

Abstract
The x-ray crystal structures of several important vitamin B12 binding proteins that have been solved in recent years have enhanced our current understanding in the vitamin B12 field. These structurally diverse groups of B12 binding proteins perform various important biological activities, both by transporting B12 as well as catalyzing various biological reactions. An in-depth comparative analysis of these structures was carried out using PDB coordinates of a carefully chosen database of B12 binding proteins to correlate the overall folding of the molecule with phylogeny, the B12 interactions, and with their biological function. The structures of these proteins are discussed in the context of this comparative analysis.
1. Introduction
Vitamin B12 (also known as Cobalamin; Cbl; B12) is a cobalt- containing heterocyclic compound and is essential for the growth and development of many eukaryotes and prokaryotes organisms. However, it is not possible for higher organisms to synthesize B12 and must be absorbed from food sources. The history of B12 dates back to the 1920s, with the discovery of the connection between B12 and pernicious anemia [1, 2]. The x-ray crystallographic structure determination of B12 was achieved in 1956 by Dorothy Hodgkin and co-workers [3], and led to the award of the Nobel Prize in Medicine. The structure of vitamin B12 is shown in figure 1. There are several excellent reviews written on the enzymes catalyzed by vitamin B12 and of the non-enzymatic vitamin B12 binding proteins [4–8]. The current study is limited to structures of the B12 binding proteins and enzymes, as determined by the method of X-ray crystallography. All the available B12 binding proteins in the PDB database (www.pdb.org) were analyzed and fifteen proteins were selected for the study, using the following criteria: (a) as one of the important aims of this study is to investigate B12 interactions with proteins and enzymes, similar proteins with different ligands for cobalt ion are also included in the dataset. (b) if more than one x-ray model was available for any protein, parameters like model quality, resolution were applied to choose the best one among them. The one exception to this criterion involved the mammalian transport protein, Transcobalamin II (Transcobalamin, TC) where both bovine and human models were analyzed (refer section 3.2). (c) mutants of any enzymes or proteins were omitted from the analysis. Table-1 lists the protein structures selected for analysis in this study.
Figure 1.
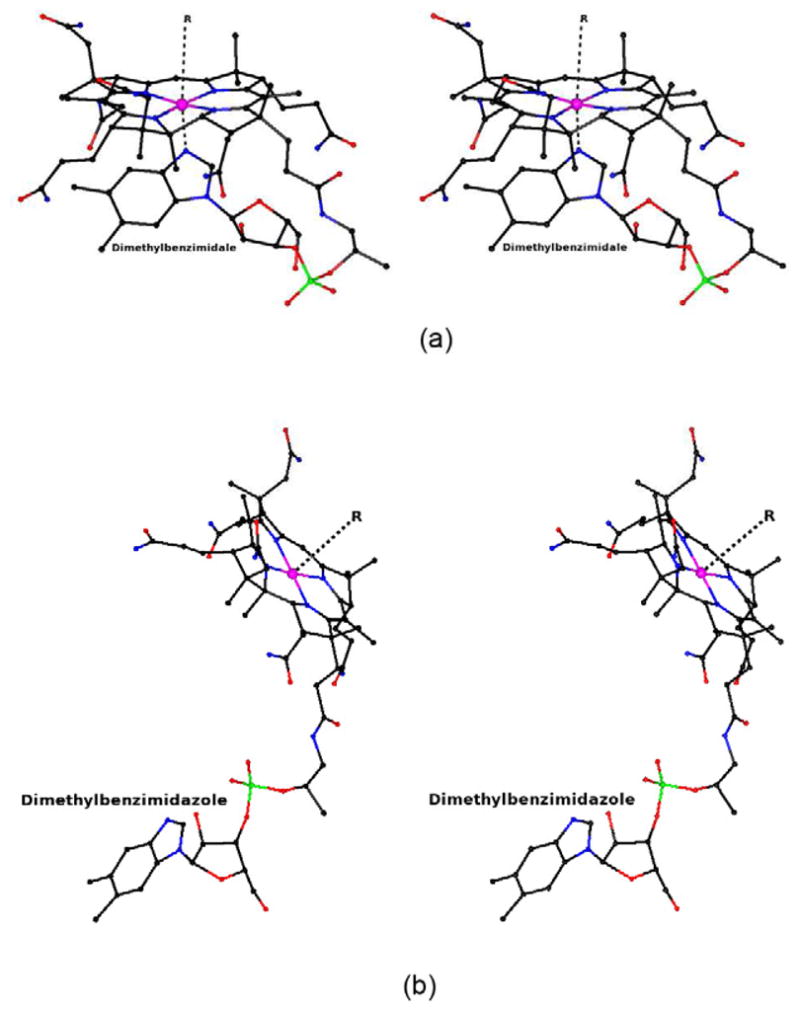
Stereoview of vitamin B12 (Cobalamin; Cbl; B12); The cobalt ion is shown as a pink sphere. Thecarbon, oxygen, nitrogen and phosphate are shown in black, red, blue and green colors respectively.B12 can adopt a base-on (top) or base-off (bottom) conformation. In the base-on conformation, thedimethylbenzimidazole group acts as a ligand for cobalt (top). Cobalt can form a sixth coordinatedligand and is represented as R=CN, OH, CH3 or Deoxyadenosyl.
Table 1.
List of proteins with B12 ligands and cobalt oxidation statea
| B12-binding proteins | PDB code | Resolution (Å) | B12 conformation | 5th ligand | 6th ligand | Co- oxidation state |
|---|---|---|---|---|---|---|
| Human Intrinsic factor | 2PMV | 2.6 | Base-on/His off | DMB – α-side of corrin ring | None- β-side is empty | +2 |
| Human Intrinsic factor-Truncated Cubilin | 3KQ4 | 3.3 | Base-on/His off | DMB – α-side of corrin ring | None | +2 |
| Human Transcobalaminb | 2BB5 | 3.2 | Base-on/His off | DMB- α-side of corrin ring | His - β-side of corrin ring | +2 ~50% and +3 ~50% |
| Bovine Transcobalamin | 2BB6 | 2.0 | Base-on/His on | DMB- α-side of corrin ring | His - β-side of corrin ring | +3 |
| Methylmalonyl-CoA mutase | 1REQ | 2.0 | Base-off | His– α-side of corrin ring | Water | +2 |
| Glutamate mutase-CN | 1CCW | 1.6 | Base-off | His– α-side of corrin ring | CN (disordered) | +2 ~50% and +3 ~50% |
| Glutamate mutase-CH3 | 1CB7 | 2.0 | Base-off | His– α-side of corrin ring | CH3 (disordered) | +2 ~50% and +3 ~50% |
| Glycerol dehydratase | 1IWP | 2.1 | Base-on | DMB – α-side of corrin ring | Nil (CN not visible in e- density map) | +2 |
| Diol dehydratase –CN | 1EGM | 1.85 | Base-on | DMB – α-side of corrin ring | CN(partly visible (no coordinate)) | +2 |
| Diol dehydratase –Adeninylpenyl-Cbl | 1EEX | 1.70 | Base-on | DMB – α-side of corrin ring | AdeninylpenylCbl | +3 |
| Ribonucleotide Reductase | 3O0O | 1.9 | Base-on | DMB – α-side of corrin ring | 5′-deoxyadenosine | +3 |
| Methionine synthase | 1BMT | 3.0 | Base-off/His on | His– α-side of corrin ring | CH3 | +3 |
| Methyltransferase Complex | 4DJF | 3.03 | Base-off/His off | Empty | CH3 | +2 |
| BtuF | 1N2Z | 2.0 | Base-on | DMB – α-side of corrin ring | Cl− | +3 |
| BtuB | 1NQH | 3.1 | Base-on | DMB – α-side of corrin ring | CN | +3 |
DMB: dimethylbenzimidazole
x-ray structural interpretation regarding His coordination bond to cobalt ion is ambiguous
The database of B12 binding proteins can be divided into three groups. The first group deals with B12 transport in mammals and consists of Intrinsic Factor (IF), IF with its truncated receptor, cubilin, and transcobalamin [9–11]. The second group deals with B12 transport in E. Coli. BtuB, BtuF and BtuCD are involved in this process [12–16]. BtuB and BtuCD are membrane proteins and BtuCD belongs to the family of ABC transporters. BtuCD was not included in the analysis as it lacks B12 in its X-ray model. The third group can be broadly classified as B12 dependent enzymes and may be divided into three sub-groups as follows [4]. Methionine synthase along with the recently determined methyltransferase complex, both of which are involved in the transfer of a methyl group from methyltetrahydrofolate to homocysteine, form the first sub-group [17–19]. The second subgroup consists of mutases and dehydratases [20–25] and is characterized by the homolytic/heterolytic cleavage of the C-Co bond, subsequent generation of a substrate radical followed by the rearrangement in substrate by breaking and reforming the C-C bond. Ribonucleotide reductase (RNR) forms the third sub-group [26, 27], in which catalysis is initiated by 5′-deoxyadenosyl radical formation which leads to elimination of the 2′-hydroxyl group of the substrate.
The conformation of B12 is also examined in this analysis. B12 in proteins is known to exist in two different conformations namely “base-on” and “base-off”(Fig 1; Table 1), though free B12 always adopts “base-on” conformation (Fig. 1a) [12, 28]. In the base-on conformation, the 5,6-dimethylbenzimidazole (DMB) group at the tail of B12 provides the fifth ligand for the cobalt from the α-side of corrin ring. The DMB group moves away from the corrin ring in the base-off conformation (Fig. 1b). In some of the B12 proteins with B12 in the base-off conformation, the movement of DMB group is compensated by the histidine of the polypeptide chain as a fifth ligand. A few B12 binding proteins have a sixth ligand for cobalt such as deoxyadenosyl, CN, OH or CH3.
This review summarizes the current literature describing the x-ray crystallographicstudies of B12 binding proteins with biological significance and structural features along withdatabase analysis. A phylogeny study on B12 binding proteins was carried out and the results werecompared with overall folding of the protein molecule. An in-depth analysis of various types ofinteractions such as hydrogen bonds, van der Waals interactions and electrostatic interactionsbetween the B12 and protein was carried out and a correlation between these interactions and theaffinity of B12 with protein is summarized. Also, an attempt has been made to correlate the type ofamino acids (aliphatic, aromatic, polar, non-polar, acidic and basic)[29] at the binding site with the affinity towards B12.
Some of the B12-binding proteins are the targets of drug development programs. Forexample, the transport proteins IF and TC in mammals have been targeted for delivery of smallmolecules or proteins such as insulin by attaching it to B12 [9, 11, 30–35]. It is very important tounderstand the kind of interactions B12 could have with the proteins to design an effective mode ofdrug delivery. A database study like the current one could help to identify commonalities among thetypes of interactions, and thereby help to advance such drug research.
