

Summary
Master transcription factors Oct4, Sox2 and Nanog bind enhancer elements and recruit Mediator to activate much of the gene expression program of pluripotent embryonic stem cells (ESCs). We report here that the ESC master transcription factors form unusual enhancer domains at most genes that control the pluripotent state. These domains, which we call super-enhancers, consist of clusters of enhancers that are densely occupied by the master regulators and Mediator. Super-enhancers differ from typical enhancers in size, transcription factor density and content, ability to activate transcription, and sensitivity to perturbation. Reduced levels of Oct4 or Mediator cause preferential loss of expression of super-enhancer-associated genes relative to other genes, suggesting how changes in gene expression programs might be accomplished during development. In other more differentiated cells, super-enhancers containing cell type-specific master transcription factors are also found at genes that define cell identity. Super-enhancers thus play key roles in the control of mammalian cell identity.
Introduction
Transcription factors typically regulate gene expression by binding cis-acting regulatory elements known as enhancers and recruiting coactivators and RNA Polymerase II (RNA Pol II) to target genes (Lelli et al., 2012; Ong and Corces, 2011). Enhancers are segments of DNA that are generally a few hundred base pairs in length and are typically occupied by multiple transcription factors (Carey, 1998; Levine and Tjian, 2003; Panne, 2008; Spitz and Furlong, 2012).
Much of the transcriptional control of mammalian development is due to the diverse activity of transcription factor-bound enhancers that control cell type-specific patterns of gene expression (Bulger and Groudine, 2011; Hawrylycz et al., 2012; Maston et al., 2006). Between 400,000 and 1.4 million putative enhancers have been identified in the mammalian genome by using a variety of high-throughput techniques that detect features of enhancers such as specific histone modifications (Bernstein et al., 2012; Thurman et al., 2012). The number of enhancers that are active in any one cell type has been estimated to be in the tens of thousands and enhancer activity is largely cell type-specific (Bernstein et al., 2012; Heintzman et al., 2009; Shen et al., 2012; Visel et al., 2009; Yip et al., 2012).
In embryonic stem cells (ESCs), control of the gene expression program that establishes and maintains ESC state is dependent on a remarkably small number of master transcription factors (Ng and Surani, 2011; Orkin and Hochedlinger, 2011; Young, 2011). These transcription factors, which include Oct4, Sox2 and Nanog, bind to enhancers together with the Mediator coactivator complex (Kagey et al., 2010). The Mediator complex facilitates the ability of enhancer-bound transcription factors to recruit RNA Pol II to the promoters of target genes (Borggrefe and Yue, 2011; Conaway and Conaway, 2011; Kornberg, 2005; Malik and Roeder, 2010) and is essential for maintenance of ESC state and embryonic development (Ito et al., 2000; Kagey et al., 2010; Risley et al., 2010).
ESCs are highly sensitive to reduced levels of Mediator. Indeed, reductions in the levels of many subunits of Mediator cause the same rapid loss of ESC-specific gene expression as loss of Oct4 and other master transcription factors (Kagey et al., 2010). It is unclear why reduced levels of Mediator, a general coactivator, can phenocopy the effects of reduced levels of Oct4 in ESCs.
Interest in further understanding the importance of Mediator in ESCs led us to further investigate enhancers bound by the master transcription factors and Mediator in these cells. We found that much of enhancer-associated Mediator occupies exceptionally large enhancer domains and that these domains are associated with genes that play prominent roles in ESC biology. These large domains, or super-enhancers, were found to contain high levels of the key ESC transcription factors Oct4, Sox2, Nanog, Klf4 and Esrrb, to stimulate higher transcriptional activity than typical enhancers, and to be exceptionally sensitive to reduced levels of Mediator. Super-enhancers were found in a wide variety of differentiated cell types, again associated with key cell type-specific genes known to play prominent roles in control of their gene expression program. These results indicate that super-enhancers drive genes essential for cell identity in many mammalian cell types.
Results
Large genomic domains occupied by master transcription factors and Mediator in ESCs
Previous studies have shown that co-occupancy of ESC genomic sites by the Oct4, Sox2 and Nanog transcription factors is highly predictive of enhancer activity (Chen et al., 2008) and that Mediator is typically associated with these sites (Kagey et al., 2010). We generated high-quality ChIP-Seq datasets for Oct4, Sox2 and Nanog (OSN) in murine ESCs and identified 8,794 sites that are co-occupied by these three transcription factors to annotate enhancers in ESCs (Table S1, Data S1). Inspection of enhancers at several genes that have prominent roles in ESC biology revealed an unusual feature: a large domain containing clusters of constituent enhancers (Figure 1). While the vast majority of enhancers spanned DNA segments of a few hundred base pairs (Figure 1A), some portions of the genome contained clusters of enhancers spanning as much as 50kb (Figure 1B). We found that ESC enhancers can be divided into two classes based on Mediator levels: one class comprised the vast majority of enhancers and the other encompassed 231 large enhancer domains (Figure 1C). Approximately 40% of the Mediator signal associated with enhancers was found in these 231 enhancer domains. The key features of the 231 domains containing high levels of Mediator, which we call super-enhancers, are 1) they span DNA regions whose median length is an order of magnitude larger than the typical enhancer and 2) they have levels of Mediator that are at least an order of magnitude greater than those at the typical enhancer (Figure 1D).
Figure 1. Enhancers and super-enhancers in ESCs.

A,B) ChIP-Seq binding profiles (reads per million per base pair) for ESC transcription factors Oct4, Sox2, and Nanog (OSN), and the Mediator coactivator (Med1) at the Gck and miR-290-295 loci in ESCs. Gene models are depicted below the binding profiles. Enhancer bars and scale bars are depicted above the binding profiles.
C) Distribution of Mediator ChIP-Seq signal (total reads) across the 8,794 ESC enhancers. Mediator occupancy is not evenly distributed across the enhancer regions, with a subset of enhancers (the 231 super-enhancers) containing exceptionally high amounts of Mediator.
D) Metagenes of Mediator ChIP-Seq density (reads per million per base pair) across the 8,563 typical enhancers and the 231 super-enhancers. Metagenes are centered on the enhancer region (703 base pairs for typical enhancers and 8.7kb for super-enhancers), with 3kb surrounding each enhancer region. ChIP-Seq fold difference for enhancer features Mediator, H3K27ac, H3K4me1, and DNaseI hypersensitivity at super-enhancers versus typical enhancers are displayed below the metagenes. Fold difference at enhancers refers to the mean ChIP-Seq signal (total reads) at super-enhancers divided by the mean ChIP-Seq signal at typical enhancers. Fold difference at enhancer constituents refers to the mean ChIP-Seq density (reads per million per base pair) at super-enhancer constituents divided by the mean ChIP-Seq density at typical enhancer constituents. See also Figure S1A and Data S1.
E) Distribution of Mediator, H3K27ac, H3K4me1, DNaseI hypersensitivity, and OSN normalized ChIP-Seq signal across a subset of the 8,794 ESC enhancers. For each enhancer feature, the plot was normalized by dividing the ChIP-Seq signal at each ESC enhancer by the maximum ChIP-Seq signal. The enhancers on the X-axis are ranked for each enhancer feature independently (i.e., the exact enhancer at position 8000 is different for each enhancer feature). The x- and y-axes are adjusted so that the differences in the distribution of each of the enhancer features can be visualized. See also Figure S1B.
