

Abstract
The pathophysiology of cerebral ischemia is traditionally understood in relation to reductions in cerebral blood flow (CBF). However, a recent reanalysis of the flow-diffusion equation shows that increased capillary transit time heterogeneity (CTTH) can reduce the oxygen extraction efficacy in brain tissue for a given CBF. Changes in capillary morphology are typical of conditions predisposing to stroke and of experimental ischemia. Changes in capillary flow patterns have been observed by direct microscopy in animal models of ischemia and by indirect methods in humans stroke, but their metabolic significance remain unclear. We modeled the effects of progressive increases in CTTH on the way in which brain tissue can secure sufficient oxygen to meet its metabolic needs. Our analysis predicts that as CTTH increases, CBF responses to functional activation and to vasodilators must be suppressed to maintain sufficient tissue oxygenation. Reductions in CBF, increases in CTTH, and combinations thereof can seemingly trigger a critical lack of oxygen in brain tissue, and the restoration of capillary perfusion patterns therefore appears to be crucial for the restoration of the tissue oxygenation after ischemic episodes. In this review, we discuss the possible implications of these findings for the prevention, diagnosis, and treatment of acute stroke.
Keywords: acute ischemic stroke, capillary transit time heterogeneity (CTTH), cerebrovascular reserve capacity (CVRC), penumbra, reperfusion injury, stroke risk factors
Introduction
The pathophysiology of cerebral ischemia is traditionally understood in relation to specific cerebral blood flow (CBF) thresholds. The term ischemia (from Greek: isch—restriction; aimía—blood) therefore refers to a reduction in CBF that causes the cessation of neuronal electrical activity in experimental ischemia and the sudden appearance of focal neurologic symptoms in patients. The corresponding CBF threshold is ∼20 mL/100 mL per minute, both in humans and across a number of animal species.1 Below CBF levels of 8 to 12 mL/100 mL per minute, oxygen supplies no longer suffice to fuel vital cell functions such as the maintenance of ion homeostasis across cell membranes, and brain tissue therefore suffers permanent damage, infarction, within minutes.2 At CBF values above this level, ischemic tissue may survive for several hours and regain function if CBF is normalized. Such impaired, yet salvageable tissue is referred to as the ischemic penumbra3, 4 and characterized by elevated oxygen extraction fraction (OEF).4 The concept of an ischemic penumbra that can be salvaged by early vessel recanalization has been highly successful in that placebo-controlled trials have shown a reduction in neurologic deficits in acute stroke patients treated by intravenous recombinant tissue plasminogen activator during the first hours after symptom onset.5, 6, 7, 8
Resting CBF in human gray matter is in the range of 40 to 70 mL/100 mL per minute and the etiology of ischemic stroke is therefore linked to conditions that can cause significant reductions in regional CBF. Before the stroke, most stroke patients have had combinations of large-vessel stenoses, small vessel disease (SVD), and a propensity to form either cardiac emboli or arterial thrombi in relation to atherosclerotic lesions9—see Figure 1. Moreover, the normal regulation of CBF to meet the changing metabolic needs of brain tissue is often disturbed in conditions that predispose to stroke, such as aging, hypertension, diabetes, and hypercholesterolemia.10 This condition, neurovascular dysfunction, is characterized by the suppression of the normal CBF increases during neural activity, and it is seemingly caused by the production of superoxide anions at the level of the vascular endothelium.10, 11 The superoxide anions form reactive oxygen species (ROS) and react with nitric oxide (NO) to form peroxynitrite.12 The resulting NO depletion disrupts the normal regulation of vessel tone, while peroxynitrite disturbs smooth muscle cell contractility13 and inactivates tissue plasminogen activator.11 In addition, the depletion of NO and the increased production of ROS initiate a proinflammatory state in the vessel wall, characterized by platelet aggregation, leukocyte adhesion to the vascular endothelium, and vascular wall remodeling and damage.14, 15 While the origin of neurovascular dysfunction remains unknown, this phenomenon is considered to be an early, critical feature in the development of the vascular wall damage in both atherosclerosis and SVD, and in the increased thrombogenicity that ultimately leads to acute stroke.16
Figure 1.

Vascular changes in conditions that predispose to stroke. The figure illustrates the three levels at which the vascular system is affected in conditions predisposing to stroke. The schematic drawing is modified from a figure by Lanza and Crea.111 At the level of conduit vessels (A), blood flow can be restricted by atherosclerotic lesions (red arrow). At the level of resistance vessels (B), small vessel disease causes thickening of the vessel wall (red arrows). More peripherally, the capillary wall undergoes morphological changes, such as thickened basement membrane, pericapillary fibrosis, and pericyte loss in aging and a number of conditions predisposing to stroke (A, right)—see also Table 1. Images from Farkas et al.112 Ischemia can induce irreversible constrictions of capillary pericytes (B, right) and thereby disturb capillary flow patterns—from Yemisci et al.18
Changes in vascular morphology and function before the development of stroke are not limited to blood vessels that affect CBF. In fact, the morphology and function of cerebral capillaries undergo profound changes both in conditions that predispose to stroke (see Table 1 and references therein) and during cerebral ischemia17—see Figure 1. The role of these capillary changes in the etiology and the pathophysiology of stroke remains uncertain. In theory, widespread capillary embolisms or pericyte constrictions18, 19 could block the passage of blood and explain the occasional lack of tissue reperfusion after recanalization therapy—the ‘no-reflow phenomenon'. Before stroke, however, the changes listed in Table 1 are all characteristic of perfused capillaries and would therefore be expected to disturb, rather than block, the passage of erythrocytes. Capillary flow patterns have been speculated to affect local oxygen delivery,20 and according to studies by direct microscopy in animals,21, 22 and by perfusion magnetic resonance imaging in human stroke,23, 24, 25 capillary flow patterns do undergo profound changes in cerebral ischemia.
Table 1. Changes in capillary wall morphology in conditions known to predispose to stroke.
| Risk factor | Changes in capillary morphology | Reference |
|---|---|---|
| Aging | Variable capillary diameters, increased tortuosity, twisting, and looping of capillaries. Thickening basement membranes with inclusions. Pericapillary fibrosis. Pericyte loss (human) | 101 102 |
| Hypertension | Pericyte degeneration, swelling of endothelium, and surrounding astrocyte endfeet. Thickened basement membrane (animal models) | 103 104 |
| Diabetes | Loss of pericytes and thickening of capillary basement membrane (animal models). Thickening of basement membrane (humans) | 104, 105 106, 107 |
| Smoking | Endothelial cell damage, subendothelial edema (peripheral arterial and arteriolar endothelium) | 108 |
| Alcoholism | Swelling of pericapillary astrocytic endfeet | 109 |
| Prior stroke | Pericyte constrictions (animal models) | 18, 110 |
Increases in capillary transit time heterogeneity (CTTH) were recently shown to reduce the maximum achievable OEF (OEFmax) for a given CBF and tissue oxygen tension.26 This effect is caused by an increasing proportion of erythrocytes passing through the capillary at transit times too short to permit proper oxygen extraction. Due to this effective ‘arteriolo-venular shunting,' disturbances in capillary flow patterns can affect the way in which CBF can support the metabolic needs of brain tissue. Accordingly, disturbances in the capillary bed could contribute to the abnormal CBF responses observed in neurovascular dysfunction and to the metabolic derangement in the ischemic penumbra, where tissue survival depends on effective oxygen extraction to compensate for the reduced perfusion.4
In this review, we use the extended model of tissue oxygen availability26 to analyze how the coupling of CBF to the metabolic needs of the tissue changes as a result of disturbances in capillary transit time patterns. We assume that the changes in capillary morphology listed in Table 1 give rise to increases in CTTH and then derive how CBF must be adjusted to secure the metabolic needs of brain tissue:
For moderate increases in CTTH, OEFmax decreases. CBF levels must therefore increase to meet the metabolic needs of the tissue.
For larger increases in CTTH, OEFmax becomes critically low during increases in CBF. As a result, tissue oxygen tension decreases, and normal CBF responses must be attenuated to permit a more efficient oxygen extraction.
CTTH can reach a critical threshold, at which the metabolic needs of resting brain tissue can no longer be met by compensatory changes in CBF. The CBF level that provides optimal oxygenation at this CTTH threshold is 21 mL/100 mL per minute—similar to the ‘classical' ischemic CBF threshold.
We then discuss these model predictions in relation to the possible origins of neurovascular dysfunction and acute stroke symptoms, and in relation to the prevention, diagnosis, and treatment of acute stroke.
The relation between CBF and tissue oxygen availability: the role of capillaries and blood
The relation between CBF and the availability of oxygen in brain tissue is traditionally derived from work by Christian Bohr, Seymour S Kety, Christian Crone, and Eugene M Renkin.27 The formalism, now referred to as the ‘flow-diffusion' or ‘Bohr–Kety–Crone–Renkin' (BKCR) equation, uses the extraction properties for freely diffusible substances, as they pass through single capillaries, to describe the entire tissue.27 This generalization assumes, however, that all capillaries within a given tissue volume are identically perfused. This precondition is rarely met in tissue: for example, the flux of erythrocytes through cortical brain capillaries is highly inhomogeneous during rest.28, 29, 30 David Chesler, coauthor of a study on the effects of capillary flow heterogeneity on tissue outcome after acute ischemic stroke in humans,23 first noted that for a given CBF, the BKCR equation for single capillaries predicts that the introduction of any degree of heterogeneity of capillary flows will lead to a reduction in tissue oxygen availability compared with the BKCR equation's predictions for tissue.23 Figure 2 illustrates how the heterogeneity of erythrocyte velocities, which occurs either naturally or as a result of disturbances in the capillary wall or in blood cell morphology, can reduce the extraction of oxygen from blood. As illustrated by the figure, both white blood cell (WBC) and erythrocyte dimensions exceed the average capillary diameter, and these cells must therefore undergo deformation to enter and pass the capillaries. Changes in the size, number, and endothelial adhesion of blood cells are known to affect the distribution of erythrocyte flows across the capillary bed.31 Both low-grade inflammation and infections increase the adhesion among blood cells and endothelium and the viscosity of leukocyte cytoplasm. This has been shown to disturb capillary flow patterns and lead to ‘shunting' of erythrocytes through the capillary bed.31 Meanwhile, the shedding of the arteriolar and capillary endothelial glycocalyx in inflammation has been shown to change capillary resistance and microcirculatory hemodynamics.32 These conditions would therefore be expected to increase to the level of CTTH for erythrocytes and reduce the efficacy of oxygen extraction by the tissue.
Figure 2.