2. Methodology
The atomic coordinates of B12 binding proteins were obtained from the PDB database(www.pdb.org). Hydrogen atoms weregenerated using the program PHENIX [36] forall the chosen structures. Each structure was analyzed and its symmetrically related molecules weregenerated using the program COOT [37]. As oneof the aims was to analyze the interactions of B12 with the protein, a PDB file with residues≤10Å around B12 molecule for each structure was generated using COOT [37]. The hydrogen bonds were calculated using the programsCCP4 [38], HBAT [39], CCP4MG [40] and the results were compared. The “Motifs and Sites” server ofEBI (http://www.ebi.ac.uk) was used for ligand site interactions and compared with resultsfrom above listed programs. The hydrogen bond parameters are defined as d(X……A)≤ 3.6Å, d(H……A) ≤ 3.0Å and < (X-H…..A)≥ 90° where d is the distance, X is the hydrogen bond donor, H is hydrogen atom andA is the hydrogen bond acceptor [41–43]. The solvent accessible surface area calculation usinga solvent-sphere probe radius of 1.4 Ǻ [44] was performed using AREAIMOL of CCP4 [38] and CCP4MG [40]. TheKm or Kd value of the protein-B12 complexes were taken from the referenceslisted in Table 2 and the references quoted therein. Allatoms that were not involved in hydrogen bonds but within 3.9Å were considered as van derWaals interactions [45].
Table 2.
Salient features of B12 binding proteins
| Cbl-binding proteins | PDB code | Overall folding | Cbl-interacting residues by conventional hydrogen bonds | e Cbl-interacting residuesby vander waals interactions | f Cbl-interacting residuesby electrostatic interaction | Cbl-interacting residues by C-H….X Hydrogen bonds | b SAA of B12 on binding(%) | Km/Kd | dBav(Å2) for B12/Bav (Å2) for correspondingprotein | Referencea |
|---|---|---|---|---|---|---|---|---|---|---|
| Human Intrinsic factor | 2PMV | Two domain (α and β), α-domainα6/α6 helical barrel. β-domain – mainly β-strands | Asp153, Asp204, Gln252, Ser347, Trp348, Val352, Trp368, Phe370,Leu377 | Gly72, His73, Ser112, Thr115, Tyr206, Ser207, Val351, Tyr367, Gln369,Thr370, Gly380, | Thr346 | Thr70, Asp153, Thr346, Trp348, Gly349, Leu350, Trp368, Leu377, Asn378,Glu379, Tyr399 | 19 | Kd=1pM | 42.2/55.8 | [1] |
| Human Intrainic factor-Truncated Cubilin | 3KQ4 | Intrinsic factor: Two domain (α and β),α-domain α6/α6 helical barrel. β-domain – mainlyβ-strands. Cubilin: Beta sandwich with a jelly-roll fold. |
Asp171, Asp222, Gln270, Ser365, Trp366, Val370, Trp386, Phe388,Leu395, Tyr417 | Gly90, His91, Leu94, Tyr133, Tyr224, Ser225, Leu228, Gly367, Val369,Asn396, Glu397, | Ser130, Asp171, Thr364, Trp386, Leu395 | 19 | Kd=1pM | 66.5/85.4 | [2] | |
| Human Transcobalamin | 2BB5 | Two domain (α and β), α-domainα6/α6 helical barrel. β-domain – mainly β-strands. | Gln86, Thr134, Gln138, Asp176, Asn224, Ser227, Gln273, Leu358, Leu363,Trp377, Leu379, Leu387 | Tyr137, His172, His173, Tyr226, Met270, Ser357, Gly360, Tyr362,Phe376, Gly390, Trp409 | Gly85, Asp176, Ser359, Pro361, Trp377, Leu388 | 7 | Kd=0.005pM | 22.8/20.4 | [3] | |
| Bovine Transcobalamin | 2BB6c | Two domain (α and β), α-domainα6/α6 helical barrel. β-domain – mainly β-strands. | Gln86, Thr134, Gln138, Asp179, Asn227, Ser230, Gln276, Leu363, Leu368,Trp382, Val384, Leu392, Gln393 | Val136, Tyr137, Tyr229, Met273, Phe367, Asn370, Phe381, Gln383,Gly395, | Trp414 | Gly85, Asp179, Ser362, Ser364, Gly365, Pro366, Trp382, Gln393 | 7 | Kd=0.2pM | 15.0/36.3 | [3] |
| Methylmalonyl CoA mutase | 1REQ | Two chains (α & β). Both α and βchains has two domains. N-terminal domain β/α barrel and c-terminalα/β domain | Tyr89, Phe117, Ala139, Val206, Arg207, Gly333, Leu336, Glu370, Ala373,Gly609, Asp611, Arg612, Gly613, Leu657, Gly686, Ser665, Tyr705, Thr709 | Ile61, Ala116, Leu119, Tyr243, His244, Typ334, Ala371, Leu374, Gln454,Leu602, His610, Gly653, Ala658, Gly659, Gly685, | Glu247, Glu370, Tyr621, Leu657, Tyr705, Thr706, Thr709 | 3.7 | Km=35nM | 31.4/37.8 | [4] | |
| Glutamate mutase-CN | 1CCW | Heterotetrameric molecule with ε2σ2.σ subunit contains α/β domain, five parallel strands encased by 6α-helices. ε subunit consists of TIM barrel (α/β)8. | Chain A: Ser13, Cys15, Ala17, Ser61, Leu63, Asn93, Val95, Gly97, ChainB: Pro180, His329, Glu330, Ile334 | Chain A: His16, Val18, Gly19, Ile22, Leu23, Gly65, Gly91, Gly92,Val96, Tyr117, Pro123 Chain B: Thr94, Arg100, Tyr181, Gly296, Ala331, Gly333, Pro410,Phe471 |
Chain A: Asp14 | Chain A: Cys15, Leu63, Tyr64, Thr121 Chain B: Thr220, Met294,Gly295, Glu330 |
1.0 | Km=5.8μM | 10.2/21.0 | [5] |
| Glutamate mutase – CH3 | 1CB7 | Heterotetrameric molecule with ε2σ2.σ subunit contains α/β domain, five parallel strands encased by 6α-helices. ε subunit consists of TIM barrel (α/β)8. | Chain A: Ser13, Cys15, Ala17, Ser61, Leu63, Asn93, Val95,Gly97 Chain B: Pro180, Phe297, His329, Glu330, Gly333, Ile334 |
Chain A: His16, Val18, Gly19, Ile22, Leu23, Gly65, Gly91, Gly92,Val96, Tyr117, Chain B: Thr94, Arg100, Tyr181, Gly296, Lys326, Ala331, Phe471 | Chain A: Asp14, Thr121 | Chain A: Cys15, Leu63, Tyr64, Thr121 Chain B: Thr220, Met294,Gly295, Glu330 |
3.8 | Kd=5.8μM | 13.3/23.5 | [5] |
| Glycerol dehydratase | 1IWP | Dimeric form of heterotrimer(αβγ)2. | Chain A: Thr173, Ala177, Ser203, Glu206, Thr223, Asp235,Met374 Chain B: Asp79, Lyz102, Thr104, Asn117, Ser122, Ala124 |
Chain A: Gly175, Val204, Tyr227, Met269, Cys303, Phe375,Ala376 Chain B: Leu46, Val80, Leu120, Gln123, Pro125, Val159, Phe163, Met164, Arg160,Ala167 |
Chain A: Leu268; Chain B: Ser81 | Chain A: Thr173, Val174, Ser302, Gln337, Met374, Phe375, Chain B:Ser81, Leu115, Ser122 | 4.6 | Km- 8nM | 21.9/39.1 | [6] |
| Diol dehydratase – CNCbl | 1EGM | Dimeric form of heterotrimer(αβγ)2. | Chain A: Thr172, Glu205, Thr222, Ser224, Asp234, Met373 ChainB: Asp112, Lyz135, Thr137, Asn150, Pro155, Gln156, Ala157 Ser200 |
Chain A: Met268, Cys302, Phe304, Ala375 Chain B: Val113,Leu153, Pro158 Arg193, Tyr196 |
Chain A: Gln267 | Chain A: Ser301, Gln267, Gln336, Chain B: Leu148, Pro155, Ser200 | 11.7 | Km=0.26mM | 17.9/19.9 | [7] |
| Dial dehydratase – Adeninylpenyl-Cbl | 1EEX | Dimeric form of heterotrimer(αβγ)2. | Chain A: Val173, Glu205, Thr222, Ser224, Asp234, Gly261, Ser299,Ser301, Met373, Chain B: Asp112, Lys135, Thr137, Asp150, Pro155, Gln156, Ala157, Ser200 | Chain A: Thr259, Ser260, Met268, Val300, Phe374, Ala375 ChainB: Leu153, Arg193, Tyr196 |
Chain A: Gln267 | Chain A: Ser224, Val225, Ser301, Gln336, Gln267, Chain B: Leu148,Pro155 | 4.6 | Km=0.0018mM | 12.4/14.5 | [7] |
| Ribonucleotide Reductase | 3O0O | 10-stranded α/β barrel | Arg208, Gly294 | Pro490, Phe508, Thr598, Asn600, Thr626, Tyr628 | Arg208, Cys322, Gly492, Asn496 | 8.4 | Kd=0.3μM | 70.3/74.6 | [8] | |
| Methionine synthase | 1BMTc | Two domain protein. N-terminal fragment forms helical bundle whileC-terminal forms Rossmann folding | Glu694, Gly756, Val758, Asp760, Ile761, Ser804, Thr808, Ala860 | Met701, Val704, Gly705, Phe708, Leu715, Val718, Ala722, Met725,Ile751, Gly762, Ile765, Val766, Gly802, Leu806, Ile807, Leu831, Gly833, Gly834, Ala835, Val857,Asn859 | Asp757 | Met698, Leu803, Gln858, Thr863 | 19.9 | Km=5μM | 6.2/25.6 | [9] |
| Methyltransferase complex | 4DJF | Methyltransferase(MeTr): Tim barrel and Rossmann fold Corrinoidiron-sulphur protein(CFeSP): Tim barrel and Rossmann fold. MeTr exist as homodimer with CFeSPbound on both side. |
Chain B: Asn199, Gln202, Asn203, Chain C: Thr340, Thr346, Gly370,Ser372, Val373, Ala433 | Chain B: Val168 Chain C: Pro318, Tyr338, Leu371, Leu374,Ala378, Ile406, Pro408 |
Chain B: Gln202 Chain C: Val339, Thr340, Thr346, Ser372,Thr375, Asp379, Pro430, Arg431 |
9.5 | Km=2mM (CFeSP) | 120.7/131.8 | [10] | |
| BtuF | 1N2Z | two structurally similar domain with Rossman like fold | Ala32, Trp66, Asp242, Arg246 | Pro31, Tyr50, Trp85, Gly88, Phe162, Phe168, Trp196, Ser241, | Gly87, Glu245 | 31.3 | Kd-15nM | 23.8/27.8 | [11] | |
| BtuB | 1NQH | a hutch domain (a four stranded β-sheet) surrounded by a22-stranded β-barrel | Leu63, Ser65, Asn72, Val90, Ser91, Asn185, Ala231, Thr289, Arg497,Tyr531 | Asn57, Gln62, Ala88, Gly89, Tyr229, Asn276, Tyr579 | Tyr531 | 27.6 | Kd=0.3nM | 53.1/24.3 | [12] |
Includes the references therein.