F) ChIP-Seq binding profiles for Oct4, Sox2, Nanog, Klf4, and Esrrb at the Gck and miR-290-295 loci in ESCs.
G) Metagenes of Oct4, Sox2, Nanog, Klf4 and Esrrb ChIP-Seq density across the constituent enhancers within the 8,563 typical enhancers and the 231 super-enhancer regions. Each metagene is centered on a constituent enhancer with 2kb surrounding the constituent enhancer region.
H) Box plots of Oct4, Sox2, Nanog, Klf4 and Esrrb ChIP-Seq density at constituent enhancers within the 8,563 typical enhancers and the 231 super-enhancers. P-values (Oct4= 0.012, Nanog= 10−4, Sox2= 0.11, Klf4= 10−34, Esrrb= 10−25) were calculated using a two-tailed t-test.
I) Table depicting transcription factor binding motifs enriched at constituent enhancers within super-enhancer regions relative to genomic background, and associated p-values. CTCF and c-Myc are not enriched.
J) Left panel, Box plot depicting the number of Oct4, Sox2, or Nanog binding motifs at constituent enhancers within typical enhancers and constituent enhancers within super-enhancers. Right panel, Box plot depicting the number of Klf4 or Esrrb binding motifs at constituent enhancers within typical enhancers and constituent enhancers within super-enhancer regions. P-values (Oct4/Sox2/Nanog= 0.36, Klf4/Esrrb= 10−45) were calculated using a two-tailed t-test.
Further characterization of the ESC super-enhancer regions revealed that they contain many features of typical enhancers but at a considerably larger scale (Figure 1D, Figure S1A). Previous studies have shown that nucleosomes with the histone modifications H3K27ac and H3K4me1 are enriched at active enhancers (Creyghton et al., 2010; Rada-Iglesias et al., 2011). Based on ChIP-Seq data, the levels of these histone modifications at the super-enhancers exceed those at the typical enhancers by at least an order of magnitude (Figure 1D). These high levels of histone modifications are due both to the size of the domain and the density of occupancy at constituent enhancers (Figure 1D). Similar results were obtained for DNase I hypersensitivity (Figure 1D, Figure S1A), another feature of enhancers (Dunham et al., 2012). We compared the relative ability of ChIP-Seq data for OSN, Mediator, H3K27ac, H3K4me1, as well as DNaseI hypersensitivity data to distinguish super-enhancers from typical enhancers (Extended Experimental Procedures and Figure S1B). We found that Mediator performed optimally, although each of these enhancer features could be used to some degree to distinguish super-enhancers from typical enhancers (Figure 1E, Figure S1B).
To investigate whether the super-enhancers have features that might further distinguish them from typical enhancers, we examined ChIP-Seq data for 18 different transcription factors, histone modifications, chromatin regulators, as well as DNaseI hypersensitivity (Table S2). The most striking difference was in the occupancy of transcription factors Klf4 and Esrrb (Figure 1F–H). While the levels of Oct4, Sox2 and Nanog were similar in constituent enhancers within typical enhancers and super-enhancers (p-val.= 0.012, 10−4, and 0.11, respectively), the levels of Klf4 and Esrrb showed considerably higher occupancy at the constituent enhancers of super-enhancer domains (p-val.<10−34 and 10−25, respectively)(Figure 1G,H). Thus, super-enhancers are not simply clusters of typical enhancers, but are particularly enriched in Klf4 and Esrrb, which have previously been shown to play important roles in the ESC gene expression program and in reprogramming of somatic cells to induced pluripotent stem (iPS) cells (Feng et al., 2009; Festuccia et al., 2012; Jiang et al., 2008; Martello et al., 2012; Percharde et al., 2012; Takahashi and Yamanaka, 2006).
To gain additional insights into the mechanisms involved in super-enhancer formation, we studied the frequency of known transcription factor binding motifs in these and other regions of the genome. We found that constituent enhancers within super-enhancer regions were significantly enriched for sequence motifs bound by Oct4, Sox2, Nanog, Klf4 and Esrrb, but not for motifs bound by other transcription factors expressed in ESCs such as CTCF and c-Myc (Figure 1I). The sequence motifs for Oct4, Sox2 and Nanog showed similar levels of enrichment at typical enhancers and constituent enhancers within super-enhancer domains, but motifs for Klf4 and Esrrb were significantly enriched in the constituent enhancers within super-enhancers (p-val.<10−45)(Figure 1J). These data indicate that ESC super-enhancers are large clusters of enhancers that can be distinguished from typical enhancers by the presence of the transcription factors Klf4 and Esrrb and exceptional levels of Mediator, and indicate that these domains are formed as a consequence of binding of specific master transcription factors to dense clusters of their binding site sequences.
Super-enhancers are associated with key ESC identity genes
Enhancers tend to loop to and associate with adjacent genes in order to activate their transcription (Ong and Corces, 2011). Most of these interactions occur within a distance of ~50kb of the enhancer, although many can occur at greater distances up to several megabases (Sanyal et al., 2012). Previous studies have utilized various methods to assign enhancers to their target genes, including proximity, enhancer-promoter unit assignments (EPUs), and genome-wide interactions discovered by chromosome conformation capture techniques (Dixon et al., 2012; Shen et al., 2012; Whyte et al., 2012). We initially used proximity to assign 231 super-enhancers to 210 genes (Table S1), because the super-enhancers tend to overlap the genes to which they were associated. These super-enhancer proximity assignments were highly consistent (95% agreement) with EPU assignments (Table S3). In addition, 93% of the super-enhancer-promoter pairs identified by proximity occur within the same topological domains defined by Hi-C (Figure 2A, Table S3). Furthermore, for three of these genes (Oct4, Nanog, and Lefty1), interactions between portions of the super-enhancer and the target promoter were previously demonstrated using chromatin conformation capture (3C)(Kagey et al., 2010).
Figure 2. Super-enhancers are associated with key ESC pluripotency genes.
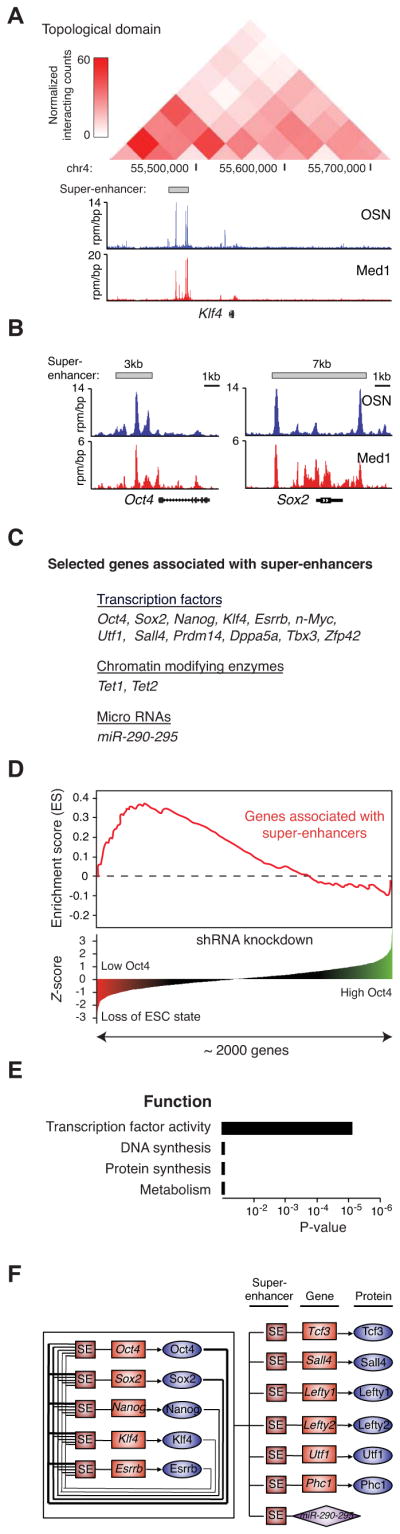
A) ChIP-Seq binding profiles for OSN and Med1 at the Klf4 locus in ESCs. Previously described Hi-C interaction frequency (normalized interaction counts), and genomic coordinates comprising a portion of a topological domain (Dixon et al., 2012), are indicated above the binding profiles.
