), Cyclin D3 (DCS22), MYC, phospho-Zap70 (Tyr319)/Syk (Tyr352)(65E4), phospho-Lck (Tyr505), LAT, phospho-LAT (Tyr191), p44/42 MAPK (137F5), phospho-p44/42 MAPK (Thr202/Tyr204)(D13.14.4E), AMPK (F6), phospho-AMPK (Thr172)(40H9), phospho-ACC (Ser79) were from Cell Signaling.
Chromatin immunoprecipitation analysis
The method is described elsewhere34. Briefly, 2×107 cells were fixed and sonicated. Precleared lysates were incubated overnight at 4 °C with polyclonal anti-SREBP1 and anti-SREBP2, or control rabbit IgG (Cell Signaling). Immunocomplex was collected and real-time PCR was performed as described above. Primer sequences are available upon request.
Gene expression analysis
RNA isolated from littermate control and Scap-deficient CD8+ T cells and hybridized to Affymetrix GeneChip Mouse Genome 430 2.0 Array. Genes were filtered for differential expression across the samples by requiring an ANOVA p-value less than 0.001. For heatmap display, genes were mean centered, normalized and clustered using a Pearson correlation coefficient metric and pairwise complete-linkage. Pathway enrichment analysis was done with DAVID 20, 21. Transcription factor analysis was generated through the use of IPA (Ingenuity Systems, www.ingenuity.com).
Metabolic assays
Cellular ATP levels were measured from the same number of live cells using ATP determination kit according to the manufacture’s protocol (Molecular Probes). Background was measured using buffer without cells and subtracted from the values of each cell sample. For mitochondrial mass, cells were incubated with 50 nM MitoTracker green (Molecular Probes) at 37 °C for 30 min. The oxygen consumption rate and extracellular acidification rate were measured with a XF24 analyzer according to the manufacture’s recommendations 35. Mixing, waiting and measure times were 2, 2, 4 minutes, respectively. Test compounds were obtained from Sigma and injected during the assay at the following final concentration: 0.2 μM oligomycin, 1 μM FCCP, 0.75 μM Rotenone/Myxothiazol. Glucose, glutamine, and lactate concentrations in culture media were measured using bioprofile analyzer (Nova Biomedical).
Cholesterol loading
Cholesterol:MβCD complex containing 40 mg of cholesterol/g was purchased from Sigma (C4951). Treatment concentration was based on cholesterol weight. Cells were activated with or without cholesterol:MβCD in 10% FBS for indicated time.
GC-MS
Purified T cells were collected at ex vivo or after 24 hours of activation with plate-bound anti-CD3 in combination with soluble CD28 and washed twice with PBS. Fatty acids extractions and analyses were performed as previously described 36 with the modifications described below. Triheptadecanoin (Nu-chek Prep, T-155) was used as the internal standard for fatty acids. Ergosterol (Sigma 17130-U) was used as the internal standard for cholesterol. Internal standards were added to trizol. Cholesterol was extracted from organic phase with petroleum ether after saponification but before acidification and fatty acid extraction. Extracted cholesterol was trimethylsilylated with BSTFA and TMCS (Sigma 33155-U). Extracted fatty acids were derivatized into their methyl ester form with methanolic boron trifluoride. Derivatized cholesterol and ergosterol were monitored at m/z 456-467 and 468 respectively. Derivatized myristate, palmatate, heptadecanoate, and sterate were monitored at m/z 241-250, 269-278, 284, and 297-306 respectively. Data was collected on an Agilent 5975C MSD connected to an Agilent 7890 Gas Chromatograph with the Phenomenex ZB-MR-1 column. Settings and oven programs are available upon request. Area under the curve quantitation for selected ions was performed on Chemstation software.
LCMV infection, MHC I tetramer and intracellular cytokine staining
Mice were infected intraperitoneally (i.p.) with 2 × 104 plaque forming units (PFU) of LCMV-Armstrong. Virus stocks were prepared and viral titers were quantified as described previously 37. Splenocytes were stained directly ex vivo with LCMV-DbGP33-41 or LCMV-DbGP396-404 specific tetramers and for surface expression of CD8. MHC tetramers were obtained from the NIH Tetramer Core Facility. To analyze cytokine expression, splenocytes were stimulated for 5 hours with 2 μg/ml of the MHC class I restricted LCMV-GP33-41 or NP396-404 peptide in the presence of 50 U/ml recombinant murine IL-2 (R&D Systems) and 1mg/ml brefeldin A (Sigma-Aldrich). Cells were stained for surface expression of CD8, then fixed, permeabilized and stained with antibodies to TNF, IFN-γ and IL-2 (BioLegend). Flow cytometric analysis was performed using an LSR II or the FACSVerse (Becton Dickinson) and analyzed using FlowJo software (Treestar).