SAA – Solvent accessibility area
2BB6 and 1BMT have Histidine (His175 and His758 respectively) attached covalently to the Corrinring.
B-factor calculation for the protein carried out only with the chain(s) with which B12 isinteracting.
residues interacting with B12 only through van der waals interactions are listed.
electrostatic interaction: as observed in EBI-motifs and sites web site at http://www.ebi.ac.uk
F.S. Mathews, M.M. Gordon, Z. Chen, K.R. Rajashankar, S.E. Ealick, D.H. Alpers, N. Sukumar,Crystal structure of human intrinsic factor: cobalamin complex at 2.6-A resolution, Proceedings ofthe National Academy of Sciences of the United States of America 104 (2007) 17311–17316.
C.B. Andersen, M. Madsen, T. Storm, S.K. Moestrup, G.R. Andersen, Structural basis for receptorrecognition of vitamin-B(12)-intrinsic factor complexes, Nature 464 (2010) 445–448.
J. Wuerges, G. Garau, S. Geremia, S.N. Fedosov, T.E. Petersen, L. Randaccio, Structural basisfor mammalian vitamin B12 transport by transcobalamin, Proceedings of the National Academy ofSciences of the United States of America 103 (2006) 4386–4391.
F. Mancia, N.H. Keep, A. Nakagawa, P.F. Leadlay, S. McSweeney, B. Rasmussen, P. Bosecke, O.Diat, P.R. Evans, How coenzyme B12 radicals are generated: the crystal structure ofmethylmalonyl-coenzyme A mutase at 2A resolution, Structure 4 (1996) 339–350.
R. Reitzer, K. Gruber, G. Jogl, U.G. Wagner, H. Bothe, W. Buckel, C. Kratky, Glutamate mutasefrom Clostridium cochlearium: the structure of a coenzyme B12-dependent enzyme provides newmechanistic insights, Structure 7 (1999) 891–902.
M. Yamanishi, M. Yunoki, T. Tobimatsu, H. Sato, J. Matsui, A. Dokiya, Y. Iuchi, K. Oe, K. Suto,N. Shibata, Y. Morimoto, N. Yasuoka, T. Toraya, The crystal structure of coenzyme B12-dependentglycerol dehydratase in complex with cobalamin and propane-1,2-diol, European journal ofbiochemistry 269 (2002) 4484–4494.
J. Masuda, N. Shibata, Y. Morimoto, T. Toraya, N. Yasuoka, How a protein generates a catalyticradical from coenzyme B(12): X-ray structure of a diol-dehydratase-adeninylpentylcobalamin complex,Structure 8 (2000) 775–788.
K.M. Larsson, D.T. Logan, P. Nordlund, Structural basis for adenosylcobalamin activation inAdoCbl-dependent ribonucleotide reductases, ACS chemical biology 5 (2010) 933–942.
C.L. Drennan, S. Huang, J.T. Drummond, R.G. Matthews, M.L. Lidwig, How a protein binds B12: A3.0 A X-ray structure of B12-binding domains of methionine synthase, Science 266 (1994)1669–1674.
Y. Kung, N. Ando, T.I. Doukov, L.C. Blasiak, G. Bender, J. Seravalli, S.W. Ragsdale, C.L.Drennan, Visualizing molecular juggling within a B12-dependent methyltransferase complex, Nature 484(2012) 265–269.
E.L. Borths, K.P. Locher, A.T. Lee, D.C. Rees, The structure of Escherichia coli BtuF andbinding to its cognate ATP binding cassette transporter, Proceedings of the National Academy ofSciences of the United States of America 99 (2002) 16642–16647.
D.P. Chimento, A.K. Mohanty, R.J. Kadner, M.C. Wiener, Substrate-induced transmembrane signalingin the cobalamin transporter BtuB, Nature structural biology 10 (2003) 394–401.
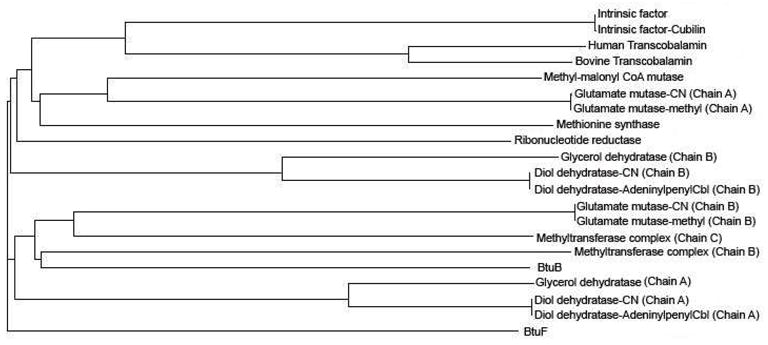
The sequences of the selected proteins (Table 1)for phylogeny study were obtained from the submitted PDB coordinates (www.pdb.org). The multi-sequence alignment was generated usingClustal-Omega web server at EMBL-EBI (www.ebi.ac.uk), and was submitted for the construction of the phylogram tree. Thephylogram tree was constructed (Fig. 2) using ClustalW2 webserver at EMBL-EBI.
Figure 2.
Phylogenetic tree of B12 binding proteins. The sequence of each protein was taken from thesubmitted PDB coordinates.
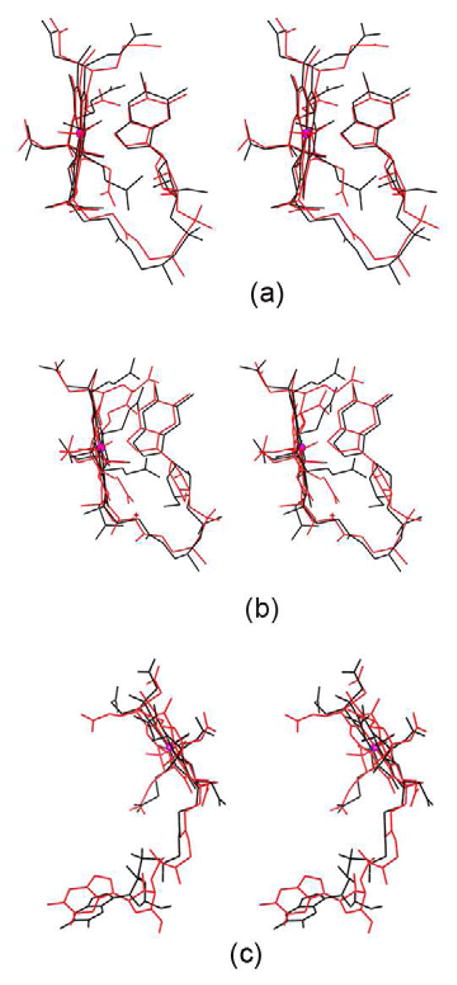
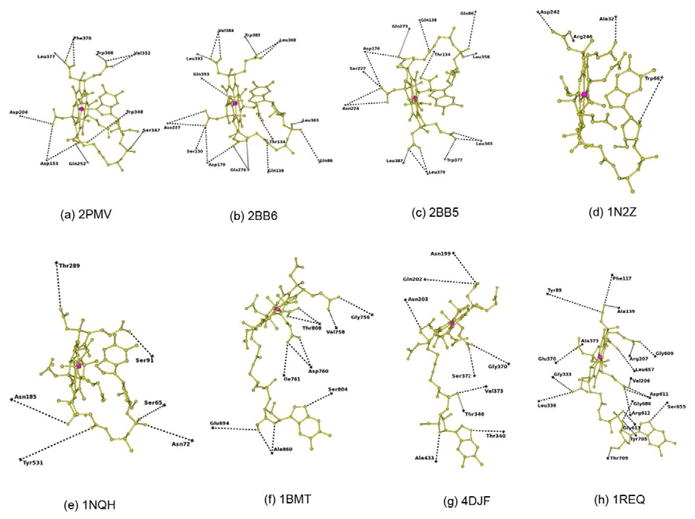
In B12 binding site interaction figures (Fig. 3),each residue position was marked as cross based on their Cα atom position. Forthe sake of clarity, the residues forming conventional hydrogen bonds (N-H…X andO-H…X hydrogen bonds) alone are represented in these figures. Table 1 lists each of the proteins analyzed with its PDB code, resolution,B12 conformation, ligands details along with the oxidation state of the cobalt ion. Table 2 lists the overall folding, residues involved in hydrogen bonding,van der Waals and electrostatic interactions, as well as the solvent accessible area, Kmor Kd value and Baverage value of B12 as well as corresponding protein chain.All the figures were generated using CCP4MG [40].
Figure 3.
Hydrogen bonding interactions at the B12 binding site. The position of each residue is marked asa cross with reference to their Cα atom. B12 is shown as ball and stick in goldcolor. Co ion is shown as a sphere in pink. The PDB id of each structure is added below each figurefor identification.
3. Structural features and B12 binding site analysis of B12-protein complexes
3.1 Phylogenetic analysis on B12 binding proteins
The phylogram tree on the B12 binding proteins is shown in Figure 2. The tree shows that all the transport proteins in mammals are evolutionallycloser. However, E. Coli transport proteins BtuB and BtuF are evolutionally diverse and BtuB iscloser to methyltransferase and glutamate mutase than to BtuF. The phylogeny study indicates thatthe individual chains of most of the B12 dependent enzymes are evolutionally diverse. For example,in the case of glutamate mutase, chain A is evolutionally closer to methylmalonyl-CoA mutase andmethionine synthase while the chain B is closer to both chains of methyltransferase complex andBtuB. Individual chains of glycerol dehydratase and diol dehydratase also show evolution diversity,similar to glutamate mutase (Fig. 2). The B12 interacts withboth the chains in these enzymes.
3.2 B12 transport proteins in mammals
The absorption of vitamin B12 in mammals is complex and any problem during the processmay lead to its deficiency. The delivery of B12 to the tissues from the digestive tract involvesthree successive transport proteins and their cellular receptors [9]. The process begins when B12 is bound to haptocorrin (HC), aglycoprotein produced in the digestive tract. After degradation of the HC-B12 complex in the uppersmall intestine, B12 is bound to intrinsic factor (IF), another glycoprotein that mediates thetranslocation of B12 across ileal mucosal enterocytes. Cubilin, which is located at the luminal sideof the ileal mucosal cells binds the IF-B12 complex and leads to its internalization. Cubilin is amulti-domain protein with 27 similar CUB domains [10,46–49]. The binding site for IF-B12 is located at CUB domains 5–8 of cubilin;finally, the released B12 binds to transcobalamin that mediates the final delivery of B12 into thecirculation and in turn into all other cells [11,50–52]. The phylogram tree shows that mammalian transport proteins are evolutionaryvery close to each other (Fig. 2).