B) ChIP-Seq binding profiles for OSN and Med1 at the Oct4 and Sox2 loci in ESCs. Gene models are depicted below the binding profiles.
C) List of selected genes associated with super-enhancers and playing prominent roles in ESC biology.
D) Super-enhancers are associated with genes encoding transcription factors, coactivators and chromatin regulators important for maintenance of ESC state. The gene set enrichment analysis (GSEA) of super-enhancer-associated genes reveals these genes encode regulators whose shRNA knockdown most impact Oct4 expression and ESC state. Knockdown of genes important for ESC identity causes Oct4 loss, which is reflected by a negative Z-score. P-value (10−2) was calculated as part of the GSEA analysis.
E) Gene ontology (GO) functional categories for super-enhancer-associated genes. Genes encoding factors important for DNA synthesis, protein synthesis, and metabolism were not enriched. See also Figure S2.
F) Schematic diagram of a small portion of the ESC core regulatory circuitry. Genes encoding the master transcription factors are themselves driven by super-enhancers. These master factors form an interconnected feedback loop and regulate their own expression, as well as the expression of other transcription factors and non-coding RNAs that play prominent roles in ESC biology.
The set of super-enhancer-associated genes contained nearly all genes that have been implicated in control of ESC identity (Table S1). They included genes encoding the master ESC transcription factors Oct4, Sox2 and Nanog (Figure 2B, Table S1). They also included genes encoding most other transcription factors implicated in control of ESC identity, as well as genes encoding DNA-modifying enzymes and miRNAs that feature prominently in the control of the ESC gene expression program (Figure 2C). For example, Klf4 and Esrrb play important roles in ESC biology and can facilitate reprogramming (Feng et al., 2009; Festuccia et al., 2012; Jiang et al., 2008; Martello et al., 2012; Percharde et al., 2012; Takahashi and Yamanaka, 2006). The products of the Tet genes are associated with most active promoters and are responsible for global 5-hydroxymethylation of DNA in ESCs (Wu et al., 2011; Yu et al., 2012a). The miR-290-295 locus produces the most abundant miRNAs in ESCs (Calabrese et al., 2007) and is essential for embryonic survival (Medeiros et al., 2011).
Previous studies have identified genes encoding a broad range of transcription factors, coactivators and chromatin regulators that are necessary for maintenance of the ESC state (Ding et al., 2009; Fazzio et al., 2008; Hu et al., 2009; Kagey et al., 2010). To further investigate the extent to which super-enhancer-associated genes are involved in control of ESC state, we compared the set of super-enhancer-associated genes to the genes in a short hairpin RNA (shRNA) knockdown screen involving 2,000 regulators, which included most transcription factors and chromatin regulators encoded in the mouse genome (Kagey et al., 2010). We found that the majority of genes encoding transcription factors, coactivators and chromatin regulators whose knockdown most profoundly caused loss of ESC state are associated with super-enhancers (p-val.<10−2) (Figure 2D). This further supports the notion that super-enhancer-associated genes encode many regulators that are key to establishing and maintaining ESC state.
Genes encoding transcription factors were the predominant class of super-enhancer-associated genes based on analysis of gene ontology functional categories (Figure 2E). In contrast, super-enhancers were not found to be associated with housekeeping genes (Figure S2). The ESC master transcription factors Oct4, Sox2 and Nanog have previously been shown to form an inter-connected autoregulatory loop, where all three factors bind as a group to the promoters of each of their own genes and form the core regulatory circuitry of ESCs (Boyer et al., 2005; Loh et al., 2006). The discovery of Klf4 and Esrrb at super-enhancers, and evidence that Klf4 and Esrrb play important roles in the ESC gene expression program and in reprogramming of somatic cells to iPS cells (Feng et al., 2009; Takahashi and Yamanaka, 2006) suggest that this autoregulatory loop should be expanded to include Klf4 and Esrrb (Figure 2F).
Functional attributes of super-enhancers
Super-enhancer-associated genes are generally expressed at higher levels than genes associated with typical enhancers (p-val.<10−5)(Figure 3A, Figure S3A, Table S4), suggesting super-enhancers drive high level expression of their target genes. To test whether super-enhancers confer stronger enhancer activity than typical ESC enhancers, we cloned DNA fragments from these elements into luciferase reporter constructs that were subsequently transfected into ESCs. Constituent enhancer segments within the super-enhancers, defined as a 600–1,400 base pair region with a single peak of Oct4/Sox2/Nanog occupancy, generated higher luciferase activity relative to single peaks from typical enhancers (3.8 fold higher; p-val.= 0.02)(Figure 3B). These results are consistent with the idea that super-enhancers and their components help drive high levels of transcription of the key genes that control ESC identity.
Figure 3. Super-enhancers confer high transcriptional activity and sensitivity to perturbation.
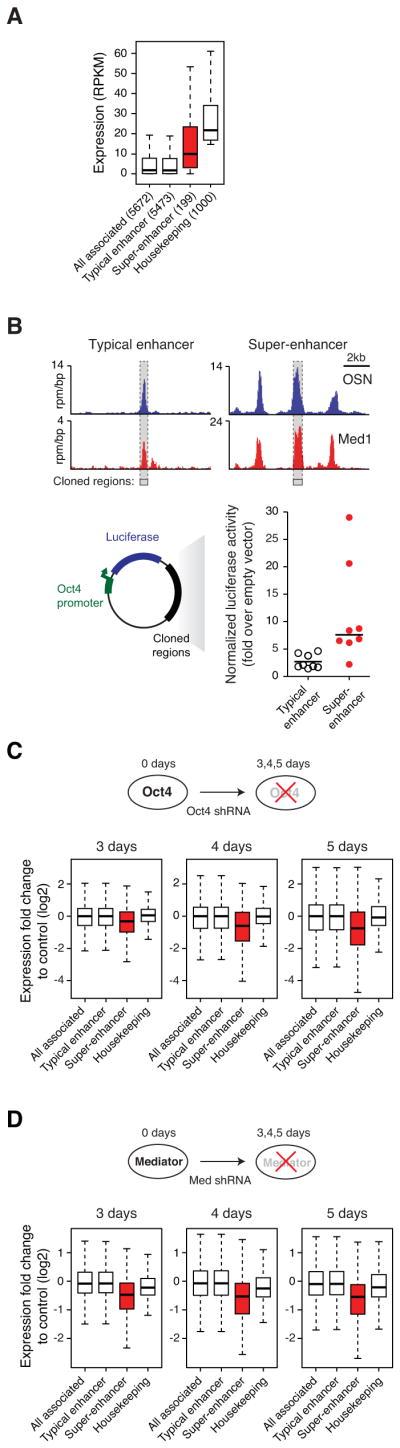
A) Box plots of expression (reads per kilobase of exon per million mapped reads (RPKM)) from typical enhancer-, super-enhancer-, and all enhancer-associated genes, as well as the top 1,000 highest expressed housekeeping genes. The number of genes belonging to each category for which we have expression data is denoted. P-value (10−5) was calculated using a two-tailed t-test. See also Figure S3A and Table S4.