Classical Bohr–Kety–Crone–Renkin (BKCR) flow-diffusion relation for oxygen. The classical BKCR curve shows the maximum amount of oxygen that can diffuse from a single capillary into tissue, for a given flow CMRO2max. The curve shape predicts three important properties of CMRO2max parallel-coupled capillaries: first, the curve slope decreases toward high perfusion values, making vasodilation increasingly inefficient as a means of improving tissue oxygenation toward high perfusion rates. This property is reflected at the macroscopic level in that CBF increases are generally several-fold larger than the corresponding increase in oxygen utilization.113 The resulting blood ‘hyperoxygenation' is thought to explain the blood oxygen level-dependent (BOLD) contrast mechanism.114 Second, if erythrocyte flows differ among capillary paths (case B) instead of being equal (case A), the net tissue oxygen availability declines. This is seen by using the BKCR curve to determine the net tissue oxygen availability resulting from the individual flows in case (B). The resulting net tissue oxygen availability is the weighted average of the oxygen availabilities for the two flows, labeled B in the plot. Note that the resulting tissue oxygen availability will always be less than that of the homogenous case, labeled A. Conversely, homogenization of capillary flows during hyperemia has the opposite effect, and serves to compensate for the first property. Third, if erythrocyte flows are hindered (rather than continuously redistributed) along single capillary paths (as indicated by slow-passing white blood cell (WBC) and rugged capillary walls) upstream vasodilation is likely to amplify the redistribution losses, as erythrocytes are forced through other branches at very high speeds, with negligible net oxygenation gains. CBF, cerebral blood flow; CMRO2max, cerebral metabolic rate of oxygen.
The extended BKCR equation
The BKCR equation was recently extended to take the CTTH of erythrocytes into account. The model is described in detail in Jespersen and Østergaard26 and briefly summarized here: the model consists of (1) a probability density function (PDF) h(τ) to describe the distribution of capillary transit times and (2) an expression 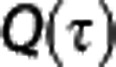 to describe the extraction of oxygen for a single capillary with transit time τ.
to describe the extraction of oxygen for a single capillary with transit time τ. 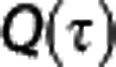 is analogous to the BKCR equation, except that is takes the oxygen binding properties of oxygen and the tissue oxygen concentration into account. From these two expressions, OEFmax for the entire capillary bed can be obtained by summing the single capillary contributions, weighted by their transit time distribution,
is analogous to the BKCR equation, except that is takes the oxygen binding properties of oxygen and the tissue oxygen concentration into account. From these two expressions, OEFmax for the entire capillary bed can be obtained by summing the single capillary contributions, weighted by their transit time distribution,
 |
We followed earlier work33 and approximated the PDF of capillary transit times by a gamma variate function with shape parameters α and β, h(τ, α, β). The hemodynamics of tissue is characterized by these two parameters, noting that the capillary mean transit time, MTT, equals αβ while CTTH is quantified by the standard deviation of capillary transit times, 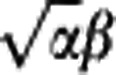 . We recall that MTT equals CBV/CBF,34 where CBV is the capillary blood volume.
. We recall that MTT equals CBV/CBF,34 where CBV is the capillary blood volume.
To model 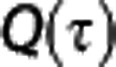 , we considered a three-compartment model consisting of tissue, blood plasma, and hemoglobin and assumed that the current of oxygen across the capillary membrane is proportional to the difference between plasma oxygen concentration (Cp) and tissue oxygen concentration (Ct). The differential equation for total oxygen concentration C as a function of the fractional distance
, we considered a three-compartment model consisting of tissue, blood plasma, and hemoglobin and assumed that the current of oxygen across the capillary membrane is proportional to the difference between plasma oxygen concentration (Cp) and tissue oxygen concentration (Ct). The differential equation for total oxygen concentration C as a function of the fractional distance 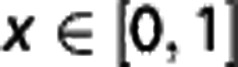 along a capillary with flow f and volume V then reads35, 36
along a capillary with flow f and volume V then reads35, 36
 |
assuming steady state (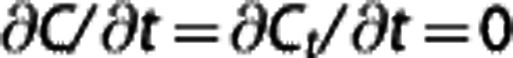 ), and equal forward and reverse rate constants k. Note that the capillary transit time τ is identical to V/f.
), and equal forward and reverse rate constants k. Note that the capillary transit time τ is identical to V/f.
The binding of oxygen to hemoglobin was approximated by the phenomenological Hill equation:
 |
where CB is the concentration of bound oxygen, B=0.1943 mL/mL is the maximum amount of oxygen bound to hemoglobin, P is oxygen partial pressure in plasma, P50=26 mmHg is the oxygen pressure required for half saturation, and h=2.8 is the Hill coefficient.
Neglecting the contribution of plasma oxygen to the total oxygen content (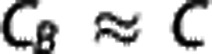 ), Equation (2) then yields a general equation for oxygen concentration as a function of the normalized distance x along a capillary with transit time τ35, 36
), Equation (2) then yields a general equation for oxygen concentration as a function of the normalized distance x along a capillary with transit time τ35, 36
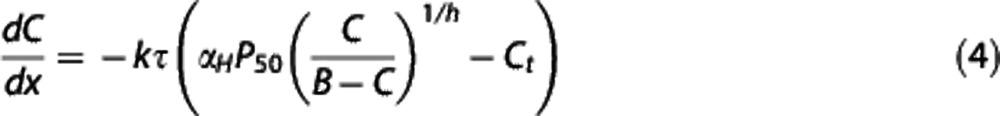 |
where  .35
.35
We note that the single capillary BKCR equation, given by the familiar exponential expression 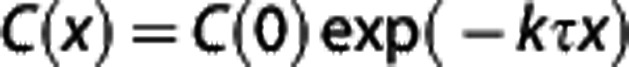 27, 37 generalizes to
27, 37 generalizes to
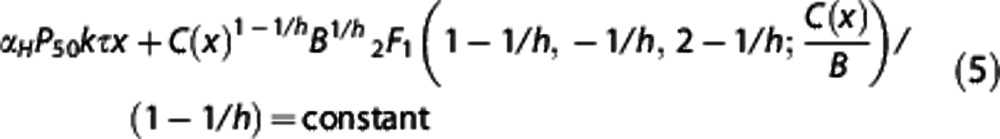 |
when one models the extraction of hemoglobin-bound oxygen by tissue with a fixed oxygen tension. The constant on the right-hand side is determined by the initial value, 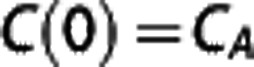 and
and 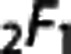 is a hypergeometric function.38 For a given tissue oxygen tension Ct, the differential Equation (4) was solved to yield the single capillary extraction fraction Q=1–C(1)/C(0) as a function of kτ.
is a hypergeometric function.38 For a given tissue oxygen tension Ct, the differential Equation (4) was solved to yield the single capillary extraction fraction Q=1–C(1)/C(0) as a function of kτ.
Finally, for a steady-state condition characterized by a certain tissue oxygen tension PtO2, MTT, and CTTH, the corresponding OEFmax can be determined from
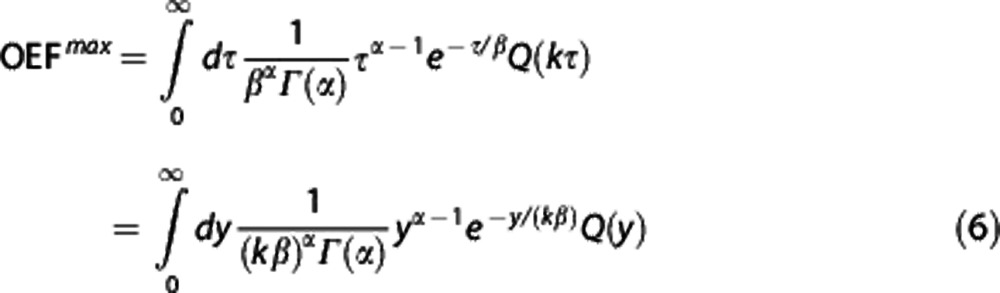 |
with k as the only unknown parameter. We fixed to k=118/second to yield a resting OEFmax=0.3 for transit time data reported in Stefanovic et al.39
The upper limit on cerebral metabolic rate of oxygen is now given by CMRO2max=CBF × Ca × OEFmax, where the arterial oxygen concentration Ca was set to Ca=19 mL/100 mL. To express CBF in terms of MTT in this relation, we assumed CBV to remain constant (1.6 mL/100 mL) during changes in CBF, and therefore for capillary transit times to be inversely proportional to CBF.26
As a consequence of the dependence of OEFmax on capillary transit time patterns, changes in CTTH alter the effective capillary surface area available for diffusion considerably. The apparent permeability-surface area product PS can be determined from OEFmax by using the expression 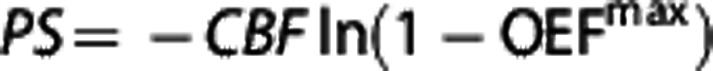 .26
.26
Metabolic effects of CTTH changes under physiologic conditions—and in disease
The extended BKCR equation predicts that the observed homogenization of capillary transit times observed during cortical activation40 and hypercapnia41 is in fact crucial to maintain efficient oxygen extraction during the accompanying increase in CBF. While the metabolic effects of reduced CBF in ischemia are well described, the additional effects of changing capillary flow patterns on cerebral oxygen availability before, during, and after suffering an acute stroke, remain unknown. Below, we used the extended BKCR equation to model this phenomenon, and refer to a condition of (1) elevated CTTH and (2) inability to homogenize capillary flows during episodes of increased CBF as capillary dysfunction. The top panel in Figure 3 shows the starting point of the analysis, namely the gradual increase in CTTH over time which we predicted to accompany aging and other conditions predisposing to stroke. The hemodynamic adaptations that are necessary to compensate for this CTTH increase to maintain sufficient tissue oxygen availability are then described according to three characteristic stages. Finally, we discuss these stages with respect to the vulnerability of tissue to reductions in CBF, to increases in CTTH, and to incomplete restoration of arterial and capillary flow patterns after recanalization therapy.
Figure 3.

Changes in cerebral blood flow (CBF) and tissue oxygen tension that must accompany increasing levels of capillary dysfunction to maintain tissue oxygen availability before stroke. The figure displays the adaptations of CBF (second panel from the top) and PtO2 (third panel from the top) that are necessary to maintain tissue oxygen availability as capillary transit time heterogeneity (CTTH) levels (top panel) gradually increase, both during rest (upper graph in each panel) and during functional activation (lower graph in each panel). These changes are hypothesized to occur in relation to conditions that predispose to stroke—cf. Table 1—years before symptoms develop. The most important hemodynamic change occurs toward the end of Stage 1 when the increase in tissue oxygen availability during hyperemia is no longer sufficient to meet the metabolic needs of the tissue. Therefore, vasodilation during episodes of increased metabolic demands must be attenuated. This transition is hypothesized to mark the onset of neurovascular dysfunction, and of increased oxidative damage due to the accompanying release of reactive oxygen species (ROS). Later, vasodilation induced during rest must also be attenuated to maintain tissue oxygen availability above the needs of resting tissue. This is hypothesized to mark the onset of detectable reductions in cerebrovascular reserve capacity. As CTTH continues to increase, oxygen availability can be secured by attenuated CBF responses and more efficient blood-tissue concentration gradients—see text. This results in increasing maximum oxygen extraction fraction (OEFmax) values. The gradual reduction in metabolic reserve capacity is indicated in the lower panel: As CTTH increases, net oxygen extraction is increasingly limited by CBF as the oxygen extraction fraction approaches unity. In the stroke prone state, minor reductions in CBF or increases in CTTH are therefore predicted to result in neurologic symptoms as maximum cerebral metabolic rate of oxygen (CMRO2max) approaches the actual, metabolic needs of the tissue. HIF-1, hypoxia-inducible transcription factor 1.