The structures of IF and TC have been determined by X-ray crystallographic methods[9, 11, 53]. Subsequently, the complex of IF with its truncatedreceptor cubilin have been determined [10].These structures provide important details about the transport of B12 in mammals.
3.2.1 IF-B12 complex
The three-dimensional structure of the recombinant glycoprotein human IF in complex withB12 consists of α and β domains [9,53]. The α-domain is organized as anα6/α6 helical barrel and the β-domain contains pre-dominantlyβ-sheet structures (Fig. 4a). The B12 molecule is boundto IF at the interface between these two domains in the “base-on” conformation. TheIF-B12 model consists of one sugar binding site at Asn395 that accommodated two N-acetyl-glucosamine(NAG) molecules although non-crystallographic data predicted glycosylation at four sites[54]. However, it is possible thatrecombinant IF is not able to support these alternate glycosylation sites.
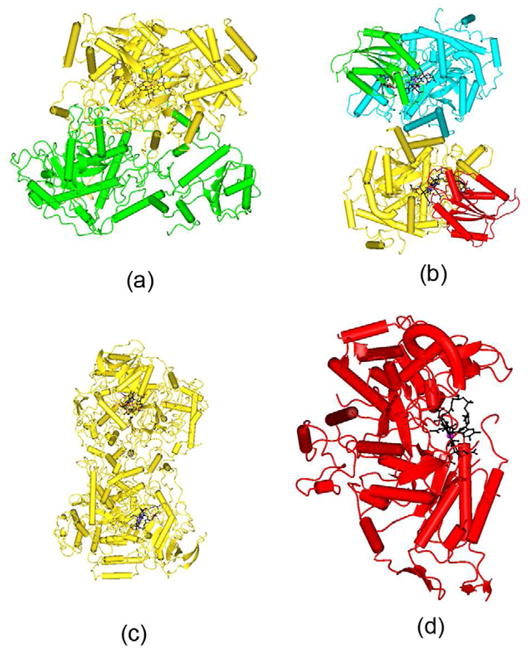
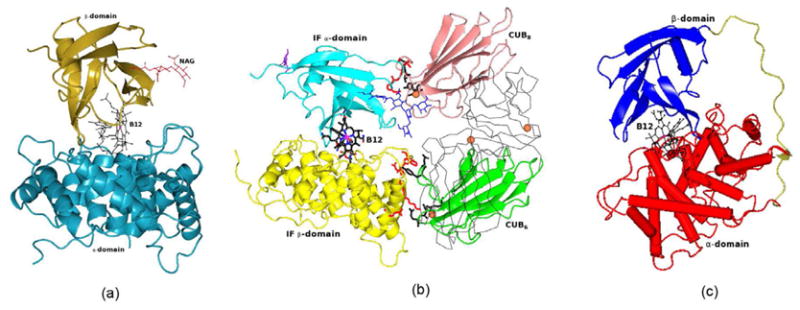
Figure 4.
(a) Intrinsic Factor – B12 complex: Ribbon diagram shows the α-domain in blue andβ-domain in gold with cobalamin in black color as ball and stick. Cobalt ion is shown as asphere in pink color. Sugar molecules are shown as sticks in red color (b) Intrinsic Factor(IF)-B12-truncated cubilin complex: Ribbon diagram shows the α-domain in yellow andβ-domain in cyan with B12 shown as a thick bond in black. The CUB6 is shown ingreen and CUB8 is in salmon color. The CUB5 and CUB7 which are notinvolved in any interaction with IF-B12 complex are shown as Cα-trace and inblack. IF residues involved in interaction with CUB domains are shown in red color, while CUBresidues are shown in black color and as thick bonds. The sugar molecules are shown as sticks inblue colors. The Ca and Co ions are shown as spheres in pink and salmon colors, respectively. (c)Worms/tubes diagram of Transcobalamin. The α-domain is in red and β-domain in bluewith B12 in black color as ball and stick. The link region between α and β domain isshown as gold color. The cobalt ion is shown as a sphere in pink.
3.2.2 IF-B12-truncated cubilin complex
The x-ray structure of the IF-B12-truncated cubilin complex contains CUB domain5–8 of cubilin that interact with IF-B12 complex (Fig.4b) [10]. The structure of IF-B12 doesnot undergo any conformational change upon binding of CUB domains [9, 10]. The B12 is bound inbase-on conformation at the interface between the α and β domains of IF. Theα-domain of IF interacts with CUB6 and the β-domain of IF interacts withthe CUB8 domain. Cub5 and Cub7 do not interact with IF but help toposition CUB6 and CUB8 for IF-B12 interaction. B12 itself does not interactwith any of the CUB domains directly and is at least ~13Å away from them. However, bindingof B12 makes IF more compact [9] and in turnenables it to bind to the CUB domains of cubilin [10]. The model consists of two sugar binding sites at Asn334 and Asn413. Theglycosylation site at Asn413 is ordered, containing two N-acetyl-glucosamine (NAG) and threemannoses, as observed in IF-B12 structure (numbered as Asn395) where two NAG molecules were modeled[9]. The model reveals that one of thedominant interactions of IF-B12 with cubilin is electrostatic pairing of the basic residues Arg/Lysof IF with Ca2+ coordinating acidic residues Asp/Glu [10]. In addition to the electrostatic interactions, a few hydrogenbonds and van der Waals stabilize the interactions between them.
3.2.3. TC-B12 Complex
The x-ray structure of TC-B12 complexes from human and bovine source are available(Fig. 4c) [11]. Of the three proteins involved in B12 transport, TC is the onlynon-glycosylated protein. Bovine and human TC has 73% sequence identity between them. TC isa two domain protein, with the α-domain made of α6-α6barrel and the β-domain predominantly β-strand, similar to IF. The linker region inthe human TC between α and β domain is three residues shorter than in bovine TC.
3.2.4 B12 binding site in mammalian transport proteins
The details of the B12 interaction with IF and TC have been described (Fig. 3a, b & c and Table 2)[9, 11]. All the conventional hydrogen bonds observed in IF-B12 model are conserved inIF-B12-Cubilin model (Fig. 3a and Table 2). In addition, the C-terminal residue, tyrosine forms a weakhydrogen bond in IF-B12-Cubilin. The sequence identity between IF and human and bovine TC is27% and 29% respectively. B12 in IF, IF-truncated cubilin and TC exists in thebase-on conformation and is bound at the interface between the α and β domains.There is an extensive network of hydrogen bonds involving B12 with protein residues and watermolecules. As the structure of bovine TC was solved at high resolution and the sequence homologybetween bovine and human TC is 73% with a rmsd of 1.2Å[11], a comparison was carried out between bovine TC and human IF[9]. IF and TC adopt similar overallconformations, yet significant differences exist between them, especially in the α-domain[9]. The histidine provides the sixth ligandfor B12 in bovine TC in contrast to B12 in IF. The solvent accessibility of B12 in IF is reduced to~19% compared to ~7% in TC (Table 2). Thebinding site of IF is dominated by negatively charged residues in contrast to TC where it isdominated by neutral residues. The comparison of Kd value for these structures confirmsthat B12 binds more strongly with TC compared with IF. On comparing the B-factor of B12 in thesetransport proteins (Table 2), it is clear again that TC hasmore strongly bound B12 both in bovine and human with respect to IF-B12 and IF-B12-cubilin. Also,the binding site for B12 in IF is broad and open on both sides compared with TC. The absence of theHis coordination bond and the wider binding site at IF enables B12 to move in and out more freelycompared with TC and that may account for the higher B-factor of B12, with important implicationsfor binding and dissociation of the B12 to these two transport proteins. The superposition of B12 inIF and TC is shown in figure 5a. There are significantdifferences in the conformation adopted by the branched side chains of corrin ring which it turnshow that how the B12 can use these to adapt to different environments.
Figure 5.
Stereoview of superposition of B12 in (a) Mammalian transport proteins IF (black) and TC (red)(b) E. Coli transport proteins BtuF (black) and BtuB (red) and (c) Methionine synthase (red) andMethyltransferase complex (black)
3.3 B12 transport proteins in E. Coli
These transport proteins are well characterized crystallographically with thedetermination of the x-ray structure of the inner membrane ABC transporter (BtuCD) [15], outer membrane protein (BtuB) [14], and the periplasmic binding protein (BtuF) which captures B12after it is transported across the membrane by BtuB [12, 13]. The structure of BtuCD was solvedwithout B12 [15]. But B12 binding was modeledbased on in-vitro docking; how B12 might translocate from BtuF to BtuCD and subsequently though theinner membrane was discussed [12]. Thecurrent study focuses only on the structure of BtuB and BtuF, as they were determined in complexwith B12. The phylogram tree shows that BtuB is evolutionally closer to methyltransferase complex,compared to BtuF (Fig. 2).
3.3.1 BtuF
The structure of E. coli BtuF was solved by the Rees group in 2002 (Fig. 6a) [12]. Thefunction of this protein is to bind B12 after it passes through the outer membrane via BtuB, and todeliver it to the periplasmic surface of the ABC transporter, BtuCD. It has two domains; the overallfolding of each domain is like a Rossmann fold (Fig. 6a). Thetwo domains are connected by a strong backbone α-helix over the length of the whole proteinrather than by a weak linker as observed in the B12 transport proteins of mammals (Fig. 4). B12 is bound at the interface between two domains in the base-onconformation.
Figure 6.
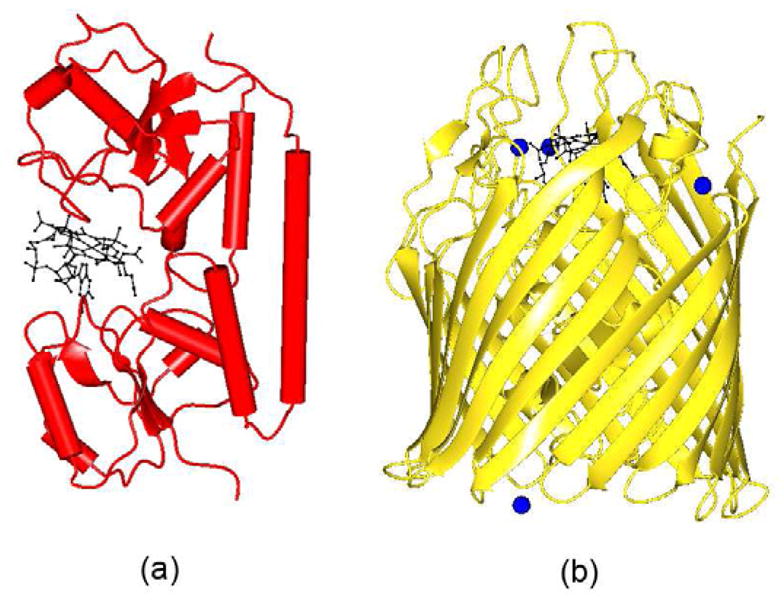
(a) Worms/tubes diagram of BtuF. The B12 is shown as ball and stick in black color (b) Ribbondiagram of BtuB. The Ca2+ ions are shown as spheres in blue color. B12 is shownas ball and stick in black color.