B) Upper panel, ChIP-Seq binding profiles for OSN and Med1 at the Sgk1 typical enhancer and Esrrb super-enhancer loci. Grey bars with dashed borders indicates cloned regions. Lower panel, Plot of luciferase activity 24 hours post transfection, normalized to a transfected control plasmid. Enhancers neighboring selected genes were cloned into reporter plasmids containing the Luciferase gene regulated by the Oct4 promoter, and subsequently transfected into ESCs. P-value (0.02) was calculated using a two-tailed t-test. See also Figure S3B.
C) Upper panel, Schematic diagram of ESCs transduced with shRNAs against Oct4. Lower panel, Box plots of fold change expression in differentiating ESCs (3, 4 and 5 days after transduction) relative to control ESCs transduced with shRNAs against GFP. P-values (day 3= 10−5, day 4=10−8, day 5= 10−10) were calculated using a two-tailed t-test. See also Figure S3C.
D) Upper panel, Schematic diagram of ESCs transduced with shRNAs against the Mediator subunit Med12. Lower panel, Box plots of fold change expression in Mediator-depleted ESCs (3, 4, and 5 days after transduction) relative to control ESCs. P-values (day 3= 10−11, day 4=10−11, day 5= 10−13) were calculated using a two-tailed t-test.
To obtain clues to the factors that contribute to the higher activity of individual enhancer elements within super-enhancers, we determined whether the levels of particular transcription factors at the enhancer elements, based on ChIP-Seq data for the genomic locus, correlated with the levels of luciferase activity in the reporter assays. The presence of Klf4 and Esrrb were correlated with high levels of luciferase activity (Figure S3B). Thus, Klf4 and Esrrb, which are especially enriched in super-enhancers (Figure 1G), may contribute to the superior activity of the enhancer elements from super-enhancers in these reporter assays.
We next investigated whether the functional attributes of super-enhancers might account for the observation that reduced levels of either Oct4 or Mediator have very similar effects on the ESC gene expression program and cause the same rapid loss of ESC state (Kagey et al., 2010). Enhancers typically function through cooperative and synergistic interactions between multiple transcription factors and coactivators (Carey, 1998; Carey et al., 1990; Giese et al., 1995; Kim and Maniatis, 1997; Thanos and Maniatis, 1995). The transcriptional output of enhancers with large numbers of transcription factor binding sites can be more sensitive to changes in transcription factor concentration than those with smaller numbers of binding sites (Giniger and Ptashne, 1988; Griggs and Johnston, 1991). We therefore hypothesized that super-enhancer-associated genes may be more sensitive to perturbations in the levels of enhancer-binding factors than genes associated with normal enhancers. We carried out two tests of this model.
In ESCs, reducing the levels of Oct4 leads to loss of ESC-specific gene expression and differentiation. If super-enhancer-associated genes are more sensitive to loss of master transcription factors than other genes, then a reduction in Oct4 levels should cause a preferential loss of super-enhancer-associated gene expression. To test this idea, we reduced the levels of Oct4 transcription using shRNAs, which leads to activation of the trophectoderm master transcription factor Cdx2 and cellular differentiation (Figure 3C)(Deb et al., 2006; Niwa et al., 2005; Strumpf et al., 2005). Oct4 depletion results in changes in cellular morphology consistent with ESC differentiation by 5 days (Figure S3C). We analyzed gene expression 3, 4 and 5 days after Oct4 depletion, and observed super-enhancer-associated genes suffered an earlier and more profound reduction in the levels of transcripts than those associated with typical enhancers (p-val.<10−5, 10−8, and 10−10, respectively)(Figure 3C). These results indicate that the transcriptional output of ESC super-enhancer-associated genes is rapidly and preferentially reduced during differentiation.
If super-enhancer-associated genes are more sensitive to loss of coactivators than other genes, then a reduction in levels of Mediator subunits should preferentially affect expression of super-enhancer-associated genes. When the levels of Mediator were reduced using shRNAs in ESCs, the most pronounced effects on gene expression were observed at super-enhancer-associated genes (p-val.<10−11, 10−11, and 10−13, respectively)(Figure 3D). In summary, these results indicate that reducing the levels of Oct4 and Mediator lead to more profound effects on expression of super-enhancer-associated genes than on other active genes with typical enhancers. These results may thus account for the observation that loss of Oct4 and loss of Mediator subunits have similar effects on ESC state (Kagey et al., 2010).
Super-enhancers in B cells
We investigated whether the super-enhancers found in ESCs had similar counterparts in differentiated cells. We annotated 13,814 enhancers using ChIP-Seq data for the master transcription factor PU.1 in murine progenitor B (pro-B) cells (Table S5)(DeKoter and Singh, 2000; Nutt and Kee, 2007). Previous studies have shown that occupancy of pro-B genomic sites by PU.1 is predictive of enhancer activity (Abujarour et al., 2010; Wlodarski et al., 2007). We found that genome-wide occupancy of the master transcription factor PU.1 and Mediator were highly correlated (Figure 4A, Figure S4). When the levels of Mediator were plotted against enhancers ranked by ChIP-Seq signal, the enhancers in these cells fell into two classes, as was observed for ESCs (Figure 4B). The pro-B cells had 395 large domains that shared key characteristics with the super-enhancers found in ESCs: they spanned DNA domains whose median length is an order of magnitude larger than the typical enhancer, and they had levels of Mediator that are at least an order of magnitude greater than those at the typical enhancer (Figure 4C). Nearly 40% of all Mediator signal observed at enhancers was associated with the super-enhancer domains in pro-B cells.
Figure 4. Super-enhancers in pro-B cells.

A) ChIP-Seq binding profiles for PU.1, and Med1 at the Foxo1 locus in pro-B cells.
B) Distribution of Mediator ChIP-Seq density across the 13,814 pro-B enhancers, with a subset of enhancers (the 395 super-enhancers) containing exceptionally high amounts of Mediator. See also Figure S4.
C) Metagenes of Mediator density across the typical and super-enhancers in proB cells. Metagenes are centered on the enhancer region (422 base pairs for typical enhancers, and 15.4kb for super-enhancers), with 3kb surrounding each enhancer region. ChIP-Seq fold difference for Mediator at super-enhancers versus typical enhancers is displayed below the metagenes.
D) Table depicting transcription factor binding motifs enriched at constituent enhancers within super-enhancer regions relative to genomic background and associated p-values. CTCF and Zfx are not enriched.