Adoptive cell transfer
CD8+ T cells or total T cells were purified from Scapfl/fl and control littermate mice or Thy1.1 mice spleens as above. Purified cells were labeled with CFSE and 1-5 × 106 cells/mouse were injected via retro-orbital into RAG-deficient host or irradiated host 1 day earlier with 600 rad. After 6 days, host spleen and LN cells were analyzed by flow cytometry. For gene expression analysis, Thy1.1+ T cells were sorted and real-time PCR was performed as described above.
OVA peptide-IFA immunization
Forty-eight hours after the OT-I cell transfer, mice were either left unimmunized or immunized with total of 300 μg of OVA peptide mixed in IFA into 3 sites in upper back and 3 sites in lower back (n=3 per group). Spleen and lymph nodes were harvested 24 hours later and CD3+CD8+ cells were analyzed on flow cytometry.
BrdU labeling
BrdU flow kit was purchased from BD Bioscience. BrdU (2 mg/mouse) was injected to Cd4-Cre/Scapflox/flox and littermate control mice intraperitoneally. Peripheral blood was collected twice a week and mononuclear cells were stained and analyzed on flow cytometry according to manufacture’s protocol.
Statistical analysis
Statistical significance was determined with the two-tailed unpaired Student’s t-test unless otherwise indicated.
Supplementary Material
Acknowledgments
We thank A Do, L Pang, A Armijo, C Radu for technical assistance, and P Tontonoz and M Day for critical feedback on the manuscript. These studies were supported by the National Institutes of Health (AI093768 to S.J.B., AI082975 and AI085043 to D.G.B.), Jonsson Comprehensive Cancer Center Foundation/UCLA seed grant (S.J.B), the National Center for Research Resources (S10RR026744 to L.V. and K.R.), Jonsson Cancer Center Foundation/UCLA fellowship (Y.K.), National Cancer Institute of the National Institutes of Health (T32-CA009120-36 to K.J.W), and USPHS National Research Service Award (T32-GM008469 to J.P.A.) and the UCLA Graduate Division (J.P.A.).
Footnotes
Author contributions
Y.K. designed, did and analyzed most of the experiments and wrote the manuscript; H.E., M.B.H., L.V., K.J.W., J.P.A., B.M.M. E.K. and E.B.W. did experiments and analyzed data; T.F.O. provided materials and intellectual input; and S.J.B. provided overall coordination for the conception, design and supervision of the study and wrote the manuscript (with input from T.F.O., T.G.G., K.R. and D.G.B.).
Accession codes
Gene expression omnibus (GEO): GSE44261, GSM1081398, GSM1081399, GSM1081400, GSM1081401, GSM1081402, GSM1081403, GSM1081404, GSM1081405, GSM1081406, GSM1081407, GSM1081408, GSM1081409.
References
- 1.Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66:8927–8930. doi: 10.1158/0008-5472.CAN-06-1501. [DOI] [PubMed] [Google Scholar]
- 2.Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nature reviews. Immunology. 2005;5:844–852. doi: 10.1038/nri1710. [DOI] [PubMed] [Google Scholar]
- 3.Maciver NJ, et al. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leukoc Biol. 2008;84:949–957. doi: 10.1189/jlb.0108024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen HW, Heiniger HJ, Kandutsch AA. Relationship between sterol synthesis and DNA synthesis in phytohemagglutinin-stimulated mouse lymphocytes. Proceedings of the National Academy of Sciences of the United States of America. 1975;72:1950–1954. doi: 10.1073/pnas.72.5.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rathmell JC, Elstrom RL, Cinalli RM, Thompson CB. Activated Akt promotes increased resting T cell size, CD28-independent T cell growth, and development of autoimmunity and lymphoma. European journal of immunology. 2003;33:2223–2232. doi: 10.1002/eji.200324048. [DOI] [PubMed] [Google Scholar]
- 6.Carr EL, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol. 2010;185:1037–1044. doi: 10.4049/jimmunol.0903586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang R, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bensinger SJ, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell. 2008;134:97–111. doi: 10.1016/j.cell.2008.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michalek RD, et al. Estrogen-related receptor-alpha is a metabolic regulator of effector T-cell activation and differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18348–18353. doi: 10.1073/pnas.1108856108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chakrabarti R, Engleman EG. Interrelationships between mevalonate metabolism and the mitogenic signaling pathway in T lymphocyte proliferation. J Biol Chem. 1991;266:12216–12222. [PubMed] [Google Scholar]
- 11.Geyeregger R, et al. Liver X receptors interfere with cytokine-induced proliferation and cell survival in normal and leukemic lymphocytes. Journal of leukocyte biology. 2009;86:1039–1048. doi: 10.1189/jlb.1008663. [DOI] [PubMed] [Google Scholar]