Stage I. Increased CTTH reduces oxygen extraction. To compensate, CBF must increase
Figure 4 illustrates how the hemodynamics of erythrocytes and the tissue oxygen tension, PtO2, affect the extraction of oxygen from blood. According to the extended BKCR equation,26 the extraction of oxygen from the capillary bed is determined partly by the MTT for erythrocytes across all capillaries and partly by the heterogeneity of these transit times, CTTH. Recall that CTTH is parameterized by the standard deviation of a gamma variate capillary transit time distribution throughout the manuscript26 and that MTT is given as the ratio of the CBV to CBF. The diagonal arrows in Figures 4A and 4B indicate typical measured changes in MTT and CTTH during functional hyperemia.26 The combined reductions in MTT and CTTH during functional hyperemia appear to be crucial to the increase in oxygen availability that accompanies hyperemia.26
Figure 4.

Effects of mean transit time (MTT), capillary transit time heterogeneity (CTTH), and oxygen tension on oxygen extraction. (A, B) The x axis position is indicated by the value of both MTT (bottom x axis) and cerebral blood flow (CBF) (top x axis). (A) Contour plot of maximum oxygen extraction fraction (OEFmax) as a function of these parameters and the CTTH at a tissue oxygen tension of PtO2=26 mm Hg.115 The value of OEFmax is indicated by a color at the corresponding location in the (MTT, CTTH) plane. The OEFmax value corresponding to this color or location in the contour plot is most easily derived from the OEFmax values indicated on the two nearest solid black lines, which display OEFmax iso-contours. Note that the oxygen extraction efficacy always increases with increasing MTT and with decreasing CTTH. The image inserts show schematic capillary beds with homogenous capillary transit times (lower left insert) and with CTTH typical of resting brain (upper right insert). The arrow indicates the changes in MTT and CTTH that occur during functional activation: The reduction in CTTH that occurs in parallel with the arteriolar dilation partly maintains the oxygen extraction efficacy during hyperemia. The maximum supported cerebral metabolic rate of oxygen (CMRO2max) is shown in units of mL/100 mL per minute in (B). Recall that this figure is derived from (A) by multiplying its OEFmax values by Ca and CBF, assuming that the capillary transit times of erythrocytes are inversely proportional to CBV.26 Note that CMRO2max, and thereby tissue oxygen availability, always increases with decreasing flow heterogeneity. The physiologic CBF response to functional activation tends to reduce OEFmax in normal tissue, both due to the shorter MTT and due to a higher PtO2.26 Hemodynamic states above the yellow line in (B) are unique in that increases in their CBF (reductions in MTT) will lead to states of lower tissue oxygen availability: these states are referred to as having malignant CTTH. The horizontal arrow indicates the metabolic consequence of increased CBF without concomitant reductions in CTTH: in the absence of any homogenization of capillary flows, vasodilation would not lead to a net increase in the availability of oxygen in tissue. The origin of this paradox effect is illustrated by the insert in the upper right of (A). The oxygenation of blood along capillary paths in this insert was modeled according to in vivo erythrocyte velocity recordings in resting rat brain26, 39 and the single-capillary Bohr–Kety–Crone–Renkin (BKCR) model, and then indicated as a color code, ranging from fully oxygenated (left) to levels well below that of mixed venular blood (right) along some capillary paths—cf. also Figure 3 in Jespersen and Østergaard.26 Note that, along some capillary paths, flow velocities are so high that little oxygen is extracted from blood. In normal brain, hyperemia is accompanied by a reduction in CTTH (lower left insert in A). This homogenization decreases the number of capillary paths with high flow and limited oxygen extraction, causing net oxygen availability to increase as a function of CBF. If this heterogeneity persist during hyperemia, for example due to changes in capillary morphology or blood rheology, the effective shunting of oxygenated blood can become critical. As discussed in the text, vasodilation may in fact be attenuated by hypoxia-sensitive mechanisms as hemodynamic states approach this critical limit. Paradoxically, the neurovascular dysfunction and the reduced CVRC observed in conditions predisposing to stroke may therefore reflect adaptations to prevent critical hypoxia in cases of high CTTH. (C) This panel shows how tissue oxygen metabolism can be secured when CBF increases no longer lead to increased tissue oxygen availability. The figure shows net oxygenation as a function of tissue oxygen tension and CTTH for fixed MTT (CBF=60 mL/100 mL per minute; MTT 1.4 seconds) to illustrate how tissue metabolism, by reducing tissue oxygen tension, can facilitate net oxygen extraction in cases where capillary dysfunction cause critical levels of ‘shunting'—provided that CBF responses are attenuated. Note that a decrease in oxygen tension of 5 mm Hg can support a CMRO2 increase of 20%. This corresponds roughly to the additional energy requirements of neuronal firing.
According to the extended BKCR equation, elevated CTTH is expected to reduce OEFmax—see Figure 4A. To maintain constant oxygenation in response to a reduction in OEFmax, resting and activity-related CBF rates must therefore increase. Accordingly, increased CBF levels, both during rest and during functional activation, are predicted to be characteristic of Stage I—see the panels of relative CBF levels in Figure 3. As CTTH increases further, however, increases in CBF can no longer maintain sufficient tissue oxygenation during episodes of functional activation. This phenomenon marks the beginning of Stage II.
Stage II. Decreasing tissue oxygen tension and the need to attenuate CBF responses
One of the most important predictions of the extended BKCR equation is that as CTTH increases, OEFmax for a given tissue oxygen tension can become so low that the activity-related increase in CBF is no longer an efficient means of improving tissue oxygenation. Instead, suppression of normal CBF responses and reduced tissue oxygen tension becomes the only means by which sufficient tissue oxygen availability can be secured.26 The tissue oxygen tension, PtO2, determines the oxygen concentration gradient between blood and tissue, and thereby limits the fraction of oxygen which can be extracted, OEFmax. Tissue metabolism constantly removes oxygen from the tissue. If the extraction of oxygen from blood becomes limited by elevated CTTH, then tissue oxygen consumption will therefore reduce PtO2 until the blood-tissue concentration gradient—and thereby OEFmax—is so high that the net oxygen extraction again meets the metabolic needs of the tissue. The need to suppress CBF responses as CTTH increases, and the importance of low tissue oxygen tension for the net extraction of oxygen, is illustrated in Figures 4B and 4C. For a typical cerebral tissue oxygen tension, Figure 4B displays a contour plot of the CMRO2max that can be supported for any combination of MTT and CTTH. As illustrated in the figure, an increase in CBF (decrease in MTT), without a concomitant reduction in CTTH, can lead to a hemodynamic state (value of MTT and CTTH) in which the availability of oxygen is reduced in comparison with the initial hemodynamic state. We refer to states such as these as malignant CTTH.26 In Figure 4B, these states correspond to combinations of CTTH and MTT that are situated above the yellow line for a given tissue oxygen tension.
States of malignant CTTH can be understood either as conditions with critical reductions in the apparent PS product of oxygen or as conditions where a critical proportion oxygenated blood is effectively shunted through the capillary bed. The latter is illustrated by the inserts in Figure 4A: in the resting, normal brain, some proportion of erythrocytes pass through the capillary bed at transit times so short that little oxygen is extracted (upper right insert). In normal brain, CTTH reductions during hyperemia (lower left insert) permit efficient oxygen extraction, and a net increase in oxygen availability (CMRO2max), during hyperemia. As changes in capillary or blood morphology accumulate (cf. Figure 2), transit times may not be sufficiently homogenized during hyperemia. In malignant CTTH (for a given tissue oxygen tension), an incremental increase in CBF will force erythrocytes through capillaries at transit times that are too fast to permit proper oxygen extraction—causing a reduction in OEFmax that outweighs the benefits of the increased supply of oxygenated blood.
In conditions where increased CBF would result in malignant CTTH, the extended BKCR equation predicts that the lowering of tissue oxygen tension becomes the only means by which oxygen availability can be maintained. As illustrated in Figure 4C, this mechanism is efficient if CBF is kept low to maximize OEFmax. In the figure, CMRO2max is plotted as a function of tissue oxygen tension and CTTH for a CBF value that is typical of resting brain tissue. Note that the additional oxygen requirements for neuronal firing, typically 15% to 20%, correspond to the increase in CMRO2max which results from a modest reduction in tissue oxygen tension. Therefore, when CBF remains suppressed, the increased blood-tissue oxygen concentration gradients that accompany the increased oxygen metabolism during functional activation can facilitate increases in OEF that are sufficient to support the additional energy needs of the tissue.26
Do we in fact observe cases in which CBF responses are suppressed and OEF elevated—in the absence of upstream large vessel abnormalities that might limit blood flow? Reductions in cerebrovascular reserve capacity (CVRC) are typically characterized either by increased resting OEF or by reduced CBF responses to standardized vasodilatory stimuli such as acetazolamide,42 hypercapnia, or functional activation.43 In the setting of cerebrovascular disease, reductions in CVRC are traditionally taken to reflect the extent to which the blood supply to the brain is limited by either large vessel stenosis or SVD. According to the predictions above, reduced CVRC and neurovascular dysfunction are also features of capillary dysfunction, in that the attenuation of CBF responses is a necessary adaptation to maintain tissue oxygen availability in conditions where CTTH is elevated. The extent to which capillary dysfunction predates the neurovascular dysfunction and the reduced CVRC observed in conditions predisposing to stroke such as aging, hypertension, and diabetes11, 44, 45 remains to be established.
Stage III. The stroke-prone patient. Ischemic thresholds
In stage III, the continued increase in CTTH causes CMRO2max to decrease toward the actual metabolic needs of resting brain tissue, cf. Figure 3. In Figure 5, the green surface corresponds to the metabolic rate of resting brain tissue, CMRO2=2.5 mL/100 mL per minute, measured in the contralateral hemisphere of patients with focal ischemia.46 The surface is created by displaying the 2.5 mL/100 mL per minute iso-contour in Figure 4B for all values of the tissue oxygen tension between 0 and 25 mm Hg. Thereby, the interior of the green half-cone represents hemodynamic conditions which can support metabolic needs above that of resting tissue. The red plane marks the boundary of malignant CTTH, to the left of which vasodilation would reduce tissue oxygen availability.
Figure 5.

Metabolic thresholds. The green iso-contour surface corresponds to the metabolic rate of contralateral tissue in patients with focal ischemia.46 The red plane marks the boundary, left of which vasodilation reduces tissue oxygen availability (malignant capillary transit time heterogeneity (CTTH)). The maximum value CTTH can attain at a PtO2 of 25 mm Hg if oxygen availability is to remain above that of resting tissue is indicated by the label A. As CTTH increases further, a critical limit is reached as PtO2 approaches zero—label B. At this stage, the metabolic needs of tissue cannot be supported unless mean transit time (MTT) is prolonged to a threshold of ∼4 seconds, corresponding to cerebral blood flow (CBF)=21 mL/100 mL per minute. If MTT increases instead, owing to limitations in blood supply, then the threshold corresponding to resting contralateral tissue is reached at MTT values of roughly 6.3 seconds, or CBF=13 mL/100 mL per minute, provided CTTH is negligible or moderate (label C). If, however, CTTH increases as a result of the reduction in CBF or because of per-ischemic changes in capillary patency (cf. Figure 1), then tissue oxygen availability cannot be maintained above the metabolic needs of resting tissue for CBF values below 21 mL/100 mL per minute. The blue arrows indicate how tissue hypoperfusion and capillary dysfunction causes MTT and CTTH to change, and tissue oxygen availability to approach the metabolic requirements of resting brain tissue (the green iso-contour).