E) Left panel, Box plot depicting the number of PU.1, Ebf1 or Foxo1 binding motifs at constituent enhancers within typical enhancers and constituent enhancers within super-enhancers. Right panel, Box plot depicting the number of E2A binding motifs at constituent enhancers within typical enhancers and constituent enhancers within super-enhancer regions. P-values (PU.1/Ebf1/Foxo1= 10−5, E2A= 10−22) were calculated using a two-tailed t-test.
F) List of selected genes associated with super-enhancers and playing prominent roles in B cell biology.
G) Box plots of expression from typical enhancer-, super-enhancer-, and all enhancer-associated genes in pro-B cells. The number of genes belonging to each category for which we have expression data is denoted. P-value (10−6) was calculated using a two-tailed t-test.
We studied the frequency of DNA sequences bound by pro-B transcription factors in super-enhancers and in other regions of the genome. Constituent enhancers within super-enhancer regions were significantly enriched for clusters of sequence motifs bound by PU.1, as well as for a set of other transcription factors that have been implicated in control of B cells (Figure 4D,E). The transcription factors with sequence motif enrichment in the super-enhancer domains included Ebf1, E2A and Foxo1, which have previously been shown to be important for control of B cells (Lin et al., 2010). The sequence motif for E2A was significantly more enriched at super-enhancer constituents relative to typical enhancer constituents (p-val.< 10−22)(Figure 4E). E2A is essential for pro-B cell development during B cell lymphopoiesis (Kwon et al., 2008). These findings are consistent with those obtained for ESCs, where DNA sequence motifs for the master transcription factors were enriched in closely spaced clusters.
We next identified genes associated with super-enhancers in pro-B cells and found that many of these are prominent regulators of B cell identify (Figure 4F). For example, super-enhancer-associated genes in pro-B cells included Foxo1 and Inpp5d. In common lymphoid progenitors, Foxo1 acts in concert with Ebf1 to specify B-cell fate as part of a positive feedback loop (Mansson et al., 2012), while the lipid metabolizing enzyme encoded by Inpp5d, SHIP1, dephosphorylates proteins to regulate the B-cell antigen receptor (BCR) signaling response (Alinikula et al., 2010). As in ESCs, the genes associated with super-enhancers in pro-B cells were expressed at higher levels than those associated with typical enhancers (p-val.<10−6)(Figure 4G, Table S5).
Super-enhancers are cell type-specific and mark key cell identity genes
To further investigate whether super-enhancers are a general feature of mammalian cells, we extended the study of these elements to a range of other cell types where the key transcription factors that control cell state are well defined (Figure 5). We found that the master transcription factors of mouse myotubes (MyoD), T helper (Th) cells (T-Bet) and macrophages (C/EBPα) also bind large domains with clusters of enhancers (Figure S5A,B), and these large domains are associated with genes that feature predominantly in the biology of these cells (Figure 5A, Figure S5C, Table S6). In myotubes, for example, a super-enhancer is associated with the gene encoding MyoD, which is a master regulator of skeletal muscle and the first factor shown to reprogram fibroblasts into muscle cells (Tapscott, 2005; Weintraub et al., 1989). In Th cells, a super-enhancer is associated with the gene Tcf7 that encodes T cell factor 1 (Tcf-1), which is critical for the production of T cells during hematopoiesis (Staal and Sen, 2008; Xue and Zhao, 2012; Yu et al., 2012b). In macrophages, a super-enhancer is associated with the gene encoding the extracellular matrix glycoprotein Thbs-1, which is involved in scavenger recognition of apoptotic cells by macrophages (Savill et al., 1992). These results support the notion that the key transcription factors controlling cell state bind to clusters of enhancers that are associated with specific genes that are key to cell identity.
Figure 5. Super-enhancers are generally associated with key cell identity genes.

A) ChIP-Seq binding profiles for master transcription factors (OSN in ESCs; PU.1 in pro-B cells; MyoD in myotubes; T-bet in Th cells; C/EBPα in macrophages), at the Esrrb, Inpp5d, Myod1, Tcf7 and Thbs-1 loci. See also Figure S5A,B.
B) Venn diagrams of typical enhancer-associated and super-enhancer-associated genes in ESCs (blue border), pro-B cells (green border) and myotubes (orange border).
C) Chow-Ruskey diagrams of typical enhancer-associated and super-enhancer-associated genes in ESCs (blue border), pro-B cells (green border), myotubes (orange border), Th cells (brown border) and macrophages (purple border). Color of the borders around each intersection corresponds to the cell types whose genes overlap. The red circle in the middle represents the overlap of all 5 cell types. Lighter shades of red, orange, and yellow represent the overlap of fewer cell types. Area of each intersection is proportional to number of genes within the intersection.
D) Super-enhancers are associated with genes playing prominent roles in cell type-specific biology. The top 10 gene ontology terms of the genes associated with super-enhancers in ESCs, pro-B cells, myotubes, Th cells and macrophages are displayed. P-values corresponding to each of the gene ontology terms are displayed as a color bar, with color scale bar denoted below. See also Figure S5C,D.
The set of enhancers that are bound by transcription factors and control transcription in any one cell type can promote expression of both cell type-specific genes and genes that are active in multiple cell types (Bernstein et al., 2012; Shen et al., 2012; Yip et al., 2012). The super-enhancer elements identified in ESCs, pro-B cells, myotubes, Th cells and macrophages spanned domains that were almost entirely cell type-specific (Figure 5A, Figure S5D) and the genes associated with these elements were highly cell type-specific relative to typical enhancer-associated genes (Figure 5B,C). These results are consistent with the idea that super-enhancers are formed by the binding of key transcription factors to clusters of binding sites that are associated with genes controlling unique cellular identities.
If super-enhancers generally form at genes whose functions are associated with cell identity, we might expect super-enhancer-associated genes to be defining of cell type. When gene ontology analysis was conducted using the set of genes associated with super-enhancers in each cell type, we found that the top 10 most significant biological process terms obtained for each cell type were remarkably descriptive of each cells’ specific function (Figure 5D). This result suggests that super-enhancer-associated genes may be valuable biomarkers for cell identity.
Discussion
We have identified exceptionally large enhancer domains that are occupied by master transcription factors and associated with genes encoding key regulators of cell identity. In ESCs, these super-enhancers consist of clusters of enhancer elements that are formed by the binding of key transcription factors and the Mediator coactivator complex. The ESC super-enhancers differ from typical enhancers in size, transcription factor density and content, ability to activate transcription and sensitivity to perturbation. Super-enhancers are found in a wide variety of other cell types, where they are associated with key cell type-specific genes known to play prominent roles in their biology. These results implicate super-enhancers in the control of mammalian cell identity.
Super-enhancer formation appears to occur as a consequence of binding of large amounts of master transcription factors to clusters of DNA sequences that are relatively abundant across these large domains. The ESC transcription factors Oct4, Sox2, Nanog, Klf4 and Esrrb have DNA binding motifs that are enriched in super-enhancer domains. Super-enhancers are not simply clusters of typical enhancers, but are particularly enriched in Klf4 and Esrrb, which have previously been shown to play important roles in the ESC gene expression program and in reprogramming of somatic cells to iPS cells (Feng et al., 2009; Festuccia et al., 2012; Jiang et al., 2008; Martello et al., 2012; Percharde et al., 2012; Takahashi and Yamanaka, 2006). Furthermore, super-enhancer-associated genes are highly sensitive to reduced levels of enhancer-bound factors and cofactors. We speculate that the signals that naturally cause ESCs to differentiate may exploit this sensitivity of super-enhancer-associated genes to facilitate transitions to new gene expression programs.