- 12.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. The Journal of clinical investigation. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimomura I, Shimano H, Korn BS, Bashmakov Y, Horton JD. Nuclear sterol regulatory element-binding proteins activate genes responsible for the entire program of unsaturated fatty acid biosynthesis in transgenic mouse liver. J Biol Chem. 1998;273:35299–35306. doi: 10.1074/jbc.273.52.35299. [DOI] [PubMed] [Google Scholar]
- 14.Yang K, Neale G, Green DR, He W, Chi H. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nature immunology. 2011;12:888–897. doi: 10.1038/ni.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shimano H, et al. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J Clin Invest. 1996;98:1575–1584. doi: 10.1172/JCI118951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimano H, et al. Elevated levels of SREBP-2 and cholesterol synthesis in livers of mice homozygous for a targeted disruption of the SREBP-1 gene. J Clin Invest. 1997;100:2115–2124. doi: 10.1172/JCI119746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsuda M, et al. SREBP cleavage-activating protein (SCAP) is required for increased lipid synthesis in liver induced by cholesterol deprivation and insulin elevation. Genes & development. 2001;15:1206–1216. doi: 10.1101/gad.891301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He HT, Lellouch A, Marguet D. Lipid rafts and the initiation of T cell receptor signaling. Seminars in immunology. 2005;17:23–33. doi: 10.1016/j.smim.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 19.Janes PW, Ley SC, Magee AI, Kabouridis PS. The role of lipid rafts in T cell antigen receptor (TCR) signalling. Seminars in immunology. 2000;12:23–34. doi: 10.1006/smim.2000.0204. [DOI] [PubMed] [Google Scholar]
- 20.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 21.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dang CV, Kim JW, Gao P, Yustein J. The interplay between MYC and HIF in cancer. Nature reviews. Cancer. 2008;8:51–56. doi: 10.1038/nrc2274. [DOI] [PubMed] [Google Scholar]
- 23.Tamas P, et al. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. The Journal of experimental medicine. 2006;203:1665–1670. doi: 10.1084/jem.20052469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levental I, Grzybek M, Simons K. Greasing their way: lipid modifications determine protein association with membrane rafts. Biochemistry. 2010;49:6305–6316. doi: 10.1021/bi100882y. [DOI] [PubMed] [Google Scholar]
- 25.Castoreno AB, et al. Transcriptional regulation of phagocytosis-induced membrane biogenesis by sterol regulatory element binding proteins. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:13129–13134. doi: 10.1073/pnas.0506716102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. The Journal of experimental medicine. 1984;160:521–540. doi: 10.1084/jem.160.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldrath AW, Bogatzki LY, Bevan MJ. Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. The Journal of experimental medicine. 2000;192:557–564. doi: 10.1084/jem.192.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freed-Pastor WA, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244–258. doi: 10.1016/j.cell.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo D, et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Science signaling. 2009;2:ra82. doi: 10.1126/scisignal.2000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Windt GJ, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Araki K, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pearce EL, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duvel K, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tone Y, et al. OX40 gene expression is up-regulated by chromatin remodeling in its promoter region containing Sp1/Sp3, YY1, and NF-kappa B binding sites. J Immunol. 2007;179:1760–1767. doi: 10.4049/jimmunol.179.3.1760. [DOI] [PubMed] [Google Scholar]
- 35.Wu M, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. American journal of physiology. Cell physiology. 2007;292:C125–136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- 36.Boren J, et al. Gleevec (STI571) influences metabolic enzyme activities and glucose carbon flow toward nucleic acid and fatty acid synthesis in myeloid tumor cells. The Journal of biological chemistry. 2001;276:37747–37753. doi: 10.1074/jbc.M105796200. [DOI] [PubMed] [Google Scholar]
- 37.Brooks DG, Teyton L, Oldstone MB, McGavern DB. Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral infection. Journal of virology. 2005;79:10514–10527. doi: 10.1128/JVI.79.16.10514-10527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.