The label A in Figure 5 shows the theoretical maximum for the increase in CTTH (indicated by a broken line parallel to the MTT axis) that can be sustained by brain tissue at a PtO2 level of 25 mm Hg before neurologic symptoms ensue. As described in the section above, tissue oxygen tension will gradually decrease as CTTH approaches this threshold, permitting normal tissue function to be preserved, provided that CBF responses are attenuated. As CTTH increases further, however, CTTH reaches a critical limit (label B), where oxygen tension cannot be reduced further, and any changes in CBF will reduce tissue oxygen availability below the needs of resting tissue. Note that as CTTH increases, MTT must be gradually prolonged to a threshold of ∼4 seconds, corresponding to CBF=21 mL/100 mL per minute, to meet the metabolic needs of the tissue. In other words, to compensate for increased CTTH, resting CBF must decrease to maintain sufficient oxygen availability in tissue. As CBF reaches 21 mL/100 mL per minute, the metabolic rate of resting brain tissue can no longer be maintained, and neurologic symptoms are therefore predicted to occur. Note that if CBF suddenly decreases due to a vessel occlusion, the critical MTT threshold is 6.3 seconds, or CBF=13 mL/100 mL per minute, provided CTTH is negligible or moderate (label C). Studies suggest that CTTH increases in proportion to MTT during reductions in perfusion pressure.21, 22, 26 The critical CBF threshold for brain function in the case of vessel occlusion or systemic hypotension is therefore likely to occur somewhere between 13 and 21 mL/100 mL per minute. As a result, the extended BKCR model predicts that stroke symptoms can be caused by reduced CBF, increased CTTH, or both, at a ‘universal' CBF threshold similar to the traditional ischemic threshold reported in clinical and experimental studies.1
Stroke triggers
The extended BKCR equation predicts that CTTH has profound effects on tissue oxygen availability and therefore that disturbances of capillary flow patterns can trigger a critical lack of oxygen—with the possibility of associated acute stroke symptoms. In the presence of capillary dysfunction, both moderate reductions in CBF and slight increases in CTTH are predicted to be potential hemodynamic sources of critical reductions in tissue oxygen availability (cf. Figure 5) and thereby of neurologic symptoms characteristic of an ischemic stroke or transitory ischemic attack. Notably, the model predicts that critical reductions in oxygen availability due to elevated CTTH may be accompanied by compensatory hypoperfusion to optimize tissue oxygenation. The extent to which an increase in CTTH can trigger an acute stroke, or be the source of specific lesion types such as lacunar and microinfarcts47 in its own right, remains unknown. Common to such lesions, however, is the prediction that they would differ from ‘traditional' thromboembolic strokes only by the absence of an upstream vascular occlusion.
It is well established from both radiologic and pathologic studies that acute ischemic stroke often occur in relation to either an embolus from a remote source or the rupture of an atherosclerotic plaque in the wall of the carotid or cerebral vessels. The release of material from the soft plaque core into the blood stream precipitates the aggregation of platelets and the formation of a thrombus that can cause the occlusion of downstream vessels.16 In support of this mechanism, vessel occlusions are found at some level proximal to the second division of the anterior, middle, and posterior cerebral arteries in 28% to 46% of stroke patients who receive computerized tomography angiography on arrival to the hospital.48 The frequency of vascular occlusions at the time of symptom onset is difficult to ascertain in that the blood's own thrombolytic system is thought to dissolve some thrombi during the early phases of the disease. For example, patients who spontaneously recover from their ischemic symptoms within 24 hours of symptom onset, having suffered what is defined as transitory ischemic attack, only show signs of large vessel occlusions in 13% of cases48 but evidence of tissue damage in up to 50%.49
If exacerbation of capillary dysfunction acts as a stroke mechanism, then some strokes would be expected to be triggered by, for example, infections during which the number of activated WBCs is elevated: activated neutrophils have been observed to disturb the transit of blood through the microcirculation by increasing the adhesion of leukocytes to the endothelium, by temporary capillary plugging owing to their stiffer cytoplasm, and by the release of vasoactive products.31 The incidence of stroke is indeed increased in the 3 days after acute infections; an effect ascribed to increase in WBC count.50 Across ischemic stroke patients in general, one-third are believed to have suffered (predominantly bacterial) infections within 1 week of stroke onset.51, 52, 53 The winter months have the highest incidence of influenza and secondary bacterial infections. During these months, stroke mortality is 20% higher than during summer. This seasonal variation is especially pronounced in the old54 and strongly related to neutrophil count and self-reported cough/cold.55 The causal relation between the incidence of infections and of acute stroke is supported by findings that influenza vaccination in the elderly significantly reduces the number of stroke deaths.56, 57, 58 It is also thought that infections can trigger plaque rupture and thereby thromboembolic events.59 The extent to which increased CTTH in relation to increased blood viscosity can trigger stroke symptoms in the absence of a thromboembolic event, however, remains hypothetical.
Tissue injury during ischemia
The mechanisms that lead to infarction of penumbral tissue remain poorly understood. Notably, tissue death is not believed to be an inevitable consequence of the ischemia per se.10 The extended BKCR equation predicts that deteriorating capillary flow patterns during hypoxia/ischemia can cause CTTH to increase, and tissue oxygen availability to decrease, even for a constant CBF level. Direct observations of erythrocyte velocities during cerebral hypotension and ischemia show progressive disturbances in the distribution of erythrocyte velocities in the capillary bed.21, 22 The extended BKCR equation predicts that these disturbances can lead to tissue damage beyond that predicted by the level of CBF reduction alone. This is consistent with the results of clinical studies in which the fate of tissue which showed early deviations of capillary flow heterogeneity from that of normal tissue was assessed by follow-up magnetic resonance imaging scans. In all of these studies, abnormal capillary perfusion patterns in the acute phase of stroke were shown to predict subsequent infarction with higher accuracy than MTT, which is the traditional marker of hypoperfusion in acute ischemic stroke.23, 24, 25 Capillary flow disturbances in hypoperfused tissue may be secondary to the reduction in perfusion pressure, but some evidence suggest that they may be related to the ensuing tissue damage: acute ischemia has been shown to result in the constriction of cerebral pericytes, seemingly due to increased levels of oxidative and nitrosative stress.18, 60 In addition, ischemia-related damage to the basement membrane and to endothelial cells leads to disruptions of the blood–brain barrier and the development of vasogenic edema.61 The compression of capillaries by pericapillary edema and by the swelling of nearby damaged cells would be expected to further increase CTTH.
The identification of reversible elevations in CTTH could be of therapeutic importance as this phenomenon could represent a target for improving tissue oxygenation, independent of thrombolytic therapy. The acute management of hydration, blood viscosity, infections, and pericapillary edema (blood–brain barrier integrity) would therefore be predicted to improve tissue oxygenation and provide neuroprotection, irrespective of parallel recanalization therapy. In fact, dehydration is found during admission in >60% of acute stroke patients, and associated with poor outcome.62 The extent to which dehydration, via its effect on capillary flow patterns, acts as a stroke trigger (see previous section) and reduces tissue oxygen availability in the ischemic penumbra, however, remains unclear.
Reperfusion
Animal studies have shown that 3 hours of focal ischemia followed by 3 hours of reperfusion in rat brain produce more damage than 6 hours of continuous ischemia without reperfusion.63 It has been estimated that 72% of the tissue damage observed after 2 to 5 hours of ischemia–reperfusion in the rat brain is caused by reperfusion injury.64 Reperfusion injury is likely to be caused in part by incomplete reperfusion of the microvasculature. The no-reflow phenomenon and its therapeutic implications were recently reviewed by Dalkara et al.19 When calculating CMRO2max values by the extended BKCR equation, no-reflow enters as a reduction in the effective capillary volume available for oxygen extraction. The CMRO2max values displayed in Figure 4B must therefore be multiplied by the fraction of open capillaries (zero in case of complete occlusion) to correct for capillary occlusions. The extended BKCR model predicts, however, that even if capillaries are fully reperfused, tissue oxygen availability still depends on their ability to permit homogenous perfusion patterns. In other words, both CBF and CTTH must be restored in order for tissue reperfusion to restore tissue oxygen availability to its prestroke level. In view of the stroke mechanisms described above, recanalization must therefore be accompanied by the reversal of any pericyte constrictions, any capillary compression that occurred during the ischemic/hypoxic period, and any capillary flow disturbances resulting from the lysis of upstream clots. In turn, any residual increases in CTTH resulting from the ischemic episode are predicted to elicit either relative hyperperfusion (if CTTH is restored to levels characteristic of Stage I, but higher than that of unaffected tissue) or suppressed CBF (if CTTH is restored to levels characteristic of Stage II or III) as vessels adjust to achieve optimal tissue oxygenation. In the latter case, the suppression of CBF is predicted to be mediated by local ROS production and accompanied by relative tissue hypoxia (cf. Phase II). Capillary disturbances that persist after ischemia can therefore add to the oxidative and inflammatory tissue damage which is characteristic of reperfusion injury.10, 65
The extended BKCR model also implies that the timing of the restoration of CTTH after recanalization is crucial. If CTTH is not immediately restored on reperfusion, then OEFmax remains low for short MTT, and the sudden restoration of flow through fully dilated arteries and arterioles could therefore, paradoxically, cause sudden and severe tissue hypoxia and tissue damage. Acute stroke patients who are recanalized by intraarterial thrombolysis often develop large areas of hyperperfusion (mean CBF 2 to 3 times greater than in contralateral, homologous tissue). This luxury perfusion syndrome66 is associated with a twofold increase in the incidence of infarction at day 7.67 The extent to which this hyperperfusion indicates a reduction in OEFmax owing to incompletely restored CTTH, a failure to suppress the extreme upstream vasodilation, or poor tissue oxygen utilization owing to mitochondrial damage,10, 65 remains unknown. The rapid increase in perfusion pressure that results from recanalization would in itself be expected to augment the dilation of capillaries to restore homogenous capillary flows. This mechanical effect may contribute to the beneficial effects of repeated episodes of arterial occlusion/reperfusion after cerebral ischemia in animal models, so-called ischemic postconditioning.68
Discussion
The extended BKCR model extends the previous concept of an ischemic penumbra defined in relation to specific CBF thresholds to also take into account the effects of CTTH on tissue oxygen availability. In the resulting extended penumbra model, the acute ischemic stroke may be preceded by changes in capillary morphology and in blood viscosity. These changes cause CTTH to increase, and the coupling between CBF and the metabolic needs of the tissue must therefore adapt to maintain brain function. The metabolic significance of capillary flow disturbances is predicted to require adaptations that are consistent with the common findings of neurovascular dysfunction and of gradual reductions in CVRC observed in most patients with conditions that predispose to stroke. Notably, the extended penumbra model predicts that stroke symptoms are the result of a focal reduction in CMRO2max to levels below the metabolic needs of the tissue, as a result of reductions in CBF (as in the ‘classical' penumbra model), as a result of elevated CTTH levels, or both. The model predicts that the condition(s) that led to the stroke may have reduced CMRO2max considerably by the time it occurs. This property suggests that some strokes may be the result of small hemodynamic perturbations rather than sudden reductions in CBF of 50% or more as implied by the classical penumbra concept. Lastly, the model predicts that in addition to the restoration of CBF, the restoration of capillary flow patterns is a key aspect in restoring the tissue oxygen availability to levels above the metabolic needs of the tissue. In theory, capillary flow patterns may be improved independent of clot lysis, and this property of the model may therefore prove relevant for the management of all stroke patients, including the prehospitalization phase and in those patients who do not fulfill criteria for thrombolytic therapy. See also the recent review by Dalkara et al.19
From Capillary Dysfunction to Neurovascular Dysfunction—How?