3.3.2 BtuB
The structure of BtuB with and without CN-B12 has been determined crystallographically[14]. The apo BtuB structure was determinedat 2Å resolution while BtuB-Ca2+-B12 and BtuB-Ca2+were determined at 3.1Å and 3.3Å resolution respectively. The structure of BtuBconsists of a “hutch” domain formed by a four stranded β-sheet, surroundedby a 22-stranded β-barrel (Fig. 6b). B12 adopts thebase-on conformation and is bound in the hutch domain. It also contains functionally activeCa2+ ions which plays important role in B12 binding.
3.3.3 B12 binding site in the E. coli transport proteins
A comparison of the B12 binding site in BtuF and BtuB indicated that the number ofresidues interacting with B12 through conventional hydrogen bonds is 4 and 10 respectively (Table 2; Fig. 3d & e). TheKm value for BtuB and BtuF is 0.3nM and 15nM respectively; thus B12 binds to BtuB ~50times stronger than to BtuF. However, the B-factor value of B12 in BtuB and BtuF indicates that B12binds more strongly with BtuF compared to BtuB (Table 2). TheB12 binding site of BtuF contains several water molecules that interact with B12 but due to lowerresolution (3.1Å), the x-ray model of BtuB does not contain any water molecules. A hydrogenbonding analysis indicates that the phosphate of B12 forms several hydrogen bonds in BtuB does notform any in BtuF. A comparison of other hydrogen bonding interactions of B12 with BtuF and BtuBindicates a completely different pattern, except at N40 of B12 which forms hydrogen bonds withacidic residue in both the structures. At least three strong hydrogen bonds formed by B12 withprotein residues of BtuB are replaced with waters at BtuF. B12 in BtuF is involved in interactionwith one aliphatic, aromatic, acidic and basic aminoacid residues, while in BtuB interaction occurswith six polar, three aliphatic and an aromatic residue. In other words, the B12 environment in BtuFis charged while polar residues dominate in the B12 binding site of BtuB. The difference in theinteractions between BtuB and BtuF suggests that the stronger binding of B12 with BtuB is necessaryto mediate passage through the membrane. BtuF may not require such stronger interactions for itsfunction. It also provides interesting example of B12 on its interactions between membrane andsoluble proteins. The B-factor of B12 in BtuF and BtuB is 23.8Å2 and53.1Å2 and their protein average B-factor is 27.8Å2 and24.3Å2 respectively. It indicates that more mobility of B12 in BtuB compared withBtuF. It is not surprising as B12 need to pass through the membrane through BtuB, but needs to staytogether with BtuF till it passed over to BtuCD.
The superposition of B12 in BtuB and BtuF is shown in figure 5b. Again, it shows that branched side chains of corrin ring in BtuB and BtuF helpB12 to adapt to difference situations.
3.4 B12 dependent enzymes
3.4.1 Methionine synthase and Methyltransferase complex
The vitamin B12-dependent methyltransferase catalyzes methyl transfer in awide range of biological processes both in prokaryotes and eukaryotes [4]. During the reaction, the methyl group from methyltetrahydrofolateis used to form methyl-cobalamin, and the methyl group then is transferred to homocysteine toproduce methionine. The phylogram reveals that methyltransferase complex is closer to chain B ofglutamate mutase and BtuB compared to methionine synthease (Fig.2).
3.4.1.1 Methionine synthase
Methionine synthase consists of four well-defined modules [55–57]. The first twomodules involved in the binding of homocysteine and methyltetrahydrofolate. The third module helpsto incorporate the B12 cofactor and the fourth binds the adomet, necessary for the reactivation ofthe Co(II)balamin form of the enzyme. The x-ray structure of the cobalamin binding module ofmethionine synthase was determined about two decades ago [17]. It is composed of two domains, a N-terminal domain of 98kD and C-terminaldomain of 38kD. The N-terminal fragment forms a helical bundle and the C-terminal fragment formsfive stranded α/β (Rossmann) fold (Figure 7a).B12 binds at the interface between the two domains in a base-off conformation. The structurerepresents the “resting” state, as B12 is covered by a four helix bundle called thecapping domain to protect it from unwanted chemical reactions.
Figure 7.
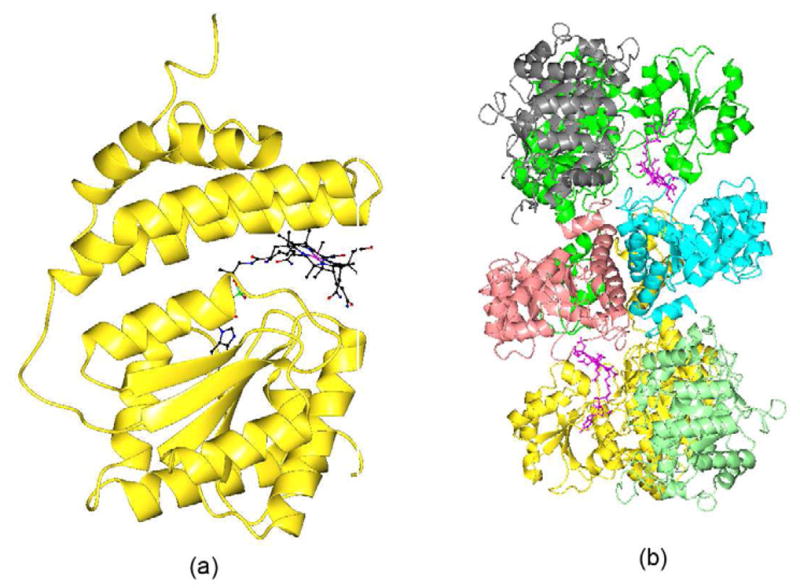
(a) Ribbon diagram of Methionine Synthase. B12 is shown as ball and stick. (b) Ribbon diagram ofMethyltransferase complex. The homodimer of MeTr is shown in pink and cyan color. The small subunitof CFeSP is shown in grey and light green. The large subunit of CFeSP is shown in green and goldcolor. The B12 is shown in ball and stick and in pink color.
3.4.1.2 Methyltransferase complex
Drennan and co-workers recently determined the structure of a methyltransferase complexcomposed of a corrinoid iron-sulphur protein(CFeSP) and the methyl transferase (MeTr) fromacetogenic bacteria (Fig. 7b) [19]. This complex is involved in the catalysis of Wood-Ljundahlcarbon fixation pathway for growth on CO2 as the sole carbon source [58]. The multi-subunit enzyme undergoes significantstructural rearrangements to alternatively activate, protect (resting position), and executecatalysis on the reactive B12 co-factor. The two molecules of the iron-sulphur protein interact withMeTr on both sides (Fig. 7b). Each CFeSP contains a small andlarge subunit. The B12 is bound in the base-off conformation at each CFeSP molecule. The largesubunit of CFeSP has three domains while the small subunit is folded as a TIM barrel which is usedas a B12 cap. The domain which binds B12 in both methionine synthase and the iron-sulfur proteinadopts conformation similar to the Rossmann fold. The B12 domain in this complex represents an“active” state in which the capping domain has moved away from B12 and prepared itfor catalysis.
3.4.1.3 B12 binding site in methionine synthase and methyltransferase complex
The structures of methionine synthase [17] and the methyltransferase complex [19] offer an opportunity to analyze the differences at the B12 binding site, as theformer represents the “resting” state and the latter the “active”state, although they perform the function in different organisms. The complex structure has all themodules necessary for transferring the methyl group and reveals that the B12 domain has to flip~18Å to complete the biological process. The superposition of B12 in methionine synthaseand methyltransferase complex is shown in figure 5c. Incomparison with E. coli and mammalian transport proteins, the B12 exist in base-off conformation.The base-off conformation leads to more flexibility in using tail of the B12 to adapt to differentenvironments. It is clearly reflected in the difference in orientation of DMB group at the tailalong with difference in the conformation of the branched side chains of the corrin ring (Fig. 5c). The number of residues involved in conventional hydrogenbonds is almost equal, with most of them either polar or non-polar amino acids (Table 2; Fig. 3f&g). The number ofresidues forming C-H…X hydrogen bonds is twice as many in methyltransferase complex asformed in methionine synthase. Binding of B12 is stronger in methionine synthease(Km=5μM) compared to methyltransferase complex(Km=2mM) although the presence of large number of C-H…X hydrogen bondscould enhance the binding strength in the latter. The B12 of methionine synthase is surrounded bymore charged residues than in the methyltransferase complex. The phosphate group that forms a singleintra hydrogen bond in methionine synthase forms two additional hydrogen bonds in themethyltransferase complex. The hydrogen bonds with two charged residues in themethionine synthase are replaced by hydrogen bonds with polar/nonpolar residues in themethyltransferase complex.
3.4.2 Mutases and Dehydratases
3.4.2.1 Methylmalonyl-coenzyme A mutase
Methylmalonyl-coenzyme A mutase (MCM) is present in both eukaryotic and prokaryoticorganisms. In bacteria, it is involved in the fermentation of pyruvate to propionate while inmammals it participates in the conversion of odd-chain fatty acids and branched chain amino acidsvia propionyl-CoA to succinyl-CoA for further degradation [59, 60].
The structure of methylmalonyl-coenzyme A mutase (MCM) from Propionibacteriumshermani was determined at 2Å resolution (Figure8a). It has a molecular weight of 150kD and the structure is an αβheterodimer. Each chain contains two domains and exhibits a (β/α)8 fold[20]. The B12 is bound to the molecule in thebase-off conformation and interact only with the residues of the α-chain.
Figure 8.
(a) Worms/tubes diagram of Methylmalonyl-CoA mutase. The α-chain and β-chain areshown in gold and green color respectively. B12 is shown as ball and stick in black color (b)Worms/tubes diagram of Glutamate mutase. The σ subunit is shown in red and green colors,while the ε subunit is shown in gold and cyan colors. B12 is shown as ball and stick inblack color (c) Worms/tubes diagram of Diol dehydrate. B12 is shown as ball and stick in blackcolor. (d) Worms/tubes diagram of ribonucleotide reductase. B12 is shown as ball and stick in blackcolor.