Remarkably, the genes encoding the ESC master transcription factors are themselves driven by super-enhancers, forming a feedback loop where the key transcription factors regulate their own expression (Figure 2F). Earlier studies identified a portion of this interconnected autoregulatory loop, consisting of the genes encoding Oct4, Sox2 and Nanog, but were unaware of the unusual enhancer structure associated with genes in this regulatory loop (Boyer et al., 2005; Loh et al., 2006). The formation of super-enhancers at these genes is also of interest because it suggests that super-enhancers may generally identify genes that are important for control of cell identity and, in some cases, capable of reprogramming cell fate. Indeed, we found evidence for super-enhancers associated with genes that control cell identity in a wide range of cell types and some of these genes do encode factors that have been demonstrated to reprogram cell fate.
We found that super-enhancers can be identified by searching for clusters of binding sites for enhancer-binding transcription factors, and they can be distinguished from typical enhancers by occupancy of cofactors or enhancer-associated surrogate marks such as histone H3K27ac or DNaseI hypersensitivity. Previous studies have noted that many different ESC transcription factors can bind to sites called multiple transcription factor-binding loci (Chen et al., 2008; Kim et al., 2008), but these loci differ from super-enhancers and are associated with different genes. Other studies have also identified large genomic domains involved in gene control, but have not noted that genes encoding the key regulators of cell state are generally driven by super-enhancers. For example, large control regions with clusters of transcription factor binding sites or DNaseI hypersensitivity sites have been described for the IgH enhancer (~20kb), the Th cell receptor (~11.5kb), the β-globin enhancer (~16kb) and others (Diaz et al., 1994; Forrester et al., 1990; Grosveld et al., 1987; Madisen and Groudine, 1994; Michaelson et al., 1995; Orkin, 1990). It is possible that previous studies did not note large domains of enhancer activity associated with key cell identity genes because most existing algorithms typically seek evidence for factor binding or DNaseI hypersensitivity within small regions of the genome. There are, however, algorithms that are designed to identify large domains (Ernst and Kellis, 2010; Filion et al., 2010; Hon et al., 2008; Thurman et al., 2012), and the algorithm we describe here should be useful for further discovery of super-enhancers and other large domains.
The presence of super-enhancers at key cell identity genes provides new insights into transcriptional control of mammalian cells. The evidence described here indicates that mammalian genomes have evolved clusters of DNA sequences near genes encoding key drivers of cell state. These clusters are bound by a combination of key transcription factors to form cell type-specific super-enhancers and in this fashion control the gene expression programs associated with specific cell identities.
The concept of super-enhancers may facilitate mapping of the regulatory circuitry of many different cell types comprising mammals. Discovering how thousands of transcription factors co-operate to control gene expression programs in the vast number of cells in vertebrates is a highly complex undertaking. If only a few hundred super-enhancers dominate control of the key genes that establish and maintain cellular identity, however, it may be possible to create basic models that describe the key features of transcriptional control of cell state.
Experimental Procedures
Cell Culture
V6.5, murine ESCs were grown on irradiated murine embryonic fibroblasts (MEFs). Cells were grown under standard ESC conditions as described previously (Whyte et al., 2012). Cells were grown on 0.2% gelatinized (Sigma, G1890) tissue culture plates in ESC media; DMEM-KO (Invitrogen, 10829-018) supplemented with 15% fetal bovine serum (Hyclone, characterized SH3007103), 1000 U/ml LIF (ESGRO, ESG1106), 100 uM nonessential amino acids (Invitrogen, 11140-050), 2 mM L-glutamine (Invitrogen, 25030-081), 100 U/ml penicillin, 100 ug/ml streptomycin (Invitrogen, 15140-122), and 8 nl/ml of 2-mercaptoethanol (Sigma, M7522).
ChIP-Seq
ChIP was carried out as described previously (Boyer et al., 2005). Additional details are provided in the Extended Experimental Procedures. ChIP-Seq of Mediator was generated using a Med1 antibody (Bethyl Labs A300-793A, Lot #A300-793A-2).
Illumina Sequencing and Library Generation
Purified ChIP DNA was used to prepare Illumina multiplexed sequencing libraries. Libraries for Illumina sequencing were prepared following the Illumina TruSeq™ DNA Sample Preparation v2 kit protocol with exceptions described in the Extended Experimental Procedures.
Luciferase Expression Constructs
A minimal Oct4 promoter was amplified from mouse genomic DNA and cloned into the XhoI and HindIII sites of the pGL3 basic vector (Promega). Enhancer fragments were subsequently cloned into the BamHI and SalI sites of the pGL3-pOct4 vector. The v6.5 murine ESCs were transfected using Lipofectamine 2000 (Invitrogen). The pRL-SV40 plasmid (Promega) was cotransfected as a normalization control. Cells were incubated for 24 hours, and luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega). The genomic coordinates of the cloned fragments are found in Table S7.
Data Analysis
All ChIP-Seq datasets were aligned using Bowtie (version 0.12.2) (Langmead et al., 2009) to build version MM9 of the mouse genome, or HG18 of the human genome. The GEO Accession ID for aligned and raw data is GSE44288 (www.ncbi.nlm.nih.gov/geo/). Datasets used in this manuscript can be found in Table S8.
We developed a simple method to calculate the normalized read density of a ChIP-Seq dataset in any region. ChIP-Seq reads aligning to the region were extended by 200 base pairs, and the density of reads per base pair (bp) was calculated. The density of reads in each region was normalized to the total number of million mapped reads producing read density in units of reads per million mapped reads per base pair (rpm/bp)
We used the MACS version 1.4.1 (Model based analysis of ChIP-Seq) (Zhang et al., 2008) peak finding algorithm to identify regions of ChIP-Seq enrichment over background. A p-value threshold of enrichment of 10−9 was used for all datasets.
Enhancers were defined as regions of ChIP-Seq enrichment for transcription factor(s). In order to accurately capture dense clusters of enhancers, we allowed regions within 12.5kb of one another to be stitched together.
The methods for identifying and characterizing super-enhancers, as well as assignment of enhancers to genes, are fully described in the Extended Experimental Procedures.
Supplementary Material
Research Highlights.