A key aspect of the extended penumbra model is the prediction that neurovascular dysfunction can be viewed as an adaptation to downstream elevations in CTTH. Neurovascular dysfunction has been most thoroughly studied in animal models of Angiotensin II-mediated hypertension.69 The suppression of CBF responses elicited by Angiotensin II is thought to be mediated by increased levels of ROS and the depletion of NO levels in the vessel walls.70, 71 Notably, the neurovascular dysfunction even precedes the development of high blood pressure.72
Angiotensin II constricts a large proportion of retinal capillary pericytes in vitro via calcium channel-dependent mechanisms.73, 74 Angiotensin II infusion is therefore likely to disturb capillary flow patterns, and thus to increase CTTH, in vivo. The way in which such capillary dysfunction could elicit the observed ROS production at the level of the arteriolar endothelium is less clear. Capillary dysfunction would be expected to cause episodes of low tissue oxygen tension during upstream vasodilation as a result of the poorer oxygen extraction. Such episodes of relative tissue hypoxia lead to the activation of hypoxia-inducible transcription factors (HIF). HIF-1 has been shown to upregulate the levels of nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX-2),75 which is a major source of ROS in the brain vasculature.76 Capillary dysfunction could therefore elicit upstream ROS production, and hence trigger neurovascular dysfunction, via hypoxia-sensitive mechanisms. This is consistent with the finding that the ROS which are involved in Angiotensin II-related neurovascular dysfunction are indeed derived from NOX-2.70, 71
Neurovascular dysfunction is also thought to lead to more long-term vascular changes. The removal of endothelial NO and the increased production of free radicals cause long-term remodeling, damage, and inflammation localized to the vessel wall.14, 15 In particular, vascular smooth muscle cells in upstream arteries and arterioles degenerate and develop abnormal narrowing of their luminal diameters.14, 15 The resulting vessel wall stiffness and the permanent reductions in vessel diameter would be expected to mechanically attenuate the CBF responses, which normally accompany functional activation and conditions such as hypercapnia and hypoxia. Paradoxically, these changes in vessel wall properties may contribute to the development of upstream vessel disease while at the same time being protective in the sense that they reduce the ROS production which would otherwise result from the tissue hypoxia elicited by hyperemic episodes from the onset of Stage II.
Therapeutic Implications
The extended penumbra model predicts that neurovascular dysfunction is in fact an adaptation, which serves to avoid increases in CBF that would otherwise elicit relative tissue hypoxia. This implies that patients with capillary dysfunction tolerate hyperemia poorly: increased CBF would be predicted to cause episodes of cerebral hypoxia, paralleled by excessive oxidative stress and accelerated vascular damage as part of the intrinsic attempts to block the high blood flow. Obstructive sleep apnoea is associated with periods of severe nocturnal hypercapnia and hypoxemia, both of which cause substantial increases in CBF in the normal brain. Obstructive sleep apnoea patients show reduced CVRC compared with controls, particularly in the morning,77 consistent with the prediction that neurovascular dysfunction is an adaptive response that serves to attenuate nocturnal cerebral hyperemia. The potential therapeutic implications of this principle are underscored by the finding that the reduced CVRC can be reversed to the levels of normal controls by treatment with continuous positive airway pressure,77 and that the increased incidence of both fatal and nonfatal strokes in obstructive sleep apnoea can be reduced by continuous positive airway pressure treatment.78
The ROS production and the NO depletion observed in neurovascular dysfunction may be beneficial in that these factors reduce the diameter of arteries and arterioles. Meanwhile, ROS also constricts capillary pericytes,18 as does NO depletion,79, 80 Such ‘downstream' effects would be expected to increase CTTH further, unless NO levels at the capillary level can be maintained. As tissue oxygen tension decreases, the risk of capillary NO depletion may become even higher, as oxygen is a crucial substrate for NO production via NO synthases.81 NO depletion at the capillary level could therefore lead to a vicious cycle by causing further tissue hypoxia, further attenuation of upstream vessel tone, and so forth. This vicious cycle is counteracted by the release of NO from erythrocytes as they unload oxygen in oxygen-deprived tissue.82 Also, the upregulation of HIF-1 in hypoxic tissue activates nuclear factor κB83 and thereby the production of inducible nitric oxide synthase and large amounts of NO in the tissue.10 The extent to which this mechanism exerts a protective effect on capillary tone and tissue oxygenation by preserving capillary level NO is unclear. Capillary NO levels may be modified in ways that do not require oxygen as a substrate, namely by administration of nitrite, which are major sources of NO in mammals.84. Indeed, early administration of nitrite has been shown to reduce the production of ROS and increase the tissue survival in ischemia–reperfusion.85 Increased NO levels have also been implicated in the protective effects of ischemic preconditioning; the application of sublethal ischemia to mitigate increased tolerance to subsequent ischemic episodes.86, 87
Diagnostic Implications
The detection of emboli sources in the heart and aorta, and the demonstration of stenoses or vulnerable plaques in carotid and intracranial vessels is a crucial part of the management of stroke risk factors, transitory ischemic attack, and acute stroke. The extended penumbra model suggests that in addition to heart disease, large vessel disease, and SVD, the extent of capillary dysfunction may also be of importance to the risk of developing an acute stroke. In principle, the severity of capillary dysfunction can be determined by assessing CVRC in relation to the routine management of most of the conditions listed in Table 1. Given the model's prediction that elevated OEF and reduced CBF responses to vasodilators are features of both capillary dysfunction and severe vascular stenosis or occlusions, diagnostic means of directly showing capillary dysfunction could be crucial. In particular, the quantification of concomitant capillary dysfunction in patients with symptomatic carotid stenosis could help predict the relative benefits of revascularization and aggressive management of cardiovascular risk factors,88 of which the latter is more likely to affect capillary dysfunction.
The demonstration of a thrombus in an intracranial vessel would be expected to rule out the possibility that a patient developed acute stroke in relation to exacerbation of severe capillary dysfunction. While patients with a confirmed vascular occlusion may have significant capillary dysfunction and thus tolerate residual elevations in CTTH after recanalization poorly, this radiologic finding would be expected to increase the chances that the patient belongs to a category of patients in which the lysis or removal of a blood clot would result in sizeable elevation of CMRO2max. This is consistent with reports of better neurologic outcomes in patients with acute radiologic signs of vessel occlusion and successful thrombolytic therapy.89, 90 Meanwhile, patients with capillary dysfunction are predicted to have been exposed to low brain tissue oxygen tension for an extended period of time. This could activate protective pathways that improve their tolerance to periods of ischemia in later life, much as in experimental preconditioning.87
The considerations above suggest that it may be of both diagnostic and prognostic value to quantify CTTH and MTT in patients suffering from conditions that predispose to stroke, and in both unaffected and hypoperfused tissue in stroke patients. CTTH and MTT can be measured by monitoring the clearance of intravascular contrast agents as part of standard perfusion-weighted magnetic resonance imaging, perfusion computed tomography, or contrast-enhanced transcranial ultrasound examinations.91, 92 Current perfusion algorithms quantify the severity of tissue hypoperfusion in terms of metrics that attempt to capture various aspects of delayed contrast passage through affected brain regions. Theoretically, the shape of the tissue bolus passage curve depends on both MTT and CTTH, and the derived metrics are therefore likely to reflect their values in one way or another. Accordingly, all of these metrics are capable of predicting the risk of subsequent infarction in acute stroke patients with some degree of certainty,93, 94, 95, 96 although most lack a direct physiologic interpretation.97 Figure 6 illustrates how the so-called Tmax parameter96 is biased by the value of CTTH and OEFmax.
Figure 6.

Digital phantom simulation of the inherent bias of the Tmax parameter by the values of mean transit time (MTT) and capillary transit time heterogeneity (CTTH). (A) Forty-nine squares with ‘true' maximum oxygen extraction fraction (OEFmax) values corresponding to combinations of seven MTT values and seven CTTH values, cf. Figure 4A, are shown. Each square consists of 14-by-14 voxels, with associated concentration time curves characteristic of the tissue bolus passage which would result from the vascular retention caused by the corresponding values of MTT and CTTH. Random noise characteristic of magnetic resonance perfusion raw data was then added at each noise free, simulated data point, and the tissue curve analyzed by a customized algorithm, which determine OEFmax based on CTTH and MTT.92 (B) This panel shows OEFmax values estimated by this algorithm. They are seen to correspond well with the true values in panel A. We also determined the corresponding Tmax value using two common singular value decomposition based deconvolution techniques.116, 117 (C, D) Maps of Tmax derived from the delay-corrected (C) and delay un-corrected (D) singular value decomposition algorithms. Note that the Tmax maps contain an incidental bias that mimics OEFmax.
Limitations to the Extended BKCR Model
When first modeling the effects of CTTH on tissue oxygen availability, we chose the widely accepted gamma variate PDF to model the distribution of capillary transit times.26 This choice permits us to represent CTTH by a single parameter, namely the standard deviation of capillary transit times, and thereby to provide a first, intuitive understanding of the effects of capillary flow patterns on tissue oxygen availability by means of two- and three-dimensional plots such as those shown in Figures 4 and 5. Needless to say, the original BKCR equation can be generalized to take into account the heterogeneity of capillary transit times by using any number of PDFs to represent their distribution. For most PDF choices, however, this would involve the inclusion of additional parameters (in addition to the standard deviation of capillary transit times) to describe CTTH and thereby render the resulting model more difficult to interpret and visualize. In subjects or animal models with age- and risk factor-related changes in capillary morphology and topology, the distribution of capillary transit times is likely to be extremely complex and therefore difficult to model with analytical PDFs. In future studies of the metabolic effect of CTTH, models based on actual, measured transit time distributions may therefore prove more informative. In the analysis of the metabolic effects of CTTH presented here, our conclusions depend little on the actual distribution of transit times in tissue. Rather, our analysis depends on the finding that increased CTTH tends to reduce tissue oxygen availability and that this phenomenon can lead to conditions in which increases in CBF fail to improve tissue oxygen availability—dubbed malignant CTTH. We have previously shown that both the dependency of tissue oxygen availability on CTTH and the malignant CTTH phenomenon are consequences of the well-accepted BKCR model, rather than of the PDF chosen for modeling purposes, cf. Figure 2 in the paper by Jespersen and Østergaard.26 We therefore believe that the analysis presented here can serve as a general rationale for considering capillary flow patterns as an integral part of stroke pathogenesis, stroke diagnosis, and stroke management.