3.4.2.2 Glutamate mutase
Glutamate mutase (Glm) is involved in the first step of glutamate fermentation togenerate NH4, CO2, acetate and molecular hydrogen. The catalysis proceedsthrough the homolysis of the Co-C bond of B12. The structure of Glm from Clostridiumcochlearium in complex with CN-B12 and CH3-B12 has been determined at1.6Å and 2 Å resolution respectively [21], and both adopt similar folding. Both complexes contain one heterotetramericmolecule of ε2σ2 in the asymmetric unit along with B12 (Figure 8b). The ε subunit adopts a TIM barrel motif while theσ subunit folds as α/β domain with a β-sheet consisting of fiveparallel strands encased by a six α-helices. The B12 is bound to each εσsubunit pair and is in the base-off conformation.
3.4.2.3 Diol dehydratase and Glycerol dehydratase
Diol dehydratase is involved in the catalysis of 1,2, diols to aldehydes, through thehomolytic cleavage of the Co-C bond of B12. The structure of diol dehydratase in complex with B12and substrate 1,2-propanediol was determined at 2.2Å [24]. The molecule exists as a dimer of heterotrimers(αβγ)2 (Figure 8c). Theα-subunit contains a (β/α)8 barrel. The B12 is bound at theinterface between α and β subunit and is in base-on configuration.
Glycerol dehydrase catalyze the same reaction as diol dehydrase but plays a differentrole in the metabolism of bacteria [23].Glycerol dehydrase shows more affinity for the R-isomer of 1,2-diols than the (S)-isomer, whereasthe diol dehydratase does not show any such preference. It also has more affinity for B12 than dioldehydratase (8nM vs 0.26mM). However, the overall structure of glycerol dehydratase is very similarto diol dehydratase (Figure 8c and Table 2).
3.4.3 Ribonucleotide reductase
Ribonucleotide reductases (RNRs) are essential for DNA replication and repair in all theorganisms and it catalyzes the conversion of the ribonucleotides to deoxyribonucleotides[61, 62]. The RNRs are divided into three major classes based on their radical generationmechanisms [5, 63]. The Class II RNRs are different from other classes as no accessory proteins areinvolved in the catalytic mechanism, and radical formation involves homolytic cleavage of the Co-Cbond of B12 [64]. The literature containscrystal structures of class II RNRs in apo form, in complex with B12, and in complex with B12 andsubstrate [26, 27]. There are two independent molecules in the asymmetric unit and each binds to aB12 molecule (Figure 8d). The overall folding of the moleculeis a ten-stranded α/β barrel. The B12 molecule is bound in the base-onconformation.
3.4.4 B12 binding site in mutases, dehydratases and ribonucleotide reductase
In most of the B12 binding enzymes, B12 interact with more than one polypeptide chains.The conventional hydrogen bond interactions of B12 with mutases, dehydratases and ribonucleotidesare shown in figures 3h–o. Comparison of the B12interactions in these enzymes indicates that ribonucleotide reductase (RNR) interacts with the leastnumber of residues of any B12 binding proteins, involving just two residues (Table 2; Fig. 3o). But, formation ofC-H…X hydrogen bonds with four residues could enhance the B12 binding strength. B12 in therest of the enzyme group has intensive interactions with its partner protein. The B12 interacts withseveral water molecules both at corrin ring and tail parts. The Km or Kd forthe B12 dependent enzymes are in the μM/mM range (Table2). The B-factor of B12 in enzymes are in the range of 10–32Å2,which indicates that B12 is strongly bound and very stable in these enzyme complexes except in RNRwhere the B-factor is ~70Å2. The solvent accessibility of B12 reduced in theranges 1–5% for B12 in most of the enzymes (Table2).
Further analysis in RNR shows that all the interactions are at the head of B12 (i.e.corrin ring) leaving the tail portion completely free of any interactions. The Km for RNRis 0.3μM, which indicates a tight complex formation compared to most of the other enzymessuch as diol dehydratase, glycerol dehydratase and glutamate mutase. It is expected that such astrong complex formation should be reflected in formation of a number of hydrogen bonds similar toother enzymes. The structure clearly shows that B12 in RNR is not in an optimum position forcatalysis and may represent a pre-enzymatic reaction state. The B-factor of 70Å2for B12 supports this observation.
The Km value of diol dehydratase-CN and diol dehydratase-adeninylpenylCbl is 0.26mM and0.002mM respectively. The CN is disordered in diol dehydratase complex while adeninylpenylCbl iswell ordered (Fig. 8c and Table2). On comparing these two complexes (Table 2; Figs. 3m and n), it is clear that adeninylpenylCbl contributes to theformation of complex by forming several hydrogen bonds. It could be one of the reasons for tightercomplex formation with respect to CN complex.
3.5 Role of the C-H…O hydrogen bonds in B12 binding
When the role of hydrogen bonds in biological system is discussed, generally it would berestricted to N-H….X and O-H….X hydrogen bonds. C-H….X hydrogen bonds areusually thought of as weak hydrogen bond as they involve an average energy of 1–2 Kcal/molas compared to N-H…..X and O-H….X hydrogen bonds with 2–10 Kcal/mole[65–67]. However, even though C-H….X are weak in nature, they can collectivelyinfluence the structure and function of protein[68]. In the case of B12 binding proteins, there are several C-H….X hydrogenbonds formed between B12 and protein. The number of residues forming such C-H…X hydrogenbonds varies widely as observed in the conventional hydrogen bonds, ranging from 1 in BtuB to 11 inIntrinsic factor (Table 2). The methyltransferase complex andmethionine synthase provide an example of the C-H…X hydrogen bond role in stabilization.B12 in Methyltransferase complex forms C-H…X bonds with nine residues but in methioninesynthease with only 4 residues, compared with involving equal number of residues in conventionalhydrogen bonds formation. This indicates that C-H….X hydrogen bonding in methyltransferasecomplex might play a role in increasing strength of complex formation and thereby plays a role instructure stabilization.
4 Summary
4.1 Overall folding
The molecular architecture of the B12 binding proteins is diverse (Table 2). It varies from a two domain protein to multi-domain proteins. Inmammalian transport proteins, it adopts the two domain configuration, with a large α-domainand small β-domain. The transport proteins in E. Coli adopt completely different foldingwith respect to mammalian transport proteins. In BtuF, it still adopts two domains but with equalsize and Rossmann like folding. In BtuB, it is a small “hutch” domain surrounded bya 22 stranded β-barrel. In the B12 dependent enzymes, the protein folding is generallymulti-domains.
The phylogenetic analysis of B12 binding proteins indicate that BtuB is notevolutionally closer to BtuF, but to methyltransferase complex. Similarly, methyltransferase complexis not closer to methionine synthase, and the latter is closer to chain A of glutamate mutase. Bycomparing the overall folding with function of the individual proteins, proteins involved in aspecific biological function adopt similar conformations. For example, IF and TC adopt similarfolding as their sole function is to transport B12. But, in the case of the E. Coli transportproteins, BtuB serves as a receptor for the E and A colicins and for bacteriophage BF23, in additionto transport of B12 [69, 70]. However, it appears that the function of BtuF is to protect B12and prevent it from escaping into the periplasm, prior to passing it on to BtuCD [12]. As their functional capabilities differ, the foldingalso differs. Even though the methionine synthase and methyl transferase complex are involved in asimilar biological process, the latter has more functional capabilities [17–19]. It isinteresting to note that multi-functional proteins BtuB and methyltransferase are evolutionallycloser (Fig. 2). An evolutionally divergent polypeptide chainsfor these B12 dependent enzymes indicates that these enzymes would have developed additionalcapabilities for biological functions over different periods of evolution. However, the methioninesynthase, BtuF and the B12 binding domain of the methyltransferase complex adopt Rossmann folding.It indicates that even though the proteins are evolutionally diverse, the B12 binding portion of theproteins tend to adopt similar conformation to possible extent. It should be noted that theseenzymes involves in different kind of chemical reactions though they need B12 as a catalyst. As theB12 binding proteins are involved in various biological activities that place them at variouslocations both in prokaryotic and eukaryotic organisms, it is reasonable for them to have such adiverse overall folding.
4.2 Analysis of the B12 binding site
The crystal structure of B12 binding proteins was determined at resolutions ranging from1.6Å to 3.3Å (Table 1), enabling theenvironment around the B12 molecule to be studied in detail. In most of the complexes, B12 binds atthe interface between two domains although it also binds at other sites based on the overall foldingof the molecule, and the function of the protein. B12 adopts either base-on or base-off conformation(Table 1). In the cases where B12 exists in a base-offconformation, it adopts two distinct configurations namely “base-off/Histidine-on”and “base-off/Histidine off”. In the mammalian transport protein, transcobalamin,the B12 configuration is unique, as His serves as a 6th ligand from the β-side ofthe corrin ring in contrast to others [11].The configuration could be termed as base-on/His-on. The B12 in methionine synthase adopts abase-off/His-on conformation and the B12 in methyltransferase adopts a base-off/His-off conformation[17, 19]. Further analysis reveals the absence of histidine residue anywhere near the B12molecule of the methyltransferase complex. The crystal structure of the B12 domain of methioninesynthase is described as the “resting” position while the B12 binding domain ofmethyltransferase complex represents the active position [19]. In addition to these major configuration differences, B12 uses its functionalgroups around the corrin ring to interact with protein residues, where preferred residues would beat different locations based on the conformation adopted by protein chains. The comparison of theB12 conformation in the mammalian transport proteins IF and TC, and E. Coli transport proteins BtuBand BtuB and in the methionine synthase/methyl transferase complex (Fig. 5) clearly shows that B12 efficiently uses its branched side chains at corrin ring andits tail to adopt to different kind of environments. The cobalt ion in B12 exits in +2 or+3 oxidation state. The B12 dependent enzymes have the cobalt ion either in the +2or +3 oxidation state, although it has been postulated that cobalt was reduced to the+2 state during x-ray data collection [9,11].
An extensive analysis of the types of interactions between B12 and protein indicatesthat most of the interactions involve either hydrogen bonds or vander waals interactions (Fig. 3; Table 2). The numberof residues involved in conventional hydrogen bonding varies widely. The number of residuesinteracting with B12 in ribonucleotide reductase and BtuF are as low as two and four residuesrespectively while the methylmalonyl-CoA mutase has as high as 18 residues. Most of the residuesinvolved in the hydrogen bonds are either polar or non-polar amino acids. However, acidic, aromaticand aliphatic residues are together involved in significant interactions with B12 (Fig. 3; Table 2). Basic residues havevery low propensity for forming hydrogen bonds with B12. Several residues of the polypeptide chainare involved in the vander Waals interaction with the B12 molecule. In addition to theseinteractions, one or two amino acids are involved in electrostatic interactions in most of theB12-dependent enzymes.