Master transcription factors form “super-enhancers” at key cell identity genes
Super-enhancers span large domains and employ a large fraction of Mediator
Super-enhancers drive cell type-specific gene expression programs
Acknowledgments
We thank Tom Volkert, Jennifer Love, Sumeet Gupta, and Jeong-Ah Kwon at the Whitehead Genome Technologies Core for Solexa sequencing; Lee M. Lawton, Jessica Reddy, Ana D’Alessio and Jasmine M. De Cock for experimental assistance; and Alla A. Sigova, Alan C. Mullen, Roshan M. Kumar and members of the Young lab for helpful discussion. This work was supported by the National Institutes of Health grants HG002668 (RAY) and CA146445 (R.A.Y., T.L.). R.A.Y. is a founder, and D.A.O. and P.B.R. have become employees, of Syros Pharmaceuticals.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abujarour R, Efe J, Ding S. Genome-wide gain-of-function screen identifies novel regulators of pluripotency. Stem Cells. 2010;28:1487–1497. doi: 10.1002/stem.472. [DOI] [PubMed] [Google Scholar]
- Alinikula J, Kohonen P, Nera KP, Lassila O. Concerted action of Helios and Ikaros controls the expression of the inositol 5-phosphatase SHIP. Eur J Immunol. 2010;40:2599–2607. doi: 10.1002/eji.200940002. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borggrefe T, Yue X. Interactions between subunits of the Mediator complex with gene-specific transcription factors. Semin Cell Dev Biol. 2011;22:759–768. doi: 10.1016/j.semcdb.2011.07.022. [DOI] [PubMed] [Google Scholar]
- Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144:327–339. doi: 10.1016/j.cell.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese JM, Seila AC, Yeo GW, Sharp PA. RNA sequence analysis defines Dicer’s role in mouse embryonic stem cells. Proc Natl Acad Sci U S A. 2007;104:18097–18102. doi: 10.1073/pnas.0709193104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey M. The enhanceosome and transcriptional synergy. Cell. 1998;92:5–8. doi: 10.1016/s0092-8674(00)80893-4. [DOI] [PubMed] [Google Scholar]
- Carey M, Leatherwood J, Ptashne M. A potent GAL4 derivative activates transcription at a distance in vitro. Science. 1990;247:710–712. doi: 10.1126/science.2405489. [DOI] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Conaway RC, Conaway JW. Function and regulation of the Mediator complex. Curr Opin Genet Dev. 2011;21:225–230. doi: 10.1016/j.gde.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb K, Sivaguru M, Yong HY, Roberts RM. Cdx2 gene expression and trophectoderm lineage specification in mouse embryos. Science. 2006;311:992–996. doi: 10.1126/science.1120925. [DOI] [PubMed] [Google Scholar]
- DeKoter RP, Singh H. Regulation of B lymphocyte and macrophage development by graded expression of PU.1. Science. 2000;288:1439–1441. doi: 10.1126/science.288.5470.1439. [DOI] [PubMed] [Google Scholar]
- Diaz P, Cado D, Winoto A. A locus control region in the T cell receptor alpha/delta locus. Immunity. 1994;1:207–217. doi: 10.1016/1074-7613(94)90099-x. [DOI] [PubMed] [Google Scholar]
- Ding L, Paszkowski-Rogacz M, Nitzsche A, Slabicki MM, Heninger AK, de Vries I, Kittler R, Junqueira M, Shevchenko A, Schulz H, et al. A genome-scale RNAi screen for Oct4 modulators defines a role of the Paf1 complex for embryonic stem cell identity. Cell Stem Cell. 2009;4:403–415. doi: 10.1016/j.stem.2009.03.009. [DOI] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol. 2010;28:817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazzio TG, Huff JT, Panning B. An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell. 2008;134:162–174. doi: 10.1016/j.cell.2008.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B, Jiang J, Kraus P, Ng JH, Heng JC, Chan YS, Yaw LP, Zhang W, Loh YH, Han J, et al. Reprogramming of fibroblasts into induced pluripotent stem cells with orphan nuclear receptor Esrrb. Nat Cell Biol. 2009;11:197–203. doi: 10.1038/ncb1827. [DOI] [PubMed] [Google Scholar]
- Festuccia N, Osorno R, Halbritter F, Karwacki-Neisius V, Navarro P, Colby D, Wong F, Yates A, Tomlinson SR, Chambers I. Esrrb is a direct Nanog target gene that can substitute for Nanog function in pluripotent cells. Cell Stem Cell. 2012;11:477–490. doi: 10.1016/j.stem.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, Brugman W, de Castro IJ, Kerkhoven RM, Bussemaker HJ, et al. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell. 2010;143:212–224. doi: 10.1016/j.cell.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrester WC, Epner E, Driscoll MC, Enver T, Brice M, Papayannopoulou T, Groudine M. A deletion of the human beta-globin locus activation region causes a major alteration in chromatin structure and replication across the entire beta-globin locus. Genes Dev. 1990;4:1637–1649. doi: 10.1101/gad.4.10.1637. [DOI] [PubMed] [Google Scholar]
- Giese K, Kingsley C, Kirshner JR, Grosschedl R. Assembly and function of a TCR alpha enhancer complex is dependent on LEF-1-induced DNA bending and multiple protein-protein interactions. Genes Dev. 1995;9:995–1008. doi: 10.1101/gad.9.8.995. [DOI] [PubMed] [Google Scholar]
- Giniger E, Ptashne M. Cooperative DNA binding of the yeast transcriptional activator GAL4. Proc Natl Acad Sci U S A. 1988;85:382–386. doi: 10.1073/pnas.85.2.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griggs DW, Johnston M. Regulated expression of the GAL4 activator gene in yeast provides a sensitive genetic switch for glucose repression. Proc Natl Acad Sci U S A. 1991;88:8597–8601. doi: 10.1073/pnas.88.19.8597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosveld F, van Assendelft GB, Greaves DR, Kollias G. Position-independent, high-level expression of the human beta-globin gene in transgenic mice. Cell. 1987;51:975–985. doi: 10.1016/0092-8674(87)90584-8. [DOI] [PubMed] [Google Scholar]
- Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, van de Lagemaat LN, Smith KA, Ebbert A, Riley ZL, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489:391–399. doi: 10.1038/nature11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon G, Ren B, Wang W. ChromaSig: a probabilistic approach to finding common chromatin signatures in the human genome. PLoS Comput Biol. 2008;4:e1000201. doi: 10.1371/journal.pcbi.1000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Kim J, Xu Q, Leng Y, Orkin SH, Elledge SJ. A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev. 2009;23:837–848. doi: 10.1101/gad.1769609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, Yuan CX, Okano HJ, Darnell RB, Roeder RG. Involvement of the TRAP220 component of the TRAP/SMCC coactivator complex in embryonic development and thyroid hormone action. Mol Cell. 2000;5:683–693. doi: 10.1016/s1097-2765(00)80247-6. [DOI] [PubMed] [Google Scholar]
- Jiang J, Chan YS, Loh YH, Cai J, Tong GQ, Lim CA, Robson P, Zhong S, Ng HH. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat Cell Biol. 2008;10:353–360. doi: 10.1038/ncb1698. [DOI] [PubMed] [Google Scholar]
- Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Chu J, Shen X, Wang J, Orkin SH. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, Maniatis T. The mechanism of transcriptional synergy of an in vitro assembled interferon-beta enhanceosome. Mol Cell. 1997;1:119–129. doi: 10.1016/s1097-2765(00)80013-1. [DOI] [PubMed] [Google Scholar]