Concluding remarks
The analysis presented here suggests that conditions that predispose to stroke can lead to neurovascular dysfunction, vascular oxidative stress, and relative tissue hypoxia well in advance of any cerebrovascular accidents. In a recent publication, we argued that such changes are also highly conducive to neurodegenerative changes and memory impairment.98 Changes in capillary morphology and neurovascular function may in fact represent important common denominators for conditions that increase both the risk of developing acute stroke and the risk of developing dementia.69 The overlap in the etiopathogenesis of these clinically diverse conditions is emphasized by the finding that cognitive decline is a predictor of acute stroke99 and that the rate of decline in memory function is related to both the risk of stroke and the risk of poor stroke outcome.100
The presence of age- or risk factor-related changes in capillary morphology and of neurovascular dysfunction constitutes key differences between human stroke and animal models of ischemia and reperfusion injury. This difference could affect the extent to which neuroprotective strategies developed in animal models with normal regulation of CTTH show similar effects when applied in human stroke.
The authors declare no conflict of interest.
Footnotes
This study was supported by the Danish National Research Foundation (CFIN; LØ, SNJ, KM, RA, NH, KDH, CZS, KN, and LRR), the Danish Ministry of Science, Technology and Innovation's University Investment Grant (MINDLab; LØ, SNJ, KM, IKM, KYJ, AT, JUB, RA, NKI, CC, PvW-M, LRR, MBH, SLB, and MGD), the European Union's 6th Framework Program (I-Know; LØ, IKM, NH, KN, LRR, JP, SP, JS, T-HC, SS, JF, and NN) and the Tryg Foundation (KDH and GA). This work was presented in part at Brain 2011, the XXVth International Symposium on Cerebral Blood Flow, Metabolism, and Function, Barcelona, Spain, 25–28 May 2011.
References
- Moustafa RS, Baron JC.Perfusion Thresholds in Cerebral IschemiaIn: Donnan GA, Baron JC, Davis SM, Sharp FR, (eds).. The Ischemic Penumbra Informa Healthcare USA, Inc.: New York, USA; 200731–36. [Google Scholar]
- Astrup J, Symon L, Branston NM, Lassen NA. Cortical evoked potential and extracellular K+ and H+ at critical levels of brain ischemia. Stroke. 1977;8:51–57. doi: 10.1161/01.str.8.1.51. [DOI] [PubMed] [Google Scholar]
- Astrup J, Siesjo BK, Symon L. Thresholds in cerebral ischemia—the ischemic penumbra. Stroke. 1981;12:723–725. doi: 10.1161/01.str.12.6.723. [DOI] [PubMed] [Google Scholar]
- Donnan GA, Baron JC, Davis SM, Sharp FR.The ischemic penumbra: overview, definition, and criteriaIn: Donnan GA, Baron JC, Davis SM, Sharp FR, (eds).. The Ischemic Penumbra Informa Healthcare USA, Inc.: New York; 20077–20. [Google Scholar]
- Albers GW, Thijs VN, Wechsler L, Kemp S, Schlaug G, Skalabrin E, et al. Magnetic resonance imaging profiles predict clinical response to early reperfusion: the diffusion and perfusion imaging evaluation for understanding stroke evolution (DEFUSE) study. Ann Neurol. 2006;60:508–517. doi: 10.1002/ana.20976. [DOI] [PubMed] [Google Scholar]
- Davis SM, Donnan GA, Parsons MW, Levi C, Butcher KS, Peeters A, et al. Effects of alteplase beyond 3 hour after stroke in the Echoplanar Imaging Thrombolytic Evaluation Trial (EPITHET): a placebo-controlled randomised trial. Lancet Neurol. 2008;7:299–309. doi: 10.1016/S1474-4422(08)70044-9. [DOI] [PubMed] [Google Scholar]
- Nagakane Y, Christensen S, Brekenfeld C, Ma H, Churilov L, Parsons MW, et al. EPITHET: positive result after reanalysis using baseline diffusion-weighted imaging/perfusion-weighted imaging co-registration. Stroke. 2011;42:59–64. doi: 10.1161/STROKEAHA.110.580464. [DOI] [PubMed] [Google Scholar]
- Mishra NK, Albers GW, Davis SM, Donnan GA, Furlan AJ, Hacke W, et al. Mismatch-based delayed thrombolysis: a meta-analysis. Stroke. 2010;41:e25–e33. doi: 10.1161/STROKEAHA.109.566869. [DOI] [PubMed] [Google Scholar]
- Amarenco P, Bogousslavsky J, Caplan LR, Donnan GA, Hennerici MG. New approach to stroke subtyping: the A-S-C-O (phenotypic) classification of stroke. Cerebrovasc Dis. 2009;27:502–508. doi: 10.1159/000210433. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Davisson RL. Hypertension and cerebrovascular dysfunction. Cell Metab. 2008;7:476–484. doi: 10.1016/j.cmet.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM. Nitric oxide neurotoxicity. J Chem Neuroanat. 1996;10:179–190. doi: 10.1016/0891-0618(96)00148-2. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah K, Qureshi SU, Johnson M, Parikh N, Schulz PE, Kunik ME. Does use of antihypertensive drugs affect the incidence or progression of dementia? A systematic review. Am J Geriatr Pharmacother. 2009;7:250–261. doi: 10.1016/j.amjopharm.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Schulz E, Jansen T, Wenzel P, Daiber A, Munzel T. Nitric oxide, tetrahydrobiopterin, oxidative stress, and endothelial dysfunction in hypertension. Antioxid Redox Signal. 2008;10:1115–1126. doi: 10.1089/ars.2007.1989. [DOI] [PubMed] [Google Scholar]
- Jackson SP. Arterial thrombosis-insidious, unpredictable and deadly. Nat Med. 2011;17:1423–1436. doi: 10.1038/nm.2515. [DOI] [PubMed] [Google Scholar]
- Gursoy-Ozdemir Y, Yemisci M, Dalkara T. Microvascular protection is essential for successful neuroprotection in stroke. J Neurochem. 2012;123 (Suppl 2:2–11. doi: 10.1111/j.1471-4159.2012.07938.x. [DOI] [PubMed] [Google Scholar]
- Yemisci M, Gursoy-Ozdemir Y, Vural A, Can A, Topalkara K, Dalkara T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med. 2009;15:1031–1037. doi: 10.1038/nm.2022. [DOI] [PubMed] [Google Scholar]
- Dalkara T, Arsava EM. Can restoring incomplete microcirculatory reperfusion improve stroke outcome after thrombolysis. J Cereb Blood Flow Metab. 2012;32:2091–2099. doi: 10.1038/jcbfm.2012.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuschinsky W, Paulson OB. Capillary circulation in the brain. Cerebrovasc Brain Metab Rev. 1992;4:261–286. [PubMed] [Google Scholar]
- Tomita Y, Tomita M, Schiszler I, Amano T, Tanahashi N, Kobari M, et al. Moment analysis of microflow histogram in focal ischemic lesion to evaluate microvascular derangement after small pial arterial occlusion in rats. J Cereb Blood Flow Metab. 2002;22:663–669. doi: 10.1097/00004647-200206000-00004. [DOI] [PubMed] [Google Scholar]
- Hudetz AG, Feher G, Weigle CG, Knuese DE, Kampine JP. Video microscopy of cerebrocortical capillary flow: response to hypotension and intracranial hypertension. Am J Physiol. 1995;268:H2202–H2210. doi: 10.1152/ajpheart.1995.268.6.H2202. [DOI] [PubMed] [Google Scholar]
- Østergaard L, Sorensen AG, Chesler DA, Weisskoff RM, Koroshetz WJ, Wu O, et al. Combined diffusion-weighted and perfusion-weighted flow heterogeneity magnetic resonance imaging in acute stroke. Stroke. 2000;31:1097–1103. doi: 10.1161/01.str.31.5.1097. [DOI] [PubMed] [Google Scholar]
- Simonsen CZ, Rohl L, Vestergaard-Poulsen P, Gyldensted C, Andersen G, Østergaard L. Final infarct size after acute stroke: prediction with flow heterogeneity. Radiology. 2002;225:269–275. doi: 10.1148/radiol.2251011249. [DOI] [PubMed] [Google Scholar]
- Perkio J, Soinne L, Østergaard L, Helenius J, Kangasmaki A, Martinkauppi S, et al. Abnormal intravoxel cerebral blood flow heterogeneity in human ischemic stroke determined by dynamic susceptibility contrast magnetic resonance imaging. Stroke. 2005;36:44–49. doi: 10.1161/01.STR.0000150495.96471.95. [DOI] [PubMed] [Google Scholar]
- Jespersen SN, Østergaard L. The roles of cerebral blood flow, capillary transit time heterogeneity and oxygen tension in brain oxygenation and metabolism. J Cereb Blood Flow Metab. 2012;32:264–277. doi: 10.1038/jcbfm.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renkin EM. B. W. Zweifach Award lecture. Regulation of the microcirculation. Microvasc Res. 1985;30:251–263. doi: 10.1016/0026-2862(85)90057-3. [DOI] [PubMed] [Google Scholar]
- Pawlik G, Rackl A, Bing RJ. Quantitative capillary topography and blood flow in the cerebral cortex of cats: an in vivo microscopic study. Brain Res. 1981;208:35–58. doi: 10.1016/0006-8993(81)90619-3. [DOI] [PubMed] [Google Scholar]
- Villringer A, Them A, Lindauer U, Einhaupl K, Dirnagl U. Capillary perfusion of the rat brain cortex. An in vivo confocal microscopy study. Circ Res. 1994;75:55–62. doi: 10.1161/01.res.75.1.55. [DOI] [PubMed] [Google Scholar]
- Kleinfeld D, Mitra PP, Helmchen F, Denk W. Fluctuations and stimulus-induced changes in blood flow observed in individual capillaries in layers 2 through 4 of rat neocortex. Proc Natl Acad Sci USA. 1998;95:15741–15746. doi: 10.1073/pnas.95.26.15741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni MC, Schmid-Schonbein GW. Mechanisms and consequences of cell activation in the microcirculation. Cardiovasc Res. 1996;32:709–719. [PubMed] [Google Scholar]
- Lipowsky HH, Gao L, Lescanic A. Shedding of the endothelial glycocalyx in arterioles, capillaries and venules and its effect on capillary hemodynamics during inflammation. Am J Physiol Heart Circ Physiol. 2011;30:H2235–H2245. doi: 10.1152/ajpheart.00803.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RB, Raymond GM, Bassingthwaighte JB. Modeling blood flow heterogeneity. Ann Biomed Eng. 1996;24:352–372. doi: 10.1007/BF02660885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GN. Researches on the circulation time in organs and on the influences which affect it. Parts I-III. J Physiol. 1894;15:1–89. doi: 10.1113/jphysiol.1893.sp000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Watabe H, Kudomi N, Kim KM, Enmi J, Hayashida K, et al. A theoretical model of oxygen delivery and metabolism for physiologic interpretation of quantitative cerebral blood flow and metabolic rate of oxygen. J Cereb Blood Flow Metab. 2003;23:1314–1323. doi: 10.1097/01.WCB.0000090506.76664.00. [DOI] [PubMed] [Google Scholar]