The weaker correlation between Km and number of hydrogen bonds in theribonucleotide reductase complex reveals that it may not be an optimum complex for catalysis. Thevalue of solvent accessibility of B12 for enzymes (Table2) indicates that there is no need for embrace of B12 for catalysis. The comparison betweensixth ligands CN and adeninylpenylCbl complexes of diol dehydratase shows that adeninylpenylCblhelps to form tighter complex by forming several hydrogen bonds.
Both conventional (N-H….X and O-H…..X) and weak C-H…..X hydrogenbonds play an important role in the interactions of B12 with protein. It is interesting to note thatthe number of hydrogen bonds varies significantly between B12 and protein involved in differentbiological activities. B12 use its ability to form C-H…X hydrogen bonds to improve complexstability with the protein in addition to conventional hydrogen bond formation, as observed in themethyltransferase complex, Intrinsic factor etc. All these factors again clearly indicate that B12is amazingly adaptive to different environments, polar, non-polar or charged. B12 can use its heador tail parts, especially the corrin ring, phosphate group or DMB group to interact with its partnerprotein and form a stable complex. Thus, B12 can use the available functional groups at an optimumlevel for the interactions rather than using all possible interactions at once.
Highlights.
Molecular architecture of the B12 binding proteins is diverse.
Interaction of B12 with proteins in polar, non-polar and charged environments.
B12 uses its functional groups at an optimum level for interactions with proteins
C-H…O hydrogen bonds play an important role in B12-protein interactions
B12 transport proteins in E. Coli shows adoptability of B12 between membrane and solubleproteins.
Acknowledgments
The work is supported by award GM103403 from the National Institute of GeneralMedical Sciences at the NIH. I thank Professor Scott Mathews and Professor David Alpers ofWashington University, St. Louis and Professor Steve Ealick of Cornell University, Ithaca forhelpful discussions.
5. Abbreviations and Definitions
- rmsd
root mean square deviations
- Km
Michaelis constant represent substrate concentration when half of the active sites are filled
- Kd
dissociation constant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has beenaccepted for publication. As a service to our customers we are providing this early version of themanuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proofbefore it is published in its final citable form. Please note that during the production processerrors may be discovered which could affect the content, and all legal disclaimers that apply to thejournal pertain.
References
- 1.Whipple GH, Robscheit-Robbins FS. Blood regeneration in severe anemia: Favorable influence of liver, heart and skeletalmuscle in diet. Am J Physiol. 1926;72:408–418. [Google Scholar]
- 2.Minot GR, Murphy WP. Landmark article (JAMA 1926)-Treatment of pernicious anemia by a special diet. ByGeorge R. Minot and William P. Murphy. Jama. 1983;250:3328–3335. doi: 10.1001/jama.250.24.3328. [DOI] [PubMed] [Google Scholar]
- 3.Hodgkin DC, Kamper J, Mackay M, Pickworth J, Trueblood KN, White JG. Structure of vitamin B12. Nature. 1956;178:64–66. doi: 10.1038/178064a0. [DOI] [PubMed] [Google Scholar]
- 4.Ludwig ML, Mathews RG. Structure based perspectives on B12-dependent enzymes. Annual review of biochemistry. 1997;66:269–313. doi: 10.1146/annurev.biochem.66.1.269. [DOI] [PubMed] [Google Scholar]
- 5.Nordlund P, Reichard P. Ribonucleotide reductases. Annual review of biochemistry. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 6.Banerjee R, Ragsdale SW. The many faces of vitamin B12: catalysis by cobalamin-dependentenzymes. Annual review of biochemistry. 2003;72:209–247. doi: 10.1146/annurev.biochem.72.121801.161828. [DOI] [PubMed] [Google Scholar]
- 7.Banerjee R, Gherasim C, Padovani D. The tinker, tailor, soldier in intracellular B12 trafficking. Current opinion in chemical biology. 2009;13:484–491. doi: 10.1016/j.cbpa.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nielsen MJ, Rasmussen MR, Andersen CB, Nexo E, Moestrup SK. Vitamin B(12) transport from food to the body’s cells-a sophisticated,multistep pathway. Nat Rev Gastroenterol Hepatol. 2012;9:345–354. doi: 10.1038/nrgastro.2012.76. [DOI] [PubMed] [Google Scholar]
- 9.Mathews FS, Gordon MM, Chen Z, Rajashankar KR, Ealick SE, Alpers DH, Sukumar N. Crystal structure of human intrinsic factor: cobalamin complex at 2.6-Aresolution. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:17311–17316. doi: 10.1073/pnas.0703228104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andersen CB, Madsen M, Storm T, Moestrup SK, Andersen GR. Structural basis for receptor recognition of vitamin-B(12)-intrinsic factorcomplexes. Nature. 2010;464:445–448. doi: 10.1038/nature08874. [DOI] [PubMed] [Google Scholar]
- 11.Wuerges J, Garau G, Geremia S, Fedosov SN, Petersen TE, Randaccio L. Structural basis for mammalian vitamin B12 transport bytranscobalamin. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:4386–4391. doi: 10.1073/pnas.0509099103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borths EL, Locher KP, Lee AT, Rees DC. The structure of Escherichia coli BtuF and binding to its cognate ATP bindingcassette transporter. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:16642–16647. doi: 10.1073/pnas.262659699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Locher KP, Borths E. ABC transporter architecture and mechanism: implications from the crystal structuresof BtuCD and BtuF. FEBS letters. 2004;564:264–268. doi: 10.1016/S0014-5793(04)00289-3. [DOI] [PubMed] [Google Scholar]
- 14.Chimento DP, Mohanty AK, Kadner RJ, Wiener MC. Substrate-induced transmembrane signaling in the cobalamin transporterBtuB. Nature structural biology. 2003;10:394–401. doi: 10.1038/nsb914. [DOI] [PubMed] [Google Scholar]
- 15.Locher KP, Lee AT, Rees DC. The E. coli BtuCD structure: a framework for ABC transporter architecture andmechanism. Science. 2002;296:1091–1098. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- 16.Rees DC, Johnson E, Lewinson O. ABC transporters: the power to change. Nature reviews. 2009;10:218–227. doi: 10.1038/nrm2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drennan CL, Huang S, Drummond JT, Matthews RG, Lidwig ML. How a protein binds B12: A 3.0 A X-ray structure of B12-binding domains of methioninesynthase. Science. 1994;266:1669–1674. doi: 10.1126/science.7992050. [DOI] [PubMed] [Google Scholar]
- 18.Drennan CL, Matthews RG, Ludwig ML. Cobalamin-dependent methionine synthase: the structure of a methylcobalamin-bindingfragment and implications for other B12-dependent enzymes. Curr Opin Struct Biol. 1994;4:919–929. doi: 10.1016/0959-440x(94)90275-5. [DOI] [PubMed] [Google Scholar]
- 19.Kung Y, Ando N, Doukov TI, Blasiak LC, Bender G, Seravalli J, Ragsdale SW, Drennan CL. Visualizing molecular juggling within a B12-dependent methyltransferasecomplex. Nature. 2012;484:265–269. doi: 10.1038/nature10916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mancia F, Keep NH, Nakagawa A, Leadlay PF, McSweeney S, Rasmussen B, Bosecke P, Diat O, Evans PR. How coenzyme B12 radicals are generated: the crystal structure ofmethylmalonyl-coenzyme A mutase at 2A resolution. Structure. 1996;4:339–350. doi: 10.1016/s0969-2126(96)00037-8. [DOI] [PubMed] [Google Scholar]
- 21.Reitzer R, Gruber K, Jogl G, Wagner UG, Bothe H, Buckel W, Kratky C. Glutamate mutase from Clostridium cochlearium: the structure of a coenzymeB12-dependent enzyme provides new mechanistic insights. Structure. 1999;7:891–902. doi: 10.1016/s0969-2126(99)80116-6. [DOI] [PubMed] [Google Scholar]
- 22.Shibata N, Nakanishi Y, Fukuoka M, Yamanishi M, Yasuoka N, Toraya T. Structural rationalization for the lack of stereospecificity in coenzymeB12-dependent diol dehydratase. The Journal of biological chemistry. 2003;278:22717–22725. doi: 10.1074/jbc.M301513200. [DOI] [PubMed] [Google Scholar]
- 23.Yamanishi M, Yunoki M, Tobimatsu T, Sato H, Matsui J, Dokiya A, Iuchi Y, Oe K, Suto K, Shibata N, Morimoto Y, Yasuoka N, Toraya T. The crystal structure of coenzyme B12-dependent glycerol dehydratase in complex withcobalamin and propane-1,2-diol. European journal of biochemistry. 2002;269:4484–4494. doi: 10.1046/j.1432-1033.2002.03151.x. [DOI] [PubMed] [Google Scholar]
- 24.Shibata N, Masuda J, Tobimatsu T, Toraya T, Suto K, Morimoto Y, Yasuoka N. A new mode of B12 binding and the direct participation of a potassium ion in enzymecatalysis: X-ray structure of diol dehydratase. Structure. 1999;7:997–1008. doi: 10.1016/s0969-2126(99)80126-9. [DOI] [PubMed] [Google Scholar]
- 25.Masuda J, Shibata N, Morimoto Y, Toraya T, Yasuoka N. How a protein generates a catalytic radical from coenzyme B(12): X-ray structure of adiol-dehydratase-adeninylpentylcobalamin complex. Structure. 2000;8:775–788. doi: 10.1016/s0969-2126(00)00164-7. [DOI] [PubMed] [Google Scholar]
- 26.Larsson KM, Logan DT, Nordlund P. Structural basis for adenosylcobalamin activation in AdoCbl-dependent ribonucleotidereductases. ACS chemical biology. 2010;5:933–942. doi: 10.1021/cb1000845. [DOI] [PubMed] [Google Scholar]
- 27.Sintchak MD, Arjara G, Kellogg BA, Stubbe J, Drennan CL. The crystal structure of class II ribonucleotide reductase reveals how anallosterically regulated monomer mimics a dimer. Nature structural biology. 2002;9:293–300. doi: 10.1038/nsb774. [DOI] [PubMed] [Google Scholar]
- 28.Jogl G, Wang X, Mason SA, Kovalevsky A, Mustyakimov M, Fisher Z, Hoffman C, Kratky C, Langan P. High-resolution neutron crystallographic studies of the hydration of the coenzymecob(II)alamin. Acta Cryst. D67:584–591. doi: 10.1107/S090744491101496X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cantor C, Schimmel P. Biophysical Chemistry Part I. W.H. Freeman and Company; San Francisco: 1980. [Google Scholar]