- Kornberg RD. Mediator and the mechanism of transcriptional activation. Trends Biochem Sci. 2005;30:235–239. doi: 10.1016/j.tibs.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Kwon K, Hutter C, Sun Q, Bilic I, Cobaleda C, Malin S, Busslinger M. Instructive role of the transcription factor E2A in early B lymphopoiesis and germinal center B cell development. Immunity. 2008;28:751–762. doi: 10.1016/j.immuni.2008.04.014. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelli KM, Slattery M, Mann RS. Disentangling the many layers of eukaryotic transcriptional regulation. Annu Rev Genet. 2012;46:43–68. doi: 10.1146/annurev-genet-110711-155437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, Tjian R. Transcription regulation and animal diversity. Nature. 2003;424:147–151. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- Lin YC, Jhunjhunwala S, Benner C, Heinz S, Welinder E, Mansson R, Sigvardsson M, Hagman J, Espinoza CA, Dutkowski J, et al. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol. 2010;11:635–643. doi: 10.1038/ni.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- Madisen L, Groudine M. Identification of a locus control region in the immunoglobulin heavy-chain locus that deregulates c-myc expression in plasmacytoma and Burkitt’s lymphoma cells. Genes Dev. 1994;8:2212–2226. doi: 10.1101/gad.8.18.2212. [DOI] [PubMed] [Google Scholar]
- Malik S, Roeder RG. The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat Rev Genet. 2010;11:761–772. doi: 10.1038/nrg2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansson R, Welinder E, Ahsberg J, Lin YC, Benner C, Glass CK, Lucas JS, Sigvardsson M, Murre C. Positive intergenic feedback circuitry, involving EBF1 and FOXO1, orchestrates B-cell fate. Proc Natl Acad Sci U S A. 2012;109:21028–21033. doi: 10.1073/pnas.1211427109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martello G, Sugimoto T, Diamanti E, Joshi A, Hannah R, Ohtsuka S, Gottgens B, Niwa H, Smith A. Esrrb is a pivotal target of the Gsk3/Tcf3 axis regulating embryonic stem cell self-renewal. Cell Stem Cell. 2012;11:491–504. doi: 10.1016/j.stem.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maston GA, Evans SK, Green MR. Transcriptional regulatory elements in the human genome. Annu Rev Genomics Hum Genet. 2006;7:29–59. doi: 10.1146/annurev.genom.7.080505.115623. [DOI] [PubMed] [Google Scholar]
- Medeiros LA, Dennis LM, Gill ME, Houbaviy H, Markoulaki S, Fu D, White AC, Kirak O, Sharp PA, Page DC, et al. Mir-290–295 deficiency in mice results in partially penetrant embryonic lethality and germ cell defects. Proc Natl Acad Sci U S A. 2011;108:14163–14168. doi: 10.1073/pnas.1111241108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson JS, Giannini SL, Birshtein BK. Identification of 3′ alpha-hs4, a novel Ig heavy chain enhancer element regulated at multiple stages of B cell differentiation. Nucleic Acids Res. 1995;23:975–981. doi: 10.1093/nar/23.6.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Surani MA. The transcriptional and signalling networks of pluripotency. Nat Cell Biol. 2011;13:490–496. doi: 10.1038/ncb0511-490. [DOI] [PubMed] [Google Scholar]
- Niwa H, Toyooka Y, Shimosato D, Strumpf D, Takahashi K, Yagi R, Rossant J. Interaction between Oct3/4 and Cdx2 determines trophectoderm differentiation. Cell. 2005;123:917–929. doi: 10.1016/j.cell.2005.08.040. [DOI] [PubMed] [Google Scholar]
- Nutt SL, Kee BL. The transcriptional regulation of B cell lineage commitment. Immunity. 2007;26:715–725. doi: 10.1016/j.immuni.2007.05.010. [DOI] [PubMed] [Google Scholar]
- Ong CT, Corces VG. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet. 2011;12:283–293. doi: 10.1038/nrg2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkin SH. Globin gene regulation and switching: circa 1990. Cell. 1990;63:665–672. doi: 10.1016/0092-8674(90)90133-y. [DOI] [PubMed] [Google Scholar]
- Orkin SH, Hochedlinger K. Chromatin connections to pluripotency and cellular reprogramming. Cell. 2011;145:835–850. doi: 10.1016/j.cell.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panne D. The enhanceosome. Curr Opin Struct Biol. 2008;18:236–242. doi: 10.1016/j.sbi.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Percharde M, Lavial F, Ng JH, Kumar V, Tomaz RA, Martin N, Yeo JC, Gil J, Prabhakar S, Ng HH, et al. Ncoa3 functions as an essential Esrrb coactivator to sustain embryonic stem cell self-renewal and reprogramming. Genes Dev. 2012;26:2286–2298. doi: 10.1101/gad.195545.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risley MD, Clowes C, Yu M, Mitchell K, Hentges KE. The Mediator complex protein Med31 is required for embryonic growth and cell proliferation during mammalian development. Dev Biol. 2010;342:146–156. doi: 10.1016/j.ydbio.2010.03.019. [DOI] [PubMed] [Google Scholar]
- Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savill J, Hogg N, Ren Y, Haslett C. Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. The Journal of clinical investigation. 1992;90:1513–1522. doi: 10.1172/JCI116019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13:613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- Staal FJ, Sen JM. The canonical Wnt signaling pathway plays an important role in lymphopoiesis and hematopoiesis. Eur J Immunol. 2008;38:1788–1794. doi: 10.1002/eji.200738118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strumpf D, Mao CA, Yamanaka Y, Ralston A, Chawengsaksophak K, Beck F, Rossant J. Cdx2 is required for correct cell fate specification and differentiation of trophectoderm in the mouse blastocyst. Development. 2005;132:2093–2102. doi: 10.1242/dev.01801. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tapscott SJ. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development. 2005;132:2685–2695. doi: 10.1242/dev.01874. [DOI] [PubMed] [Google Scholar]
- Thanos D, Maniatis T. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell. 1995;83:1091–1100. doi: 10.1016/0092-8674(95)90136-1. [DOI] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub H, Tapscott SJ, Davis RL, Thayer MJ, Adam MA, Lassar AB, Miller AD. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc Natl Acad Sci U S A. 1989;86:5434–5438. doi: 10.1073/pnas.86.14.5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Bilodeau S, Orlando DA, Hoke HA, Frampton GM, Foster CT, Cowley SM, Young RA. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature. 2012;482:221–225. doi: 10.1038/nature10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarski P, Zhang Q, Liu X, Kasprzycka M, Marzec M, Wasik MA. PU.1 activates transcription of SHP-1 gene in hematopoietic cells. The Journal of biological chemistry. 2007;282:6316–6323. doi: 10.1074/jbc.M607526200. [DOI] [PubMed] [Google Scholar]
- Wu H, D’Alessio AC, Ito S, Xia K, Wang Z, Cui K, Zhao K, Sun YE, Zhang Y. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389–393. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue HH, Zhao DM. Regulation of mature T cell responses by the Wnt signaling pathway. Annals of the New York Academy of Sciences. 2012;1247:16–33. doi: 10.1111/j.1749-6632.2011.06302.x. [DOI] [PubMed] [Google Scholar]
- Yip KY, Cheng C, Bhardwaj N, Brown JB, Leng J, Kundaje A, Rozowsky J, Birney E, Bickel P, Snyder M, et al. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol. 2012;13:R48. doi: 10.1186/gb-2012-13-9-r48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RA. Control of the embryonic stem cell state. Cell. 2011;144:940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, Li X, Dai Q, Shen Y, Park B, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012a;149:1368–1380. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Zhou X, Steinke FC, Liu C, Chen SC, Zagorodna O, Jing X, Yokota Y, Meyerholz DK, Mullighan CG, et al. The TCF-1 and LEF-1 transcription factors have cooperative and opposing roles in T cell development and malignancy. Immunity. 2012b;37:813–826. doi: 10.1016/j.immuni.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.