- Mintun MA, Lundstrom BN, Snyder AZ, Vlassenko AG, Shulman GL, Raichle ME. Blood flow and oxygen delivery to human brain during functional activity: theoretical modeling and experimental data. Proc Natl Acad Sci USA. 2001;98:6859–6864. doi: 10.1073/pnas.111164398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crone C. The permeability of capillaries in various organs as determined by use of the ‘Indicator Diffusion' Method. Acta Physiol Scand. 1963;58:292–305. doi: 10.1111/j.1748-1716.1963.tb02652.x. [DOI] [PubMed] [Google Scholar]
- Arfken GB, Weber HJ. Mathematical Methods for Physicists, Sixth Edition: A Comprehensive Guide. Elsevier Academic Press: Boston, USA; 2005. p. 1182. [Google Scholar]
- Stefanovic B, Hutchinson E, Yakovleva V, Schram V, Russell JT, Belluscio L, et al. Functional reactivity of cerebral capillaries. J Cereb Blood Flow Metab. 2008;28:961–972. doi: 10.1038/sj.jcbfm.9600590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte ML, Wood JD, Hudetz AG. Cortical electrical stimulation alters erythrocyte perfusion pattern in the cerebral capillary network of the rat. Brain Res. 2003;963:81–92. doi: 10.1016/s0006-8993(02)03848-9. [DOI] [PubMed] [Google Scholar]
- Hudetz AG, Biswal BB, Feher G, Kampine JP. Effects of hypoxia and hypercapnia on capillary flow velocity in the rat cerebral cortex. Microvasc Res. 1997;54:35–42. doi: 10.1006/mvre.1997.2023. [DOI] [PubMed] [Google Scholar]
- Vorstrup S, Henriksen L, Paulson OB. Effect of acetazolamide on cerebral blood flow and cerebral metabolic rate for oxygen. J Clin Invest. 1984;74:1634–1639. doi: 10.1172/JCI111579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers WJ. Cerebral hemodynamics in ischemic cerebrovascular disease. Ann Neurol. 1991;29:231–240. doi: 10.1002/ana.410290302. [DOI] [PubMed] [Google Scholar]
- Maeda H, Matsumoto M, Handa N, Hougaku H, Ogawa S, Itoh T, et al. Reactivity of cerebral blood flow to carbon dioxide in hypertensive patients: evaluation by the transcranial Doppler method. J Hypertens. 1994;12:191–197. [PubMed] [Google Scholar]
- Lavi S, Gaitini D, Milloul V, Jacob G. Impaired cerebral CO2 vasoreactivity: association with endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2006;291:H1856–H1861. doi: 10.1152/ajpheart.00014.2006. [DOI] [PubMed] [Google Scholar]
- Sette G, Baron JC, Mazoyer B, Levasseur M, Pappata S, Crouzel C. Local brain haemodynamics and oxygen metabolism in cerebrovascular disease. Positron emission tomography. Brain. 1989;112 (Pt 4:931–951. doi: 10.1093/brain/112.4.931. [DOI] [PubMed] [Google Scholar]
- Smith EE, Schneider JA, Wardlaw JM, Greenberg SM. Cerebral microinfarcts: the invisible lesions. Lancet Neurol. 2012;11:272–282. doi: 10.1016/S1474-4422(11)70307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WS, Lev MH, English JD, Camargo EC, Chou M, Johnston SC, et al. Significance of large vessel intracranial occlusion causing acute ischemic stroke and TIA. Stroke. 2009;40:3834–3840. doi: 10.1161/STROKEAHA.109.561787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purroy F, Begue R, Quilez A, Pinol-Ripoll G, Sanahuja J, Brieva L, et al. The California, ABCD, and unified ABCD2 risk scores and the presence of acute ischemic lesions on diffusion-weighted imaging in TIA patients. Stroke. 2009;40:2229–2232. doi: 10.1161/STROKEAHA.108.537969. [DOI] [PubMed] [Google Scholar]
- Elkind MS. Why now? Moving from stroke risk factors to stroke triggers. Curr Opin Neurol. 2007;20:51–57. doi: 10.1097/WCO.0b013e328012da75. [DOI] [PubMed] [Google Scholar]
- Macko RF, Ameriso SF, Barndt R, Clough W, Weiner JM, Fisher M. Precipitants of brain infarction. Roles of preceding infection/inflammation and recent psychological stress. Stroke. 1996;27:1999–2004. doi: 10.1161/01.str.27.11.1999. [DOI] [PubMed] [Google Scholar]
- Grau AJ, Buggle F, Becher H, Zimmermann E, Spiel M, Fent T, et al. Recent bacterial and viral infection is a risk factor for cerebrovascular ischemia: clinical and biochemical studies. Neurology. 1998;50:196–203. doi: 10.1212/wnl.50.1.196. [DOI] [PubMed] [Google Scholar]
- Zurru MC, Alonzo C, Brescacin L, Romano M, Camera LA, Waisman G, et al. Recent respiratory infection predicts atherothrombotic stroke: case-control study in a Buenos Aires healthcare system. Stroke. 2009;40:1986–1990. doi: 10.1161/STROKEAHA.108.535559. [DOI] [PubMed] [Google Scholar]
- Sheth T, Nair C, Muller J, Yusuf S. Increased winter mortality from acute myocardial infarction and stroke: the effect of age. J Am Coll Cardiol. 1999;33:1916–1919. doi: 10.1016/s0735-1097(99)00137-0. [DOI] [PubMed] [Google Scholar]
- Woodhouse PR, Khaw KT, Plummer M, Foley A, Meade TW. Seasonal variations of plasma fibrinogen and factor VII activity in the elderly: winter infections and death from cardiovascular disease. Lancet. 1994;343:435–439. doi: 10.1016/s0140-6736(94)92689-1. [DOI] [PubMed] [Google Scholar]
- Lavallee P, Perchaud V, Gautier-Bertrand M, Grabli D, Amarenco P. Association between influenza vaccination and reduced risk of brain infarction. Stroke. 2002;33:513–518. doi: 10.1161/hs0202.102328. [DOI] [PubMed] [Google Scholar]
- Nichol KL, Nordin J, Mullooly J, Lask R, Fillbrandt K, Iwane M. Influenza vaccination and reduction in hospitalizations for cardiac disease and stroke among the elderly. N Engl J Med. 2003;348:1322–1332. doi: 10.1056/NEJMoa025028. [DOI] [PubMed] [Google Scholar]
- Grau AJ, Fischer B, Barth C, Ling P, Lichy C, Buggle F. Influenza vaccination is associated with a reduced risk of stroke. Stroke. 2005;36:1501–1506. doi: 10.1161/01.STR.0000170674.45136.80. [DOI] [PubMed] [Google Scholar]
- Elkind MS. Inflammatory mechanisms of stroke. Stroke. 2010;41:S3–S8. doi: 10.1161/STROKEAHA.110.594945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppiatt CM, Howarth C, Mobbs P, Attwell D. Bidirectional control of CNS capillary diameter by pericytes. Nature. 2006;443:700–704. doi: 10.1038/nature05193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon I, Kim EH, del Zoppo GJ, Heo JH. Ultrastructural and temporal changes of the microvascular basement membrane and astrocyte interface following focal cerebral ischemia. J Neurosci Res. 2009;87:668–676. doi: 10.1002/jnr.21877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowat A, Graham C, Dennis M. Dehydration in hospital-admitted stroke patients: detection, frequency, and association. Stroke. 2012;43:857–859. doi: 10.1161/STROKEAHA.111.640821. [DOI] [PubMed] [Google Scholar]
- Yang GY, Betz AL. Reperfusion-induced injury to the blood-brain barrier after middle cerebral artery occlusion in rats. Stroke. 1994;25:1658–1664. doi: 10.1161/01.str.25.8.1658. [DOI] [PubMed] [Google Scholar]
- Aronowski J, Strong R, Grotta JC. Reperfusion injury: demonstration of brain damage produced by reperfusion after transient focal ischemia in rats. J Cereb Blood Flow Metab. 1997;17:1048–1056. doi: 10.1097/00004647-199710000-00006. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Lassen NA. The luxury-perfusion syndrome and its possible relation to acute metabolic acidosis localised within the brain. Lancet. 1966;2:1113–1115. doi: 10.1016/s0140-6736(66)92199-4. [DOI] [PubMed] [Google Scholar]
- Kidwell CS, Saver JL, Mattiello J, Starkman S, Vinuela F, Duckwiler G, et al. Diffusion-perfusion MRI characterization of post-recanalization hyperperfusion in humans. Neurology. 2001;57:2015–2021. doi: 10.1212/wnl.57.11.2015. [DOI] [PubMed] [Google Scholar]
- Zhao H. Ischemic postconditioning as a novel avenue to protect against brain injury after stroke. J Cereb Blood Flow Metab. 2009;29:873–885. doi: 10.1038/jcbfm.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol. 2006;100:328–335. doi: 10.1152/japplphysiol.00966.2005. [DOI] [PubMed] [Google Scholar]
- Kazama K, Anrather J, Zhou P, Girouard H, Frys K, Milner TA, et al. Angiotensin II impairs neurovascular coupling in neocortex through NADPH oxidase-derived radicals. Circ Res. 2004;95:1019–1026. doi: 10.1161/01.RES.0000148637.85595.c5. [DOI] [PubMed] [Google Scholar]
- Girouard H, Park L, Anrather J, Zhou P, Iadecola C. Cerebrovascular nitrosative stress mediates neurovascular and endothelial dysfunction induced by angiotensin II. Arterioscler Thromb Vasc Biol. 2007;27:303–309. doi: 10.1161/01.ATV.0000253885.41509.25. [DOI] [PubMed] [Google Scholar]
- Capone C, Faraco G, Park L, Cao X, Davisson RL, Iadecola C. The cerebrovascular dysfunction induced by slow pressor doses of angiotensin II precedes the development of hypertension. Am J Physiol Heart Circ Physiol. 2011;300:H397–H407. doi: 10.1152/ajpheart.00679.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsugi T, Chen Q, Anderson DR. Contractile responses of cultured bovine retinal pericytes to angiotensin II. Arch Ophthalmol. 1997;115:1281–1285. doi: 10.1001/archopht.1997.01100160451011. [DOI] [PubMed] [Google Scholar]
- Kawamura H, Kobayashi M, Li Q, Yamanishi S, Katsumura K, Minami M, et al. Effects of angiotensin II on the pericyte-containing microvasculature of the rat retina. J Physiol. 2004;561:671–683. doi: 10.1113/jphysiol.2004.073098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan G, Khan SA, Luo W, Nanduri J, Semenza GL, Prabhakar NR. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J Cell Physiol. 2011;226:2925–2933. doi: 10.1002/jcp.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorce S, Krause KH. NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal. 2009;11:2481–2504. doi: 10.1089/ars.2009.2578. [DOI] [PubMed] [Google Scholar]