- 30.Clardy SM, Allis DG, Fairchild TJ, Doyle RP. Vitamin B12 in drug delivery: breaking through the barriers to a B12 bioconjugatepharmaceutical. Expert opinion on drug delivery. 2011;8:127–140. doi: 10.1517/17425247.2011.539200. [DOI] [PubMed] [Google Scholar]
- 31.Allis DG, Fairchild TJ, Doyle RP. The binding of vitamin B12 to transcobalamin(II); structural considerations forbioconjugate design--a molecular dynamics study. Molecular bioSystems. 2010;6:1611–1618. doi: 10.1039/c003476b. [DOI] [PubMed] [Google Scholar]
- 32.Viola-Villegas N, Rabideau AE, Bartholoma M, Zubieta J, Doyle RP. Targeting the cubilin receptor through the vitamin B(12) uptake pathway: cytotoxicityand mechanistic insight through fluorescent Re(I) delivery. Journal of medicinal chemistry. 2009;52:5253–5261. doi: 10.1021/jm900777v. [DOI] [PubMed] [Google Scholar]
- 33.Petrus AK, Vortherms AR, Fairchild TJ, Doyle RP. Vitamin B12 as a carrier for the oral delivery of insulin. ChemMedChem. 2007;2:1717–1721. doi: 10.1002/cmdc.200700239. [DOI] [PubMed] [Google Scholar]
- 34.Chalasani KB, Russell-Jones GJ, Jain AK, Diwan PV, Jain SK. Effective oral delivery of insulin in animal models using vitamin B12-coated dextrannanoparticles. J Control Release. 2007;122:141–150. doi: 10.1016/j.jconrel.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 35.Chalasani KB, Russell-Jones GJ, Yandrapu SK, Diwan PV, Jain SK. A novel vitamin B12-nanosphere conjugate carrier system for peroral delivery ofinsulin. J Control Release. 2007;117:421–429. doi: 10.1016/j.jconrel.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 36.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structuredetermination. Acta Cryst. 2002;D58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.CCP4. Collaborative Computational Project Number 4. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 39.Tiwari A, Panigrahi SK, Desiraju GR. Hydrogen Bond Analysis Tool (HBAT) School of Chemistry, University of Hyderabad; Hyderabad 500 046, India: [Google Scholar]
- 40.Potterton L, McNicholas S, Krissinel E, Gruber J, Cowtan K, Emsley P, Murshudov GN, Cohen S, Perrakis A, Noble M. Developments in the CCP4 molecular-graphics project. Acta Cryst. 2004;D60:2288–2294. doi: 10.1107/S0907444904023716. [DOI] [PubMed] [Google Scholar]
- 41.Panigrahi SK, Desiraju GR. Strong and weak hydrogen bonds in the protein-ligand interface. Proteins. 2007;67:128–141. doi: 10.1002/prot.21253. [DOI] [PubMed] [Google Scholar]
- 42.Nagendra HG, Sukumar N, Vijayan M. Role of water in plasticity, stability, and action of proteins: the crystalstructures of lysozyme at very low levels of hydration. Proteins. 1998;32:229–240. doi: 10.1002/(sici)1097-0134(19980801)32:2<229::aid-prot9>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 43.Sukumar N, Biswal BK, Vijayan M. Structures of orthorhombic lysozyme grown at basic pH and its low-humidityvariant. Acta Cryst. 1999;D55:934–937. doi: 10.1107/s0907444998015522. [DOI] [PubMed] [Google Scholar]
- 44.Lee B, Richards FM. The interpretation of protein structures: estimation of staticaccessibility. Journal of molecular biology. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 45.Luscombe NM, Laskowski RA, Thornton JM. Amino acid-base interactions: a three-dimensional analysis of protein-DNAinteractions at an atomic level. Nucleic acids research. 2001;29:2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moestrup SK, Kozyraki R, Kristiansen M, Kaysen JH, Rasmussen HH, Brault D, Pontillon F, Goda FO, Christensen EI, Hammond TG, Verroust PJ. The intrinsic factor-vitamin B12 receptor and target of teratogenic antibodies is amegalin-binding peripheral membrane protein with homology to developmental proteins. The Journal of biological chemistry. 1998;273:5235–5242. doi: 10.1074/jbc.273.9.5235. [DOI] [PubMed] [Google Scholar]
- 47.Fyfe JC, Madsen M, Hojrup P, Christensen EI, Tanner SM, de la Chapelle A, He Q, Moestrup SK. The functional cobalamin (vitamin B12)-intrinsic factor receptor is a novel complexof cubilin and amnionless. Blood. 2004;103:1573–1579. doi: 10.1182/blood-2003-08-2852. [DOI] [PubMed] [Google Scholar]
- 48.Fedosov SN, Fedosova NU, Berglund L, Moestrup SK, Nexo E, Petersen TE. Composite organization of the cobalamin binding and cubilin recognition sites ofintrinsic factor. Biochemistry. 2005;44:3604–3614. doi: 10.1021/bi047936v. [DOI] [PubMed] [Google Scholar]
- 49.Moestrup SK, Verroust PJ. Megalin- and cubilin-mediated endocytosis of protein-bound vitamins, lipids, andhormones in polarized epithelia. Annu Rev Nutr. 2001;21:407–428. doi: 10.1146/annurev.nutr.21.1.407. [DOI] [PubMed] [Google Scholar]
- 50.Li N, Seetharam S, Seetharam B. Genomic structure of human transcobalamin II: comparison to human intrinsic factorand transcobalamin I. Biochemical and biophysical research communications. 1995;208:756–764. doi: 10.1006/bbrc.1995.1402. [DOI] [PubMed] [Google Scholar]
- 51.Brada N, Gordon MM, Wen J, Alpers DH. Transfer of cobalamin from intrinsic factor to transcobalamin II. J Nutr Biochem. 2001;12:200–206. doi: 10.1016/s0955-2863(00)00129-7. [DOI] [PubMed] [Google Scholar]
- 52.Fedosov SN, Petersen TE, Nexo E. Binding of cobalamin and cobinamide to transcobalamin from bovinemilk. Biochemistry. 1995;34:16082–16087. doi: 10.1021/bi00049a023. [DOI] [PubMed] [Google Scholar]
- 53.Sukumar N, Mathews FS, Gordon MM, Ealick SE, Alpers DH. Post crystallization Analysis of the Irreproducibility of the Human IntrinsicFactor-Cobalamin Complex Crystals. Crystal growth & design. 2009;9:348–351. doi: 10.1021/cg800509f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dieckgraefe BK, Seetharam B, Banaszak L, Leykam JF, Alpers DH. Isolation and structural characterization of a cDNA clone encoding rat gastricintrinsic factor. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:46–50. doi: 10.1073/pnas.85.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Banerjee RV, Johnston NL, Sobeski JK, Datta P, Matthews RG. Cloning and sequence analysis of the Escherichia coli metH gene encodingcobalamin-dependent methionine synthase and isolation of a tryptic fragment containing thecobalamin-binding domain. Journal of biological chemistry. 1989;264:13888–13895. [PubMed] [Google Scholar]
- 56.Drummond JT, Huang S, Blumenthal RM, Matthews RG. Assignment of enzymatic function to specific protein regions of cobalamin-dependentmethionine synthase from Escherichia coli. Biochemistry. 1993;32:9290–9295. doi: 10.1021/bi00087a005. [DOI] [PubMed] [Google Scholar]
- 57.Goulding CW, Postigo D, Matthews RG. Cobalamin-dependent methionine synthase is a modular protein with distinct regionsfor binding homocysteine, methyltetrahydrofolate, cobalamin, and adenosylmethionine. Biochemistry. 1997;36:8082–8091. doi: 10.1021/bi9705164. [DOI] [PubMed] [Google Scholar]
- 58.Ragsdale SW, Pierce E. Acetogenesis and the Wood-Ljungdahl pathway of CO(2) fixation. Biochimica et biophysica acta. 2008;1784:1873–1898. doi: 10.1016/j.bbapap.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Retey J, Lynen F. The absolute configuration of methylmalonyl-CoA. Biochemical and biophysical research communications. 1964;16:358–361. doi: 10.1016/0006-291x(64)90040-3. [DOI] [PubMed] [Google Scholar]
- 60.Eggerer H, Stadtmann ER, Overath P, Lynen F. On the mechanism of the rearrangement of methylmalonyl CoA to succinyl CoA catalyzedby the cobalamine coenzyme. Biochemische Zeitschrift. 1960;333:1–9. [PubMed] [Google Scholar]
- 61.Jordan A, Reichard P. Ribonucleotide reductases. Annual review of biochemistry. 1998;67:71–98. doi: 10.1146/annurev.biochem.67.1.71. [DOI] [PubMed] [Google Scholar]
- 62.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 63.Eklund H, Uhlin U, Farnegardh M, Logan DT, Nordlund P. Structure and function of the radical enzyme ribonucleotide reductase. Progress in biophysics and molecular biology. 2001;77:177–268. doi: 10.1016/s0079-6107(01)00014-1. [DOI] [PubMed] [Google Scholar]
- 64.Tamao Y, Blakley RL. Direct spectrophotometric observation of an intermediate formed fromdeoxyadenosylcobalamin in ribonucleotide reduction. Biochemistry. 1973;12:24–34. doi: 10.1021/bi00725a005. [DOI] [PubMed] [Google Scholar]
- 65.Wahl MC, Sundaralingam M. C-H…O hydrogen bonding in biology. Trends Biochem Sci. 1997;22:97–102. doi: 10.1016/s0968-0004(97)01004-9. [DOI] [PubMed] [Google Scholar]
- 66.Desiraju GR, Steiner T. The weak hydrogen bond in structural chemistry and biology. Oxford University Press; New York: 1999. [Google Scholar]
- 67.Domagala M, Grabowski SJ, Urbaniak K, Mlostori G. Role of C–H···S andC–H···N Hydrogen Bonds in Organic Crystal Structures – TheCrystal and Molecular Structure of3-Methyl-2,4-diphenyl-(1,3)-thiazolidine-5-spiro-2′-adamantane and3-Methyl-2,4,5,5-tetraphenyl-(1,3)-thiazolidine. J Phys Chem. 2003;A107:2730–2736. [Google Scholar]
- 68.Sukumar N, Mathews FS, Langan P, Davidson VL. Role of Protein Dynamics in electron transfer - A joint X-ray and Neutron Study onAmicyanin. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:6817–6822. doi: 10.1073/pnas.0912672107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Di Masi DR, White JC, Schnaitman CA, Bradbeer C. Transport of vitamin B12 in Escherichia coli: common receptor sites for vitamin B12and the E colicins on the outer membrane of the cell envelope. Journal of bacteriology. 1973;115:506–513. doi: 10.1128/jb.115.2.506-513.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bradbeer C, Woodrow ML, Khalifah LI. Transport of vitamin B12 in Escherichia coli: common receptor system for vitamin B12and bacteriophage BF23 on the outer membrane of the cell envelope. Journal of bacteriology. 1976;125:1032–1039. doi: 10.1128/jb.125.3.1032-1039.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]