- Diomedi M, Placidi F, Cupini LM, Bernardi G, Silvestrini M. Cerebral hemodynamic changes in sleep apnea syndrome and effect of continuous positive airway pressure treatment. Neurology. 1998;51:1051–1056. doi: 10.1212/wnl.51.4.1051. [DOI] [PubMed] [Google Scholar]
- Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet. 2005;365:1046–1053. doi: 10.1016/S0140-6736(05)71141-7. [DOI] [PubMed] [Google Scholar]
- Haefliger IO, Zschauer A, Anderson DR. Relaxation of retinal pericyte contractile tone through the nitric oxide-cyclic guanosine monophosphate pathway. Invest Ophthalmol Vis Sci. 1994;35:991–997. [PubMed] [Google Scholar]
- Haefliger IO, Anderson DR. Oxygen modulation of guanylate cyclase-mediated retinal pericyte relaxations with 3-morpholino-sydnonimine and atrial natriuretic peptide. Invest Ophthalmol Vis Sci. 1997;38:1563–1568. [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen FB. The dual roles of red blood cells in tissue oxygen delivery: oxygen carriers and regulators of local blood flow. J Exp Biol. 2009;212:3387–3393. doi: 10.1242/jeb.023697. [DOI] [PubMed] [Google Scholar]
- Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364:656–665. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Faassen EE, Bahrami S, Feelisch M, Hogg N, Kelm M, Kim-Shapiro DB, et al. Nitrite as regulator of hypoxic signaling in mammalian physiology. Med Res Rev. 2009;29:683–741. doi: 10.1002/med.20151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung KH, Chu K, Ko SY, Lee ST, Sinn DI, Park DK, et al. Early intravenous infusion of sodium nitrite protects brain against in vivo ischemia-reperfusion injury. Stroke. 2006;37:2744–2750. doi: 10.1161/01.STR.0000245116.40163.1c. [DOI] [PubMed] [Google Scholar]
- Terpolilli NA, Moskowitz MA, Plesnila N. Nitric oxide: considerations for the treatment of ischemic stroke. J Cereb Blood Flow Metab. 2012;32:1332–1346. doi: 10.1038/jcbfm.2012.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8:398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers WJ, Clarke WR, Grubb RL, Videen TO, Adams HP, Derdeyn CP, et al. Extracranial-intracranial bypass surgery for stroke prevention in hemodynamic cerebral ischemia: the Carotid Occlusion Surgery Study randomized trial. JAMA. 2011;306:1983–1992. doi: 10.1001/jama.2011.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiebach JB, Al-Rawi Y, Wintermark M, Furlan AJ, Rowley HA, Lindsten A, et al. Vascular occlusion enables selecting acute ischemic stroke patients for treatment with desmoteplase. Stroke. 2012;43:1561–1566. doi: 10.1161/STROKEAHA.111.642322. [DOI] [PubMed] [Google Scholar]
- De Silva DA, Churilov L, Olivot JM, Christensen S, Lansberg MG, Mlynash M, et al. Greater effect of stroke thrombolysis in the presence of arterial obstruction. Ann Neurol. 2011;70:601–605. doi: 10.1002/ana.22444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouridsen K, Friston K, Hjort N, Gyldensted L, Østergaard L, Kiebel S. Bayesian estimation of cerebral perfusion using a physiological model of microvasculature. Neuroimage. 2006;33:570–579. doi: 10.1016/j.neuroimage.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Mouridsen K, Østergaard L, Christensen S, Jespersen SN. Proceedings of the International Society for Magnetic Resonance in Medicines 19th Annual Meeting and Exhibition. Montréal: Canada; 2011. Reliable estimation of capillary transit time distributions at voxel-level using DSC-MRI; p. 3915. [Google Scholar]
- Butcher KS, Parsons M, MacGregor L, Barber PA, Chalk J, Bladin C, et al. Refining the perfusion-diffusion mismatch hypothesis. Stroke. 2005;36:1153–1159. doi: 10.1161/01.str.0000166181.86928.8b. [DOI] [PubMed] [Google Scholar]
- Christensen S, Mouridsen K, Wu O, Hjort N, Karstoft H, Thomalla G, et al. Comparison of 10 perfusion MRI parameters in 97 sub-6-hour stroke patients using voxel-based receiver operating characteristics analysis. Stroke. 2009;40:2055–2061. doi: 10.1161/STROKEAHA.108.546069. [DOI] [PubMed] [Google Scholar]
- Takasawa M, Jones PS, Guadagno JV, Christensen S, Fryer TD, Harding S, et al. How reliable is perfusion MR in acute stroke? Validation and determination of the penumbra threshold against quantitative PET. Stroke. 2008;39:870–877. doi: 10.1161/STROKEAHA.107.500090. [DOI] [PubMed] [Google Scholar]
- Olivot JM, Mlynash M, Thijs VN, Kemp S, Lansberg MG, Wechsler L, et al. Optimal Tmax threshold for predicting penumbral tissue in acute stroke. Stroke. 2009;40:469–475. doi: 10.1161/STROKEAHA.108.526954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calamante F, Christensen S, Desmond PM, Østergaard L, Davis SM, Connelly A. The physiological significance of the time-to-maximum (Tmax) parameter in perfusion MRI. Stroke. 2010;41:1169–1174. doi: 10.1161/STROKEAHA.110.580670. [DOI] [PubMed] [Google Scholar]
- Østergaard L, Aamand R, Gutierrez-Jimenez E, Ho Y-L, Blicher JU, Madsen SM, et al. The capillary dysfunction hypothesis of Alzheimer's disease. Neurobiol Aging. 2013;34:1018–1031. doi: 10.1016/j.neurobiolaging.2012.09.011. [DOI] [PubMed] [Google Scholar]
- Elkins JS, Knopman DS, Yaffe K, Johnston SC. Cognitive function predicts first-time stroke and heart disease. Neurology. 2005;64:1750–1755. doi: 10.1212/01.WNL.0000161850.01792.77. [DOI] [PubMed] [Google Scholar]
- Wang Q, Capistrant BD, Ehntholt A, Glymour MM. Long-term rate of change in memory functioning before and after stroke onset. Stroke. 2012;43:2561–2566. doi: 10.1161/STROKEAHA.112.661587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell MA, Ball MJ. Morphometric comparison of hippocampal microvasculature in ageing and demented people: diameters and densities. Acta Neuropathol. 1981;53:299–318. doi: 10.1007/BF00690372. [DOI] [PubMed] [Google Scholar]
- Kalaria RN. Cerebral vessels in ageing and Alzheimer's disease. Pharmacol Ther. 1996;72:193–214. doi: 10.1016/s0163-7258(96)00116-7. [DOI] [PubMed] [Google Scholar]
- Tagami M, Nara Y, Kubota A, Fujino H, Yamori Y. Ultrastructural changes in cerebral pericytes and astrocytes of stroke-prone spontaneously hypertensive rats. Stroke. 1990;21:1064–1071. doi: 10.1161/01.str.21.7.1064. [DOI] [PubMed] [Google Scholar]
- Junker U, Jaggi C, Bestetti G, Rossi GL. Basement membrane of hypothalamus and cortex capillaries from normotensive and spontaneously hypertensive rats with streptozotocin-induced diabetes. Acta Neuropathol. 1985;65:202–208. doi: 10.1007/BF00686999. [DOI] [PubMed] [Google Scholar]
- McCuskey PA, McCuskey RS. In vivo and electron microscopic study of the development of cerebral diabetic microangiography. Microcirc Endothelium Lymphatics. 1984;1:221–244. [PubMed] [Google Scholar]
- Johnson PC, Brendel K, Meezan E. Thickened cerebral cortical capillary basement membranes in diabetics. Arch Pathol Lab Med. 1982;106:214–217. [PubMed] [Google Scholar]
- Reske-Nielsen E, Lundbæk K, Rafaelsen OJ. Pathological changes in the central and peripheral nervous system of young long-term diabetics. I. Diabetic encephalopathy. Diabetologia. 1965;1:233–241. doi: 10.1007/BF01257917. [DOI] [PubMed] [Google Scholar]
- Mayhan WG, Sharpe GM. Nicotine impairs histamine-induced increases in macromolecular efflux: role of oxygen radicals. J Appl Physiol. 1998;84:1589–1595. doi: 10.1152/jappl.1998.84.5.1589. [DOI] [PubMed] [Google Scholar]
- Karwacka H. Ultrastructural and biochemical studies of the brain and other organs in rats after chronic ethanol administration. I. Electronmicroscopic investigations of the morphologic elements of the blood-brain barrier in the rat after ethanol intoxication. Exp Pathol (Jena) 1980;18:118–126. doi: 10.1016/s0014-4908(80)80061-5. [DOI] [PubMed] [Google Scholar]
- Dalkara T, Gursoy-Ozdemir Y, Yemisci M. Brain microvascular pericytes in health and disease. Acta Neuropathol. 2011;122:1–9. doi: 10.1007/s00401-011-0847-6. [DOI] [PubMed] [Google Scholar]
- Lanza GA, Crea F. Primary coronary microvascular dysfunction: clinical presentation, pathophysiology, and management. Circulation. 2010;121:2317–2325. doi: 10.1161/CIRCULATIONAHA.109.900191. [DOI] [PubMed] [Google Scholar]
- Farkas E, de Vos RA, Donka G, Jansen Steur EN, Mihaly A, Luiten PG. Age-related microvascular degeneration in the human cerebral periventricular white matter. Acta Neuropathol. 2006;111:150–157. doi: 10.1007/s00401-005-0007-y. [DOI] [PubMed] [Google Scholar]
- Fox PT, Raichle ME. Focal physiological uncoupling of cerebral blood flow and oxidative metabolism during somatosensory stimulation in human subjects. Proc Natl Acad Sci USA. 1986;83:1140–1144. doi: 10.1073/pnas.83.4.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raichle ME, Mintun MA. Brain work and brain imaging. Annu Rev Neurosci. 2006;29:449–476. doi: 10.1146/annurev.neuro.29.051605.112819. [DOI] [PubMed] [Google Scholar]
- Ndubuizu O, LaManna JC. Brain tissue oxygen concentration measurements. Antioxid Redox Signal. 2007;9:1207–1219. doi: 10.1089/ars.2007.1634. [DOI] [PubMed] [Google Scholar]
- Wu O, Østergaard L, Weisskoff RM, Benner T, Rosen BR, Sorensen AG. Tracer arrival timing-insensitive technique for estimating flow in MR perfusion-weighted imaging using singular value decomposition with a block-circulant deconvolution matrix. Magn Reson Med. 2003;50:164–174. doi: 10.1002/mrm.10522. [DOI] [PubMed] [Google Scholar]
- Østergaard L, Weisskoff RM, Chesler DA, Gyldensted C, Rosen BR. High resolution measurement of cerebral blood flow using intravascular tracer bolus passages. Part I: Mathematical approach and statistical analysis. Magn Reson Med. 1996;36:715–725. doi: 10.1002/mrm.1910360510. [DOI] [PubMed] [Google Scholar]